

Abstract
The binding of the S component (LukS-PV) from the bicomponent staphylococcal Panton-Valentine leucocidin to human polymorphonuclear neutrophils (PMNs) and monocytes was determined using flow cytometry and a single-cysteine substitution mutant of LukS-PV. The mutant was engineered by replacing a glycine at position 10 with a cysteine and was labeled with a fluorescein moiety. The biological activity of the mutant was identical to that of the native protein. It has been shown that LukS-PV has a high affinity for PMNs (Kd = 0.07 ± 0.02 nM, n = 5) and monocytes (Kd = 0.020 ± 0.003 nM, n = 3) with maximal binding capacities of 197,000 and 80,000 LukS-PV molecules per cell, respectively. The nonspecifically bound molecules of LukS-PV do not form pores in the presence of the F component (LukF-PV) of leucocidin. LukS-PV and HlgC share the same receptor on PMNs, but the S components of other staphylococcal leukotoxins, HlgA, LukE, and LukM, do not compete with LukS-PV for its receptor. Extracellular Ca2+ at physiological concentrations (1 to 2 nM) has only a slight influence on the LukS-PV binding, in contrast to its complete inhibition by Zn2+. The down-regulation by phorbol 12-myristate 13-acetate (PMA) of the binding of LukS-PV was blocked by staurosporine, suggesting that the regulatory effect of PMA depends on protein kinase C activation. The labeled mutant form of LukS-PV has proved very useful for detailed binding studies of circulating white cells by flow cytometry. LukS-PV possesses a high specific affinity for a unique receptor on PMNs and monocytes.
The so-called Panton-Valentine leucocidin (PVL) was shown to differ from hemolysins secreted by strain V8, which was isolated from a patient with chronic furunculosis (16). Gladstone and Van Heyningen (9) have reported the nonhemolytic properties of PVL. Woodin (22, 23) characterized PVL as being composed of two protein components. Epidemiological studies have demonstrated that PVL is secreted by clinical strains associated with abscesses, furuncles (5, 8), and community-acquired pneumonia (13). Genes encoding PVL were cloned and sequenced, and proteins were named LukS-PV (32,317 Da) and LukF-PV (34,386 Da). They belong to the staphylococcal bicomponent pore-forming leukotoxin family (18). PVL induces the opening of Ca2+ channels responsible for an influx of Ca2+ (19) and the formation of pores through the membrane of target cells (7).
Previous work (4) showed that the binding of LukS-PV is a prerequisite for the binding of LukF-PV and subsequent activation of polymorphonuclear neutrophils (PMNs). Binding studies by Colin et al. (4) indicated that LukS-PV had a Kd of 6 nM and showed a maximal binding capacity (Bm) of 39,000 molecules per PMN using an iodinated toxin. Several reasons prompted us to reevaluate this determination and to use a simpler, nonradioactive technique. First, the radioiodination of LukS-PV had altered its biological activity to some extent, and second, a very high PMN concentration was used (3 × 106 PMNs/ml). In addition, we chose flow cytometry, which allows both the analysis of low cell concentrations and fluorescence determinations. Furthermore, since LukS-PV does not possess any cysteine, substitution of a cysteine for a glycine was carried out by site-directed mutagenesis in order to label one leukotoxin molecule with one fluorescein. In these conditions, we could accurately analyze the binding of very low concentrations of leukotoxin to measure its apparent affinity and to characterize some of its binding properties.
MATERIALS AND METHODS
Chemical reagents.
H-89, phorbol 12-myristate 13-acetate (PMA), staurosporine, wortmannin, and salts were purchased from Sigma-Aldrich (Saint Quentin Fallavier, France); yeast extract was purchased from Oxoid (Dardilly, France); and Bacto-Casamino Acids were purchased from Difco (Becton Dickinson, Le Pont de Claix, France).
Leukotoxin purification.
Leukotoxins were produced from cultures of Staphylococcus aureus strain V8 (ATCC 49775) harvested at the stationary growth phase, as described previously (17). Briefly, the strain was grown for 17 h in YCP medium (3% [wt/vol] yeast extract, 2% [wt/vol] Bacto-Casamino Acids, 2% [wt/vol] sodium pyruvate, 0.25% [wt/vol] Na2HPO4, 0.042% [wt/vol] KH2PO4, pH 7.0) at 37°C with vigorous shaking (10). The exoproteins were concentrated after precipitation with 80% (wt/vol) ammonium sulfate and dialysis against 30 mM Na-phosphate (pH 6.5). A bulk of positively charged proteins was selected on a Sepharose SP Fast Flow chromatography plate (Pharmacia, Uppsala, Sweden) after elution in 0.5 M NaCl. The resulting proteins were then subjected to cation-exchange MonoS fast-performance liquid chromatography (Pharmacia) and to further alkyl-Superose fast-performance liquid chromatography (Pharmacia) as described previously (10). Proteins were adjusted at 0.6 mg/ml in 30 mM Na-phosphate–200 mM NaCl and stored at −80°C until utilization. Protein purification was assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
Leukocyte preparation.
PMNs, lymphocytes, and monocytes were prepared from buffy coats of healthy donors of either sex, kindly provided by the Etablissement Régional de Transfusion Sanguine de Strasbourg, France, as described previously (15). Briefly, 5 ml of injectable perfusion solution (Plasmion; Lab. Roger Bellon, Neuilly sur Seine, France) was added to 20 ml of a dilution of white-cell-enriched blood in 0.9% NaCl (1/3, vol/vol) and left to sediment for 30 min. The supernatant was centrifuged and washed in HEPES buffer (140 mM NaCl, 5 mM KCl, 10 mM glucose, 0.1 mM EGTA, 10 mM HEPES, 3 mM Tris base, pH 7.3). When only PMNs were used, they were further purified as described previously (19). Briefly, 40 ml of a dilution of blood cells in 0.9% NaCl (1/3, vol/vol) was layered on 12 ml of J Prep (Techgen International, Voisins le Bretonneux, France). After a 20-min centrifugation (800 × g), the pellet was suspended in 30 ml of 0.9% NaCl and added to 10 ml of 6% (wt/vol) dextran for a 30-min sedimentation. Thirty milliliters of the supernatant was centrifuged for 10 min at 800 × g. The pellet was suspended in HEPES buffer, and the contaminating erythrocytes were removed by a 45-s hypotonic lysis and subsequent washing in HEPES buffer. The final suspension was adjusted to 6 × 106 PMNs/ml, and 0.1% bovine serum albumin was added to prevent nonspecific adherence of leukotoxin on tube walls.
Leukotoxin mutation.
The luk-PV locus (EMBL accession no. X72700) previously cloned in pUC19 (17) was subcloned after NruI-HindIII (3.0-kb) restriction into the SmaI-HindIII-linearized and dephosphorylated shuttle plasmid pCU1 (3). This new recombinant plasmid was mutated by using dedicated complementary oligonucleotides (sense, 5′-POHCAATATTGAGAATATTGGTGATTGTGCTGAGGTAGTCAAAAGAAC-3′) to obtain LukS-PV Gly10Cys (LukSG10C). Site-directed mutagenesis was performed using the Quick Change mutagenesis kit (Stratagene, Montigny le Bretonneux, France) in the presence of 5 ng of template (5.8 kb) and 0.4 nM (each) dedicated oligonucleotides in 50 μl as recommended by the manufacturer. Temperatures for hybridization, elongation, and denaturation were 50, 68, and 95°C for 0.5, 3.5, and 1 min, respectively. Initial templates were removed by an 80-min DpnI restriction, and 80 μl of XL1 Blue Supercompetent cells (Stratagene) was transformed with 2.5 μl of the mixture as recommended. Mutated genes were sequenced (21) from Qiagen (Paris, France) plasmid preparations, and 1 μg of positive plasmids was electroporated at 1.8 kV, 200 MΩ, and 25 μF in 80 μl of S. aureus RN 4220 (r− m+ agr negative) stored at 5.0 A560 units in 10% (vol/vol) glycerol. After 1 h of regeneration into SOC medium (TY [1% {wt/vol} Tryptone, 0.5% {wt/vol} yeast extract, 0.5% {wt/vol} NaCl] plus 10% [wt/vol] 3,350-Da polyethylene glycerol, 5% dimethyl sulfoxide, 50 mM MgCl2), bacteria were plated onto TY-chloramphenicol (5 μl/ml) and incubated overnight at 37°C. Total DNA from recombinant clones was prepared as described previously (17) and electroporated under the same conditions as mentioned above into the recently described S. aureus hlg-negative strain Newman (20). Purification of the mutated protein LukSG10C was performed as described previously for the native protein (17).
Fluorescein labeling.
The mutated protein LukSG10C was labeled with fluorescein (LukSG10C*) as follows. A fivefold excess of fluorescein 5-maleimide (Molecular Probes, Eugene, Oreg.) for a 10 μM LukSG10C solution was incubated for 30 min at room temperature in 50 mM Na-phosphate–0.15 M NaCl–1 mM EDTA-Na2, pH 7.0. The coupling reaction was stopped by addition of 10 mM β-mercaptoethanol. The mixture was then desalted, the coupling yield R of LukSG10C* was determined by the ratio of the determined concentration of fluorescein (ɛ490 = 81,900 cm−1 · mol−1), and that of the protein was determined by Bradford titration (Bio-Rad, Ivry sur Seine, France). The R value was determined to be 0.95 < R < 1.
Determination of the specific fluorescence of LukSG10C*.
The specific fluorescence of LukSG10C* was determined using a PMN suspension (3 × 106 cells/ml) incubated for 30 min with 5 nM LukSG10C*. After two washes, PMN fluorescence intensity was measured with a spectrofluorometer (Deltascan TM 4000; Kontron, PTI, Montigny Le Bretonneux, France), and the autofluorescence was determined by addition of fluorescein antibody (Molecular Probes) to quench the fluorescein fluorescence. The number of LukSG10C* molecules (N) bound to PMNs per milliliter was estimated, after autofluorescence subtraction, by comparison with the fluorescence of known concentrations of free LukSG10C* added in the cuvette. Control experiments were performed to ensure that free LukSG10C* and bound LukSG10C* emitted the same fluorescence (i.e., the fluorescence intensity of different LukSG10C* concentrations was not modified by addition of PMNs) (data not shown).
Then, the PMNs were analyzed with a FACSort cytometer (Becton Dickinson) which was set so that calibrated fluorescent microbeads (Immuno-Brite; Coulter Corporation, Hialeah, Fla.) displayed an identical fluorescence intensity in each experiment. Thus, the mean PMN fluorescence intensity was expressed in standardized fluorescence units (SFU). The precise number n of PMNs in the suspension (only fluorescent cells were counted, including dead cells, which represented less than 5% of the total cells) was measured using calibrated fluorescent microbeads of known concentration given by the supplier (Flow-Count; Coulter Corporation), and the mean fluorescence intensity of PMNs, Fm*, was measured. The specific fluorescence spF* of LukSG10C* was calculated according to the following formula:
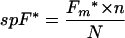 |
spF* = 2.05 × 10−3 SFU per LukSG10C* molecule and 1 SFU = 488 LukSG10C* molecules.
Flow cytometry determinations.
Flow cytometry determinations were made using a FACSort cytometer (Becton Dickinson) equipped with a 15-mW argon laser tuned to 488 nm. PMNs, monocytes, and lymphocytes were classically discriminated by forward and side light scattering, and their fluorescence was recorded according to the experiment. Variations of intracellular free Ca2+ and pore formation were determined by recording variations of the fluorescence intensity of Fluo3 loaded in PMNs and of variations of ethidium penetrating PMNs by pores, respectively, as described previously (15). Fluo3 or fluorescein fluorescence intensities were recorded in the FL1 channel (λEM = 530 nm) (in setting conditions for Fluo3, the fluorescence of LukSG10C* was negligible and did not interfere with Fluo3 fluorescence determination), and ethidium fluorescence intensity was recorded in the FL3 channel (λEM > 650 nm).
RESULTS
Leukotoxic activity of LukSG10C*.
The leukotoxic activities of the native, the nonfluorescent, and the fluorescent mutated leukotoxins were examined by studying their abilities, first, to open Ca2+ channels (19), and second, to form pores through the plasma membrane of PMNs (7). The cysteine substitution and further addition of the fluorescein moiety did not modify the activity of LukS-PV, since the intracellular Ca2+ increases induced in the presence of Ca2+ (Fig. 1A) and the pores formed in the absence of Ca2+ (Fig. 1B) by the three pairs LukS-PV–LukF-PV, LukSG10C–LukF-PV, and LukSG10C*–LukF-PV were unchanged. Consequently, the fluorescent leukotoxin mutant, LukSG10C*, was considered to be as potent as the native protein and was used to study its binding.
FIG. 1.

Flow cytometry analysis of the time course of free intracellular Ca2+ variations (A) and of pore formation (B) in human PMNs after the simultaneous addition of LukF-PV and of the wild-type or mutated LukS-PV. PMNs are either loaded with Fluo3 (A) or suspended in the presence of 4 μM ethidium bromide (B). LukS-PV, LukSG10C, and LukSG10C* were each added at a 1 nM concentration (arrow) to PMNs (5 × 105 PMNs/ml) in the presence of 30 nM LukF-PV.
Optimal concentrations of PMNs for binding studies.
The mean fluorescence intensities of increasing concentrations of PMNs were measured by flow cytometry in the presence of 0.1 or 1 nM LukSG10C*. As shown in Fig. 2, the binding of LukSG10C* per PMN was constant up to 1 × 105 PMNs/ml for 0.1 nM LukSG10C* and up to 7 × 105 PMNs/ml for 1 nM LukSG10C*. These observations influenced the choice of PMN concentrations used in this study.
FIG. 2.

Flow cytometry analysis of the binding of LukSG10C* to increasing concentrations of human PMNs. PMNs were incubated with 0.1 and 1 nM LukSG10C* for 60 and 15 min, respectively. The binding was not modified by PMN concentrations of up to 105 PMNs/ml for 0.1 nM LukSG10C* and to 7 × 105 PMNs/ml for 1 nM LukSG10C*.
Properties of binding of LukSG10C* to white blood cells. (i) PMNs.
An example of the kinetics of association between increasing concentrations of LukSG10C* and human PMNs is shown in Fig. 3. Different times were required for each concentration to reach the plateau value utilized for the evaluation of the apparent Kd and the Bm of LukSG10C* in the absence of Ca2+. The fitting of saturation curves by nonlinear regression analysis (SigmaPlot; SPSS Science, Ehrkarth, Germany) as shown in Fig. 4 allowed the calculation of the following values: Kd = 0.07 ± 0.02 nM (n = 5); Bm = 383.2 ± 62.3 SFU/PMN (n = 5), i.e., 196,900 ± 30,400 binding sites per PMN. The binding of LukSG10C* appeared very specific, since the nonspecific binding coefficient was 1.96 ± 1.28 (n = 5). The Hill coefficient was 1.03 ± 0.03 (n = 5).
FIG. 3.

Example of a flow cytometry analysis of kinetics of binding of increasing concentrations of LukSG10C* to human PMNs in the absence of Ca2+. PMN concentrations were 5 × 105 PMNs/ml for 0.5 to 10 nM LukSG10C* and 8 × 104 PMNs/ml for 0.01 to 0.2 nM LukSG10C*.
FIG. 4.

Calculation by nonlinear regression of the equilibrium constants of LukSG10C* binding from values generated from the experiment whose results are shown in Fig. 3. non spec. coef., nonspecific coefficient.
(ii) Monocytes.
The same experiment carried out with monocytes gave the following mean values: Kd = 0.020 ± 0.003 nM (n = 3); Bm = 163.4 ± 20.5 SFU/PMN (n = 3), i.e., 79,740 ± 10,004 binding sites per monocyte. The nonspecific binding was negligible, and the Hill coefficient was 1.0 ± 0.1 (n = 3).
(iii) Lymphocytes.
No measurable binding of LukSG10C* could be detected on lymphocytes.
Nonspecific binding activity.
At low LukS-PV concentrations, the nonspecific binding is not very consistent, but at high concentrations it might affect the pore formation activity of the leukotoxin. Thus, the ability of LukS-PV to nonspecifically bind to the membrane of PMNs and form pores in association with LukF-PV components was assayed by incubating increasing concentrations of LukS-PV with 30 nM LukF-PV in the presence of ethidium bromide. Figure 5 shows that pore formation was saturable and did not increase at concentrations higher than 0.5 nM, which corresponds to the binding saturation of LukS-PV.
FIG. 5.

Flow cytometry determination of the time course of the pore formation induced in human PMNs by increasing LukS-PV concentrations in the presence of LukF-PV. The pore formation is determined by the ethidium influx stimulated in 5 × 104 PMNs/ml by the addition of 0.01 to 100 nM LukS-PV and 30 nM LukF-PV.
Competition with other S components.
In order to determine the binding capacities of the other S components of the staphylococcal leukotoxin family, such as HlgA, HlgC, LukE, and LukM, for the binding site of LukS-PV, increasing concentrations of HlgA (expressed in Escherichia coli in order to avoid contaminating effects by HlgC, especially at high concentrations), HlgC, LukE, LukM, and the native protein LukS-PV were incubated for 10 min with 1 nM LukSG10C* and PMNs. The mean fluorescence intensity of PMNs measured by flow cytometry (Fig. 6) showed that HlgA and LukE were very weak competitors of LukSG10C* and that no 50% inhibitory concentration could be measured even at a concentration 2 orders of magnitude higher than that of LukSG10C*. LukM did not compete at all with LukSG10C*. Conversely, LukS-PV had the same affinity as did LukSG10C* for the receptor, which confirms that LukSG10C* possesses the same binding properties as does LukS-PV. HlgC had an even higher affinity than did LukS-PV for the binding site, and a Ki of 0.03 nM could be calculated.
FIG. 6.

Determination by flow cytometry of the competitive binding between LukSG10C* and S components (LukS-PV, HlgC, HlgA, LukM, and LukE) in human PMNs. LukSG10C* concentration, 1 nM; PMN concentration, 5 × 104 PMNs/ml; incubation time, 30 min; n = 4.
Influence of divalent cations.
It has been shown previously that less pore formation was observed in the presence of Ca2+ and Zn2+ (7). Thus, in order to determine whether this was due to an inhibition of the LukS-PV binding, 1 nM LukSG10C* was added to PMNs in the presence of increasing concentrations of Ca2+ and Zn2+. A 1 mM concentration of Ca2+ reduced the LukSG10C* binding by 5%, and 20 mM Ca2+ reduced binding by 20%, whereas 1 and 2 mM Zn2+ decreased it by 80 and 90%, respectively.
Receptor regulation.
A previous study showed that the protein kinase C (PKC) activator PMA completely inhibited the formation of pores by the pair LukS-PV–LukF-PV (O. Meunier, unpublished results). To determine whether this inhibition was due to an alteration of the binding of LukS-PV, PMNs were preincubated with increasing PMA concentrations and the binding of LukSG10C* was measured on PMNs sampled every 10 min. Figure 7 shows that the binding of LukSG10C* was inhibited by PMA in a concentration-dependent manner. To determine whether the effect of PMA on binding inhibition occurred through activation of PKC, PMNs were preincubated in the presence or absence of the PKC inhibitor staurosporine (1 mM) for 15 min prior to incubation with 10 nM PMA, and the binding of LukSG10C* was measured as done previously. Under these conditions, staurosporine markedly decreased the effect of PMA (Fig. 8) but was without effect on the binding to cells not treated with PMA. Wortmannin, a selective inhibitor of phosphoinositide 3-kinase, and H-89, a selective inhibitor of protein kinase A, had no effect on the binding in both the presence and the absence of PMA (data not shown). Taken together, these data suggest that PKC participates in the regulation of the availability of the LukS-PV receptor.
FIG. 7.

Determination by flow cytometry of the effect of PMA preincubation on the binding of LukSG10C* on human PMNs. LukSG10C* concentration, 1 nM. PMNs (5 × 105 PMNs/ml) were preincubated at each PMA concentration and sampled every 10 min. Then LukSG10C* was added 9 min before analysis (100% = binding of LukSG10C* in the absence of PMA).
FIG. 8.

Determination by flow cytometry of the inhibition by staurosporine (STA) of the effect of PMA on the binding of LukSG10C* on human PMNs. Measurements were made as described for Fig. 7. LukSG10C* concentration, 1 nM; PMA concentration, 10−8 M. PMNs (5 × 105 PMNs/ml) were preincubated for 60 min with 10−6 M staurosporine.
DISCUSSION
The binding of LukS-PV, the class S component of PVL, has been studied by flow cytometry using a single-cysteine substitution mutant form of the protein. The mutant was labeled with fluorescein. This mutant was obtained by site-directed mutagenesis of an S. aureus strain whose hlg locus was deleted and which thus did not secrete any leukotoxin component besides LukSG10C*. Furthermore, this fluorescent protein, LukSG10C*, was ideally suited for binding studies since (i) LukSG10C*, like the native protein, is capable of acting in synergy with LukF-PV to open Ca2+ channels as well as form pores through the PMN membrane and (ii) the mutant protein had the same affinity as did the native protein, as confirmed by competition experiments. In addition, the very low cell concentration used in flow cytometry (Fig. 2) is an essential requirement for determination of binding at low antigen concentrations. Thus, the present study confirms that LukS-PV binding is saturable and that LukS-PV possesses only one class of receptors but demonstrates that LukS-PV affinity is much higher than previously reported (4). In the previous study, the concentration of 3 × 106 PMNs/ml used by the authors led to the determination of an apparent Kd 2 orders of magnitude higher (6.01 nM) than the one observed in the present study. At this high PMN concentration, the binding of LukS-PVG10C* is notably decreased, especially at low concentrations of leucocidin (Fig. 2), which explains the differences observed with Kd determinations.
The LukS-PV component has a higher affinity for monocytes than for PMNs, but monocytes possess less than half the receptors of PMNs. The absence of LukS-PV binding to lymphocytes suggests that lymphocytes are not target cells for PVL (17), because they lack receptors for LukS-PV. The LukS-PV affinity is in the same order of magnitude as the affinity of cytokines for their receptors (11, 14). Moreover, the pair LukS-PV–LukF-PV induces cellular events similar to those induced by cytokines, i.e., the opening of Ca2+ channels (19), the induction of secretion (4), and the release of inflammatory mediators (12). Thus, we speculate that the receptor of LukS-PV might belong to a cytokine receptor family, but this remains to be determined.
The LukS-PV-specific binding and the pore formation are events which both saturate at the same concentrations, indicating that nonspecific binding is not efficient. Two hypotheses can be proposed: either LukF-PV binds only to the complex LukS-PV–receptor or LukS-PV and LukF-PV can together oligomerize in the membrane without opening a pore. The latter proposition is supported by results obtained by Ferreras et al. (6), who showed that LukS-PV and LukF-PV could integrate into synthetic lipidic membranes without forming pores, in contrast to the HlgA-HlgB pair from γ-hemolysin. It has been suggested that the leukotoxin pore is hexameric, consisting of three LukS-PV and three LukF-PV components (6). However, up to now, it has been unclear whether three LukS-PV components bind to three identical domains of one particular receptor or whether each LukS-PV component binds to one particular receptor. In the latter case, the LukS-PV–receptor complexes would have to assemble three by three with LukF-PV components to form heterohexameric pores.
Competition binding experiments with other S components showed that HlgC and LukS-PV share the same receptor but that HlgA, LukE, and LukM have different receptors. This implies that leukotoxins might activate PMNs through different intracellular signaling pathways and, consequently, induce release of separate inflammatory mediators.
The extracellular free Ca2+ has only a slight influence on the binding of LukS-PV, notably so at physiological concentrations. Consequently, the significant inhibition of the pore formation observed in the presence of Ca2+ (7) was not due to an inhibition of LukS-PV binding but might be due to the influence of Ca2+ on LukF-PV binding. Conversely, Zn2+ can completely inhibit the binding of LukS-PV to cells, indicating the presence of at least one structural position able to bind Zn2+.
Activation of PKC by PMA leads to a decrease in the binding to receptors by LukSG10C* which can be overcome by staurosporine. These observations suggest that LukS-PV receptors are down-regulated through a PKC-dependent pathway. Further studies are necessary to determine whether the inhibition of the binding of LukS-PV to its receptor by PMA is due to an internalization, as for fMet-Leu-Phe receptors (2), or to a shedding, as for l-selectin receptors (1).
In conclusion, LukSG10C* is a very useful tool to study the binding of LukS-PV on different white blood cells by flow cytometry. We demonstrated that LukS-PV has a high affinity for both PMNs and monocytes. LukS-PV has the same receptor as HlgC, one of the S components of the γ-hemolysin of S. aureus, but HlgA and LukE have separate receptors and LukM does not compete at all with LukS-PV. LukS-PV binding is not influenced by Ca2+ but is completely inhibited by Zn2+. The LukS-PV receptor seems to be down-regulated by PKC.
ACKNOWLEDGMENTS
We thank Viviane Finck-Barbançon for helpful comments, Daniel Keller for expert toxin purification and labeling, and Raymonde Girardot for her excellent technical assistance.
This work was supported by grant EA-1318 from the Direction de la Recherche et des Etudes Doctorales.
REFERENCES
- 1.Alexander S R, Kishimoto T K, Walchek B. Effects of selective protein kinase C inhibitors on the proteolytic down-regulation of L-selectin from chemoattractant-activated neutrophils. J Leukoc Biol. 2000;67:415–422. doi: 10.1002/jlb.67.3.415. [DOI] [PubMed] [Google Scholar]
- 2.Andersson T, Dahlgren C, Lew P D, Stendahl O. Cell surface expression of fMet-Leu-Phe receptors on human neutrophils. Correlation to changes in the cytosolic free Ca2+ and action of phorbol myristate acetate. J Clin Investig. 1987;79:1226–1233. doi: 10.1172/JCI112941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Augustin J, Rosenstein R, Wieland B, Schneider V, Schnell N, Engelke G, Entian K D, Götz F. Genetic analysis of epidermin biosynthetic genes and epidermin-negative mutants of Staphylococcus epidermidis. Eur J Biochem. 1992;204:1149–1154. doi: 10.1111/j.1432-1033.1992.tb16740.x. [DOI] [PubMed] [Google Scholar]
- 4.Colin D A, Mazurier I, Sire S, Finck-Barbançon V. Interaction of the two components of leukocidin from Staphylococcus aureus with human polymorphonuclear leukocyte membranes: sequential binding and subsequent activation. Infect Immun. 1994;62:3184–3188. doi: 10.1128/iai.62.8.3184-3188.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cribier B, Prévost G, Couppié P, Finck-Barbançon V, Grosshans E, Piémont Y. Staphylococcus aureus leukocidin: a new virulence factor in cutaneous infections? An epidemiological and experimental study. Dermatology. 1992;185:175–180. doi: 10.1159/000247443. [DOI] [PubMed] [Google Scholar]
- 6.Ferreras M, Höper F, Dalla Serra M, Colin D A, Prévost G, Menestrina G. The interaction of Staphylococcus aureus bi-component γ-hemolysins and leucocidins with cells and lipid membranes. Biochim Biophys Acta. 1998;1414:108–126. doi: 10.1016/s0005-2736(98)00160-6. [DOI] [PubMed] [Google Scholar]
- 7.Finck-Barbançon V, Duportail G, Meunier O, Colin D A. Pore formation by a two-component leukocidin from Staphylococcus aureus within the membrane of human polymorphonuclear leukocytes. Biochim Biophys Acta. 1993;1182:275–282. doi: 10.1016/0925-4439(93)90069-d. [DOI] [PubMed] [Google Scholar]
- 8.Finck-Barbançon V, Prévost G, Piémont Y. Improved purification of leukocidin from Staphylococcus aureus and toxin distribution among hospital strains. Res Microbiol. 1991;142:75–85. doi: 10.1016/0923-2508(91)90099-v. [DOI] [PubMed] [Google Scholar]
- 9.Gladstone G P, Van Heyningen W E. Staphylococcal leucocidins. Br J Exp Pathol. 1957;38:123–127. [PMC free article] [PubMed] [Google Scholar]
- 10.Gravet A, Colin D A, Keller R, Girardot R, Monteil H, Prévost G. Characterization of a novel structural member, LukE-LukD, of the bi-component staphylococcal leucotoxin family. FEBS Lett. 1998;436:202–208. doi: 10.1016/s0014-5793(98)01130-2. [DOI] [PubMed] [Google Scholar]
- 11.Guthridge A M, Stomski F C, Thomas D, Woodcock J M, Bagley C J, Berndt M C, Lopez A F. Mechanism of activation of the GM-CSF, IL-3, and IL-5 family of receptors. Stem Cells. 1998;16:301–313. doi: 10.1002/stem.160301. [DOI] [PubMed] [Google Scholar]
- 12.König B, Prévost G, Piémont Y, König W. Effects of Staphylococcus aureus leukocidins on inflammatory mediator release from human granulocytes. J Infect Dis. 1995;171:607–613. doi: 10.1093/infdis/171.3.607. [DOI] [PubMed] [Google Scholar]
- 13.Lina G, Piémont Y, Godail-Gamot F, Bes M, Peter M-O, Gauduchon V, Vandenesch F, Etienne J. Involvement of Panton-Valentine leukocidin-producing Staphylococcus aureus in primary skin infections and pneumonia. Clin Infect Dis. 1999;29:1128–1132. doi: 10.1086/313461. [DOI] [PubMed] [Google Scholar]
- 14.McCusker R H. Controlling insulin-like growth factor activity and the modulation of insulin-like growth factor binding protein and receptor binding. J Dairy Sci. 1998;81:1790–1800. doi: 10.3168/jds.S0022-0302(98)75748-0. [DOI] [PubMed] [Google Scholar]
- 15.Meunier O, Falkenrodt A, Monteil H, Colin D A. Application of flow cytometry in toxinology: pathophysiology of human polymorphonuclear leukocytes damaged by a pore-forming toxin from Staphylococcus aureus. Cytometry. 1995;21:241–247. doi: 10.1002/cyto.990210304. [DOI] [PubMed] [Google Scholar]
- 16.Panton P N, Valentine F C O. Staphylococcal toxins. Lancet. 1932;ii:506–508. [Google Scholar]
- 17.Prévost G, Bouakham T, Piémont Y, Monteil H. Characterization of a synergohymenotropic toxin produced by Staphylococcus intermedius. FEBS Lett. 1995;376:135–140. doi: 10.1016/0014-5793(95)01260-9. [DOI] [PubMed] [Google Scholar]
- 18.Prévost G, Cribier B, Couppié P, Petiau P, Supersac G, Finck-Barbançon V, Monteil H, Piémont Y. Panton-Valentine leucocidin and gamma-hemolysin from Staphylococcus aureus ATCC 49775 are encoded by distinct genetic loci and have different biological activities. Infect Immun. 1995;63:4121–4129. doi: 10.1128/iai.63.10.4121-4129.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Staali L, Monteil H, Colin D A. The staphylococcal pore-forming leukotoxins open Ca2+ channels in the membrane of human polymorphonuclear neutrophils. J Membr Biol. 1998;162:209–216. doi: 10.1007/s002329900358. [DOI] [PubMed] [Google Scholar]
- 20.Supersac G, Piémont Y, Kubina M, Prévost G, Foster T J. Assessment of the role of gamma-toxin in experimental endophthalmitis using a hlg-deficient mutant of Staphylococcus aureus. Microb Pathog. 1998;24:241–251. doi: 10.1006/mpat.1997.0192. [DOI] [PubMed] [Google Scholar]
- 21.Tabor S, Richardson C C. DNA sequence analysis with a modified bacteriophage T7 DNA polymerase. Proc Natl Acad Sci USA. 1987;84:4767–4771. doi: 10.1073/pnas.84.14.4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woodin A M. Fractionation of a leucocidin from Staphylococcus aureus. Biochem J. 1959;73:225–237. doi: 10.1042/bj0730225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woodin A M. Purification of the two components of leucocidin from Staphylococcus aureus. Biochem J. 1960;75:158–165. doi: 10.1042/bj0750158. [DOI] [PMC free article] [PubMed] [Google Scholar]