

Abstract
Low-grade Oncocytic Tumor (LOT) of the kidney is a recently described entity with poorly understood pathogenesis. Using next-generation sequencing (NGS) and complementary approaches, we provide insight into its biology. We describe 22 LOT corresponding to 7 patients presenting with a median age of 75 years (range 63–86 years) and male to female ratio 2:5. All 22 tumors demonstrated prototypical microscopic features. Tumors were well circumscribed and solid. They were composed of sheets of tumor cells in compact nests. Tumor cells had eosinophilic cytoplasm, round to oval nuclei (without nuclear membrane irregularities), focal subtle perinuclear halos and occasional binucleation. Sharply delineated edematous stromal islands were often observed. Tumor cells were positive for PAX8, negative for CD117 and exhibited diffuse and strong cytokeratin-7 expression. Six patients presented with pT1 tumors and at a median follow-up of 29 months, 4 patients were alive without recurrence (3 patients had died from unrelated causes). All tumors were originally classified as chromophobe renal cell carcinoma, eosinophilic variant (chRCC-eo). While none of the patients presented with known syndromic features, one patient with multiple bilateral LOTs was subsequently found to have a likely pathogenic germline TSC1 mutation. Somatic, likely activating, mutations in MTOR and RHEB were identified in all other evaluable LOTs. As assessed by phospho-S6 and phospho-4E-BP1, mTOR complex 1 (mTORC1) was activated across all cases but to different extent. MTOR mutant LOT typically exhibited lower levels of mTORC1 activation, possibly related to mTORC1 dimerization and the preservation of a wild-type MTOR copy (retained chromosome 1). Supporting its distinction from related entities, gene expression analyses showed that LOT clustered separately from classic chRCC, chRCC-eo and RO. In summary, converging mTORC1 pathway mutations, mTORC1 complex activation and a distinctive gene expression signature along with characteristic phenotypic features support LOT designation as a distinct entity with both syndromic and non-syndromic cases associated with an indolent course.
Keywords: Oncocytoma, eosinophilic variant of chromophobe renal cell carcinoma, RCC with cytoplasmic eosinophilia, eosinophilic tumor, unclassified oncocytic tumor, hybrid oncocytic / chromophobe RCCs, LOT, low-grade oncocytic tumor, TSC associated RCC
Introduction
The differential diagnosis of renal tumors with exclusively eosinophilic (or oncocytic) cytoplasm traditionally involves differentiating a malignant tumor, chromophobe renal cell carcinoma (chRCC), in particular the eosinophilic variant (chRCC-eo), from benign renal oncocytoma (RO)1,2. This distinction frequently poses a dilemma given their shared morphologic features especially with unifocal tumors in non-syndromic settings2. Furthermore, markers often used in the differential diagnosis, such as cytokeratin (CK) 7 (typically diffuse in chRCC and scattered in RO), are not always able to distinguish chRCC-eo, which can have rare or patchy expression. This difficulty is further compounded by a lack of definite criteria to distinguish chRCC-eo from the classic variant of chRCC.
The differential diagnosis of renal tumors with eosinophilic cytoplasm has become even more challenging with newly described entities2,3. These include tuberous sclerosis complex (TSC) associated renal cell carcinoma (RCC)4,5, hybrid oncocytic/chromophobe tumors (HOCT) in the setting of Birt-Hogg-Dubé (BHD) syndrome6, eosinophilic solid and cystic RCC (ESC RCC)7–9, MiT family translocation RCC10,11, succinate dehydrogenase (SDH)-deficient RCC12, fumarate hydratase (FH)-deficient RCC13, eosinophilic vacuolated tumors (EVT)/ RCC with eosinophilic and vacuolated cytoplasm14–17, as well as other currently unclassified RCCs18.
Recently, a distinct low-grade oncocytic tumor (LOT) was reported as a potential new entity19,20. Two publications describe LOT as unifocal small tumors (mean size 3.9 cm [range 1.1–13.5 cm]), devoid of aggressive features in non-syndromic settings19,21. Histologically, LOT have a predominantly solid architecture composed of tight nests with foci of delineated edematous stroma that contains strands of tumor cells. The tumor cells have eosinophilic/oncocytic cytoplasm, uniform round nuclei without significant irregularities, and only focal subtle perinuclear halos. They have a characteristic but unexpected immunoprofile: they are CD117 (c-kit) negative and show strong and diffuse CK7 reactivity. To date, all tumors have been reported to be associated with an indolent clinical course2,19,20,22.
We previously published a comprehensive genomic analysis of 167 primary non-clear cell RCC that included RO (n= 31), renal oncocytic neoplasm (RON) (n= 5), and chRCC (36 classic variant; 12 eosinophilic variant)23. Notably, the majority of chRCC-eo lacked characteristic DNA copy number alterations of the classic variant of chRCC. Two of the 12 chRCC-eo morphologically fit the description of LOT19,20,24 and were CD117 negative/CK7 positive. Interestingly, both tumors had a unique gene expression signature, and harbored mammalian target of rapamycin (MTOR) mutations. These results led us to hypothesize that LOT may be characterized by a unique gene expression signature and mutations of the mammalian target of rapamycin complex 1 (mTORC1) pathway (such as in MTOR, TSC1, TSC2 or RHEB). In the current study, we evaluate this notion through systematic analyses of eosinophilic tumors at our institution that fit the morphology and immunohistochemistry profiles of LOT.
Materials and methods
Case selection
The study was conducted with approval by the UT Southwestern Medical Center (UTSW) Institutional Review Board (IRB) and according to the Health Insurance Portability and Accountability Act (HIPAA) guidelines. We searched our institutional RCC database (Kidney Cancer Explorer [KCE]; sponsored by the UT Southwestern [UTSW] Kidney Cancer NIH SPORE grant, the Lyda Hill Department of Bioinformatics and the Bioinformatics Core Facility) for tumors diagnosed as chRCC, RON, and RO. We excluded diagnoses based on biopsy specimens as well as consultation cases. Between 2005 and 2016, 204 cases (99 chRCC, 20 RON, and 85 RO) were identified out of a total of 1870 renal tumors, including some that were published previously23. Archival material, where available, was retrieved (204 cases) and re-reviewed by two genitourinary (GU) pathologists (S.C and P.K), who classified these tumors as LOT based on previously described characteristic morphology and IHC (CK7 diffuse+/CD117−)19. Additional IHCs were obtained as needed during review (CK7 and CD117, CK20, cathepsin K, TFEB [transcription factor EB], TFE3 [transcription factor E3], SDHB [succinate-dehydrogenase B], and FH [fumarate hydratase]).
Clinical Data
Patient age at nephrectomy, race, sex, tumor size, AJCC TNM staging categories, histologic subtype, date of surgery, date of development of metastases, and date of last follow-up were collected from KCE. The tumors were staged based on the 2018 American Joint Committee on Cancer (AJCC) TNM classification for pathologic staging. Data in KCE were complemented through comprehensive review of clinical data and pathology slides.
Immunohistochemistry
IHC analysis was performed on representative 3–5 μm formalin-fixed paraffin-embedded (FFPE) whole tumor tissue sections. Staining for routinely used markers including Pax8 (10336-1-A P, 1:1000; Proteintech), CA9 (11071-1-AP, 1:100; Thermo Fisher), CD10 (56C6, 1:40; Dako), CK7 (M7018- OV-TL 12/30, 1:100; Dako), CK20 (7019-Ks20.8, 1:50; Dako), CD117 (A4502, 1:700; Dako), Melan A (7196-A103, 1;200; Dako), cathepsin-K (37259-3F9, 1:100; Abcam), FH (100743-J13, 1:1500; Santa Cruz), SDHB (14714-21A11, 1:100; Abcam), TFE3 (MRQ-37, 1:200; Cell Marque), TFEB (166736, 1:100; Santa Cruz), vimentin (M0725, 1:75; Dako), phospho-S6 (Ser240/244) (p-S6) (5364-D68F8, 1:300; Cell Signaling Technology) and phospho-4E-BP1 (Thr37/46) (p-4EBP1) (236B4, 1:800; Cell Signaling Technology) was performed using a Dako automated system (Agilent).
Immunoreactivity was interpreted as “negative” if < 5% tumor cells stained positively. P-S6 and p-4EBP1 expression was determined based on the percentage of tumor cells staining positive and the intensity of expression, which were multiplied.
Next generation sequencing
All hematoxylin and eosin (H&E) FFPE slides were examined to select most representative areas from 7 LOTs as well as matched normal tissues. Corresponding areas were macrodissected (LOT 3–7). Tissue was submitted to BGI Genomics for DNA extraction and whole-exome sequencing (WES). DNA libraries were sequenced to an average read depth > 100x for exome sequencing using 2 × 75-base paired-end on a HiSeq2500 platform (Illumina, San Diego, CA, USA). DNA from LOT 7 tumor and LOT 6 normal did not pass quality control. The two index cases (LOT 1–2) were previously evaluated using fresh frozen tissue with simultaneous extraction of both DNA and RNA to enable integrative genomic analyses25 and sequencing was performed as described previously23.
Sanger Sequencing
Validation of the MTOR, TSC1, RHEB mutations was carried out using PCR followed by Sanger sequencing on an ABI 3700. 20ng of patient DNA from normal and tumor samples was amplified using primers designed to capture each of the mutations in LOT 3–6. Normal tissue from LOT 6 had not passed quality control and more tissue was not available for further analysis. Oligonucleotide primer sequences are available on request.
Molecular analysis
Exome-seq reads from FASTQ files were aligned to the human reference genome GRCh38 (hg38) using the Burrows-Wheeler Aligner algorithm (BWA-MEM; version 0.7.17) set to default parameters26. BAM files were generated using Picard Tools (version 2.20.5) to add read group information and sambamba was used to mark PCR duplicates. GATK toolkit (version 4.0.7)27–29 was used to perform base quality score recalibration and local realignment around indels. MuTect230, VarScan (v2.4.0)31, Shimmer (v0.2)32, SpeedSeq (v0.1.2)33, Manta (v1.6.0), and Strelka2 (v2.9.10)34 were used to call somatic variants and small-scale insertions and deletions (indels) for each tumor. A mutation that was repeatedly called by any two of these software tools was retained. Annovar was used to annotate germline/somatic mutations and indels35. A minimum of 7 total reads in the normal sample and at least 3 variant reads in the tumor sample with a variant allele frequency (VAF) ≥ 5% were required for somatic mutation calling. Intronic, untranslated region and intergenic mutations outside splice sites were filtered out. Missense mutations predicted to be benign by both PolyPhen-2 and SIFT (with < 5% chance of inducing functional changes at the protein level) were filtered out36. Somatic variant calls were inspected using Integrated Genomics Viewer v2.3.4 (IGV; Broad Institute, MIT Harvard, Cambridge, MA, USA). Variants were classified according to the American College of Medical Genetics and Genomics (ACMG) 2015 Guidelines. COSMIC and ClinVar were used as additional databases to annotate cancer relevance and clinical potential for detected variants.
We carried out somatic copy number variation (CNV) analyses on our exome-seq data using CNVkit (https://github.com/etal/cnvkit) with default parameters on tumor-normal pairs. CNVkit uses both on- and off-target sequencing reads to calculate log2 copy ratios across the genome for each sample and improves accuracy in copy number calling by applying a series of corrections37. Arm gain or loss was called when > 50% of the chromosome exhibited copy number gain or loss38.
RNA-seq reads were aligned to the human reference genome GRCh38 (hg38) using STAR39 with the parameters “--runThreadN 48 --outSAMtype BAM Unsorted --outReandsUnmapped Fastx.” The FeatureCounts40 software program with parameters “--primary -O -t exon -g transcript_id -s 0 -T 48 --largestOverlap --minOverlap 3 --ignoreDup -p -P -B –C” was used to measure gene expression levels. The human genome annotation file employed by FeatureCounts was downloaded from the UCSC table browser under the RefSeq Gene track. Genes were kept if they had at least 10 read counts in one or more of the 141 samples evaluated (2 LOT, 35 RO [including 4 RON-favor RO], 9 chRCC-eo [including 1 RON-favor chRCC-eo], 35 classic chRCC, and 60 normal kidney). These data were integrated with 89 TCGA cases (4 LOT, 15 chRCC-eo, 46 classic chRCC, and 24 normal kidney samples) for downstream analyses. Two tumor/normal RO pairs believed to have tumor/normal labels transposed were removed from the gene expression analyses: sample 13730N, which was labeled as a normal and had a non-coding mutation [chrM:564:G->A] previously reported to occur in RO41 and its corresponding tumor (13730T), which lacked the mutation); and sample XP174N, which had a frameshift insertion [chrM:11866:A->AC] on MT-ND4 also reported in RO41 and its corresponding tumor (XP174T), which lacked the mutation. The Bioconductor packages edgeR42 (version 3.28.1) and sva (version 3.34.0) were employed to perform normalization (using TMM43) and batch effect removal (using ComBat44). EdgeR was further utilized to: (1) identify differentially expressed genes between LOT and chRCC-eo types, and (2) perform KEGG analysis of differentially expressed genes to identify significantly enriched pathways with p-values ≤ 0.05.
Results
Study Cohort
From among 204 cases between 2005 – 2016 of chRCC, RON, and RO at our institution with adequate material, we identified 22 tumors corresponded to 8 nephrectomy/partial nephrectomy cases and 7 patients that fit the morphologic description of LOT. Overall, LOT represented 4% (8 of 204 cases) of reviewed cases and 0.4% of all nephrectomies for renal tumors at our institution during the study timeframe. All 22 tumors had been originally diagnosed as chRCC-eo.
Baseline Clinical Characteristics and Clinical Course
The patient demographics and tumor characteristics are summarized in Table 1. The patients ranged from 63 to 86 years at diagnosis (mean age: 75 years). There were more females than males (M:F ratio, 2:5). None of the patients had prior documented history of a syndrome including TSC and BHD. Suspicious skin lesions, pneumothoraxes, unexplained seizures, angiomyolipomas, or other characteristic syndromic features were not documented. Three patients, however, had multiple renal masses. One patient (LOT 3) had multiple bilateral renal tumors (6 on the right and 10 on the left) (Supplementary Figure 1A–B). The patient was found to have a likely pathogenic germline variant in the TSC1 gene. The patient received therapy with an mTORC1 inhibitor, everolimus, which led to remission of several lesions and stabilization of the rest. Another patient (LOT 5) had a history of prior nephrectomy for a benign renal tumor at an outside institution, and another (LOT 7) was found to have a concurrent contralateral 2nd renal mass. Neither LOT 5, nor LOT 7, were found to have a genetic predisposition. Amongst the solitary LOTs, there was predilection for the right kidney (right: 5; left: 1). Most patients underwent partial nephrectomy. At the time of this analysis, 3 patients were deceased due to unrelated causes including LOT 1, who expired from a subsequently diagnosed contralateral metastatic clear cell RCC. The 4 remaining patients were alive with no evidence of metastatic disease at the time of last follow-up. Follow-up ranged from 3 months to 53 months (median 29 months).
Table 1.
Clinicopathologic Characteristics of LOT Cases in the Cohort
| Patient ID | Age | Sex | Presentation | Family History | Other Significant Medical History | Original Diagnosis | Nephrectomy | Laterality | Tumor Size (cm) | Focality (number) | pT | pN | Stage | Metastasis | Follow-up (months) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LOT 1 | 79 | F | Back pain | None | Contralateral metastatic ccRCC | ChRCC-eo | Partial | Right | 7.8 | Unifocal | pT2a | pNx | II | No | Deceased (26) |
| LOT 2 | 86 | F | Incidental | None | None | ChRCC-eo | Radical | Right | 6.5 | Unifocal | pT1b | pNx | I | No | ALF (3) |
| LOT 3a | 63 | F | Anemia | Father with b/l Nx for unknown reason | B/l multiple LOTs | ChRCC-eo | Partial | Right | 4.0, 2.3, 1.5, 1.4, 0.8, 0.5 | Multifocal (6) | pT1a* | pNx | I | No | ALF (37) |
| LOT 3b | ChRCC-eo | Partial | Left | 5.3, 5.2, 4.9, 3.7, 2.1, 2.0, 1.9, 1.6, 1.0, 0.9 | Multifocal (10) | pT1b* | pNx | I | No | ||||||
| LOT 4 | 69 | M | Hematuria | None | None | ChRCC-eo | Partial | Right | 2.3 | Unifocal | pT1a | pNx | I | No | ALF (29) |
| LOT 5 | 71 | F | Incidental | None | Prior contralateral nephrectomy for benign tumor | ChRCC-eo | Partial | Right | 3.8 | Unifocal | pT1a | pNx | I | No | ALF (53) |
| LOT 6 | 75 | F | Incidental | None | None | ChRCC-eo | Partial | Left | 2.4 | Unifocal | pT1a | pNx | I | No | Deceased (36) |
| LOT 7 | 84 | M | Incidental | None | Contralateral renal mass followed with AS | ChRCC-eo | Radical | Right | 6.5 | Unifocal | pT1b | pNx | I | No | Deceased (29) |
ccRCC: clear cell renal cell carcinoma; B/l: bilateral; Nx: nephrectomy; AS: active surveillance; ChRCC-eo: chromophobe renal cell carcinoma, eosinophilic variant
: pT represents the highest stage
ALF: alive at last follow-up.
Imaging Features
Computed tomography (CT) was available in 4 patients (LOT 1–4). All masses exhibited areas with intense enhancement (i.e. similar to that of renal cortex) during the corticomedullary phase likely signifying pronounced angiogenesis (Supplementary Figure 1). LOT 3 had multiple bilateral renal masses with similar appearance (Supplementary Figure 1A–B). These tumors also demonstrated irregular areas of decreased central enhancement with some degree of progressive centripetal enhancement during the excretory phase (Supplementary Figure 1B). Renal imaging confirmed the absence of classic angiomyolipoma features (i.e. lacking macroscopic fat) in all cases evaluated.
Pathologic Features
Grossly, the tumors were noted to be well circumscribed, solid, tan to brown, and homogenous on cut surface. Tumors ranged from 0.5 to 7.8 cm in size (mean: 3.1 cm). Areas with loose myxoid stroma, similar to oncocytomas, were appreciated grossly. None of the tumors had renal sinus involvement (Table 1). AJCC pathologic T stage was pT1a in 4/8, pT1b in 3/8, and pT2a in 1/8.
A review of the H&E slides demonstrated striking morphologic similarities in all 22 tumors. All tumors were well demarcated, either devoid of or with a thin partial peripheral fibrous pseudocapsule (Figure 1A). At low power, tumors were homogenous and solid. They were composed of sheets of compact uniform small nests (Figure 1A–B). Abrupt areas of hypocellular, loose edematous stroma with scattered cords of tumor cells were present (Figure 1C). In some tumors, focal areas of hemorrhage and dilated vascular channels filled with serous or hemorrhagic fluid were also seen (Figure 1D). The tumor cells were uniform, polygonal in shape and had ill-defined cell borders (Figure 1E). They contained eosinophilic, finely granular, cytoplasm and central round to mildly oval nuclei with focal mild nuclear membrane irregularities and prominent small eosinophilic nucleoli (Figure 1E–F). Subtle perinuclear clearing, mild pleomorphism and binucleation were seen in all cases, more apparent focally in some cases. Focal scattered small clusters of lymphocytes and histocytes were also seen (generally near the loose stromal areas) (Figure 1F). Tumor necrosis, frequent or atypical mitosis, nuclear pleomorphism, cytoplasmic clearing, cytoplasmic stippling or inclusions were not identified. Typical features of classic chRCC (wrinkled, raisinoid, nuclear membrane; marked perinuclear clearing; well defined cell borders; clear cytoplasm) were not apparent. Peritumoral blood vessels were markedly thick-walled in the majority of cases (Supplementary Figure 2A). All 16 tumors in the patient with the germline TSC1 mutation (LOT 3) showed similar features with the addition of small tumorlets in the adjacent renal parenchyma (Supplementary Figure 2B). Consistent with the imaging findings, no angiomyolipomas were identified in adjacent kidney sections. A small distinct area (3.0 mm) with slightly different morphology was observed in one of the 16 tumors with large intracytoplasmic vacuoles similar to EVT (Supplementary Figure 2C–D). A small distinctive area (4.0 mm) was also seen in a LOT 4 tumor with more pronounced perinuclear clearing and nuclear membrane irregularities (Supplementary Figure 2E–F).
Figure 1. Histologic characteristics of LOT.
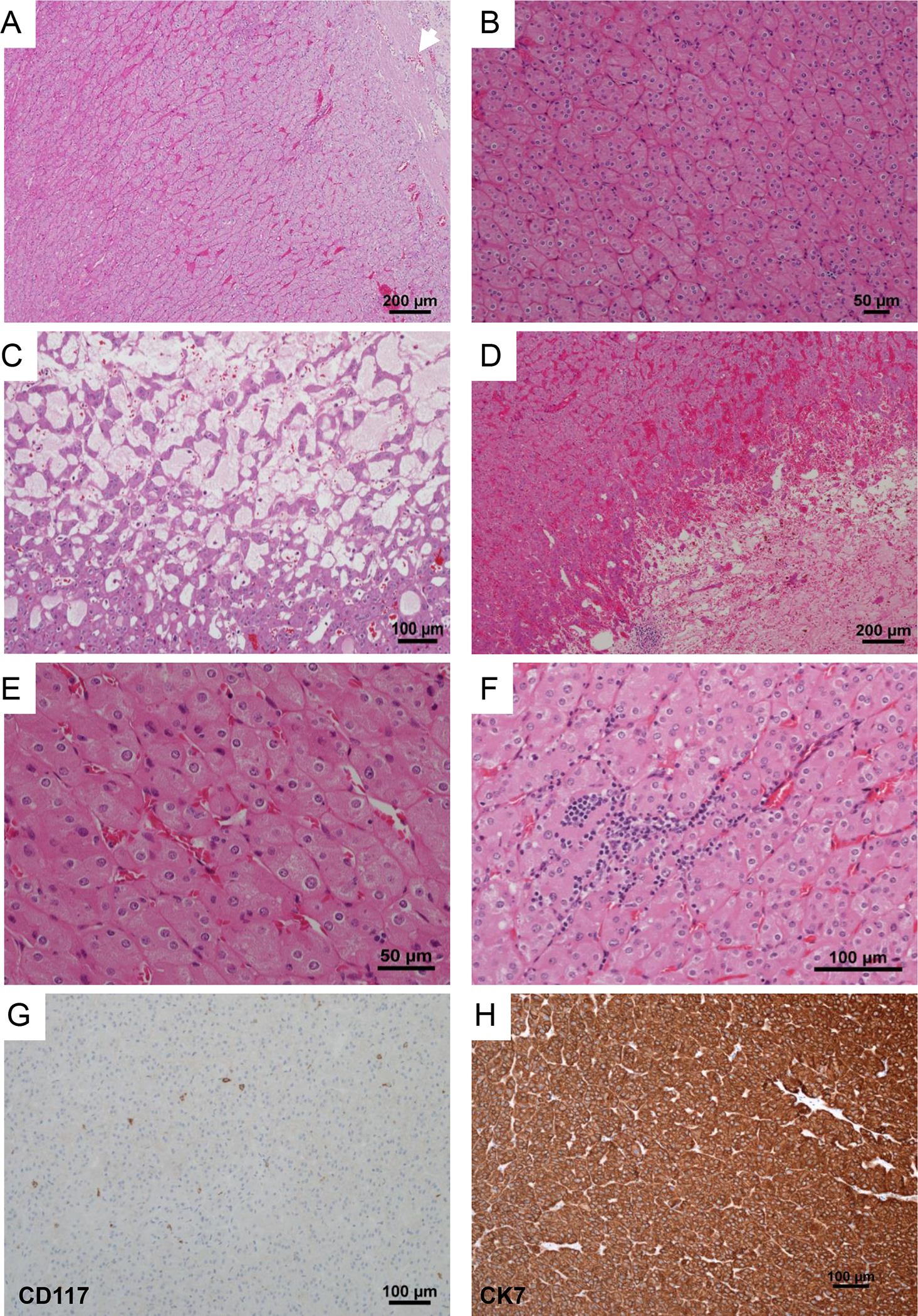
Representative H&E images from LOT reveal a well circumscribed partly encapsulated eosinophilic tumor (white arrow) (A) composed of homogenous cells arranged in sheets of tight nests (B) with focal edematous areas with loose cords of tumor cells (C), and occasional dilated vascular channels and hemorrhage (D). High-power images show that the tumors are uniformly composed of cells with abundant finely granular eosinophilic cytoplasm, indistinct cell borders, round nuclei with relatively smooth membranes, subtle perinuclear clearing and binucleation (E). Lymphoid clusters are frequent (F). Representative immunohistochemical profile of CD117 with only scattered positive mast cells (G), and cytokeratin 7, which is strong and diffusely positive (H).
Immunohistochemical Features
LOTs (including representative sections from multiple tumors in LOT 3) were interrogated with immunohistochemical (IHC) stains. All LOTs showed diffuse and strongly reactive cytokeratin 7 and were negative for CD117 (Table 2, Figure 1G–H). They were positive for PAX8, retained FH as well as SDHB, and were negative for TFE3 and TFEB. They were also negative for HMB45/Melan A, cytokeratin 20, cathepsin K, and carbonic anhydrase-IX. Whenever performed, E-cadherin was positive whereas CD10 and vimentin were negative.
Table 2.
Immunohistochemical Profile of LOT Cases in the Cohort
| Patient ID | CD117 | CK7 | P-S6 | P-4EBP1 | ||
|---|---|---|---|---|---|---|
| intensity | percentage | intensity | percentage | |||
|
| ||||||
| LOT 1 | negative | diffuse strong positive | moderate | 20 | weak-moderate | 30 |
|
| ||||||
| LOT 2 | negative | diffuse strong positive | weak-moderate | 5 | weak-moderate | 5 |
|
| ||||||
| LOT 3a* | negative | diffuse strong positive | moderate-strong | 90 | weak-moderate | 40 |
|
| ||||||
| LOT 3b* | negative | diffuse strong positive | strong | 90 | moderate | 60 |
|
| ||||||
| LOT 4 | negative | diffuse strong positive | strong | 75 | moderate | 50 |
|
| ||||||
| LOT 5 | negative | diffuse strong positive | moderate | 80 | moderate | 50 |
|
| ||||||
| LOT 6 | negative | diffuse strong positive | moderate | 30 | weak-moderate | 20 |
|
| ||||||
| LOT 7 | negative | diffuse strong positive | weak-moderate | 5 | weak | 5 |
: Representative section from a total of 5 tumors were evaluated by immunohistochemistry.
Molecular Features
Whole exome sequencing (WES) was available for 2 cases (LOT 1, 2) from our previously published study23. Among the new cases (LOT 3–7), DNA from the tumor region of LOT 7 and normal kidney of LOT 6 did not pass quality control. WES from 5 tumors with paired normal samples demonstrated mutations in mTORC1 pathway genes (MTOR, TSC1, or RHEB) in all cases (Table 3, Supplementary Figure 3). In addition, unpaired WES analyses from the LOT 6 tumor also showed an MTOR gene mutation (Table 3, Supplementary Figure 4A, 5). Variant calls were inspected using the Integrated Genomics Viewer (IGV; Supplementary Figure 4). Mutations were validated by Sanger sequencing on available tumor and normal samples (Supplementary Figure 5).
Table 3.
Molecular Alterations identified in LOT Cases in the Cohort
| Patient ID | Genomic ID | Chromosome | Start Position | End Position | Reference Sequence | Tumor Sequence | Matched Normal Sequence | Gene | Variant_Classification | Validation_Method* | Somatic_Mutation* | Amino Acid Alteration |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LOT 1 | XP238T | chr1 | 11114338 | 11114338 | A | T | A | MTOR | nonsynonymous SNV | Prior | NM_004958.3:c.7280 A>T | p.Leu2427Gln |
| LOT 2 | 17827T | chr1 | 11124516 | 11124516 | G | T | G | MTOR | nonsynonymous SNV | Prior | NM_004958.3:c.6644G>T | p.Ser2215Tyr |
| LOT 3a | KC02816-T1-FFPE | chr9 | 132902719 | 132902719 | A | AGCTT | A | TSC1 | frameshift substitution | Sangers | NM_000368.4:c.2273_2276dup | p.Arg760Serfs*2 |
| LOT 4 | KC02129-T1-FFPE | chr7 | 151490963 | 151490964 | TA | AG | TA | RHEB | nonsynonymous SNV | Sangers | NM_005614.3:c.103_104delinsCT | p.Tyr35Leu |
| LOT 5 | KC02942-T2-FFPE | chr1 | 11114380 | 11114381 | CT | AG | CT | MTOR | nonsynonymous SNV | Sangers | NM_004958.3:c.7237_7238delinsCT | p.Ser2413Leu |
| LOT 6 | KC00627-T1-FFPE | chr1 | 11157255 | 11157267 | ACTCGTGCAGTTT | A | not available | MTOR | nonframeshift substitution | Sangers | NM_004958.3:c.4354_4365del | p.Lys1452_Glu1455del |
| LOT 7 | Not pass quality check | not available | not available |
somatic mutation were confirmed by Sanger
The presence of mTORC1 pathway gene mutations across all of the samples (LOT 1, 2, 5, 6 [MTOR], LOT 3 [TSC1] and LOT 4 [RHEB]) (Table 3) suggested that mTORC1 may play a role in LOT development. To evaluate this notion further, we assessed the particular mutations. While RHEB and mTOR are positive regulators of mTORC1 and mutations would be expected to be activating, TSC1 is a negative regulator and mutations would be expected to inactivate it. A likely pathogenic germline TSC1 variant was detected in the blood (and normal kidney sample) from LOT 3. The mutation caused a substitution two nucleotides after exon 15 of the TSC1 gene (NM_000368.4:c.2208+2T>A), and was predicted to abolish the native splice donor site (Supplementary Figure 4B). In addition, a somatic frameshift TSC1 mutation (p.Arg760Serfs*2) was found in the tumor (Table 3, Supplementary Figure 4A). mTOR mutations were found in LOT 1 (mTOR p.L2427Q), LOT 2 (mTOR p.S2215Y), LOT 5 (mTOR p.S2413L) and LOT 6 (mTOR p.K1452_E1455del). Consistent with their being activating, 3 out of the 4 mTOR mutations affected a residue previously shown to be mutated in other tumors (https://www.cbioportal.org; https://cancer.sanger.ac.uk/cosmic)45,46. Furthermore, two mutations (p.L2427Q and p.S2215Y) were previously shown to activate mTORC1 in studies in vitro45,46,47. A mutation in RHEB in LOT 4 (RHEB p.Y35L) was both previously reported in other tumors (https://www.cbioportal.org) and shown to be activating in vitro45.
To evaluate the functional significance of the least characterized mTOR mutations (K1452-E1455 deletion and p.S2413L), we assessed their impact on the mTORC1 structure. We mapped the mutations onto the crystal structures of both the apo and RHEB-activated mTOR proteins (Figure 2A). Previously published cancer-associated mTOR mutations leading to kinase hyperactivation tend to occur at specific areas including the FAT hinge (i.e. A1459P) and the interface between the kinase C-lobe and FAT domain (i.e. E2419K) where they destabilize the apo state48,49,50. The K1452-E1455 deletion removes a single turn from a hinge helix in the FAT domain near the known hyperactivating A1459 mutation (Figure 2B left). The turn interacts with neighboring helices that adopt alternate conformations in the inactive and active states and the deletion would remove helical interactions from both states suggesting increased flexibility at the hinge that could lead to hyperactivation. The mTOR p.S2413L mutation resides in the kinase C-lobe at the FAT domain interface. This interface also adopts alternate conformations in the active and inactive states (Figure 2B right). Another known activating mutation (E2419K) is found in the same C-lobe helix at the FAT interface and is thought to lower the barrier to activation48,50. Overall, these data are consistent with the notion that the K1452-E1455 deletion and p.S2413L substitution are similarly activating as other previously reported mTOR mutations tested in vitro.
Figure 2. Schematic mapping of mTOR mutations in LOT samples.
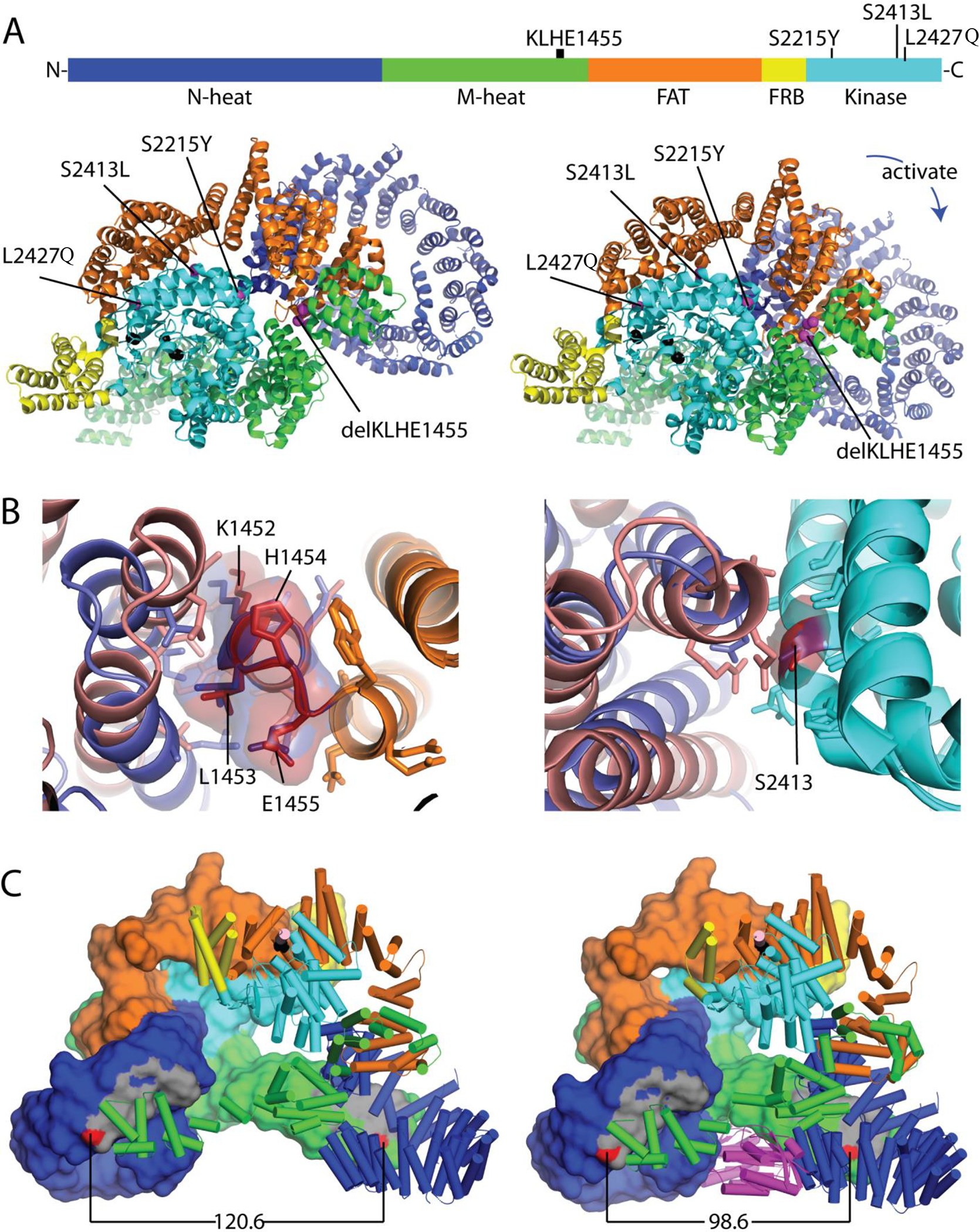
(A) Mutation positions in the mTOR protein primary structure as well as tertiary structures of apo mTORC1 (PDB: 6bcx, left) and RHEB-activated mTORC1 (PDB: 6bcu, right) with colored domains. (B) Structure context of less well characterized mTOR mutations. Left: Superimposed wild type mTOR FAT domain hinge region (orange) with area corresponding to K1452-E1455 (deleted in LOT 6) illustrating differential interactions with neighboring FAT helices in apo (slate) and RHEB activated (salmon) conformations. Residues within 4Å are in stick. Right: Superimposed kinase C-lobes (cyan) containing the S2413I mutation (LOT 5). The mutation is positioned at the FAT domain interface, which adopts alternate conformations in the active (salmon) and apo (slate) states. Residues within 4Å are in stick. (C) mTORC1 dimerization interface is incompatible with mixed dimers. Left: mTOR dimer (domains colored as above) from an apo mTORC1 structure (PDB: 6BCX) with one chain in surface rendering and the second chain in cartoon mode. The dimer interface (gray) is formed by interaction of N-heat (dark blue) and M-heat domains (green), with the distance between two residues in the dimer interface (N612 and R1161, red) indicated below. Right: RHEB (magenta)-activated mTOR dimer with closer interaction surfaces as shown by the distance between N612 and R1161 (red).
To evaluate the impact of these mutations on mTORC1 activity, we analyzed p-S6 (Ser240/244) and p-4E-BP1 (Thr37/46). All LOTs evaluated showed p-S6 expression in at least 5% tumor cells (Figure 3). Similar results were observed for p-4E-BP1 (Figure 3).
Figure 3. Evaluation of mTORC1 pathway activation in LOT.
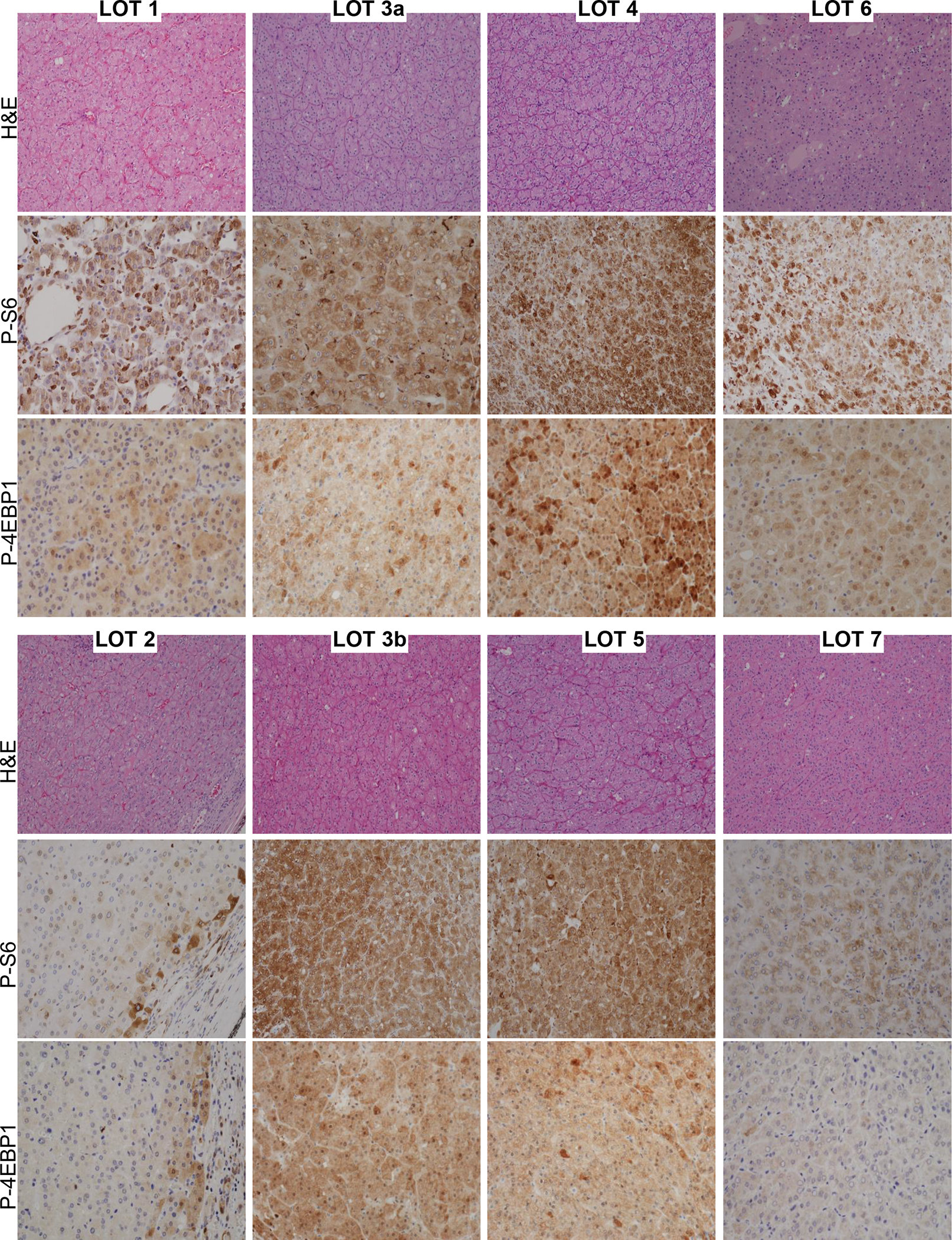
Representative H&E, p-S6, and p-4EBP1 immunohistochemical expression in LOTs. The percentage of tumor cells expressing cytoplasmic phospho-S6 and the intensity of expression is tabulated in the Table 2.
However, we observed a range of mTORC1 activation. Higher activation was observed in LOT 3, 4, and 5, but LOT 1, 2, 6 and 7 had lower levels (Figure 3). These data suggested a correlation between MTOR mutation (LOT 1, 2, 5, 6) and lower mTORC1 activity (LOT 1, 2, 6 and 7). Specifically, 3/4 cases with MTOR mutations had lower levels of mTORC1 activation (LOT 1, 2, 6). One potential explanation for the moderate p-S6 and p-4E-BP1 activation levels in these cases may be the presence of a wild-type MTOR allele and protein, in particular since mTORC1 dimerizes. NGS and Sanger studies were consistent with the presence of a wild-type MTOR allele suggesting that mutations were heterozygous. Furthermore, copy number analyses available for 3 out of the 4 cases (a normal sample is missing for LOT 6) showed two copies of chromosome 1, where MTOR is located (1p36.22) (Supplementary Figure 6). mTOR forms a dimer with two interfaces between N-heat and M-heat domains. In the apo structure, the N-heat domain is positioned in a relatively open state with respect to the rest of the protein (Figure 2C left). Upon RHEB binding, the N-heat domain rotates toward the intersection of the M-heat and FAT domains, and the kinase catalytic cleft closes to align active site residues for catalysis48,51. This rotation approximates the dimer interaction surfaces (Figure 2C right). The changes induced by RHEB binding are not compatible with a mixed dimer of activated and inactivated mTOR48. Furthermore, RHEB-induced kinase activation is cooperative in the wild-type complex48. In the absence of RHEB, mTOR could adopt a heterogenous mixture of activation states depending upon whether it is constituted by wild-type dimers, heterodimers (of wild-type and mutant) or mutant dimers. Thus, the presence of a wild-type subunit could potentially reduce the impact of hyperactivating mTOR mutations. The overall mTORC1 activation state as compared, for instance, to tumors in which mTORC1 activation is driven by a single mutant protein (with loss of the wild-type allele) or by activation upstream (such as through RHEB or TSC1/TSC2 mutations) may be reduced.
To extend the findings in our cohort, we reviewed all tumors categorized as chRCC-eo in the TCGA (KICH) project52. We used the NIH GDC Data Portal to review digital slides (www.portal.gdc.cancer.gov). While IHCs are unavailable (CK7+/CD117− status could not be confirmed), 4 out of 19 tumors morphologically resembled LOT (TCGA-KN-8437, TCGA-KM-8439, TCGA-KM-8441, TCGA-KM-8639). We assessed the mutation status of mTORC1 pathway genes in these 4 tumors. Interestingly, 2 of these tumors had pathogenic mTOR mutations (TCGA-KN-8437; p.L2427R and TCGA-KM-8441; p.I2017T) (Supplementary Figure 7A–B). TCGA-KN-8437 had a mutation in the same residue as LOT 1 in our cohort and the particular substitution, L2427R, has been shown to be a gain of function mutation in the literature18. Similarly, the mTOR mutation in TCGA-KM-8441 (p.I2017T) has been previously reported in tumors and shown to induce kinase activity in vitro53. Both tumors were reported to be diploid and without chromosome 1 loss. A third TCGA tumor had a pathogenic frameshift TSC1 mutation (TCGA-KM-8639; p.R718Pf*3). No mutations were found in mTORC1 pathway genes in the fourth tumor.
Consistently, all LOTs cases in our cohort were diploid with all 22 autosome pairs (Figure 4). None of the cases showed iconic chromosomal alterations of other RCCs, such as multiple chromosomal losses, which typify chromophobe RCC, 3p loss in clear cell RCC, or gain of chromosome 7 and 17 as seen in papillary RCC.
Figure 4. Copy number analysis in LOT.
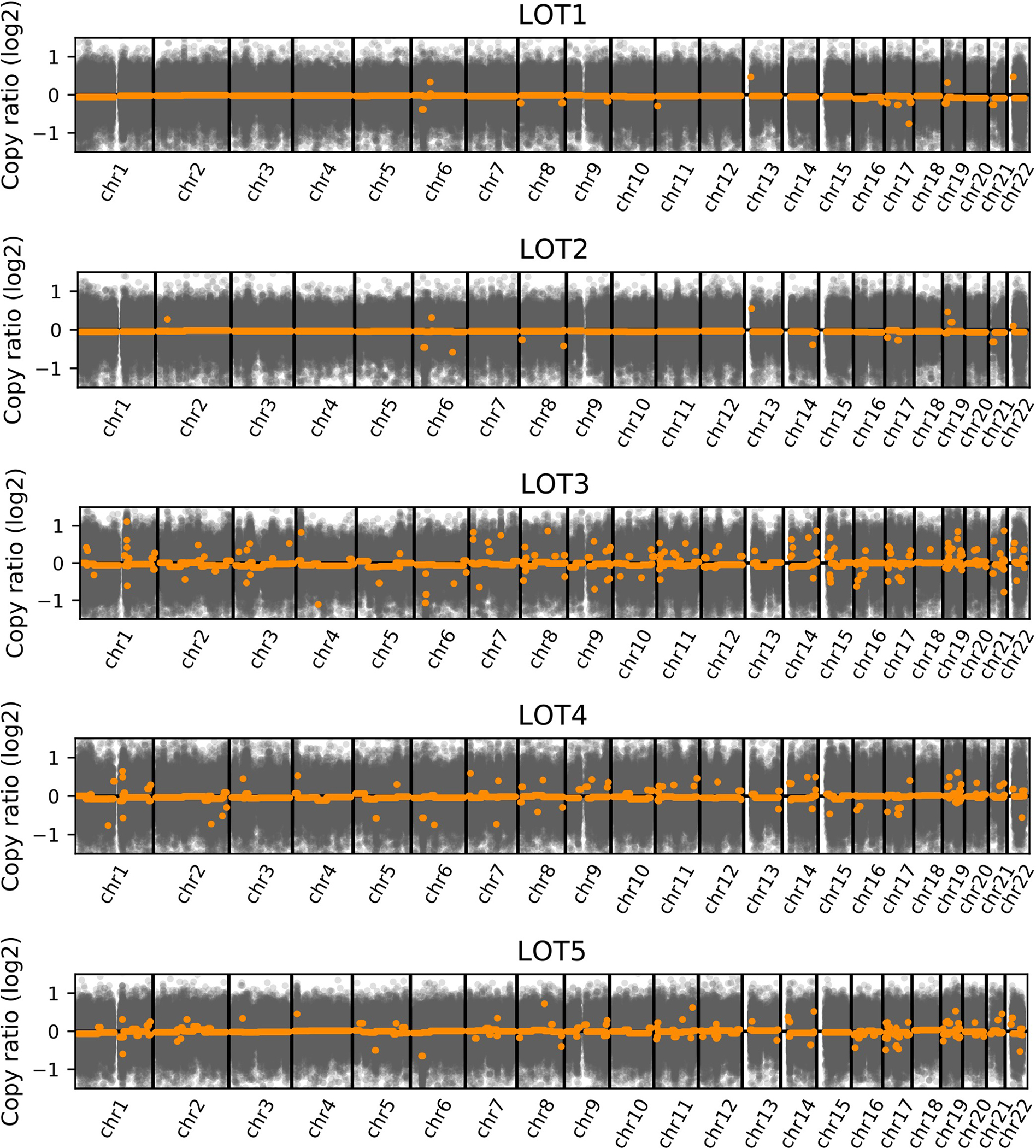
Whole-genome profiles of log2 copy ratio by CNVkit on WES in LOTs. Average log2 copy ratio estimations are marked in orange.
We next asked how LOTs relate to other RCCs and performed gene expression analyses. Whole transcriptome data was available for 6 cases (LOT 1–2 from UTSW23 and the 4 TCGA LOTs52). We compared the gene expression profiles with those from chRCC-eo (9 from UTSW including 1 RON-favor chRCC-eo23 and 15 from TCGA52), classic chRCC (35 from UTSW23 and 46 from TCGA52) and RO (33 from UTSW including 4 RON-favor RO23). In addition, normal kidney samples (60 from UTSW23 and 24 from TCGA52) were included. We generated a t-distributed stochastic neighbor embedding (t-SNE) plot, which showed tight clustering of the LOT cases, away from RO, chRCC-eo, classic chRCC and normal kidney (Figure 5A). Intriguingly, LOT clustered away even from chRCC with mTORC1 pathway gene mutations (Figure 5A).
Figure 5. RNA-seq-based clustering of LOT, RO and chRCC.
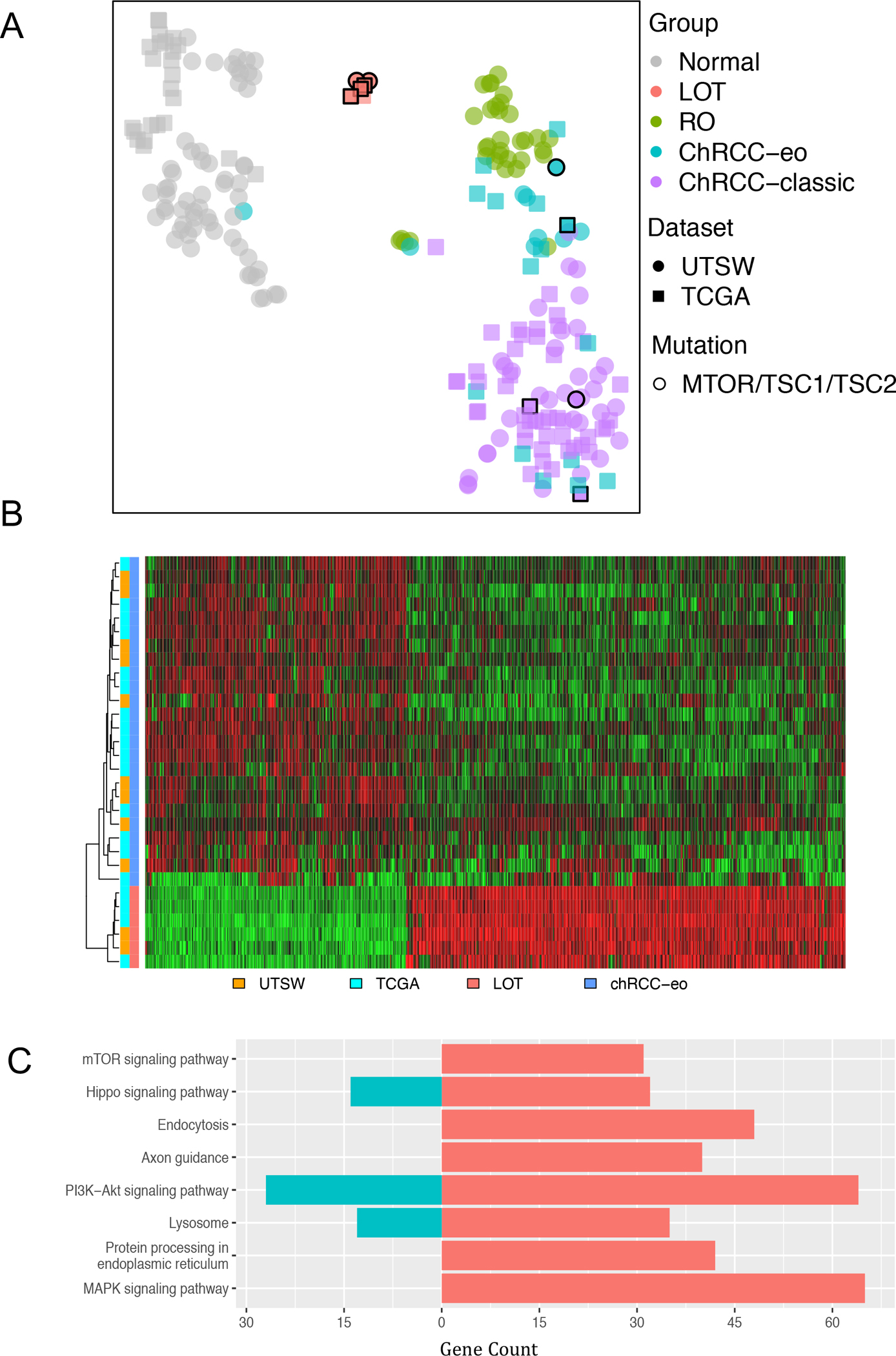
(A) t-distributed stochastic neighbor embedding (t-SNE) plot of filtered whole transcriptomic data from pooled UTSW and TCGA cohorts showing clustering of LOT samples away from classic chRCC, chRCC-eo, and RO, including chRCC with mTORC1 pathway gene mutations (highlighted with black borders). (B) Heatmap of 1674 most differentially expressed genes distinguishing LOT from chRCC-eo. (C) Select enriched pathways between LOT and chRCC-eo by KEGG analysis of DEGs with over-representation p-value ≤0.05. Up (red) and down (cyan) regulated gene counts are shown.
We then focused on chRCC-eo and LOT due to their morphologic similarity. We found 1674 differentially expressed genes (DEGs) between LOT and chRCC-eo tumor samples at a false discovery rate (FDR) ≤ 0.05 with an absolute log2 fold-change (LogFC) ≥1.5. A heatmap of these DEGs highlights the differences between LOT and chRCC-eo (Figure 5B). KEGG analysis of the DEGs between LOT and chRCC-eo samples identified 175 differentially enriched pathways (Figure 5C). Of particular interest, mTOR signaling, protein processing in endoplasmic reticulum, and MAPK signaling pathways were enriched in the LOT respective to chRCC-eo. Overall, these findings suggest that LOTs have a unique gene expression signature that sets them apart from other tumors.
Discussion
We report a comprehensive molecular profiling of a recently described renal tumor entity, low-grade oncocytic tumor (LOT) expanding our understanding of this novel disease19,21. Our results show that LOT can present in both syndromic and non-syndromic cases with uniform morphology and IHC features. LOT tumors have converging mutations in mTOR pathway genes and mTORC1 activation. Gene expression analyses show that LOT has a unique gene expression signature that differs from close morphologic mimics, chRCC-eo and RO.
Our results support and expand the morphologic and IHC features previously reported. In 2019, Trpkov et al19 reported 28 distinct LOTs from four institutional archives. All tumors had consistent morphology characterized by tight nests of bland tumor cells with eosinophilic cytoplasm and were devoid of prominent nuclear membrane irregularities. Informatively, LOTs had a unique immunoprofile with CD117 negativity and diffuse CK7 reactivity. These findings were consistently observed in 7 additional cases reported by Guo et al21 and in the 22 tumors in our series.
These initial studies reported LOTs to be single, and devoid of a syndromic association. One patient in our series had multiple bilateral LOTs and had a likely pathogenic germline TSC1 mutation. TSC syndrome can have variable penetrance and a significant percent of cases have de novo mutations with no known family history. Individuals may have a mild phenotype, particularly when the TSC1 gene is implicated54. However, it is quite likely that our LOT 3 patient had TSC as the same TSC1 germline mutation has been reported in a 3-generation family with mild clinical TSC features55. All tumors seen in this patient were indistinguishable morphologically and by IHC from other LOTs in our series. A recent study similarly reported multifocal LOTs in TSC and in end stage renal disease (ESRD)22. None of our patients had ESRD. Similarly, previous studies of kidneys in TSC patients include cases that morphologically resemble LOT histologically and by IHC (CD117−/CK7+)4,5. These findings support that LOT can occur in both the sporadic and syndromic context. Although metastases have not been reported to date in LOT, as illustrated by the benefits seen in our LOT 3 patient, rapalogs may be considered for the management of multiple syndromic LOTs. This is further supported by the observation that at least in some contexts, mTOR pathway mutations are associated with rapalog responsivenes56,57.
The classification of renal tumors is largely based on morphologic, and to a lesser extent, molecular features. LOT exhibits distinct but overlapping histologic features with chRCC-eo and RO. Using exome and transcriptome sequencing Joshi et al, identified two main subtypes of RO. Type 1 tumors were diploid and exhibited CCND1 rearrangement. In contrast, Type 2 tumors were hypodiploid with loss of chromosome 1, X/Y, and/or 14 and 2141. Type 2 tumors could progress to chRCC-eo41. Our prior work shows that most chRCC-eo are similar to RO, do not have classic chromosomal abnormalities of chRCC, and are often diploid23. In the TCGA dataset, 10/19 chRCC-eo showed the characteristic chRCC copy number pattern with loss of one copy of entire chromosomes for most chromosomes including 1, 2, 6, 10, 13, 17. In a Swiss cohort, chromosomal losses were observed in 27% of chRCC-eo58. Our data and those from others59 suggest that LOT, like RO and chRCC-eo, are largely diploid and lack the recurrent chromosomal abnormalities of classic chRCC. It is unclear, however, whether LOT is part of the spectrum of chRCC-eo and oncocytoma (RO). It is tempting to speculate a stepwise progression from RO (at least a subset) to chRCC-eo to classic chRCC especially given the link to chromosome 1 loss, which may be a driver event.
Our gene expression analyses show that LOTs have a unique expression profile that distinguishes these tumors from RO and chRCC (both classic and eosinophilic variant). These changes in gene expression likely contribute to differential protein expression (CK7, CD117). Other markers include FOXI1, which is expressed in chRCC-eo and not in LOT60. Both CD117 and FOXI1 are expressed in renal intercalated cells of distal tubules, suggesting perhaps that these cells give raise to chRCC-eo but not LOT. In fact, differences in the expression of FOXI1 mRNA in TCGA-pan RCC data allowed Tong K et al.60 to identify 5 outlier cases in TCGA (4 of which are the same that we found to resemble LOT morphologically and one additional TCGA-MH-A857 from KIRP) which they reclassified as “eosinophilic chromophobe-like renal tumors”. Similarly, Skala SL et al.61 found 2 out of their 10 institutional chRCC-eo to have low or absent FOXI1 expression. The morphology of these 2 tumors matches LOT and they were reported to have MTOR mutations, were diploid, and lacked chromosome 1 loss (Supplementary Figure 7A–B).
Our study illustrates that LOT have converging mutations in genes involved in the mTORC1 pathway (MTOR, TSC1 and RHEB). Interestingly, using target gene sequencing, Tjota et al. recently described a spectrum of renal tumors with pathogenic mutations in TSC1/TSC2/MTOR.59,62 A subset of these tumors (group 2) were CK7+ and had absent to weak (1+) CD117 expression and could represent LOT. Though it may be tempting to classify LOT as “TSC/mTOR-associated renal neoplasms”62, these represent a heterogenous group of tumors based on morphology and IHC. Somatic alterations in mTOR pathway genes have been described in several RCC23,52,61–65. We first reported the identification of mTORC1 pathway gene mutations in ccRCC in 2011, when we found mutations in TSC166. Subsequently, mTORC1 pathway gene mutations were reported in chRCC by us and others23,52,59,61. mTORC1 pathway mutations have also been reported in eosinophilic tumors including EVT and ESC RCC2. Thus, mTORC1 pathway mutations alone are not a sufficiently distinguishing feature.
In our cohort, all cases exhibited activation of mTORC1. mTOR is found in two complexes, mTORC1 and mTORC2. Our data suggest that mTORC1 may, in particular, be affected. This idea is supported by the finding of mTORC1 activation (of which p-S6 and p-4EBP1 are specific markers), as well as by mutations in TSC1 and RHEB, that specifically regulate mTORC1. The TSC complex is a heterotrimer that includes TSC1 and TSC2 as well as TBC1D7 and acts as a GTPase activating protein (GAP) for the small GTPase RHEB67. TSC complex inactivation leads to constitutive activation of RHEB. In its GTP-bound form, RHEB binds and activates mTORC1. mTORC1 plays a central role in coordinating cell growth and metabolism by taking cues from its environment such as availability of nutrients and growth factors67. For a cell to divide, it needs to promote anabolism and increase its mass. mTORC1 is a downstream mediator of several growth factors and mitogen-dependent signaling pathways that converge in the regulation of the TSC complex.
In our LOT cases, the levels of mTORC1 activation varied. Interestingly, we noted a correlation between LOT cases with mTOR mutations and lower mTORC1 activation. A potential explanation for this finding is that MTOR mutations were heterozygous and the wild-type allele appeared to be preserved. The significance of a preserved wild-type protein may be heightened by the fact that mTORC1 complexes are homodimers. This consideration is particularly interesting by comparison to EVT14,17 where MTOR activating mutations are typically accompanied by loss of chromosome 1 (and the 2nd copy of MTOR). This may contribute to explain the higher grade appearance of EVT compared to LOT. One possibility is that LOT may evolve to EVT upon loss of chromosome 1 (and the remaining MTOR wild-type allele). Thus, mTORC1 may serve as rheostat in LOT, EVT, as well as other tumors with driver mTORC1 pathway alterations and morphologic overlap such as ESC-RCC. Provocatively, and as shown in Supplementary Figure 2C–F, LOT 3 and LOT 4 showed focal distinct area that morphologically resembled EVT/chRCC-eo with stronger p-S6 expression (data not shown). Furthermore, mTORC1 pathway mutations appear to be associated with worse clinical outcomes in chRCC68. While this makes sense conceptually, most tumors described to date (and our own observations) do not show morphologic subclones suggestive of progression. As alluded to above, it is also possible that they involve different cells of origin. Thus, the extent to which these tumors are related remains to be fully determined.
In summary, our study provides a comprehensive molecular, and histopathologic analysis of low grade oncocytic tumors, a recently described renal cell neoplasm. We show that they exhibit a unique gene expression signature that distinguishes them from morphologically related tumors, as well as consistent mTORC1 pathway mutations and broaden the clinical presentation to include syndromic patients. Our data supports LOT as a unique and distinct entity of indolent clinical behavior.
Supplementary Material
Supplementary Figure 1. Imaging characteristics of LOT. Axial contrast-enhanced computed tomography (CT) images of the abdomen (LOT 3) obtained during the corticomedullary phase (A) and excretory phase (B). Multiple heterogeneous intensely enhancing masses during the corticomedullary phase (red arrows) are present. Note large irregular areas of decreased central enhancement during the corticomedullary phase (yellow arrows) which exhibit slow progressive centripetal enhancement during the excretory phase.
Supplementary Figure 2. Microscopic features of LOT. (A) Representative H&E images illustrating frequently observed thick walled peritumoral vessels. (B) Small tumorlets in non-neoplastic renal parenchyma in LOT 3 patient. Small subclone (red asterisk) with diverse morphology in one of 16 tumors in LOT 3 (C-D) and LOT 4 (E-F). Subclones are characterized by large intracytoplasmic vacuoles in LOT 3 with germline TSC1 mutation (D), and more pronounced perinuclear clearing and nuclear membrane irregularities in LOT 4 (F).
Supplementary Figure 3. Somatic mutations from whole exome sequencing (WES) analysis. Oncoplot showing number and types of selected somatic alterations in LOT samples. Frequent non-overlapping mutations were observed in mTOR pathway genes. LOT 6 had MTOR gene mutation that is not represented in the oncoplot as WES data from the corresponding normal kidney was not available.
Supplementary Figure 4. Schematic of LOT mutations by next generation sequencing. Sequencing reads with somatic (A) and germline mutations (B) as visualized with Integrated Genome Viewer.
Supplementary Figure 5. Sanger sequencing confirming the mTOR pathway gene mutations. Representative chromatograms of forward and reverse Sanger sequencing from tumor and normal tissue samples.
Supplementary Figure 6. Copy number analysis of chromosome 1 in LOT. Log2 copy ratio of chromosome 1 and MTOR neighborhood on WES in LOTs with MTOR mutations. Chromosome copy number in LOT 1, LOT 2 and LOT 5 tumor samples was called by using CNVkit (Version 0.9.5). Figure shows that the three cases consistently have average log2 ratio 0 for chromosome 1, suggesting the presence of two copies of chromosome 1. Mean b-allele frequencies approximate the expected 0.5 value. Lack of divergence from 0.5 indicates no copy neutral loss of heterozygosity. Zoom-in plots of MTOR gene neighborhood are also displayed.
Supplementary Figure 7. Somatic mutations and schematic mapping of the mTOR mutations in LOT. (A) Table with somatic mutations in the mTOR protein in 4 LOT from UTSW and 4 tumors (2 from TCGA and 2 from Skala et al61) that were morphologically consistent with LOT. (B) Structure context of mTOR mutations as shown in Figure 2A including those from TCGA and Skala et al61.
Acknowledgments:
We acknowledge the patients whose samples provided the foundation for this study and are grateful to the Kidney Cancer Program and the Clinical Data Warehouse teams for their support and assistance.
Funding Sources:
This work was supported by the NIH sponsored Kidney Cancer SPORE grant (P50CA196516) and endowment from Jan and Bob Pickens Distinguished Professorship in Medical Science and Brock Fund for Medical Science Chair in Pathology.
Footnotes
Conflicts of interest disclosures: The authors declare that they have no conflict of interest.
References
- 1.Trpkov K et al. New developments in existing WHO entities and evolving molecular concepts: The Genitourinary Pathology Society (GUPS) update on renal neoplasia. Mod Pathol 10.1038/s41379-021-00779-w (2021). [DOI] [PubMed] [Google Scholar]
- 2.Trpkov K et al. Novel, emerging and provisional renal entities: The Genitourinary Pathology Society (GUPS) update on renal neoplasia. Mod Pathol 34, 1167–1184 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Williamson SR et al. Diagnostic criteria for oncocytic renal neoplasms: a survey of urologic pathologists. Hum Pathol 63, 149–156 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Guo J et al. Tuberous sclerosis-associated renal cell carcinoma: a clinicopathologic study of 57 separate carcinomas in 18 patients. Am J Surg Pathol 38, 1457–1467 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Yang P et al. Renal cell carcinoma in tuberous sclerosis complex. Am J Surg Pathol 38, 895–909 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pavlovich CP et al. Renal tumors in the Birt-Hogg-Dube syndrome. Am J Surg Pathol 26, 1542–1552 (2002). [DOI] [PubMed] [Google Scholar]
- 7.McKenney JK, Przybycin CG, Trpkov K & Magi-Galluzzi C Eosinophilic solid and cystic renal cell carcinomas have metastatic potential. Histopathology 72, 1066–1067 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Trpkov K et al. Eosinophilic Solid and Cystic Renal Cell Carcinoma (ESC RCC): Further Morphologic and Molecular Characterization of ESC RCC as a Distinct Entity. Am J Surg Pathol 41, 1299–1308 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Trpkov K et al. Eosinophilic, Solid, and Cystic Renal Cell Carcinoma: Clinicopathologic Study of 16 Unique, Sporadic Neoplasms Occurring in Women. Am J Surg Pathol 40, 60–71 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Argani P MiT family translocation renal cell carcinoma. Semin Diagn Pathol 32, 103–113 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Srigley JR et al. The International Society of Urological Pathology (ISUP) Vancouver Classification of Renal Neoplasia. Am J Surg Pathol 37, 1469–1489 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Gill AJ et al. Succinate dehydrogenase (SDH)-deficient renal carcinoma: a morphologically distinct entity: a clinicopathologic series of 36 tumors from 27 patients. Am J Surg Pathol 38, 1588–1602 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith SC et al. A distinctive, low-grade oncocytic fumarate hydratase-deficient renal cell carcinoma, morphologically reminiscent of succinate dehydrogenase-deficient renal cell carcinoma. Histopathology 71, 42–52 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Trpkov K et al. High-grade oncocytic tumour (HOT) of kidney in a patient with tuberous sclerosis complex. Histopathology 75, 440–442 (2019). [DOI] [PubMed] [Google Scholar]
- 15.He H et al. “High-grade oncocytic renal tumor”: morphologic, immunohistochemical, and molecular genetic study of 14 cases. Virchows Arch 473, 725–738 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Chen YB et al. Somatic Mutations of TSC2 or MTOR Characterize a Morphologically Distinct Subset of Sporadic Renal Cell Carcinoma With Eosinophilic and Vacuolated Cytoplasm. Am J Surg Pathol 43, 121–131 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kapur P et al. Eosinophilic Vacuolated Tumor of the Kidney: A Review of Evolving Concepts in This Novel Subtype With Additional Insights From a Case With MTOR Mutation and Concomitant Chromosome 1 Loss. Adv Anat Pathol 10.1097/PAP.0000000000000299 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen YB et al. Molecular analysis of aggressive renal cell carcinoma with unclassified histology reveals distinct subsets. Nat Commun 7, 13131 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trpkov K et al. Low-grade oncocytic tumour of kidney (CD117-negative, cytokeratin 7-positive): a distinct entity? Histopathology 75, 174–184 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Siadat F & Trpkov K ESC, ALK, HOT and LOT: Three Letter Acronyms of Emerging Renal Entities Knocking on the Door of the WHO Classification. Cancers (Basel) 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo Q et al. Characterization of a distinct low-grade oncocytic renal tumor (CD117-negative and cytokeratin 7-positive) based on a tertiary oncology center experience: the new evidence from China. Virchows Arch 10.1007/s00428-020-02927-0 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Kravtsov O et al. Low-Grade Oncocytic Tumor of Kidney (CK7-Positive, CD117-Negative): Incidence in a Single Institutional Experience with Clinicopathological and Molecular Characteristics. Hum Pathol 10.1016/j.humpath.2021.04.013 (2021). [DOI] [PubMed] [Google Scholar]
- 23.Durinck S et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat Genet 47, 13–21 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trpkov K & Hes O New and emerging renal entities: a perspective post-WHO 2016 classification. Histopathology 74, 31–59 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Pena-Llopis S & Brugarolas J Simultaneous isolation of high-quality DNA, RNA, miRNA and proteins from tissues for genomic applications. Nat Protoc 8, 2240–2255 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DePristo MA et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43, 491–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKenna A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van der Auwera GA et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43, 11 10 11–33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cibulskis K et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31, 213–219 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koboldt DC et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22, 568–576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hansen NF, Gartner JJ, Mei L, Samuels Y & Mullikin JC Shimmer: detection of genetic alterations in tumors using next-generation sequence data. Bioinformatics 29, 1498–1503 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chiang C et al. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat Methods 12, 966–968 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saunders CT et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Wang K, Li M & Hakonarson H ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang T et al. Probability of phenotypically detectable protein damage by ENU-induced mutations in the Mutagenetix database. Nat Commun 9, 441 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Talevich E, Shain AH, Botton T & Bastian BC CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput Biol 12, e1004873 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turajlic S et al. Deterministic Evolutionary Trajectories Influence Primary Tumor Growth: TRACERx Renal. Cell 173, 595–610 e511 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Joshi S et al. The Genomic Landscape of Renal Oncocytoma Identifies a Metabolic Barrier to Tumorigenesis. Cell Rep 13, 1895–1908 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robinson MD & Oshlack A A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11, R25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson WE, Li C & Rabinovic A Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Zhao S et al. A brain somatic RHEB doublet mutation causes focal cortical dysplasia type II. Exp Mol Med 51, 84 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu J et al. Mechanistically distinct cancer-associated mTOR activation clusters predict sensitivity to rapamycin. J Clin Invest 126, 3526–3540 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lim JS et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med 21, 395–400 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Yang H et al. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 552, 368–373 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wagle N et al. Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer Discov 4, 546–553 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ghosh AP et al. Point mutations of the mTOR-RHEB pathway in renal cell carcinoma. Oncotarget 6, 17895–17910 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang H et al. mTOR kinase structure, mechanism and regulation. Nature 497, 217–223 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davis CF et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell 26, 319–330 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ohne Y et al. Isolation of hyperactive mutants of mammalian target of rapamycin. J Biol Chem 283, 31861–31870 (2008). [DOI] [PubMed] [Google Scholar]
- 54.Au KS et al. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med 9, 88–100 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Streff H et al. TSC1 Variant Associated With Mild or Absent Clinical Features of Tuberous Sclerosis Complex in a Three-Generation Family. Pediatr Neurol 110, 89–91 (2020). [DOI] [PubMed] [Google Scholar]
- 56.Wolff N et al. Sirolimus and temsirolimus for epithelioid angiomyolipoma. J Clin Oncol 28, e65–68 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Maroto P et al. Biallelic TSC2 Mutations in a Patient With Chromophobe Renal Cell Carcinoma Showing Extraordinary Response to Temsirolimus. J Natl Compr Canc Netw 16, 352–358 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Ohashi R et al. Classic Chromophobe Renal Cell Carcinoma Incur a Larger Number of Chromosomal Losses Than Seen in the Eosinophilic Subtype. Cancers (Basel) 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tjota MY, Wanjari P, Segal J & Antic T TSC/MTOR mutated eosinophilic renal tumors are a distinct entity that are CK7+/CK20-/vimentin-: a validation study. Hum Pathol 10.1016/j.humpath.2020.12.006 (2020). [DOI] [PubMed] [Google Scholar]
- 60.Tong K & Hu Z FOXI1 expression in chromophobe renal cell carcinoma and renal oncocytoma: a study of The Cancer Genome Atlas transcriptome-based outlier mining and immunohistochemistry. Virchows Arch 10.1007/s00428-020-02900-x (2020). [DOI] [PubMed] [Google Scholar]
- 61.Skala SL et al. Next-generation RNA Sequencing-based Biomarker Characterization of Chromophobe Renal Cell Carcinoma and Related Oncocytic Neoplasms. Eur Urol 78, 63–74 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tjota M et al. Eosinophilic Renal Cell Tumors With a TSC and MTOR Gene Mutations Are Morphologically and Immunohistochemically Heterogenous: Clinicopathologic and Molecular Study. Am J Surg Pathol 44, 943–954 (2020). [DOI] [PubMed] [Google Scholar]
- 63.Ricketts CJ et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep 23, 313–326 e315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cancer Genome Atlas Research, N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499, 43–49 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Linehan WM & Ricketts CJ The Cancer Genome Atlas of renal cell carcinoma: findings and clinical implications. Nat Rev Urol 16, 539–552 (2019). [DOI] [PubMed] [Google Scholar]
- 66.Kucejova B et al. Interplay between pVHL and mTORC1 pathways in clear-cell renal cell carcinoma. Mol Cancer Res 9, 1255–1265 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saxton RA & Sabatini DM mTOR Signaling in Growth, Metabolism, and Disease. Cell 168, 960–976 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roldan-Romero JM et al. Molecular characterization of chromophobe renal cell carcinoma reveals mTOR pathway alterations in patients with poor outcome. Mod Pathol 33, 2580–2590 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Imaging characteristics of LOT. Axial contrast-enhanced computed tomography (CT) images of the abdomen (LOT 3) obtained during the corticomedullary phase (A) and excretory phase (B). Multiple heterogeneous intensely enhancing masses during the corticomedullary phase (red arrows) are present. Note large irregular areas of decreased central enhancement during the corticomedullary phase (yellow arrows) which exhibit slow progressive centripetal enhancement during the excretory phase.
Supplementary Figure 2. Microscopic features of LOT. (A) Representative H&E images illustrating frequently observed thick walled peritumoral vessels. (B) Small tumorlets in non-neoplastic renal parenchyma in LOT 3 patient. Small subclone (red asterisk) with diverse morphology in one of 16 tumors in LOT 3 (C-D) and LOT 4 (E-F). Subclones are characterized by large intracytoplasmic vacuoles in LOT 3 with germline TSC1 mutation (D), and more pronounced perinuclear clearing and nuclear membrane irregularities in LOT 4 (F).
Supplementary Figure 3. Somatic mutations from whole exome sequencing (WES) analysis. Oncoplot showing number and types of selected somatic alterations in LOT samples. Frequent non-overlapping mutations were observed in mTOR pathway genes. LOT 6 had MTOR gene mutation that is not represented in the oncoplot as WES data from the corresponding normal kidney was not available.
Supplementary Figure 4. Schematic of LOT mutations by next generation sequencing. Sequencing reads with somatic (A) and germline mutations (B) as visualized with Integrated Genome Viewer.
Supplementary Figure 5. Sanger sequencing confirming the mTOR pathway gene mutations. Representative chromatograms of forward and reverse Sanger sequencing from tumor and normal tissue samples.
Supplementary Figure 6. Copy number analysis of chromosome 1 in LOT. Log2 copy ratio of chromosome 1 and MTOR neighborhood on WES in LOTs with MTOR mutations. Chromosome copy number in LOT 1, LOT 2 and LOT 5 tumor samples was called by using CNVkit (Version 0.9.5). Figure shows that the three cases consistently have average log2 ratio 0 for chromosome 1, suggesting the presence of two copies of chromosome 1. Mean b-allele frequencies approximate the expected 0.5 value. Lack of divergence from 0.5 indicates no copy neutral loss of heterozygosity. Zoom-in plots of MTOR gene neighborhood are also displayed.
Supplementary Figure 7. Somatic mutations and schematic mapping of the mTOR mutations in LOT. (A) Table with somatic mutations in the mTOR protein in 4 LOT from UTSW and 4 tumors (2 from TCGA and 2 from Skala et al61) that were morphologically consistent with LOT. (B) Structure context of mTOR mutations as shown in Figure 2A including those from TCGA and Skala et al61.