

Abstract
We report a trifluoromethylarene reductive coupling method that dramatically expands the scope of difluorobenzylic substructures accessible via C–F bond functionalization. Catalytic quantities of a Lewis base, in conjunction with a disilane reagent in formamide solvent, leads to the replacement of a single trifluoromethyl fluorine atom with a silylated hemiaminal functional group. The reaction proceeds through a difluorobenzyl silane intermediate that can also be isolated. Together, these defluorinated products are shown to provide rapid access to over 20 unique difluoroalkylarene scaffolds.
Graphical Abstract
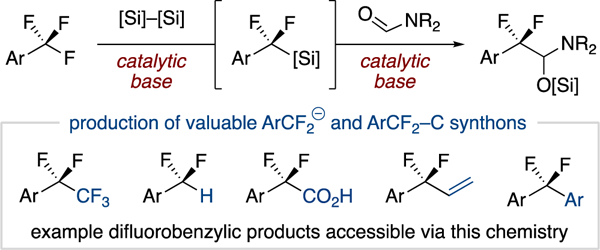
The α,α-difluorobenzylic substructure (ArCF2R) is often studied in pharmaceutical and agrochemical research as a means to modulate bioavailability and metabolic stability, amongst other potential benefits of fluorine incorporation.1 A key feature of an aromatic difluoroalkyl substituent is the structural modularity possible via the R group, allowing further optimization of a compound’s desired properties. The challenge of exploring this chemical space lies in the lack of general methods to access derivatives from a single precursor, typically requiring the synthesis of a unique reagent for each target of interest.2 For example, carbonyl deoxyfluorination3,4 and cross-coupling5 reactions are commonly used to access such motifs, but these routes first require access to the carbonyl or RCF2X coupling partner, respectively.6 Therefore, a method that can access valuable α,α-difluorobenzylic frameworks in a diversifiable fashion could greatly accelerate investigation of this substructure.7
The C–F functionalization of trifluoromethylarenes is an ideal route to α,α-difluorobenzylic compounds due to the wide availability of trifluoromethylarenes and their prevalence in late-stage settings.8 The impact of such methodology hinges on the ability to access α,α-difluorobenzylic derivatives that reflect the structural diversity found in bioactive compounds (Figure 1).9 A major challenge for the single C–F substitution of a trifluoromethyl group is the fact that the C–F bonds become weaker as defluorination proceeds10, typically resulting in overfunctionalization.11 Early efforts to address this challenge using electrochemical12 and metal13 reducing conditions are either limited to simple trifluoromethylbenzenes or are unselective. Recent reports by König, Jui, and Gouverneur use photoredox catalysis to achieve monoselective C–F reduction and hydroalkylation on a wide range of trifluoromethylarenes.14 An alternative strategy reported by Young employs frustrated Lewis pairs to form C–F substituted pyridinium and phosphonium salts, primarily used as electrophilic difluorobenzylic reagents.15,16 Despite these impressive advancements, there is still the need for a unified method that accesses a greater breadth of α,α-difluorobenzylic substructures from trifluoromethylarenes.
Figure 1.
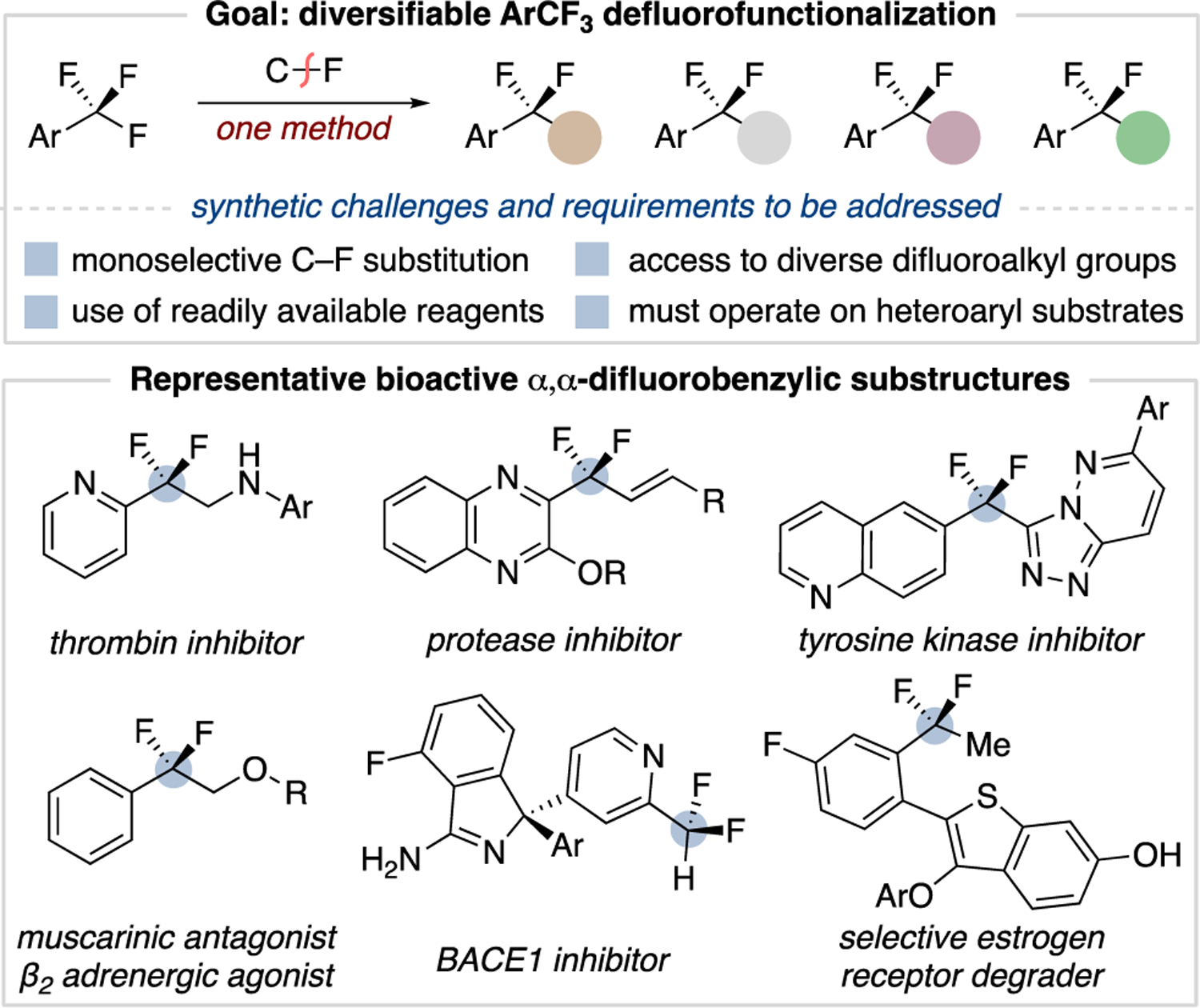
Goals for defluorofunctionalization methodology.
Our group recently reported a fluoride-initiated protocol for the selective defluoroallylation of trifluoromethylarenes using allyltrimethylsilane coupling partners (Figure 2a).17,18 While practical, this reaction can only access difluoroalkyl substituents that map onto the allyl coupling fragment. To address this limitation, we herein report the development of a base-initiated, silane-mediated, reductive coupling platform of trifluoromethylarenes (Figure 2b). This method expands the C–F transformations accessible from trifluoromethylarenes by providing a versatile silylated hemiaminal synthon that possesses the reactivity of both an aldehyde and an iminium ion. The identification of a difluorobenzyl silane as the key intermediate for the reductive coupling reaction also allowed for its isolation and use as a general difluorobenzylic pronucleophile.
Figure 2.
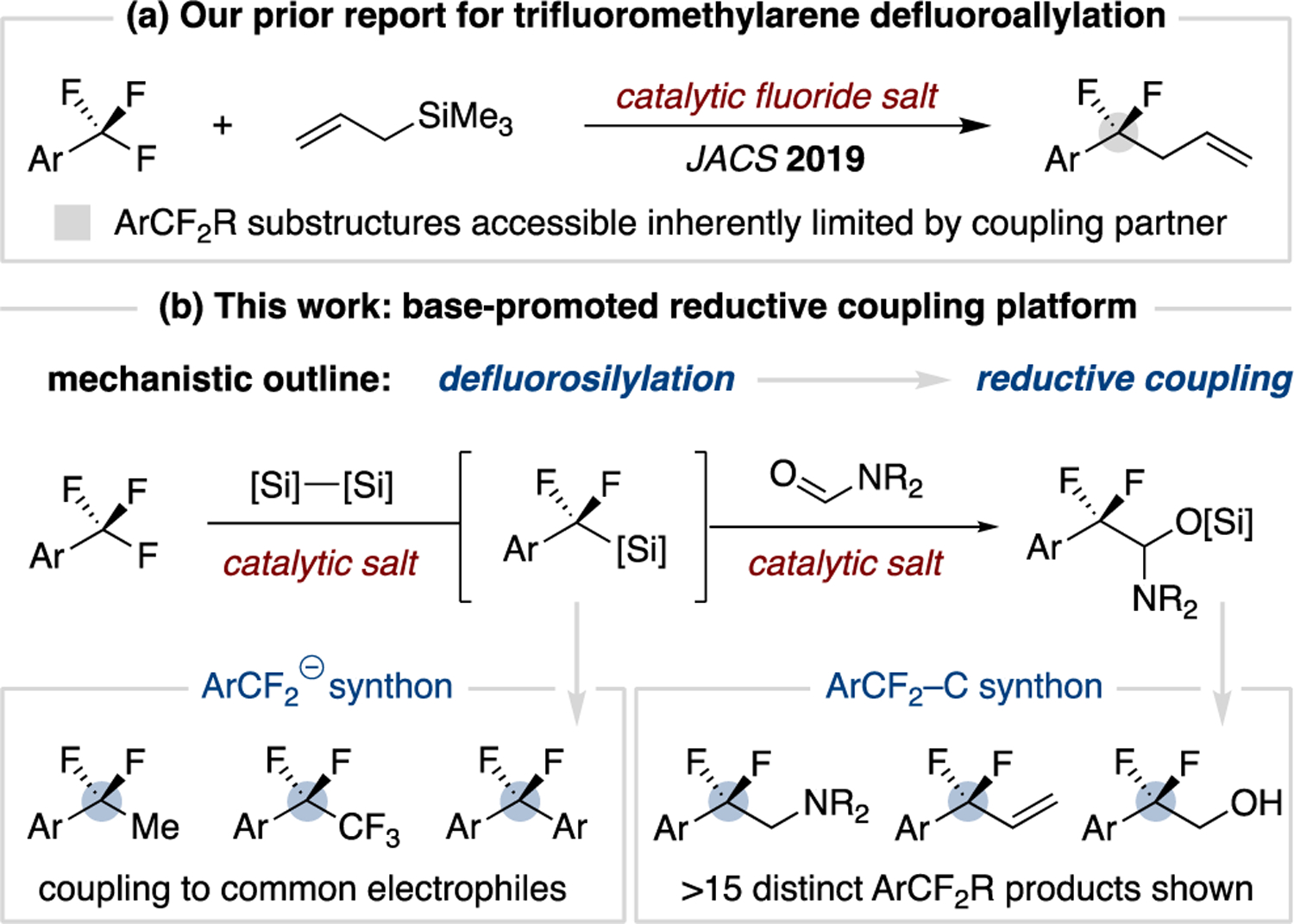
Overview of base-promoted ArCF3 functionalization.
This work began with the goal of discovering a single silane reagent that couples with trifluoromethylarenes to generate synthetically versatile difluorobenzylic products. While investigating disilane19 coupling partners, we observed that catalytic activation of commercially available tris(trimethylsilyl)silane (TTMSS) with Lewis basic salts in DMF generates silylated hemiaminal adduct 2 (Scheme 1). Given that a silylated hemiaminal could potentially serve as a branching point for a wealth of derivatization reactions, we sought to further optimize this reaction. Notably, similar silylated hemiacetal and hemiaminal adducts were proposed as intermediates in Lalic’s dual Pd/Cu-catalyzed selective trifluoromethylarene reduction protocol that is conducted with triphenylsilane in DMF.20 Strong Lewis bases, such as fluoride and alkoxide salts, provide low yields of 2, while carboxylate salts lead to significantly higher yields (entries 1–3). Ultimately, we found 18-crown-6-ligated cesium formate to be the optimal catalyst system (76% yield, entry 4). The reaction proceeds in slightly lower yield without 18-crown-6 (63%, entry 5) or using tetrabutylammonium acetate (69%, entry 6). Other commercial disilanes are less effective at promoting this reaction (entries 7 and 8). Unfortunately, all attempts to isolate product 2 via chromatography resulted in decomposition. Evaluation of other formamides led to the identification of 4-formylmorpholine (4FM) in NMP (1:1 mixture) as a satisfactory substitute for DMF, providing chromatographically stable 3 in 66% isolated yield (Scheme 1b, entry 10). Benzotrifluoride can also be used as a cosolvent (entry 11) and, for some substrates, results in improved yields (vide infra).
Scheme 1. Development of ArCF3 reductive coupling reaction.
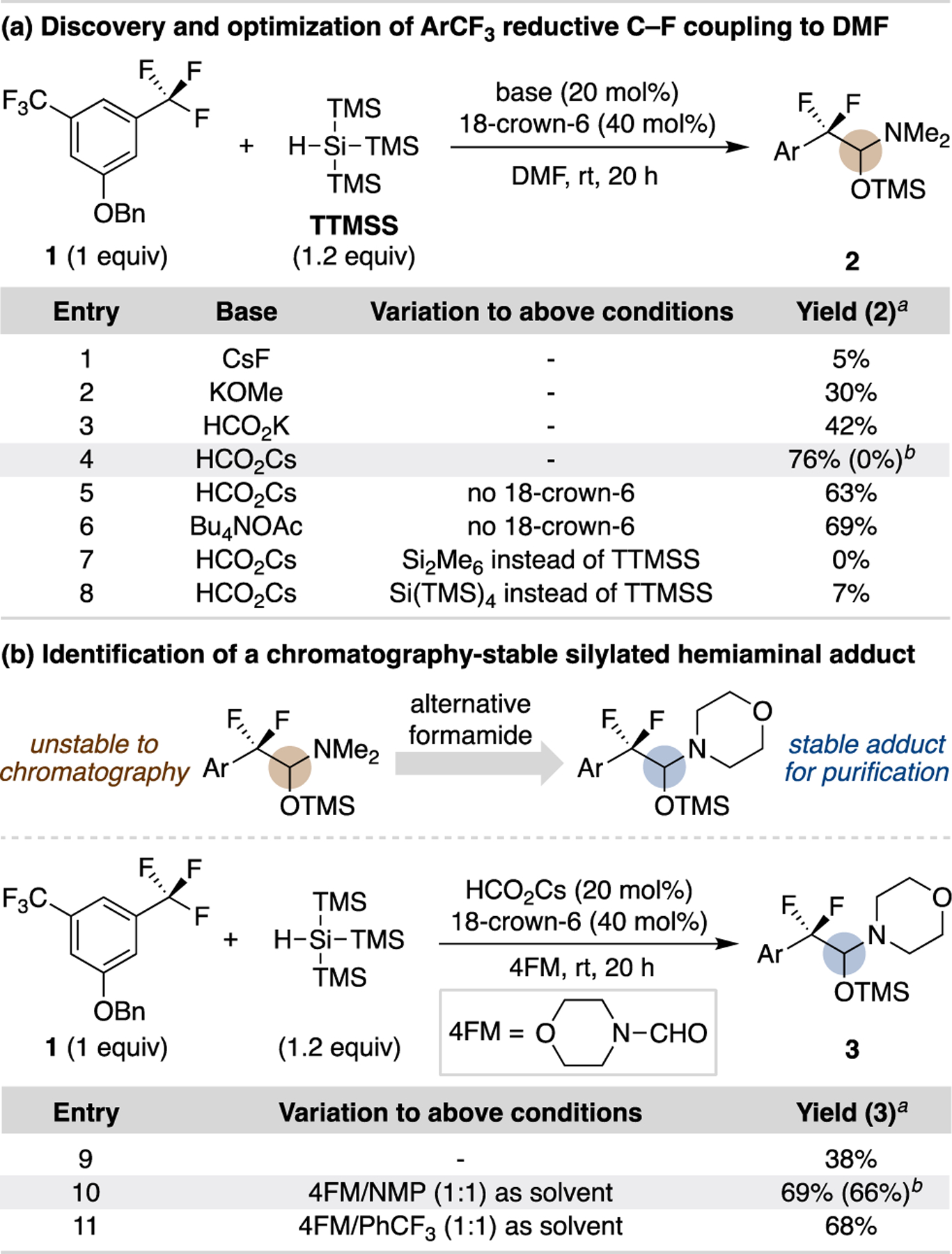
a Yields determined by 1H NMR spectroscopy. b Yields in parentheses are isolated yields by column chromatography.
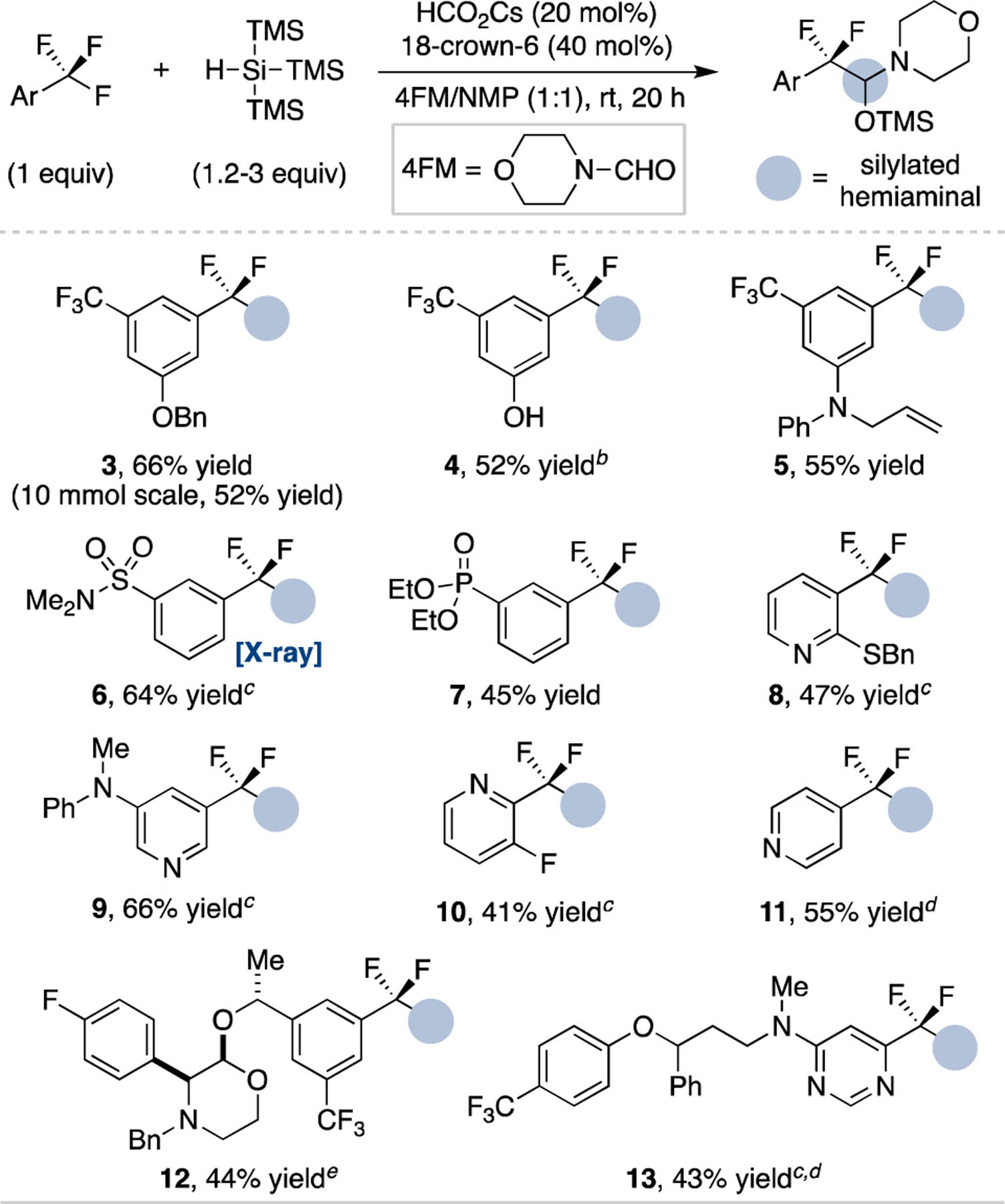
Table 1 shows representative trifluoromethylarenes that are amenable to the base-promoted coupling reaction. 1,3-Bis(trifluoromethyl)arenes are effective substrates, including when scaled to 10 mmol (3) or with free phenolic O–H (4) and terminal alkene (5) functional groups. Sulfonamide (6, characterized by X-ray crystallography) and phosphonate ester (7) aryl substituents also sufficiently activate the trifluoromethylarene towards functionalization. Heteroaryl and drug-like trifluoromethylarenes are also effective, including 2-, 3- and 4-trifluoromethylpyridines (8–11), a benzylated aprepitant precursor (12) and a fluoxetine-trifluoromethylpyrimidine derivative (13) that selectively couples at the electron-deficient heteroarene. Under the current reaction conditions, trifluoromethylbenzenes that lack an electron-withdrawing group do not react, while substrates with electrophilic functional groups (e.g. ketone) undergo competitive side reactions with the silane.21
Table 1.
Substrate Scope of Trifluoromethylarenes.a
|
Yields are of purified product on a 0.25–1 mmol scale; see Supporting Information for full details.
Isolated as adduct with NEt3.
PhCF3 used in place of NMP.
Reaction conducted at 80 °C.
Additional base (20 mol%) and TTMSS (1.2 equiv) added after 16 h.
We expected these silylated hemiaminal products to be versatile synthetic intermediates based on their resemblance to reported trifluoromethyl formamide adducts.22 The diversity of difluorobenzylic frameworks accessible from the silylated hemiaminal unit is demonstrated in Scheme 2, with sixteen transformations shown starting from product 3 (prepared on multigram scale). These reactions employ common reagents and require one purification step, with detailed procedures described in the Supporting Information (Section VII, pages S14–25). Numerous reactions can be conducted directly with the silylated hemiaminal (Scheme 2a), including cleavage to a deuterated difluoromethylarene (14), condensation to an oxime (15), condensation-dehydration to a nitrile (16), a Petasis-type styrenylation process (17), oxidation to an amide (18), reduction to a tertiary amine (19), and a Mannich-type addition reaction (20). Conversion of 3 into a hemiacetal is facile with catalytic acid in ethanol without the need for isolation.23,24 From this intermediate, many transformations are possible (Scheme 2b), including reduction (21), reductive amination (22), Wittig (23, 24), heterocycle condensation (25), Henry (26), oxidation (27), cyanide addition (28) and silylation (29) reactions.
Scheme 2. Synthetic utility of hemiaminal adduct.a.
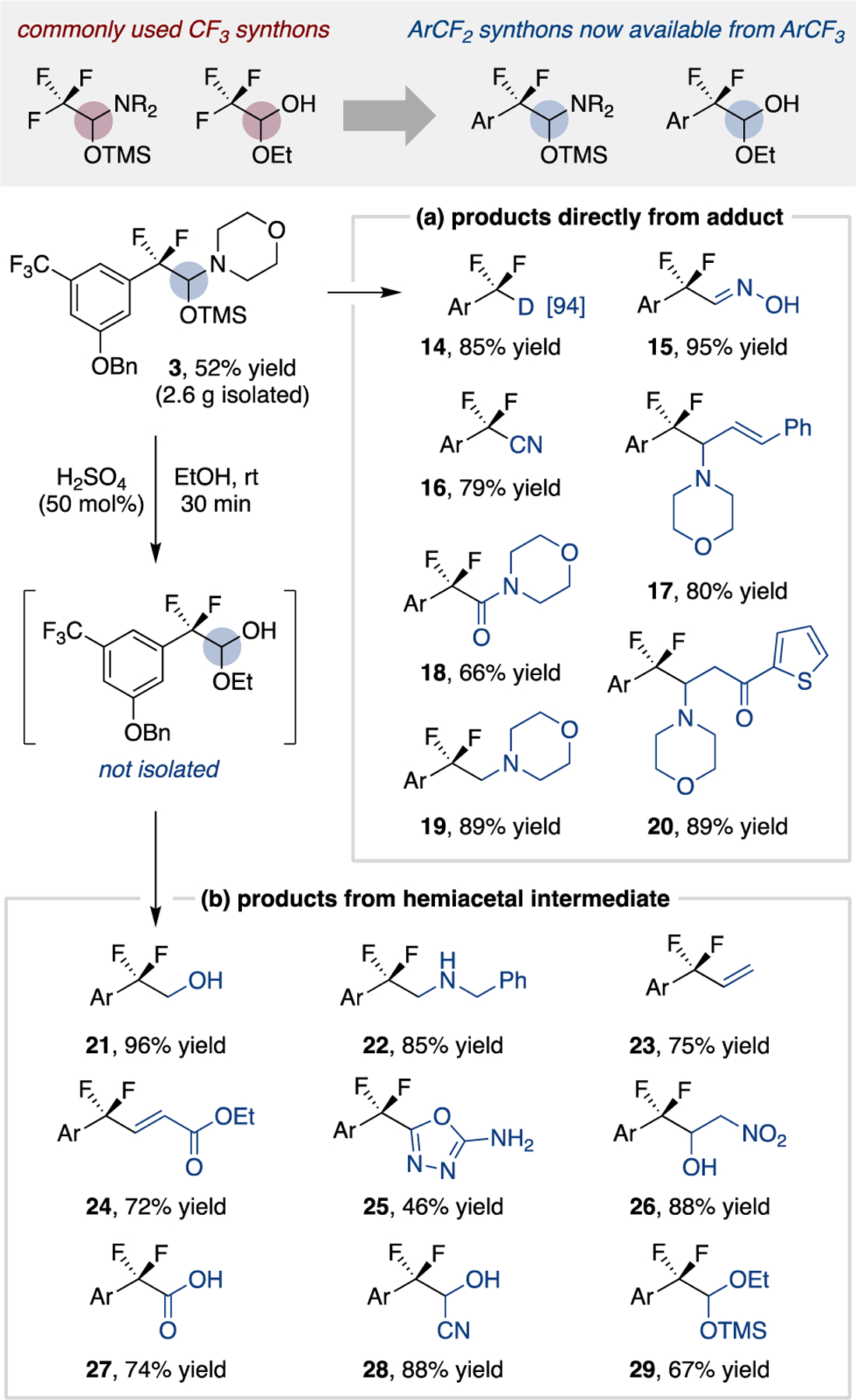
a Isolated product yields; see Supporting Information for full synthetic details for each derivatization reaction.
An additional application of this method is its use for late-stage C–F functionalization via sequenced reductive coupling and derivatization. A series of pharmaceutical derivatives and drug-like structures are shown in Scheme 3 that underwent defluorofunctionalization using one total purification step. This includes trifluoromethylaryl derivatives of apriprazole (30 and 31), fluoxetine (34) and bepotastine (35), as well as an aprepitant precursor (32 and 33) and a trifluoromethylquinoline substrate (36). These examples demonstrate the ability to modify trifluoromethyl substituents of bioactive compounds, as well as the ability to carry the typically inert trifluoromethyl group through multistep syntheses before derivatization.
Scheme 3. Divergent late-stage ArCF3 C–F functionalization.a.
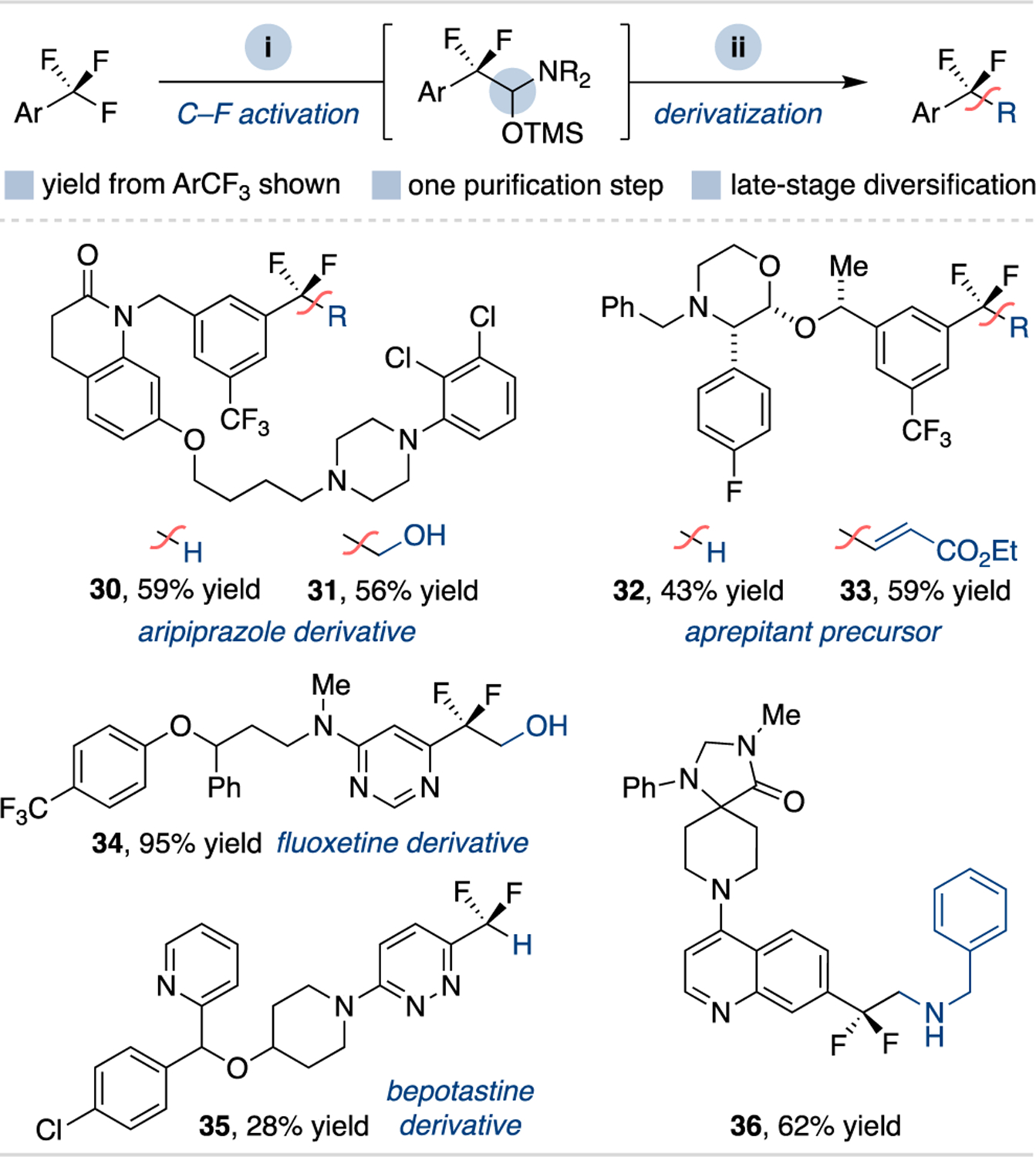
a Isolated product yields starting from trifluoromethylarene; see Supporting Information for full synthetic details for each entry.
Insight into the mechanism of this reductive coupling process first came while varying the reaction conditions. We observed the product identity to be dependent on the solvent used; in NMP, the major product is difluorobenzylsilane 37, and in MeCN, the major product is difluoromethylarene 38 (Figure 3a).25 We reasoned that formation of the benzylsilane (37) and subsequent in situ base-promoted desilylation could explain the solvent dependence.26 Subjection of benzylsilane 37 to cesium formate in MeCN or DMF provides difluoromethylarene 38 or silylated hemiaminal 2, respectively (Figure 3b).27 A profile of the model reaction in DMF shows the concurrent formation of benzylsilane 37 and the silylated hemiaminal 2, and once the trifluoromethylarene has been consumed, the remaining benzylsilane is converted to the silylated hemiaminal (Figure 3c). These observations support defluorosilylation as the key process en route to the formamide addition product 2. Each reaction of this sequence generates an anion (fluoride or oxyanion) that could regenerate the formate anion or propagate silane activation via an anionic chain process, explaining why only catalytic quantities of formate salt are required (Figure 3d).28
Figure 3.
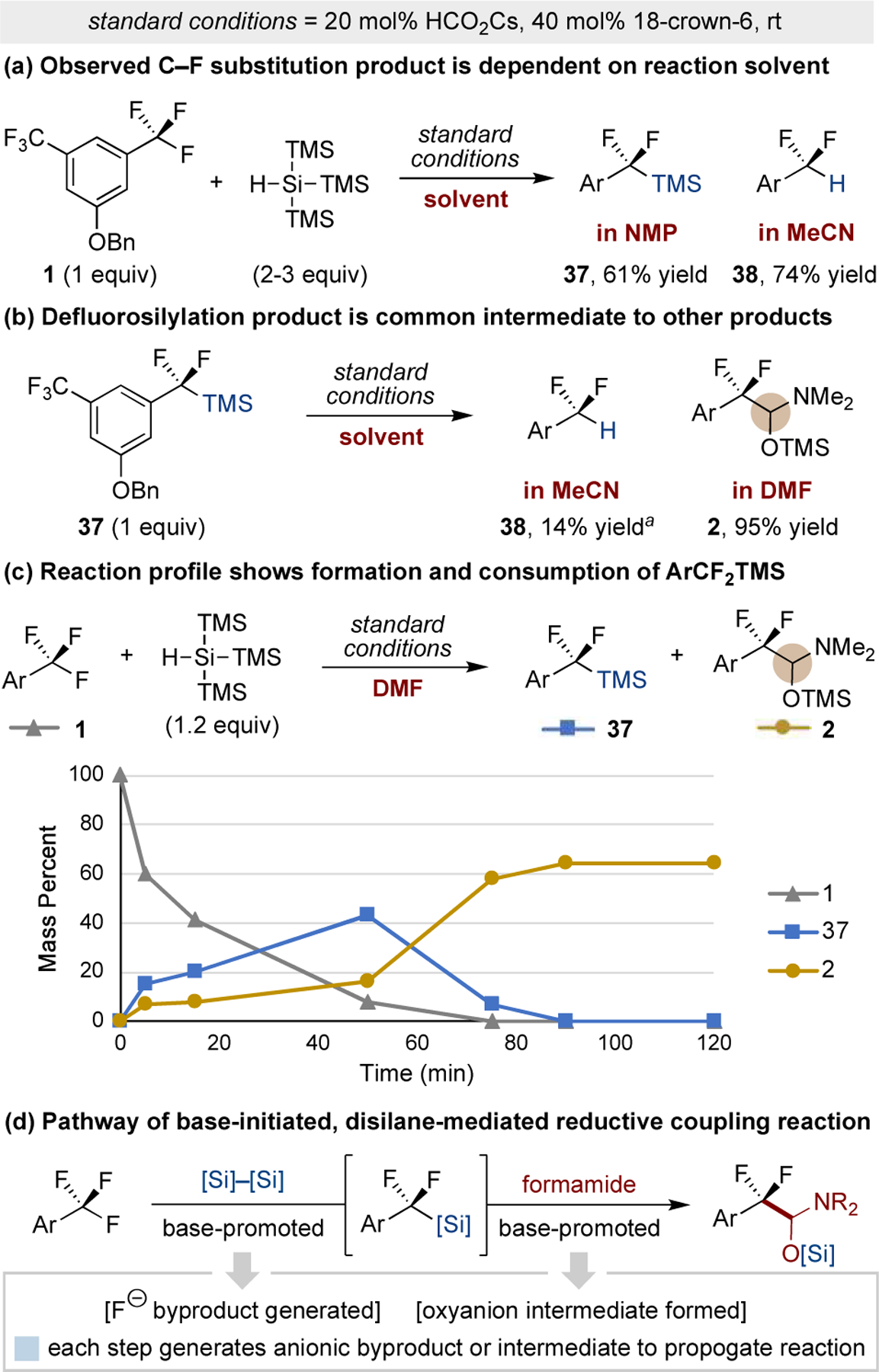
Studies and observations into reaction mechanism. Yields determined by 1H or 19F NMR spectroscopy. a Under alternative conditions, the yield of 38 is 86% if the reaction is conducted at 80 °C and 80% if CsF is used at rt in place of HCO2Cs.
Defluorosilylation likely occurs via initial formation of a silicate29 or silyl anion from TTMSS30,31, and we have obtained evidence that both TMS and HSi(TMS)2 anions may be generated under the reaction conditions.32,33 As silyl anions are known to be potent reductants34, bases35 and halophilic nucleophiles36, numerous mechanistic pathways for defluorosilylation seem plausible. Interestingly, when TTMSS is replaced with other disilane reagents (e.g. Si2Me6 or Si(TMS)4) for the model reaction, consumption of the trifluoromethylarene is observed but with little formation of the hemiaminal product.37 These comparisons indicate TTMSS strikes the right balance of Lewis acidity and capability of silyl anion generation to mediate the selective reductive coupling reaction. Details of these control studies and a discussion of possible pathways for the initiation of this reaction are provided in the Supporting Information. Investigations are underway to gain more insight into the defluorosilylation process and to identify disilanes that can activate a wider scope of trifluoromethylarenes.38,39
The discovery of a defluorosilylation pathway provides an opportunity to expand the scope of accessible C–F coupling products. Use of the α,α-difluorobenzylsilane as a masked carbanion can access derivatives that are challenging to prepare from the hemiaminal intermediates.26 The model defluorosilylation product 37 was first isolated from a reaction conducted in NMP on a 5 mmol scale in 40% yield. From 37, our recently reported fluoride-promoted protocol for benzylsilane cross-coupling to aryl nitriles can be used to generate defluoroarylation products (Scheme 4a).40 This route provides an alternative to Zhang’s recently developed light-promoted Pd-catalyzed trifluoromethylarene C–F arylation method.41
Scheme 4. Isolation and utility of α,α-difluorobenzylsilane.a.
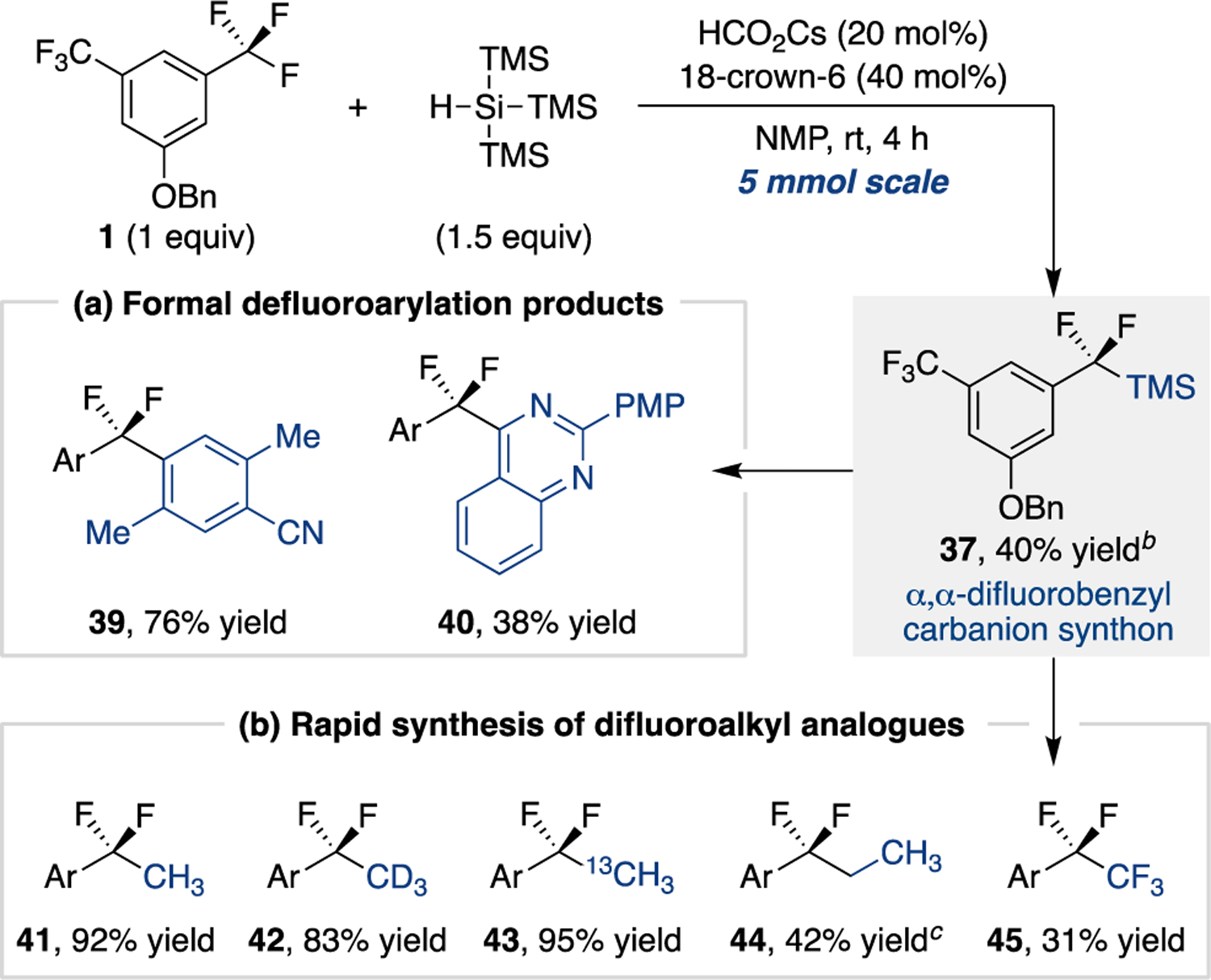
a Isolated product yields; see Supporting Information for full synthetic details for each entry. b 53% 19F NMR yield; isolated yield reduced due to coelution with protodesilylated compound 38. c 76% 1H NMR yield; isolated yield reduced due to coelution with protodesilylation side product.
We also sought to show how defluorosilylation could serve as an entry to assembling difluoroalkylarene libraries with minor structural differences (Scheme 4b). Fluoride-activation of 37 promotes facile substitution with alkyl iodides, providing the defluoromethylation product (41), its isotopologues (42 and 43), and the ethyl derivative (44). Substitution using Togni reagent II42 provides pentafluoroethyl product 45, thus accomplishing a net extension of a trifluoromethyl substituent into a pentafluoroethyl group.
In summary, this reductive coupling platform expands the scope of α,α-difluorobenzylic substructures accessible from trifluoromethylarenes to better reflect the structural diversity found in bioactive compounds (Figure 1). The reaction leverages the continuous generation of anionic intermediates to propogate a disilane-mediated defluorosilylation and formamide addition sequence. This ensemble allows a trifluoromethyl C–F bond to formally serve as a masked nucleophile, thus delivering new difluoroalkylarene synthetic linchpins.
Supplementary Material
ACKNOWLEDGMENT
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM138350. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health. We are grateful for support from Colorado State University (CSU) and the Research Corporation for Science Advancement (Cottrell Scholar Award for J.S.B). We thank the Analytical Resources Core (ARC, RRID: SCR_021758) at CSU for instrument access, training and assistance with sample analysis. We also thank Dr. Brian S. Newell of the CSU ARC for performing X-ray diffraction analyses on compounds 6 and 37.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. Experimental procedures and characterization data for all compounds (PDF).
REFERENCES
- (1).(a) For reviews on the general properties of fluorine incorporation in medicinal and agrochemical compounds, see: Gillis EP; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA Applications of Fluorine in Medicinal Chemistry. J. Med. Chem 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]; (b) Meanwell N, Fluorine A and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]; (c) Johnson BM; Shu Y-Z; Zhuo X; Meanwell NA Metabolic and Pharmaceutical Aspects of Fluorinated Compounds. J. Med. Chem 2020, 63, 6315–6386. [DOI] [PubMed] [Google Scholar]; (d) Jeschke P. Latest generation of halogen-containing pesticides. Pest Manage. Sci 2017, 73, 1053–1066. [DOI] [PubMed] [Google Scholar]
- (2).(a) For selected reviews on preparing difluorobenzylic compounds, see: Belhomme D-C; Besset T; Poisson T; Pannecoucke X Recent Progress toward the Introduction of Functionalized Difluoromethylated Building Blocks onto C(sp2) and C(sp) Center. Chem. Eur. J 2015, 21, 12836–12865. [DOI] [PubMed] [Google Scholar]; (b) Carvalho DR; Christian AH Modern approaches towards the synthesis of geminal difluoroalkyl groups. Org. Biomol. Chem 2021, 19, 947–964. [DOI] [PubMed] [Google Scholar]
- (3).Ni C; Hu M; Hu J Good Partnership between Sulfur and Fluorine: Sulfur-Based Fluorination and Fluoroalkylation Reagents for Organic Synthesis. Chem. Rev 2015, 115, 765–825. [DOI] [PubMed] [Google Scholar]
- (4).(a) Benzylic C–H fluorination is emerging as an alternative to carbonyl deoxyfluorination; see: Koperniku A; Liu H; Hurley PB Mono- and Difluorination of Benzylic Carbon Atoms. Eur. J. Org. Chem 2016, 871–886. [Google Scholar]; (b) Meanwell M; Britton R Synthesis of Heterobenzylic Fluorides. Synthesis 2018, 50, 1228–1236. [Google Scholar]; (c) Szpera R; Moseley DFJ; Smith LB; Sterling AJ; Gouverneur V The Fluorination of C–H Bonds: Developments and Perspectives. Angew. Chem. Int. Ed 2019, 58, 14824–14848. [DOI] [PubMed] [Google Scholar]
- (5).(a) For selected reviews and examples on cross-coupling reactions using functionalized difluoroalkyl reagents, see: Feng Z; Xiao Y-L; Zhang X Transition-Metal (Cu, Pd, Ni)-Catalyzed Difluoroalkylation via Cross-Coupling with Difluoroalkyl Halides. Acc. Chem. Res 2018, 51, 2264–2278. [DOI] [PubMed] [Google Scholar]; (b) Dong D-Q; Yang H; Shi J-L; Si W-J; Wang Z-L; Xu X-M Promising reagents for difluoroalkylation. Org. Chem. Front 2020, 7, 2538–2575. [Google Scholar]; (c) Xiao Y-L; Min Q-Q; Xu C; Wang R-W; Zhang X Nickel-Catalyzed Difluoroalkylation of (Hetero)Arylborons with Unactivated 1-Bromo-1,1-difluoroalkanes. Angew. Chem., Int. Ed 2016, 55, 5837–5841. [DOI] [PubMed] [Google Scholar]; (d) Ge S; Arlow SI; Mormino MG; Hartwig JF Pd-Catalyzed α-Arylation of Trimethylsilyl Enolates of α,α-Difluoroacetamides. J. Am. Chem. Soc 2014, 136, 14401–14404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) For reviews and examples of radical-based routes to difluorobenzylic products, see ref 2b and Lemos A; Lemaire C; Luxen A Progress in Difluoroalkylation of Organic Substrates by Visible Light Photoredox Catalysis. Adv. Synth. Catal 2019, 361, 1500–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhou Q; Ruffoni A; Gianatassio R; Fujiwara Y; Sella E; Shabat D; Baran PS Direct Synthesis of Fluorinated Heteroarylether Bioisosteres. Angew. Chem. Int. Ed 2013, 52, 3949–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Douglas JJ; Albright H; Sevrin MJ; Cole KP; Stephenson CRJ A Visible-Light-Mediated Radical Smiles Rearrangement and its Application to the Synthesis of a Difluoro-Substituted Spirocyclic ORL-1 Antagonist. Angew. Chem. Int. Ed 2015, 54, 14898–14902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) For recent methods that functionalize aryldifluoromethyl reagents, see: Geri JB; Wade Wolfe MM; Szymczak NK The Difluoromethyl Group as a Masked Nucleophile: A Lewis Acid/Base Approach. J. Am. Chem. Soc 2018, 140, 9404–9408. [DOI] [PubMed] [Google Scholar]; (b) Wade Wolfe MM; Shanahan JP; Kampf JW; Szymczak NK Defluorinative Functionalization of Pd(II) Fluoroalkyl Complexes. J. Am. Chem. Soc 2020, 142, 18698–18705. [DOI] [PubMed] [Google Scholar]; (c) Santos L; Panossian A; Donnard M; Vors J-P; Pazenok S; Bernier D; Leroux FR Deprotonative Functionalization of the Difluoromethyl Group. Org. Lett 2020, 22, 8741–8745. [DOI] [PubMed] [Google Scholar]
- (8).(a) Jaroschik F Picking One out of Three: Selective Single C–F Activation in Trifluoromethyl Groups. Chem. Eur. J 2018, 24, 14572–14582. [DOI] [PubMed] [Google Scholar]; (b) Yan G; Qiu K; Guo M Recent advance in the C–F bond functionalization of trifluoromethyl-containing compounds. Org. Chem. Front 2021, 8, 3915–3942. [Google Scholar]; (c) Zhao F; Zhou W; Zuo Z Recent Advances in the Synthesis of Difluorinated Architectures from Trifluoromethyl Groups. Adv. Synth. Catal 2022, 364, 234–267. [Google Scholar]
- (9). For the full structures and references for the compounds shown in Figure 1, see page S56 of the Supporting Information file.
- (10).(a) For discussions and values of C–F bond strengths, see: Luo YR Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, 2007. [Google Scholar]; (b) O’Hagan D Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev 2008, 37, 308–319. [DOI] [PubMed] [Google Scholar]; (c) Wiberg KB; Rablen PR Origin of the stability of carbon tetrafluoride: negative hyperconjugation reexamined. J. Am. Chem. Soc 1993, 115, 614–625. [Google Scholar]
- (11).(a) For selected examples and reviews of multiple C–F functionalization reactions, see: Saito K; Umi T; Yamada T; Suga T; Akiyama T Niobium(V)-catalyzed defluorinative triallylation of α,α,α-trifluorotoluene derivatives by triple C−F bond activation. Org. Biomol. Chem 2017, 15, 1767–1770. [DOI] [PubMed] [Google Scholar]; (b) Gu W; Haneline MR; Douvris C; Ozerov OV Carbon−Carbon Coupling of C(sp3)−F Bonds Using Alumenium Catalysis. J. Am. Chem. Soc 2009, 131, 11203–11212. [DOI] [PubMed] [Google Scholar]; (c) Zhu J; Pérez M; Caputo CB; Stephan DW Use of Trifluoromethyl Groups for Catalytic Benzylation and Alkylation with Subsequent Hydrodefluorination. Angew. Chem. Int. Ed 2016, 55, 1417–1421. [DOI] [PubMed] [Google Scholar]; (d) Stahl T; Klare HFT; Oestreich M Main-Group Lewis Acids for C–F Bond Activation. ACS Catal 2013, 3, 1578–1587. [Google Scholar]; (e) Shen Q; Huang Y-G; Liu C; Xiao J-C; Chen Q-Y; Guo Y Review of recent advances in C−F bond activation of aliphatic fluorides. J. Fluor. Chem 2015, 179, 14–22. [Google Scholar]
- (12).(a) Saboureau C; Troupel M; Sibille S; Périchon J Electroreductive Coupling of Trifluoromethylarenes with Electrophiles: Synthetic Applications. J. Chem. Soc., Chem. Commun 1989, 1138–1139. [Google Scholar]; (b) Clavel P; Léger-Lambert M-P; Biran C; Serein-Spirau F; Bordeau M; Roques N; Marzouk H Selective Electrosynthesis of (Trimethylsilyldifluoro)methylbenzene, a PhCF2− Precursor; Conditions for a Molar Scale Preparation without HMPA. Synthesis 1999, 5, 829–834. [Google Scholar]; (c) Clavel P; Lessene G; Biran C; Bordeau M; Roques N; Trévin S; de Montauzon D Selective electrochemical synthesis and reactivity of functional benzylic fluorosilylsynthons. J. Fluor. Chem 2001, 107, 301–310. [Google Scholar]
- (13).(a) Utsumi S; Katagiri T; Uneyama K Cu-deposits on Mg metal surfaces promote electron transfer reactions. Tetrahedron 2012, 68, 1085–1091. [Google Scholar]; (b) Utsumi S; Katagiri T; Uneyama K Mg–Cu bimetal system for selective C–F bond activation. J. Fluor. Chem 2013, 152, 84–89. [Google Scholar]; (c) Munoz SB; Ni C; Zhang Z; Wang F; Shao N; Mathew T; Olah GA; Prakash GKS Selective Late-Stage Hydrodefluorination of Trifluoromethylarenes: A Facile Access to Difluoromethylarenes. Eur. J. Org. Chem 2017, 2017, 2322–2326. [Google Scholar]
- (14).(a) For photoredox-catalyzed trifluoromethylarene C–F functionalization methods, see: Chen K; Berg N; Gschwind R; König B Selective Single C(sp3)−F Bond Cleavage in Trifluoromethylarenes: Merging VisibleLight Catalysis with Lewis Acid Activation. J. Am. Chem. Soc 2017, 139, 18444–18447. [DOI] [PubMed] [Google Scholar]; (b) Wang H; Jui NT Catalytic Defluoroalkylation of Trifluoromethylaromatics with Unactivated Alkenes. J. Am. Chem. Soc 2018, 140, 163–166. [DOI] [PubMed] [Google Scholar]; (c) Vogt DB; Seath CP; Wang H; Jui NT Selective C−F Functionalization of Unactivated Trifluoromethylarenes. J. Am. Chem. Soc 2019, 141, 13203–13211. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sap JBI; Straathof NJW; Knauber T; Meyer CF; Médebielle M; Buglioni L; Genicot C; Trabanco AA; Noël T; am Ende CW; Gouverneur V Organophotoredox Hydrodefluorination of Trifluoromethylarenes with Translational Applicability to Drug Discovery. J. Am. Chem. Soc 2020, 142, 9181–9187. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) For additional recent photoredox-based methods, see: Yuan X; Zhuang K-Q; Cui Y-S; Qin L-Z; Sun Q; Duan X; Chen L; Zhu N; Li G; Qui J-K; Guo K Photocatalytic radical defluoroalkylation of unactivated alkenes via distal heteroaryl ipso-migration. Commun. Chem 2020, 3, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Liu C; Shen N; Shang R Photocatalytic defluoroalkylation and hydrodefluorination of trifluoromethyls using o-phosphinophenolate. Nat. Commun 2022, 13, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Liu C; Li K; Shang R Arenethiolate as a Dual Function Catalyst for Photocatalytic Defluoroalkylation and Hydrodefluorination of Trifluoromethyls. ACS Catal 2022, 12, 4103–4109. [Google Scholar]
- (15).(a) Mandal D; Gupta R; Jaiswal AK; Young RD Frustrated Lewis-Pair-Mediated Selective Single Fluoride Substitution in Trifluoromethyl Groups. J. Am. Chem. Soc 2020, 142, 2572–2578. [DOI] [PubMed] [Google Scholar]; (b) Gupta R; Mandal D; Jaiswal AK; Young RD FLP-Catalyzed Monoselective C–F Functionalization in Polyfluorocarbons at Geminal or Distal Sites. Org. Lett 2021, 23, 1915–1920. [DOI] [PubMed] [Google Scholar]
- (16).(a) For monoselective C–F functionalization reactions that are facilitated by an ortho-silylium ion, see: Yoshida S; Shinomori K; Kim Y; Hosoya T Single C–F Bond Cleavage of Trifluoromethylarenes with an ortho-Silyl Group. Angew. Chem. Int. Ed 2016, 55, 10406–10409. [DOI] [PubMed] [Google Scholar]; (b) Idogawa R; Kim Y; Shinomori K; Hosoya T; Yoshida S Single C–F Transformations of o-Hydrosilyl Benzotrifluorides with Trityl Compounds as All-in-One Reagents. Org. Lett 2020, 22, 9292–9297. [DOI] [PubMed] [Google Scholar]; (c) Kim Y; Kanemoto K; Shinomori K; Hosoya T; Yoshida S Functionalization of a Single C–F Bond of Trifluoromethylarenes Assisted by an ortho-Silyl Group Using a Trityl-Based All-in-One Reagent with Ytterbium Triflate Catalyst. Chem. Eur. J 2020, 26, 6136–6140. [DOI] [PubMed] [Google Scholar]
- (17).Luo C; Bandar JS Selective Defluoroallylation of Trifluoromethylarenes. J. Am. Chem. Soc 2019, 141, 14120–14125. [DOI] [PubMed] [Google Scholar]
- (18).(a) For recently reported alternative methods for defluoroallylation of perfluoroalkylarenes, see: Sugihara N; Suzuki K; Nishimoto Y; Yasuda M Photoredox-Catalyzed C–F Bond Allylation of Perfluoroalkylarenes at the Benzylic Position. J. Am. Chem. Soc 2021, 143, 9308–9313. [DOI] [PubMed] [Google Scholar]; (b) Zhou F-Y; Jiao L Asymmetric Defluoroallylation of 4-Trifluoromethylpyridines Enabled by Umpolung C−F Bond Activation. Angew. Chem. Int. Ed 2022, 61, e202201102. [DOI] [PubMed] [Google Scholar]
- (19). We herein refer to any compound with a silicon-silicon bond as a disilane.
- (20).Dang H; Whittaker AM; Lalic G Catalytic activation of a single C–F bond in trifluoromethylarenes. Chem. Sci 2016, 7, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21). For trifluoromethylbenzenes, electron-withdrawing groups in the meta positions lead to the highest yields, although electron-withdrawing groups at any position are sufficient for activation. For example, 1,2- and 1,4-bis(trifluoromethyl)benzenes undergo reductive coupling at room temperature or 50 °C in moderate yields. Additionally, 2-, 3-, and 4-trifluoromethyl substituted pyridines perform similarly as shown in Table 1. Benzotrifluoride does not react under the standard reaction conditions. A comparison of these electronic effects is documented in the Supporting Information on page S7.
- (22).(a) For selected studies on formamide adducts of fluoroform and their synthetic utility that inspired the derivatization reactions in this work, see: Billard T; Bruns S; Langlois BR New Stable Reagents for the Nucleophilic Trifluoromethylation. 1. Trifluoromethylation of Carbonyl Compounds with N-Formylmorpholine Derivatives. Org. Lett 2000, 2, 2101–2103. [DOI] [PubMed] [Google Scholar]; (b) Blond G; Billard T; Langlois BR Reactivity of Stable Trifluoroacetaldehyde Hemiaminals. 1. An Unexpected Reaction with Enolizable Carbonyl Compounds. J. Org. Chem 2001, 66, 4826–4830. [DOI] [PubMed] [Google Scholar]; (c) Billard T; Langlois BR Reactivity of Stable Trifluoroacetaldehyde Hemiaminals. 2. Generation and Synthetic Potentialities of Fluorinated Iminiums. J. Org. Chem 2002, 67, 997–1000. [DOI] [PubMed] [Google Scholar]
- (23).(a) For representative references using fluorinated hemiacetals as synthetic intermediates, see Tsukamoto T; Kitazume T Difluorinated Maloaldehyde Derivatives as Useful Difluoromethylene-containing Building Blocks. J. Chem. Soc., Chem. Commun 1992, 540–541. [Google Scholar]; (b) Tsukamoto T; Kitazume T Preparation and Reaction of Difluorinated Malonaldehydic Acid Derivatives: a New Route to Functionalized ⍺,⍺-Difluorinated Esters and Amides. J. Chem. Soc., Perkin Trans 1, 1993, 1177–1181. [Google Scholar]; (c) Kubota T; Iijima M; Tanaka T Facile Synthesis of ⍺-Trifluoromethylated Alcohols from Trifluoroacetaldehyde Ethyl Hemiacetal. Tetrahedron Lett 1992, 33, 1351–1354. [Google Scholar]
- (24).Attempts to prepare and isolate the corresponding aldehyde were unsuccessful due to its instability. This instability mirrors that of related compounds, such as trifluoroacetaldehyde. For a discussion of trifluoroacetaldehyde instability, see: Lin P Trifluoroacetaldehyde. In Encyclopedia of Reagents for Organic Synthesis; Wiley, 2003. DOI: 10.1002/047084289X.rn00213. [DOI] [Google Scholar]
- (25). Defluorosilylation has been reported under electrochemical and metal reducing conditions for several trifluoromethylbenzenes; see refs 12b-c and 13a-b.
- (26).(a) For alternative routes to difluorobenzylic silanes and their use as nucleophilic reagents: see ref 23.; (b) Aikawa K; Maruyama K; Nitta J; Hashimoto R; Mikami K Siladifluoromethylation and Difluoromethylation onto C(sp3), C(sp2), and C(sp) Centers Using Ruppert–Prakash Reagent and Fluoroform. Org. Lett 2016, 18, 3354–3357. [DOI] [PubMed] [Google Scholar]; (c) Guidotti J; Metz F; Tordeux M; Wakselman C Preparation of (Phenyldifluoromethyl)- and (Phenoxydifluoromethyl)-silanes by Magnesium-Promoted Carbon-Chlorine Bond Activation. Synlett 2004, 10, 1759–1762. [Google Scholar]
- (27). Because the yield of 38 is low under the “standard conditions” for the experiment shown in Figure 3b, we conducted additional control experiments that could mimic the conditions typical of the defluorofunctionalization reactions in Figure 3a. Thus, use of catalytic CsF as a base, which is formed as an intermediate under the defluorosilylation reaction conditions, promotes the desilylation reactions shown in Figure 3b in high yield. These results are shown as footnote a in Figure 3 and described in the Supporting Information.
- (28). Mixing fluoride salts with trimethylsilylated carboxylates (RCO2TMS) rapidly releases the carboxylate anion and forms fluorotrimethylsilane. This observation, and the fact that use of fluoride as an initiator for the model reaction (Scheme 1, entry 1) leads to low yields, leads us to hypothesize that cesium formate is continuously generated under the reaction conditions and is acting as a true catalyst.
- (29).(a) For reviews on base activation of organosilanes, see: Chuit C; Corriu RJP; Reye C; Young JC Reactivity of penta- and hexacoordinate silicon compounds and their role as reaction intermediates. Chem. Rev 1993, 93, 1371–1448. [Google Scholar]; (b) Reich HJ Mechanism of C–Si Bond Cleavage Using Lewis Bases (n → σ*). In Lewis Base Catalysis in Organic Synthesis, Vol. 1; Wiley-VCH, 2016; pp 233–280. [Google Scholar]; (c) García-Domínguez A; Leach AG; Llyod-Jones GC In Situ Studies of Arylboronic Acids/Esters and R3SiCF3 Reagents: Kinetics, Speciation, and Dysfunction at the Carbanion–Ate Interface. Acc. Chem. Res 2022, 55, 1324–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).(a) For reviews on silyl anion generation and reactivity, see: Lickiss P D; Smith, C. M. Silicone derivatives of the metals of groups 1 and 2. Coord. Chem. Rev 1995, 145, 75–124. [Google Scholar]; (b) Tamao K; Kawachi A Silyl Anions. In Advances in Organometallic Chemistry, Vol. 38; Academic Press, 1995, 38, pp 1–58. [Google Scholar]; (c) Marschner C Chapter 7 – Silicon-Centered Anions. In Organosilicon Compounds, Vol. 1; Academic Press, 2017, pp 295–360. [Google Scholar]
- (31).(a) For reviews on the use of TTMSS, see: Giese B; Dickhaut J; Chatgilialoglu C; Wu X; Zhang Z Tris(trimethylsilyl)silane. In Encyclopedia of Reagents for Organic Synthesis; Wiley, 2022. Updated January 28, 2022. DOI: 10.1002/047084289X.rt420.pub3. [DOI] [Google Scholar]; (b) Chatgilialoglu C; Ferreri C; Landais Y; Timokhin VI Thirty Years of (TMS)3SiH: A Milestone in Radical-Based Synthetic Chemistry. Chem. Rev 2018, 118, 6516–6572. [DOI] [PubMed] [Google Scholar]
- (32).(a) It is well known that Lewis base activation of tetrakis(trimethylsilyl)silane (Si(TMS)4) can generate the Si(TMS)3 anion; see ref 30c. Similarly, it has been observed that Lewis base activation of TTMSS can generate the HSi(TMS)2 anion; see: Marschner C A New and Easy Route to Polysilanylpotassium Compounds. Eur. J. Inorg. Chem 1998, 221–226. [Google Scholar]
- (33). As control experiments, we subjected TTMSS and cesium formate in DMF at rt to various electrophiles in place of trifluoromethylarenes. We observed substitution products that most likely arise from generation of both the TMS and HSi(TMS)2 anions; see Supporting Information for full details. Thus, we cannot rule out the generation of either silyl anion under the standard reaction conditions.
- (34).(a) Sakurai H; Akane O; Umino H; Kira M Silyl anions. IV. New convenient method of producing radical anions involving one-electron transfer from trimethylsilylsodium. J. Am. Chem. Soc 1973, 95, 955–956. [Google Scholar]; (b) Hideki S; Mitsuo K; Hiroshi U Silyl Anions VII. Electron Transfer From Trimethylsilylpotassium To Benzophenone And Naphthalene. Generation Of Anion Radicals In Nonpolar Solvents Such As n-Hexane. Chem. Lett 1977, 6, 1265–1268. [Google Scholar]; (c) Smith AJ; Young A; Rohrbach S; O’Connor EF; Allison M; Wang H-S; Poole DL; Tuttle T; Murphy JA Electron-Transfer and Hydride-Transfer Pathways in the Stoltz–Grubbs Reducing System (KOtBu/Et3SiH). Angew. Chem. Int. Ed 2017, 56, 13747–13751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).For pKa values of organosilanes, see: Fu Y; Liu L; Li R-Q; Liu L; Guo Q-X First-Principal Predictions of Absolute pKa’s of Organic Acids in Dimethyl Sulfoxide Solution. J. Am. Chem. Soc 2003, 126, 814–822. [DOI] [PubMed] [Google Scholar]
- (36).(a) For representative halophilic substitution processes involving silyl anions, see: Shippey MA; Dervan PB Trimethylsilyl Anions. Direct Synthesis of Trimethylsilylbenzenes. J. Org. Chem. 1977, 42, 2654–2655. [Google Scholar]; (b) Uematsu R; Yamamoto E; Maeda S; Ito H; Taketsugo T Reaction Mechansim of the Anomalous Formal Nucleophilic Borylation of Organic Halides with Silylborane: Combined Theoretical and Experimental Studies. J. Am. Chem. Soc 2015, 137, 4090–4099. [DOI] [PubMed] [Google Scholar]
- (37). Use of other disilanes in place of TTMSS results in low yields of 2 and/or poor mass balance of 1; these studies are described in the Supporting Information. The disilane must be capable of being activated by the Lewis base to promote defluorosilylation. The inability of hexamethyldisilane, which can only generate the TMS anion, to promote the reaction in high yield suggests that the role of TTMSS constitutes more than just the generation of a TMS anion.
- (38).TTMSS is known to readily generate the Si(TMS)3 radical and engage in radical-based reactions (see ref 31). However, replacement of H in TTMSS with other substituents (e.g. alkyl or amino groups) still results in some yield of 2 for the model reaction, suggesting the Si–H bond is not critical for the reaction mechanism. Furthermore, replacement of cesium formate with radical initiators (e.g. AIBN and benzoyl peroxide) results in no reductive coupling reaction. As another control, a reaction conducted with catalytic TTMSS and stoichiometric Si(TMS)4 provided only low yields of product. Additionally, our optimization studies show that acetate bases perform similarly to formate bases, indicating that formate does not act as a reductant or radical anion precursor in this reaction; see: Hendy CM; Smith GC; Xu Z; Lian T; Jui NT Radical Chain Reduction via Carbon Dioxide Radical Anion (CO2•–). J. Am. Chem. Soc 2021, 143, 8987–8992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).For base-promoted defluorosilylation of aryl and alkyl fluorides, see: Liu X-W; Zarate C; Martin R Base-Mediated Defluorosilylation of C(sp2)–F and C(sp3)–F Bonds. Angew. Chem. Int. Ed 2019, 58, 2064–2068. [DOI] [PubMed] [Google Scholar]
- (40).(a) Reidl TW; Bandar JS Lewis Basic Salt-Promoted Organosilane Coupling Reactions with Aromatic Electrophiles. J. Am. Chem. Soc 2021, 143, 11939–11945. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) see also ref 26b. [Google Scholar]
- (41).(a) Luo Y-C; Tong F-F; Zhang Y; He C-Y; Zhang X Visible-Light-Induced Palladium-Catalyzed Selective Defluoroarylation of Trifluoromethylarenes with Arylboronic Acids. J. Am. Chem. Soc 2021, 143, 139741–13979. [DOI] [PubMed] [Google Scholar]; (b) For several examples of formal defluoroarylation using aryl boronic acid coupling partners via Young’s frustrated Lewis pair approach, see ref 15b. [Google Scholar]
- (42).Charpentier J; Früh N; Togni A Electrophilic Trifluoromethylation by Use of Hypervalent Iodine Reagents. Chem. Rev 2015, 115, 650–682 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.