

Abstract
Non-structural protein 1 (Nsp1) of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a major virulence factor and thus an attractive drug target. The last 33 amino acids of Nsp1 have been shown to bind within the mRNA entry tunnel of the 40S ribosomal subunit, shutting off host gene expression. Here, we report the solution-state structure of full-length Nsp1, which features an α/β fold formed by a six-stranded, capped β-barrel-like globular domain (N-terminal domain [NTD]), flanked by short N-terminal and long C-terminal flexible tails. The NTD has been found to be critical for 40S-mediated viral mRNA recognition and promotion of viral gene expression. We find that in free Nsp1, the NTD mRNA-binding surface is occluded by interactions with the acidic C-terminal tail, suggesting a mechanism of activity regulation based on the interplay between the folded NTD and the disordered C-terminal region. These results are relevant for drug-design efforts targeting Nsp1.
Keywords: SARS-CoV-2, Nsp1, non-structural proteins, drug targets, NMR spectroscopy, disordered protein domains
Graphical abstract
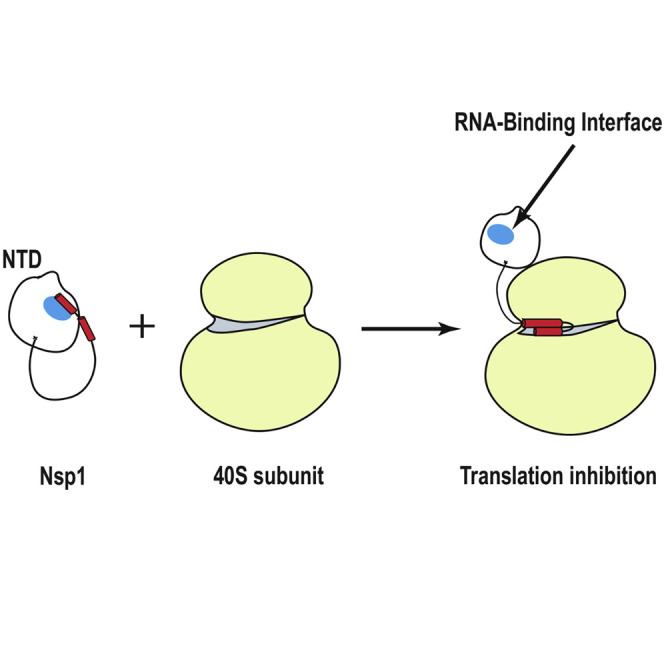
The SARS-CoV-2 protein Nsp1 is a major virulence factor due to its role in host translation shutoff and viral evasion. Wang et al. present the atomic-resolution structure of full-length Nsp1, revealing hitherto unknown features that provide the basis for unraveling its functional regulation and guiding future drug-design strategies.
Introduction
The COVID-19 pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has considerably increased the awareness of coronaviruses as a threat to human health worldwide. Seven coronaviruses—all belonging to the Alpha and Beta coronavirus genera—are known to infect mammalian species.1 Human Alpha coronaviruses such as HCoV-229E, HCoV-NL63, HCoV-OC43, and HCoV-HKU1 cause seasonal and mild respiratory tract infections with common-cold-like symptoms,2 while SARS-CoV-2, together with its homolog SARS-CoV-1, and the Middle East respiratory syndrome (MERS) CoV are established agents of life-threatening acute respiratory pathologies and severe lung disease.3 , 4
SARS-CoV-2 has an enveloped, positive-sense, single-stranded RNA genome approximately 30 kb in length. The 5′-capped and 3′-poly-adenylated genome has two terminal untranslated regions (the 5′ and 3′ UTRs) surrounding the central coding region, which is comprised of 12 open reading frames (ORFs). The first two—ORF1a and ORF1b—are directly translated from the genomic RNA, while the other 10 ORFs are translated from a set of eight subgenomic RNAs that are first generated from the genomic RNA by two rounds of transcription.5 , 6 Translation of ORF1a and ORF1b, which together span two thirds of the entire genome, produces two polyproteins that are subsequently cleaved by two viral proteases to yield 16 non-structural proteins.7 , 8 Among these, non-structural protein 1 (Nsp1), also known as the leader protein, plays a key role in suppressing host-cell protein translation and thus the host immune response to the virus.
Nsp1 inhibits host translational activity by inserting the second half of its C-terminal tail (residues 148–180; termed the C-terminal domain [CTD]; Figure 1A) into the entrance of the mRNA tunnel of the 40S ribosomal subunit. This blocks translation of host transcripts at the initiation stage and may lead to a template-dependent endonucleolytic cleavage of the mRNA transcript and, eventually, host mRNA degradation in infected cells.9 , 10 , 11 , 12 , 13 Nsp1-dependent translational inhibition represses the ability of human lung epithelial cells to respond to interferon (IFN)-β stimulation, thus inactivating virus- and IFN-dependent signaling pathways and resulting in the attenuation of the innate immune response.14 , 15
Figure 1.

Solution NMR structure of Nsp1 from SARS-CoV-2
(A) Domain structure and ribbon representation of the final water-refined ensemble of 10 NMR structures of full-length Nsp1. The short N-terminal and long C-terminal tails are largely disordered (colored in turquoise and purple, respectively). The globular NTD, comprising residues 10–127, is colored in gray. Coordinates have been deposited in the PDB with accession code 8AOU (PDB: 8AOU).
(B) Ribbon representation of the closest-to-the-mean structure for the NTD alone. The β-strands are colored in orange, the three helices are in light blue, and segments not adopting canonical secondary structure are in gray. The secondary-structure elements are labeled sequentially.
(C) Ribbon representation of Nsp1 with the hydrophobic residues that comprise the core of the β-barrel shown as sticks. The three layers are colored in green, red, and light brown. L46, which stabilizes the opening layer of β-barrel, is shown in light blue.
The viral mRNA evades Nsp1-mediated translation inhibition through as-yet unknown mechanisms that have been proposed to rely on the association between the N-terminal domain (NTD) of Nsp1 (defined here as residues 10–127; Figure 1A) and the SARS-CoV-2 5′ UTR.12 , 14 , 16 , 17 , 18 , 19 , 20 However, the viral 5′ UTR appears to have no affinity for either free Nsp1 or any component of the ribosome.20 Deletion of either the NTD or the CTD or mutations in both regions substantially reduce the impact of Nsp1 on viral load and suppression of host gene expression, underlining the significance of both the Nsp1-NTD and the C-terminal tail in sustaining the activity of Nsp1 as a major virulence factor.12 , 13 , 15 , 19 , 21 , 22 , 23
There have been several structural studies on Nsp1 from SARS-CoV-1 (PDB: 2GDT, 2HSX 24) and SARS-CoV-2 (PDB: 7K7P, 7K3N, and 7EQ4 25 , 26 , 27), but all of the corresponding structures represent only the globular core and end at residue 126 or 127. Cryoelectron microscopy (cryo-EM) studies on Nsp1 from SARS-CoV-2 in complex with the 40S ribosomal subunit have shown that the CTD forms two α-helices stably bound inside the ribosomal mRNA entry channel,12 , 13 , 17 , 28 but the structure does not extend further toward the N terminus of the protein. Recently, the nuclear magnetic resonance (NMR)-derived secondary-structure determination of full-length Nsp1 (FL-Nsp1) has shown that the C-terminal tail (residues 128–180, linker + CTD; Figure 1A) remains largely disordered in solution, although helix-forming propensity was detected in the region that forms the helical hairpin in the ribosome mRNA tunnel.29 Up to now, there has been no complete atomic-resolution structure of FL-Nsp1 from either SARS-CoV-1 or SARS-CoV-2.
Here, we report the high-resolution solution structure of FL-Nsp1 from SARS-CoV-2. The FL protein retains its globular core domain formed by the various truncated forms of the protein (NTD, residues 10–127; Figure 1A): the α/β fold comprising a six-stranded, capped β-barrel motif.24 Both the short N-terminal and long C-terminal tails (residues 1–9 and 128–180, respectively) are mostly unstructured and flexible, but the NMR spectra reveal the presence of interactions between the tails and the globular core domain. The structure reveals that—in the FL protein—the positively charged patch comprising the end of the β6-strand, which has been demonstrated to be critical for RNA binding,11 , 19 , 23 , 24 , 30 , 31 interacts with the highly acidic C-terminal tail, effectively occluding the RNA-binding surface from intermolecular interactions. From the perspective of drug design, our study indicates that the RNA-binding surface of Nsp1 is not completely accessible in the free protein and that competition with the long C-terminal tail for this binding site needs to be taken into account in the design of novel therapeutics targeting free Nsp1.
Results
Structure determination of FL-Nsp1 from SARS-CoV-2
We used NMR spectroscopy to determine the structure of FL-Nsp1 in solution. The protein was expressed with 13C,15N-labeling and purified as described previously.29 The rotational correlation time estimated from backbone 15N relaxation experiments32 , 33 indicated that Nsp1 is a monomer in solution (vide infra). Nearly complete backbone and side-chain resonance assignments were determined using standard triple-resonance NMR methods. The backbone assignment has been reported previously.29 In brief, we were able to obtain 1H and 15N assignments for 99.4% and 93.9% backbone amide groups and 13C assignments for 96.7%, 100%, and 100% of the C′, Cα, and Cβ nuclei, respectively. Amide assignments are missing for residue G127. The side-chain 1H resonances were assigned to a completeness of 98.4%. NOEs (nuclear Overhauser effects) were derived from standard 3D NOESY-15N-HSQC and NOESY-13C-HSQC spectra. The final data collection statistics and structural quality scores are shown in Tables 1 and 2 . The structure closest to the mean was chosen as the representative structure of the ensemble and used in all figures unless stated otherwise.
Table 1.
Assignment and restraint statistics
| Assignment coverage | ||||||
| Overall, by element (%) | ||||||
| H | C | N | ||||
| 98.9 | 91.6 | 75.4 | ||||
| Backbone, by nucleus type (%) | ||||||
| HN | N | C′ | Cα/Cβ | Hα | Overall | |
| 99.4 | 93.9 | 96.7 | 100 | 100 | 98.3 | |
| Side chain, by element (%) | ||||||
| H | C/N | |||||
| 98.4 | 79.3 | |||||
| Restraint counts | ||||||
| NOE distance restraints | ||||||
| Intra-res. | Sequential | Short range | Med. range | Long range | Total | |
| Unambig | 1,484 | 813 | 283 | 74 | 783 | 3,437 |
| Ambig. | 528.0 | 472.4 | 309.6 | 93.8 | 797.2 | 2,201 |
| Overall | 2,012 | 1,285.4 | 592.6 | 167.8 | 1,580.2 | 5,638 |
| Backbone dihedral restraints | ||||||
| φ | ψ | Total | ||||
| 94 | 94 | 188 | ||||
| Hydrogen-bond restraints | ||||||
| H to O | N to O | Total | ||||
| 51 | 38 | 89 | ||||
| RDC restraints (backbone N–H) | ||||||
| Tight | Loose | Total | ||||
| 73 | 16 | 89 | ||||
NOE distance restraints are classified by sequence-separation (Δi): intra-residue (Δi = 0); sequential (Δi = 1); short range (2 ≤ Δi ≤ 3); medium range (4 ≤ Δi ≤ 5); and long range (Δi > 5). Intra-res., intra-residue; Med., medium; Unambig., unambiguous; Ambig., ambiguous.
Table 2.
Structural quality parameters
| Structural RMSDs (fitting to mean) | Ensemble average (±SD) | Representative structure |
|---|---|---|
| Structural RMSDs (fitting to mean) | ||
| Backbone (Å) | 0.57 (±0.11) | 0.43 |
| Heavy atoms (Å) | 0.99 (±0.09) | 0.92 |
| RMSDs from experimental restraints | ||
| NOE distances (Å) | 0.02897 (±0.00091) | 0.02773 |
| H-bond distances (Å) | 0.0142 (±0.0029) | 0.0129 |
| Dihedral angles (°) | 0.977 (±0.087) | 1.012 |
| RDCs, tight (Hz) | 0.894 (±0.066) | 0.881 |
| RDCs, loose (Hz) | 2.92 (±0.19) | 2.63 |
| Violation counts | ||
| NOE distances >0.5 Å | 0.10 (±0.30) | 0 |
| NOE distances >0.3 Å | 10.9 (±2.3) | 7 |
| NOE distances >0.1 Å | 103.5 (±4.4) | 104 |
| H-bond distances >0.5 Å | 0 (±0) | 0 |
| H-bond distances >0.5 Å | 0 (±0) | 0 |
| H-bond distances >0.5 Å | 0.6 (±0.5) | 1 |
| Dihedral angles >10° | 0 (±0) | 0 |
| Dihedral angles >5° | 2.00 (±0.63) | 2 |
| RMSDs from idealized geometry | ||
| Bond lengths (Å) | 0.004177 (±0.000084) | 0.004083 |
| Bond angles (°) | 0.567 (±0.013) | 0.558 |
| Improper angles (°) | 1.726 (±0.056) | 1.760 |
| Ramachandran plot summary (PROCHECK) (%) | ||
| Most favored | 84.5 | 71.3 |
| Additionally allowed | 13.9 | 24 |
| Generously allowed | 1.1 | 1.3 |
| Disallowed | 0.5 | 3.3 |
The first column lists average values for the ensemble (with standard deviations in parentheses), while the second column lists the corresponding values for the representative structure (closest-to-the-mean). Structural RMSDs were calculated over residues 12–126.
The solution NMR structure of FL-Nsp1 includes a short unstructured N-terminal region (N-terminal tail) (residues 1–9), followed by a folded globular domain (NTD, residues 10–127), and finally a long and largely disordered C-terminal tail (linker + CTD, residues 128–180; Figure 1A; PDB: 8AOU). Canonical secondary-structure elements of the folded core of Nsp1 are labeled sequentially as β1, 310, α1, β2, α2, β3, β4, β5, and β6 (Figure 1B). The globular domain is defined by the NOEs and the residual dipolar couplings (RDCs) to a precision of 0.8 Å (backbone root-mean-square deviation [RMSD] calculated over residues 10–127 for the 10 lowest-energy structures). This domain features an α/β fold formed by three helices and a six-stranded β-barrel motif, which is composed of three pairs of antiparallel β-strands: β2 (51–54) and β5 (104–109); β3 (68–73) and β4 (84–91); and β1 (13–20) and β6 (116–124). α1 (residues 35–48) acts as a “cap,” covering one opening of the barrel, while the 310-helix (residues 23–25) and α2 (residues 58–63) are oriented parallel to and alongside the β-barrel. The residues that form the core of the β-barrel are predominantly hydrophobic. The side chains of residues L16, L18, V69, L88, L107, and L123 form one of the two edging layers of the β-barrel, which is also stabilized by L46 in α1 donating its side chain to the center of this layer. Residues L53, I71, V86, and V121 contribute their side chains to form the central layer of the β-barrel, while the other edging layer is comprised of residues V84 and L104 (Figure 1C).
Comparison with solution NMR structure of Nsp113−127 from SARS-CoV-1 and crystal structures of the globular core domain of Nsp1 from SARS-CoV-2
SARS-CoV-2 Nsp1 shows approximately 84% amino acid sequence identity with its homologous protein from SARS-CoV-1 (Figure S1A). Tertiary structural alignment of the two globular domains reveals a few differences, for example in the conformations of the major loops (Figure S1B). However, the conformations of loops 1 and 2 are variable in the NMR ensemble of SARS-CoV-2 Nsp1 (Figures 1A and S1C) and also differ between the representative structure of the NMR ensemble and the available crystal structures of SARS-CoV-2 Nsp1 (PDB: 7K7P, 7K3N, and 7EQ4; Figures S1D and S1E). Thus, we conclude that the differences observed between the conformations of the SARS-CoV-2 and SARS-CoV-1 loops are likely due to the internal flexibility of these loops rather than to differences in the primary sequence.
In the crystal structures of the SARS-CoV-2 Nsp1-NTD, a one-residue extension of strand β4 is associated with the appearance of an additional β-strand in loop 2, named β5 (residues 95 to 97), which, together with β4, forms a short β-hairpin25 (Figure S1E). The emergence of the extra β sheet may be due to the influence of intermolecular contacts in the crystal lattice; in solution, this loop does not appear to form a canonical β-hairpin.
Comparing the electrostatic surface of the SARS-CoV-2 Nsp1-NTD with that of SARS-CoV-1 Nsp113−127 shows that the two proteins have similar surface potentials, even in regions where the primary sequences differ the most, such as loop 1 (Figures S2A–S2C). However, some deviations between the electrostatic surfaces are brought about by the different conformations of the loops 1 and 2. One striking difference caused by the flexibility of loop 2 is the emergence of a negatively charged pocket in SARS-CoV-2 Nsp1 formed by residues S74, E87, G98, and S100 (Figures S2D–S2F). The presence of this negative pocket is highly dependent on the position of S100 in loop 2. In conformers where the S100 side chain is far from the side chains of S74 and E87, this negatively charged pocket disappears (Figures S1C and S2G). In the crystal structure of the SARS-CoV-2 Nsp1-NTD, the negative patch is still present but with reduced size.
Interactions between the Nsp1-NTD and the disordered terminal tails
Our initial NMR studies of SARS-CoV-2 Nsp1 yielded spectra whose appearances were characteristic of a protein consisting of a well-folded globular domain followed by an extended, largely disordered C-terminal tail.29 To further analyze the dynamics of the terminal regions, we performed the standard set of backbone 15N-relaxation experiments (15N R 1, 15N R 2 (R 1ρ), and {1H}15N heteronuclear NOE; Figure S3; Table S1) and analyzed the relaxation rates within the Lipari-Szabo model-free framework.34 , 35 The isotropic rotational correlation time, as calculated from the subset of R 2/R 1 ratios for amide groups with limited internal motion and without significant exchange broadening (see STAR Methods), is ∼13.5 ns, which is slightly larger than would be expected for a compact globular 20 kDa protein. The corresponding rotational diffusion tensor is only slightly anisotropic, with principal components D xx, D yy, and D zz of 1.12, 1.23, and 1.32 × 107 s−1, respectively. Figure 2A shows the backbone N–H order parameters (S 2) resulting from the model-free analysis, which confirm that the globular domain is rigid with the exception of two loops (loop 1, between β3 and β4, and loop 2, between β4 and β5), whose lower S 2 values (0.6–0.7) reveal the presence of enhanced local flexibility relative to the remainder of the core domain. The N-terminal and C-terminal tails (residues 1–9 and 128–180) are instead dynamic (S 2 values in the range 0.2–0.3). However, some regions of the C-terminal tail (130–144 and 154–163) appear to experience partial restrictions on their local flexibility, which could arise from either transient secondary-structure formation, weak interactions with the NTD, or a combination of the two. Upon interaction with the ribosome, two helices are formed comprising residues 154–163 and 171–176. In free Nsp1, backbone chemical shifts indicate a clear helical propensity only for residues 171–176,29 which, on the other hand, have very low order parameters. The backbone chemical shifts of residues 154–163 are not indicative of secondary-structure formation,29 and thus their higher-order parameters are likely due to intra-molecular interactions.
Figure 2.

The C-terminal tail is dynamic but interacts with the NTD
(A) Top: order parameters (S2) for backbone amide N–H groups derived from 15N-relaxation data. An N–H group that is totally rigid in the molecular frame has an S2 value of 1, while a group with completely unrestricted motion has an S2 value of 0. Bottom: probabilities (P(ss)) of α-helical (upward-pointing red bars) and β-strand (downward-pointing green bars) secondary structure predicted by TALOS-N from the backbone chemical shifts.
(B and C) Long-range NOE contacts (black dashed lines) between the short N-terminal tail and the NTD.
(D–F) Long-range NOE contacts between the NTD and the long C-terminal tail. The dashed lines are labeled with the corresponding distances in the closest-to-the-mean structure.
Interactions between the tails and the globular domain were revealed in the NOESY spectra (although there are no contacts between the two flexible tails themselves). Long-range NOEs were detected from the short N-terminal tail to the globular core domain between the side chains of L4 and L122 (Figure 2B) and between V5 and T78 (Figure 2C). Similarly, we could also detect three sets of unambiguous NOEs between the long flexible C-terminal tail and the globular NTD. For example, residue W161 experiences contacts with residues R43 and K125, the latter of which belongs to the conserved eight amino acid sequence (LRKxGxKG) that was shown to be important for the RNA-binding activity of Nsp1 proteins (Figure 2D).11 , 19 , 24 , 31 Residue L177, close to the very end of the CTD, contacts residue V35 of the core domain (Figure 2E). Similarly, another long-range NOE was observed between the side chains of the NTD V89 and A131 in the linker region (Figure 2F). These three sets of unambiguous NOEs represent common points of contact between the C-terminal tail and the globular NTD that are reproduced in all structures of the ensemble. In addition to these unambiguous NOEs, we could detect numerous ambiguous NOEs with contributions from proton pairs that correspond to long-range NOEs between the C-terminal tail and the globular core. The combination of the common points of contact at A131, W161, and L177 with the other more fuzzy contacts reflected in the ambiguous NOEs produces an ensemble of structures where the C-terminal tail is always close to the surface of the NTD but in which its conformation over the long stretches in between the common points of contact varies over the ensemble.
Despite the presence of these contacts between the NTD and the N- and C-terminal tails of Nsp1, the structural ensemble clearly shows that neither the secondary nor the tertiary structures of the short N-terminal and long C-terminal tail are well defined. This is in agreement with the 15N-relaxation data, which indicate that the two tails are flexible. The overall picture that emerges is of a C-terminal tail with significant local flexibility but engaging in interactions with the globular NTD. Interestingly, the CTD residues forming the two helices interacting with the 40S ribosomal subunit in the cryo-EM structure of Nsp1 appear to be mostly disordered in free Nsp1 and tend to fall back onto the surface of the Nsp1-NTD, as revealed by the points of contact at W161 and L177. While the NMR structural ensemble presented here clearly reveals the presence of multiple and widely distributed interactions between the flexible C-terminal tail and the globular NTD, the degree of flexibility of the tail backbone revealed by the relaxation data indicates that the tail samples, on a fast timescale, a wide conformational space that may be only partially represented in the NMR ensemble.
Free Nsp1 has no affinity for RNAs derived from the 5′ UTR region of the SARS-CoV-2 genome
The 40S ribosomal subunit has been suggested to be critical for mediating association of Nsp1 with the 5′ UTR of viral mRNA.20 , 23 However, Shi et al. have also reported a direct interaction of the SARS-CoV-2 5′ UTR RNA with the Nsp1-NTD.17 To test whether Nsp1 is able to bind SARS-CoV-2 5′ UTR RNA or any of its stem-loop elements, we compared the 1H,15N-heteronuclear single quantum coherence (HSQC) spectrum of isolated 15N-labeled Nsp1 with its spectra in the presence of two RNA constructs derived from the 5′ UTR: either stem-loop 1 (SL1; present at 2-fold stoichiometric excess) or SL1–4 (present at equimolar concentration). No significant changes were observed in the 1H,15N-HSQC spectra after the addition of the RNAs, suggesting that neither of these RNAs bind Nsp1 in the respective binary mixtures (Figure S4A). We next tested the binding between Nsp1 and the complete 5′ UTR RNA via an electrophoretic mobility shift assay (EMSA). The migration of 0.3, 0.6, 1.25, and 2.5 μM of the 5′ UTR RNA through a native gel was monitored after incubation with increasing amounts of Nsp1 (0-, 5-, 10-, 20-, and 40-fold excess). No significant binding was observed from this EMSA (Figure S4B), confirming that Nsp1 has no direct binary affinity for the RNAs SL1, SL1–4, or even the FL 5′ UTR RNA of SARS-CoV-2.
Importance of charged surface regions for the potential functional role of Nsp1
Several studies have indicated that Nsp1 is a major virulence factor for viral pathogenicity of SARS-CoV-2, inhibiting host gene expression by binding to the 40S ribosomal subunit through its CTD and inducing cellular mRNA degradation.9 , 10 , 16 , 19 , 30 The host mRNA associates with Nsp1 in a ribosome-dependent manner,23 , 36 and this association requires both the NTD and CTD of Nsp1. Recent cryo-EM studies have demonstrated that inhibition of host protein synthesis by Nsp1 is mediated by insertion of the end of the C-terminal tail into the entrance of the mRNA tunnel in the 40S ribosomal subunit.12 , 13 , 17 , 28 It therefore appears that two distinct functional domains of Nsp1 are required for SARS-CoV-2 to selectively inhibit host protein production: the CTD, as a general inhibitor of mRNA translation, is necessary for translational shutoff, while the NTD is required for host mRNA binding and induction of host mRNA degradation. The mode of interaction of the host mRNA with the Nsp1-NTD in the Nsp1-40S-mRNA complex is still unknown. In addition, the CTD domain is not the sole region responsible for the recruitment of Nsp1 to the 40S ribosomal subunit as both the Nsp1-NTD and the linker domain have been shown to reinforce binding.23 , 36
SARS-CoV-2 mRNAs containing the 5′ UTR are resistant to Nsp1-induced mRNA cleavage as well as to repression of translation.12 , 16 , 17 , 18 , 19 , 20 This function of the 5′ UTR of viral mRNA has been shown to depend on the ribosome-mediated association of the Nsp1 core domain with SL1 of the 5′ UTR.14 , 17 , 19 , 20 In addition, a mutant Nsp1, where the linker domain was extended beyond 20 residues, failed to support viral translation, emphasizing the importance of this spacer in the regulation of Nsp1 activity.17 Despite all these data, the precise mechanism by which SARS-CoV-2 eludes the Nsp1-mediated translation inhibition—thereby switching the host translation machinery from host to viral protein synthesis—and how the association between the Nsp1-NTD and the SARS-CoV-2 5′ UTR supports this mechanism are still unknown.
The consensus amino acid sequence (LRKxGxKG), located at the end of β6 of the Nsp1-NTD, is conserved among Beta coronaviruses (Figure S5). Its positively charged amino acids R124 and K125 have been shown to be crucial for the association of Nsp1 with viral 5′ UTR RNA, as well as with host mRNA.11 , 19 , 23 , 24 , 30 , 31 In our structure, the positively charged surface formed by this conserved sequence (Figures 3A and 3E) is partially masked by both the N- and C-terminal tails (the solvent-accessible surface area drops from 515 Å2 in Nsp110−130 to 262 Å2 in FL-Nsp1; Figure 3). While the N-terminal tail is predominantly hydrophobic, the long C-terminal tail is highly acidic, so the electrostatic potential of the accessible surface of this region in FL-Nsp1 is different from that of the masked patch beneath.
Figure 3.

Electrostatic surface potential and functional surface regions of full-length SARS-CoV-2 Nsp1
(A) Electrostatic surface potential of the closest-to-the-mean structure of the Nsp110−130, i.e., the surface potential that would be observed in the absence of the N- and C-terminal tails. The positively charged putative RNA-binding site made up of the conserved amino acid sequence (LRKxGxKG) is indicated by the black square.
(B–D) Electrostatic surface potential of the closest-to-the-mean structure of full-length Nsp1 (FL-Nsp1) shown from three different viewing-angles. The position of the conserved amino acid sequence (LRKxGxKG) is indicated as before.
(E) Surface/ribbon representation of the structural ensemble of Nsp110−130, with the surface patch corresponding to the eight-residue consensus sequence colored in yellow.
(F–H) Surface/ribbon representations of the structural ensemble of FL-Nsp1 shown from three different views. The NTD is colored gray, and the N- and C-terminal tails are colored turquoise and purple, respectively. Here, the yellow surface patch of the consensus sequence visible in (E) is largely covered by the C-terminal tail.
The interaction between the Nsp1-NTD and the C-terminal tail may have functional relevance in two ways. First, it may serve to protect the long, disordered region from either degradation or non-specific binding before it encounters the 40S ribosomal complex; the intra-molecular interaction is incompatible with binding of residues 148–180 to the ribosome mRNA entry tunnel and thus needs to be disrupted upon recruitment of Nsp1 to the ribosome. Second, the occlusion of positively charged surface patches on the NTD by the C-terminal tail may function to prevent non-specific RNA recruitment to Nsp1 away from the ribosome; this would explain why free FL-Nsp1 has no binding affinity to RNAs corresponding to SL1, SL1–4, or FL 5′ UTR of SARS-CoV-2 (Figure S4), while Shi et al. found that the Nsp1-NTD can directly bind to SARS-CoV-2 5′ UTR RNA.17 Consequently the engagement of the C-terminal tail by the ribosome would make the RNA-binding surface of Nsp1 more accessible, supporting the model proposed by Mendez et al. in which recruitment of Nsp1 by the ribosome is coupled to its recognition of RNA.23
The recognition modes of host mRNA and viral RNA by the Nsp1-NTD remain unknown, as does the explanation of why binding of the viral 5′ UTR RNA to the Nsp1-NTD promotes translation of the viral RNA while binding of the host mRNA to the Nsp1-NTD instead results in mRNA cleavage. It has been proposed that binding of the viral 5′ UTR to the Nsp1-NTD induces a large conformational change in the linker, which ultimately leads to weakening of the interaction of the CTD domain with the mRNA entry site of the 40S particle.20 , 23 Our structure supports this hypothesis. Besides R124 and K125, R99 of loop 2 was also found to be important for binding both types of RNA,23 thus identifying the surface formed by the N-terminal end of β1 and the C-terminal end of β6 and β3, as well as loop 2 as a potential RNA-binding platform (Figure 4 ). Even if this surface contains a negative patch in free Nsp1 (Figures S2D and 4), the rearrangement of loop 2 expected upon RNA binding is likely to cause a conformational change that eliminates the negative patch, as readily seen in other conformers of the ensemble (Figure S2G). In free Nsp1, this surface is flanked on one side by the linker region, as confirmed by the unambiguous NOEs seen between V89 and A131 (Figure 2F). In our structure, the linker region makes many fuzzy contacts with the Nsp1-NTD and shows reduced flexibility with respect to, for example, the residues at the very end of the C-terminal tail. Depending on the secondary and tertiary structures of the RNA binding to this surface, and the exact mode of the interaction, the linker may be either displaced from its location on the Nsp1-NTD or, at a minimum, undergo a conformational change. The nature of this displacement and/or conformational change may determine the different functional consequences of host mRNA or viral 5′ UTR RNA binding to the Nsp1-NTD. It is worth noting that when measuring the weakened binding of mRNA to Nsp1 mutants, deletion of the NTD had similar effects on the association of Nsp1 with both host and viral mRNAs (15- and 17-fold reductions in binding, respectively), while the mutations R124A/K125A and R99A had a stronger effect on the association of Nsp1 with viral mRNA (13- and 7-fold reductions for the R124A/K125A and R99A mutants, respectively) than with the host mRNA (5- and 2-fold reductions for the R124A/K125A and R99A mutants, respectively).23 These data support the hypothesis that the binding mode of the RNA to Nsp1, and the consequences of that binding event on the conformation of the linker region, is sequence-, and thus structure-, dependent. This hypothesis is also in agreement with the observation that not all host mRNAs are subject to Nsp1-induced cleavage.16
Figure 4.

The putative RNA-binding surface of SARS-CoV-2 Nsp1 is flanked by the linker region
Electrostatic potential of the RNA-binding surface defined by residues R99, R124, and R125 (shown in sticks and labeled in large font). Also shown in sticks and labeled in small font are the residues that can form a negative patch, depending on the conformation of loop 2 (S74, E87, G98, and S100), and the residues of the conserved LRKxGxKG motif. An asterisk marks the end of the linker region and the start of the CTD domain.
Discussion
We have presented the structure of FL-Nsp1, which reveals that the two highly disordered terminal tails are prone to fall back onto the surface of the globular core domain largely due to long-range and fuzzy electrostatic interactions. As a result, a large portion of the positively charged surface around the key RNA-binding residues R124 and K125 is not completely accessible in free Nsp1. This provides an explanation for the proposed coupling of RNA binding by the NTD with ribosome binding of the CTD, which necessarily disrupts the interaction of the NTD with the C-terminal tail. Furthermore, the linker region between the NTD and the CTD delineates the RNA-binding surface so that binding of RNA is likely to significantly perturb this region, which may in turn influence the CTD and its interaction with the ribosome. The precise nature of the perturbation would depend on what type of RNA binds to Nsp1, rationalizing the different fates for host mRNA and viral RNA. As ribosome-dependent binding of RNA to Nsp1 is essential for viral replication, the surface region of Nsp1 containing R124 and K125 (and also R99) is an attractive drug target. Our study shows that in free FL-Nsp1, and probably also in the ribosome-bound state, the nature of the accessible surface in this key region is different from the corresponding surface of the isolated globular domain. The structure determined here provides important insights to guide drug-design efforts that target the RNA-binding function of Nsp1.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E.coli BL21(DE3) | Laboratory collection | N/A |
| E.coli Top10 | Laboratory collection | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| 15NH4Cl | Merck (Sigma-Aldrich) | Cat # 299251 |
| 13C6-D-glucose | Merck (Sigma-Aldrich) | Cat # 389374 |
| 6xHis-TEV protease | Prepared in-house | N/A |
| DNAse | New England Biolabs | Cat #M0303S |
| Plasmid Mini Kit | Qiagen | Cat # 12123 |
| Plasmid Mega Kit | Qiagen | Cat # 12181 |
| T7 RNA polymerase | Laboratory collection | N/A |
| Deposited data | ||
| NMR Data of Nsp1 | This Study |
https://bmrb.io/ BMRB:34748 |
| NMR Structure of Nsp1 | This Study |
https://www.wwpdb.org/ PDB: 8AOU |
| Oligonucleotides | ||
| PCR forward primer: ACTGAGAATCTTTATTTTCA G GGCGCCATGGAAAGCCTG GTT CCG GGTT |
Microsynth | N/A |
| PCR reverse primer: GGTGGTGGTGGTGGTGC T CGA GTTAACCACCATTCA GTTCGCGCATCA |
Microsynth | N/A |
| Recombinant DNA | ||
| pETM-11-Nsp1 | This study | N/A |
| pHDV-5-SL1 | Wacker et al.37 | N/A |
| pHDV-5-SL1−4 | Wacker et al.37 | N/A |
| pSP64-5′UTR | Wacker et al.37 | N/A |
| Software and algorithms | ||
| NMRPipe v.10.1 | Delaglio et al.38 | https://www.ibbr.umd.edu/nmrpipe/index.html |
| CcpNmr Analysis v2.4 | Vranken et al.39 | https://ccpn.ac.uk/software/analysisassign/ |
| ARIA v2.3 | Rieping et al.40 | http://aria.pasteur.fr/ |
| Pymol | Schrödinger | https://www.pymol.org/2/ |
| Others | ||
| HisTrap HP column | GE Healthcare | GE17-5247-01 |
| HiLoad Superdex 75 16/600column | GE Healthcare | GE28-9893-33 |
| HiPrep Desalting column with Sephadex G-25 resin | Cytiva | Cytiva 17508702 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Teresa Carlomagno (t.carlomagno@bham.ac.uk).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Full-length Nsp1 was expressed in E. coli BL21(DE3) cells in LB or M9 minimal medium supplemented with 15NH4Cl and 13C6-D-glucose as required.
Method details
Construct design and protein preparation
The construct design, protein expression and purification were carried out as described previously.29
RNA transcription and purification
All RNA sequences were derived from the 5′-UTR region of the SARS-CoV-2 genome37 and comprised either the entire 5′-UTR or stem-loops 1 to 4 (SL1–4) or stem-loop 1 (SL1). The exact sequences are given in Table S2. All RNAs were generated by in-vitro transcription.
Plasmids containing DNA templates were transformed into E. coli Top10 cells; transformed cells were grown in LB medium at 37°C overnight and harvested by centrifugation at 6000 rpm and 4°C for 20 minutes. Plasmids were extracted using the Qiagen Plasmid Mega Kit (Qiagen) according to the manufacturer’s instructions and linearized with HindIII (NEB) before in-vitro transcription by T7 RNA polymerase (prepared in-house). Plasmid DNAs were then purified by phenol/chloroform/isoamyl alcohol and chloroform/isoamyl alcohol (Carl Roth) extraction and precipitated by pure ethanol and 5 M NaCl.
For the transcription of each RNA construct, the concentrations of DNA, nucleoside triphosphates (NTPs, Carl Roth), MgCl2 and T7 polymerase were optimized to maximize the yield. Large-scale transcription reactions were run for 4 to 6 hours at 37°C. RNAs were purified using preparative, denaturing polyacrylamide gels containing 7 M urea, except for the 5′-UTR, which was purified directly after transcription by size-exclusion chromatography (Superdex S200 16/60) to preserve its fold. Purity was verified using analytical denaturing polyacrylamide gels.
Electrophoretic mobility shift assays
Samples for EMSA were prepared by diluting the appropriate volumes of concentrated stock solutions (Table S3) of 5′-UTR (7.7 μM) and Nsp1 (38 and 152 μM) in EMSA buffer (50 mM sodium phosphate (pH 6.5), 50 mM NaCl, 2 mM EDTA, 2 mM DTT; prepared with nuclease-free water) to a total volume of 10 μL, followed by incubation for 30 mins at room-temperature. Four different concentrations of 5′-UTR were used (0.3, 0.6, 1.25 & 2.5 μM), with five samples prepared at each 5′-UTR concentration (corresponding to 0-, 5-, 10-, 20- and 40-fold excess Nsp1), giving 20 samples in total. 2.5 μL 5x loading-dye (50 mM Tris–HCl (pH 7.5), 0.25% xylene cyanol, 0.25% bromophenol blue, 30% glycerol) was added to each sample just prior to loading onto a 5% polyacrylamide gel, which had been pre-run for 1 h with TBE running-buffer (100 mM Tris-HCl (pH 8.3), 100 mM boric acid, 2 mM EDTA). After sample-loading, the gel was run overnight (with the same running-buffer) at 4 °C and a current of 2 mA. The gel was stained in a solution of ethidium bromide (0.25 μg/mL, 2 mins incubation), washed three times in water and then visualized using a Gel Doc XR+ gel documentation system (Bio-Rad).
NMR data acquisition
Sample conditions
Except where specified otherwise, NMR spectra were collected on uniformly 13C,15N-labelled Nsp1 samples at concentrations of 500–700 μM, dissolved in NMR buffer (50 mM sodium phosphate (pH 6.5), 200 mM sodium chloride, 2 mM dithiothreitol, 2 mM ethylene-diamine-tetra-acetic acid, 0.01% w/v sodium azide, 0.001% w/v 3-(trimethylsilyl)propane-1-sulfonate, 10% v/v D2O), and loaded into 3-mm NMR tubes (sample volume ∼180 μL). For the RNA titrations, the sodium chloride concentration in the buffer was reduced to 50 mM. All NMR spectra were measured at a temperature of 298 K.
Instrumentation
All spectra were measured on Bruker AVIII-HD spectrometers running Bruker Topspin software (v3.2), operating at 1H field-strengths of 600 MHz and 850 MHz, and equipped with inverse HCN CPP-TCI (nitrogen-cooled) and CP-TCI (helium-cooled) cryogenic probeheads, respectively.
Assignment experiments
2D 15N-HSQC spectra were recorded using States-TPPI for frequency discrimination, with water suppression achieved via a combination of WATERGATE and water flip-back pulses to preserve the water magnetization.41 , 42 Backbone resonance assignments were obtained as described previously.29 2D 1H–13C correlation spectra for sidechain assignment purposes were recorded as 13C-HSQCs with both real-time and constant-time 13C chemical-shift evolution periods.43 , 44 Separate constant-time 13C-HSQC spectra were recorded for aliphatic and aromatic CH groups, with the length of the constant-time period optimized for the respective 1 J CC coupling constants. Sidechain assignments were obtained from the following set of experiments: 3D aliphatic HC(C)H-TOCSY,45 , 46 3D aromatic HC(C)H-TOCSY, 3D aromatic (H)CCH-TOCSY, 3D H(CCCO)NH,47 , 48 3D HA(CACO)NH and 3D HBHA(CBCACO)NH, 2D (HB)CB(CD)HD and 2D (HB)CB(CDCE)HDH49 (all recorded at 600 MHz, with the exception of the aliphatic HC(C)H-TOCSY, which was recorded at 850 MHz). Stereospecific assignments of Leu and Val methyl groups were obtained from a constant-time 13C-HSQC spectrum (850 MHz) recorded on a sample prepared from cells grown in minimal medium containing a mixture of 10% 13C-glucose and 90% 12C-glucose.50
Structural restraint data
Distance restraints for structure calculation were extracted from 3D NOESY–15N-HSQC51 , 52 and 3D NOESY–13C-HSQC spectra,53 , 54 both recorded at 850 MHz. The NOESY mixing-time was 100 ms. Hydrogen-bond restraints were partly inferred from a CLEANEX experiment,55 recorded at 850 MHz as a pseudo-3D spectrum with incrementation of the CLEANEX mixing-time. Backbone amide N–H residual-dipolar-coupling (RDC) restraints were extracted from two pairs of spectra, both recorded under isotropic (unaligned) and anisotropic (aligned) conditions at 850 MHz using the same sample of 15N-labelled Nsp1: (1) 15N-HSQC and 15N-TROSY-HSQC spectra; and (2) the upfield/downfield sub-spectra generated from an IPAP-15N-HSQC experiment.56 Anisotropic conditions were achieved by addition of Pf1 filamentous bacteriophage (ASLA Biotech, Latvia) to a final concentration of 13.5 mg/mL. The formation and homogeneity of the anisotropic phase was confirmed by inspection of the D2O 2H spectrum, which showed a well-resolved doublet with a splitting of 13.0 Hz. The 15N-HSQC spectrum was recorded using semi-constant-time 15N chemical-shift-evolution together with 1H CPD (WALTZ-16).57 The 15N-TROSY-HSQC and IPAP-15N-HSQC experiments were recorded with Hα/Hβ band-selective decoupling for 15N chemical-shift evolution.58
Relaxation experiments
Relaxation experiments were measured on a solely 15N-labelled sample of Nsp1. Backbone 15N relaxation rates (R 1 and R 1ρ) were measured at 600 MHz using established proton-detected experiments based on a gradient-selected, sensitivity-enhanced, refocused 15N-HSQC sequence.32 , 33 The water signal was preserved using selective water pulses and weak bipolar gradients during the indirect chemical shift evolution time to maintain the water magnetization along the +z axis. The relaxation delays were varied between 10 ms and 1.2 s for the R 1 experiment, and between 3 ms and 120 ms for the R 1ρ experiment. In the R 1 sequence, N–H cross-relaxation pathways were suppressed by application of 1H amide-selective IBURP-1 inversion pulses59 at intervals of 5 ms during the relaxation delay. In the R 1ρ sequence, cross-relaxation was suppressed by application of between one and four 1H amide-selective inversion pulses during the 15N spin-lock relaxation period,60 and the 15N magnetization was explicitly aligned with the spin-lock field,61 which was applied at a 15N field-strength of 2.5 kHz. Backbone {1H}15N steady-state heteronuclear NOEs were measured using the standard method.62 The reference and saturated spectra were recorded in an interleaved fashion, with an inter-scan recycle delay of 6 s for both spectra. Water magnetization was preserved in the reference spectrum as described above. Saturation of the amide proton magnetization was achieved using a train of high-power 180° pulses applied at 10.9-ms intervals for the duration of the inter-scan recycle delay.63 An inter-increment delay of 18 s was added to ensure full recovery of the water magnetization at the start of each increment of the reference experiment.
NMR data processing and analysis
All NMR spectra were processed/visualized with a combination of the software packages NMRPipe v.10.139 and CcpNmr Analysis v2.4.40
CLEANEX spectrum
The final CLEANEX spectrum that was inspected in the process of deriving hydrogen-bond restraints was generated as a 2D projection over the subset of six 2D sub-spectra with mixing-times of 30.41, 40.55, 50.69, 60.82, 81.10 & 101.38 ms.
Extraction of RDCs
Two sets of backbone amide N–H RDCs, and , were calculated from the HSQC/TROSY and IPAP spectra, respectively, according to:
where the various are the 15N chemical-shift positions of the peaks in the four sub-spectra indicated in the respective superscript labels and is the 15N Larmor frequency (−86.151 MHz). Estimated errors in the individual RDCs were calculated based on the following empirical relationship for the estimated error in the frequency position of any individual peak:
where FWHH and SINO are the full-width at half-height (in Hz) and the signal-to-noise of the peak, respectively. RDCs were not calculated for amides where any of the peaks in the four sub-spectra were heavily overlapped, and the estimated error was doubled for amides where any of the peaks were partially overlapped. Final raw RDCs were calculated as averages of the and values where both were available or taken as one or the other when only one value was available. Where the final raw RDC was calculated as the average of the two values, the corresponding error was set either as the SD of the two values if this was larger than both of the estimated errors for the individual values, or otherwise as the average of the estimated errors for the individual values. Scaled RDCs used for fitting of the alignment tensor and as restraints for structure calculation were generated by scaling the raw values by the inverse of the backbone order-parameter, S NH, as derived from analysis of the relaxation data (errors were scaled accordingly, and a lower-limit of 0.25 Hz was imposed on the final set of errors for the scaled RDCs). Alignment-tensor parameters were obtained by SVD fitting64 of the scaled RDCs with the software package PALES65; RDCs from amides with order parameters below 0.95 were excluded from the fit.
Relaxation data analysis
Relaxation spectra were processed with partial Lorentzian-to-Gaussian apodization in both frequency dimensions and limited linear-prediction in the 15N dimension, and the corresponding peak intensities quantified by lineshape-fitting with the software package FuDA (D. Flemming Hansen; https://www.ucl.ac.uk/hansen-lab/fuda/). R 1 and R 1ρ rate-constants were calculated by fitting the intensity profiles to mono-exponential decay functions, also with FuDA, and {1H}15N heteronuclear NOEs (hNOEs) were calculated as the ratios of the peak-intensities in the saturated and reference sub-spectra. For subsequent analysis, the pure transverse relaxation rate-constants, R 2, were calculated from the following equation:
θ is the tip-angle of the magnetization-vector, given by:
where ν1 is the field-strength of the spin-lock and Δν is the offset relative to the frequency of the transmitter (both in Hz).
The three relaxation parameters — R 1, R 2 & {1H}15N hNOE — were analyzed within the Model-Free framework, as implemented in the software package TENSOR2.66 Briefly, in the first step, the relaxation parameters for a subset of backbone amides with restricted internal mobility and negligible residual exchange-contributions to R 2 are used to determine a global anisotropic rotational diffusion tensor. The second step yields the parameters describing the internal mobility for all backbone amides by fitting the relaxation parameters to the Model-Free34 and Extended Model-Free67 spectral-density functions, using the diffusion-tensor parameters determined in the first step. For the first step, amides with restricted internal mobility were defined according to the hNOE; only amides with hNOE values higher than 0.62 were retained. Amides with residual exchange-contributions to R 2 were identified from within the subset of those with restricted internal mobility by reference to the exchange-indicator statistic, Ex Ind, calculated as:
where T 1 = 1/R 1 and T 2 = 1/R 2. Amides for which Ex Ind < σ[Ex Ind], where σ[ Ex Ind] is the SD of the exchange-indicator statistic over the considered subset, were considered to have negligible exchange-contributions to R 2 and retained for diffusion-tensor fitting. The backbone order-parameter used for filtering the RDC data, S NH, was calculated according to:
where and are the squares of the order-parameters for fast and slow internal motions derived from the analysis of the relaxation data.
Structure calculation
Structures were calculated with the software package ARIA v2.3,40 which uses CNS v1.2168 for simulated-annealing molecular-dynamics. All restraint tables and NOESY peak-lists have been included in the BMRB submission (BMRB: 34748).
Distance restraints
Distance restraints were generated from NOE peak-heights using the ARIA internal relaxation-matrix calibration routine to account for the effects of spin-diffusion. The majority of NOE cross-peaks in the NOESY–15N-HSQC and NOESY–13C-HSQC spectra were manually assigned in the two HSQC dimensions; the NOESY dimension was left unassigned for automatic assignment by ARIA. Some heavily overlapped peaks were left unassigned in all dimensions. For each NOESY spectrum, the cross-peaks were divided into three lists: (1) peaks for which the HSQC assignments were to residues in the globular domain; (2) peaks for which the HSQC assignments were to residues in the flexible tails; and (3) peaks that were unassigned in all dimensions. For this purpose, the globular domain was taken to comprise residues 8–127, with the remaining residues (0–7 and 128–180) constituting the flexible tails. Each peak-list was converted to distance restraints using a separate calibration; the rotational correlation times required for the calibrations were set to 13.5, 4.5 and 9.0 ns for peak-lists (1), (2) and (3), respectively. The peak-heights in peak-lists (1) and (2) from the NOESY-15N-HSQC spectrum were pre-scaled prior to the ARIA run according to the peak-heights of the respective peaks in the 2D 15N-HSQC spectrum:
where and are the unscaled and scaled NOE peak-heights, respectively, is the peak-height of the corresponding amide peak in the 2D 15N-HSQC spectrum and is the average peak-height in that spectrum. Peak-heights in peak-list (3) from the NOESY–15N-HSQC spectrum and in all three peak-lists from the NOESY–13C-HSQC spectrum were unscaled.
Dihedral-angle restraints
The backbone dihedral angles φ and ψ were predicted by TALOS-N69 using the chemical shifts for the nuclei HN, NH, Hα, Cα, Cβ and C'. Dihedral-angle predictions described as “strong” or “generous” according to the TALOS-N classification system were used as dihedral-angle restraints for the structure calculation. The error-bounds for the restraints were calculated by doubling and tripling the TALOS-N estimated errors, with lower limits of 20° and 30°, for “strong” and “generous” predictions, respectively.
RDC restraints
Scaled N–H RDCs were incorporated into the structure calculation as SANI-format restraints. Only RDCs from amides in the globular domain (here defined as residues 7–128) and with backbone order-parameters, S NH, greater than 0.80 were used as restraints. The restraints were divided into two classes depending on the respective values of S NH: (1) S NH ≥ 0.95; and (2) 0.80 ≤ S NH ≤ 0.95. The SANI force-constants k cool1/k cool2 were set to 0.1/0.5 and 0.05/0.25 for classes (1) and (2), respectively (half and quarter as strong as the respective default ARIA settings). The tensor parameters (magnitude, D a, and rhombicity, R) for each structure-calculation run were calculated by SVD fitting of the scaled RDCs to the closest-to-the-mean structure of the previous run; the tensor parameters for the final run were: D a = −8.8 Hz & R = 0.20.
Hydrogen-bond restraints
Where appropriate, hydrogen-bond restraints were imposed between backbone amide and backbone carbonyl groups. The final set of hydrogen-bond restraints was derived in an iterative fashion during the structure-calculation process, by careful inspection for the presence of recognisable hydrogen-bonds in interim structures (involving amide and carbonyl groups between which no hydrogen-bond restraints had been imposed in the generation of the interim structure under inspection) and adhering to the criterion that there was no peak in the 2D CLEANEX spectrum for the corresponding amide-group. Linearity of the restraints was imposed for the subset of identified hydrogen-bonds located in regions of canonical secondary-structure.
Cis-prolines
The 13C chemical-shifts of P67-Cβ (downfield-shifted) and P67-Cγ (upfield-shifted), and the pattern of NOEs between P67-Hα/Hδ and Q66-HN/Hα were indicative of cis geometry for the amide bond between residues Q66 and P67. Therefore, a cis-geometry Q66–P67 amide-bond was imposed during the structure calculation via the standard patch to the force-field parameter-set.
Protocol settings
The following adjustments were made to the default ARIA protocol settings:
-
•
The number of calculated structures per iteration were: 50 structures for it0–it5 and 100 structures for it6–it8. Per-iteration violation analysis and calibration were done using the 10 lowest-energy structures from the previous iteration. The 10 lowest-energy structures from it8 were water-refined to generate the final structural ensemble.
-
•
The number of MD steps for each stage of the simulated-annealing were: steps_high = 30000; steps_cool1 = 15000; and steps_cool2 = 12000.
-
•
The maximum numbers of allowed contributions to an ambiguous distance restraint were increased slightly in the early iterations only: from 20 to 30 in it0 & it1, and from 20 to 25 in it2 & it3.
-
•
Chemical-shift matching tolerances for automatic assignment were set to: 1H[direct/HSQC dimension] = 0.02 ppm; 1H[indirect/NOESY dimension] = 0.04 ppm; 13C/15N = 0.2 ppm.
Quantification and statistical analysis
The NMR structure of full-length Nsp1 was determined using the materials and software listed in the key resources table. Assignment & restraint statistics and structural quality parameters are listed in Tables 1 and 2, respectively. Relaxation parameters and estimated uncertainties generated from backbone 15N relaxation experiments (15N R 1, 15N R 2 (R 1ρ) and {1H}15N heteronuclear NOE) are shown in Figure S3 and listed in Table S1. Backbone N–H order parameters (S 2) resulting from the Model-Free analysis of the relaxation data are shown in Figure 2A.
Acknowledgments
We thank Dr Andrea Graziadei (TU Berlin) for the kind gift of the Nsp1 expression plasmid and Prof. Dr. Harald Schwalbe (Goethe University Frankfurt, BMRZ) and Dr. Julia Weigand (Technical University Darmstadt) for the kind gift of the DNA template plasmids of the 5′ UTR region of the SARS-CoV-2. This work was funded by the Deutsche Forschungsgemeinschaft through grant CA294/16-1 to T.C.
Author contributions
Y.W. and J.K. performed research, analyzed data, and wrote the manuscript; S.z.L. performed research; and T.C. designed and supervised the project, analyzed data, and wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: January 6, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.str.2022.12.006.
Supplemental information
R1, R1ρ and R2 relaxation rate-constants, and {1H}15N heteronuclear NOEs (hNOE). The columns headed “σ[P]” list the associated uncertainties in the values of parameter P. For the R1, R1ρ and R2 rate-constants, the associated uncertainties are derived from the least-squares fitting of the experimental intensity-decay curves to the respective exponential-decay functions as implemented by FuDA (D. Flemming Hansen; https://www.ucl.ac.uk/hansen-lab/fuda/). For the {1H}15N heteronuclear NOEs, the associated uncertainties are calculated from the uncertainties of the peak-intensities in the saturated and reference sub-spectra (according to standard error-propagation formulae); the uncertainties in the peak-intensities themselves are derived from the least-square fitting of the spectral peak-shapes, as implemented by FuDA.
For EMSAs, samples of 5'-UTR and Nsp1 were prepared by diluting the appropriate volumes of their concentrated stock solutions.
Data and code availability
The NMR data of SARS-CoV-2 Nsp1 has been deposited at the BioMagResBank (https://bmrb.io) under accession number 34748 (BMRB: 34748). The atomic coordinates reported in this paper have been deposited in the Protein Data Bank (https://www.wwpdb.org) under accession code 8AOU (PDB: 8AOU). Accession numbers are also listed in the key resources table. Any data reported in this paper that is not included in the above depositions will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.
References
- 1.Hartenian E., Nandakumar D., Lari A., Ly M., Tucker J.M., Glaunsinger B.A. The molecular virology of coronaviruses. J. Biol. Chem. 2020;295:12910–12934. doi: 10.1074/jbc.REV120.013930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paules C.I., Marston H.D., Fauci A.S. Coronavirus infections-more than just the common cold. JAMA. 2020;323:707–708. doi: 10.1001/jama.2020.0757. [DOI] [PubMed] [Google Scholar]
- 3.Skowronski D.M., Astell C., Brunham R.C., Low D.E., Petric M., Roper R.L., Talbot P.J., Tam T., Babiuk L. Severe acute respiratory syndrome (SARS): a year in review. Annu. Rev. Med. 2005;56:357–381. doi: 10.1146/annurev.med.56.091103.134135. [DOI] [PubMed] [Google Scholar]
- 4.Tang Q., Song Y., Shi M., Cheng Y., Zhang W., Xia X.Q. Inferring the hosts of coronavirus using dual statistical models based on nucleotide composition. Sci. Rep. 2015;5:17155. doi: 10.1038/srep17155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masters P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006;66:193–292. doi: 10.1016/s0065-3527(06)66005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan J.F.W., Kok K.H., Zhu Z., Chu H., To K.K.W., Yuan S., Yuen K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020;9:221–236. doi: 10.1080/22221751.2020.1719902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khailany R.A., Safdar M., Ozaslan M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020;19:100682. doi: 10.1016/j.genrep.2020.100682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stadler K., Masignani V., Eickmann M., Becker S., Abrignani S., Klenk H.D., Rappuoli R. SARS--beginning to understand a new virus. Nat. Rev. Microbiol. 2003;1:209–218. doi: 10.1038/nrmicro775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kamitani W., Narayanan K., Huang C., Lokugamage K., Ikegami T., Ito N., Kubo H., Makino S. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc. Natl. Acad. Sci. USA. 2006;103:12885–12890. doi: 10.1073/pnas.0603144103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kamitani W., Huang C., Narayanan K., Lokugamage K.G., Makino S. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat. Struct. Mol. Biol. 2009;16:1134–1140. doi: 10.1038/nsmb.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lokugamage K.G., Narayanan K., Huang C., Makino S. Severe acute respiratory syndrome coronavirus protein nsp1 is a novel eukaryotic translation inhibitor that represses multiple steps of translation initiation. J. Virol. 2012;86:13598–13608. doi: 10.1128/JVI.01958-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schubert K., Karousis E.D., Jomaa A., Scaiola A., Echeverria B., Gurzeler L.A., Leibundgut M., Thiel V., Mühlemann O., Ban N. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 2020;27:959–966. doi: 10.1038/s41594-020-0511-8. [DOI] [PubMed] [Google Scholar]
- 13.Thoms M., Buschauer R., Ameismeier M., Koepke L., Denk T., Hirschenberger M., Kratzat H., Hayn M., Mackens-Kiani T., Cheng J., et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science. 2020;369:1249–1255. doi: 10.1126/science.abc8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banerjee A.K., Blanco M.R., Bruce E.A., Honson D.D., Chen L.M., Chow A., Bhat P., Ollikainen N., Quinodoz S.A., Loney C., et al. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell. 2020;183:1325–1339.e21. doi: 10.1016/j.cell.2020.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wathelet M.G., Orr M., Frieman M.B., Baric R.S. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J. Virol. 2007;81:11620–11633. doi: 10.1128/JVI.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang C., Lokugamage K.G., Rozovics J.M., Narayanan K., Semler B.L., Makino S. SARS coronavirus nsp1 protein induces template-dependent endonucleolytic cleavage of mRNAs: viral mRNAs are resistant to nsp1-induced RNA cleavage. PLoS Pathog. 2011;7:e1002433. doi: 10.1371/journal.ppat.1002433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi M., Wang L., Fontana P., Vora S., Zhang Y., Fu T.M., Lieberman J., Wu H. SARS-CoV-2 Nsp1 suppresses host but not viral translation through a bipartite mechanism. bioRxiv. 2020 doi: 10.1101/2020.09.18.302901. Preprint at. [DOI] [Google Scholar]
- 18.Lapointe C.P., Grosely R., Johnson A.G., Wang J., Fernández I.S., Puglisi J.D. Dynamic competition between SARS-CoV-2 NSP1 and mRNA on the human ribosome inhibits translation initiation. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2017715118. e2017715118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanaka T., Kamitani W., DeDiego M.L., Enjuanes L., Matsuura Y. Severe acute respiratory syndrome coronavirus nsp1 facilitates efficient propagation in cells through a specific translational shutoff of host mRNA. J. Virol. 2012;86:11128–11137. doi: 10.1128/JVI.01700-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tidu A., Janvier A., Schaeffer L., Sosnowski P., Kuhn L., Hammann P., Westhof E., Eriani G., Martin F. The viral protein NSP1 acts as a ribosome gatekeeper for shutting down host translation and fostering SARS-CoV-2 translation. RNA. 2020;27:253–264. doi: 10.1261/rna.078121.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benedetti F., Snyder G.A., Giovanetti M., Angeletti S., Gallo R.C., Ciccozzi M., Zella D. Emerging of a SARS-CoV-2 viral strain with a deletion in nsp1. J. Transl. Med. 2020;18:329. doi: 10.1186/s12967-020-02507-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin J.W., Tang C., Wei H.C., Du B., Chen C., Wang M., Zhou Y., Yu M.X., Cheng L., Kuivanen S., et al. Genomic monitoring of SARS-CoV-2 uncovers an Nsp1 deletion variant that modulates type I interferon response. Cell Host Microbe. 2021;29:489–502.e8. doi: 10.1016/j.chom.2021.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mendez A.S., Ly M., González-Sánchez A.M., Hartenian E., Ingolia N.T., Cate J.H., Glaunsinger B.A. The N-terminal domain of SARS-CoV-2 nsp1 plays key roles in suppression of cellular gene expression and preservation of viral gene expression. Cell Rep. 2021;37:109841. doi: 10.1016/j.celrep.2021.109841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Almeida M.S., Johnson M.A., Herrmann T., Geralt M., Wüthrich K. Novel beta-barrel fold in the nuclear magnetic resonance structure of the replicase nonstructural protein 1 from the severe acute respiratory syndrome coronavirus. J. Virol. 2007;81:3151–3161. doi: 10.1128/JVI.01939-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clark L.K., Green T.J., Petit C.M. Structure of nonstructural protein 1 from SARS-CoV-2. J. Virol. 2021;95 doi: 10.1128/JVI.02019-20. e02019-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Semper C., Watanabe N., Savchenko A. Structural characterization of nonstructural protein 1 from SARS-CoV-2. iScience. 2021;24:101903. doi: 10.1016/j.isci.2020.101903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao K., Ke Z., Hu H., Liu Y., Li A., Hua R., Guo F., Xiao J., Zhang Y., Duan L., et al. Structural basis and function of the N terminus of SARS-CoV-2 nonstructural protein 1. Microbiol. Spectr. 2021;9:e0016921. doi: 10.1128/Spectrum.00169-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan S., Peng L., Park J.J., Hu Y., Devarkar S.C., Dong M.B., Shen Q., Wu S., Chen S., Lomakin I.B., Xiong Y. Nonstructural protein 1 of SARS-CoV-2 is a potent pathogenicity factor redirecting host protein synthesis machinery toward viral RNA. Mol. Cell. 2020;80:1055–1066.e6. doi: 10.1016/j.molcel.2020.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y., Kirkpatrick J., Zur Lage S., Korn S.M., Neißner K., Schwalbe H., Schlundt A., Carlomagno T. (1)H, (13)C, and (15)N backbone chemical-shift assignments of SARS-CoV-2 non-structural protein 1 (leader protein) Biomol. NMR Assign. 2021;15:287–295. doi: 10.1007/s12104-021-10019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakagawa K., Lokugamage K.G., Makino S. Viral and cellular mRNA translation in coronavirus-infected cells. Adv. Virus Res. 2016;96:165–192. doi: 10.1016/bs.aivir.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Terada Y., Kawachi K., Matsuura Y., Kamitani W. MERS coronavirus nsp1 participates in an efficient propagation through a specific interaction with viral RNA. Virology. 2017;511:95–105. doi: 10.1016/j.virol.2017.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dayie K., Wagner G. Relaxation-rate measurements for 15N− 1H groups with pulsed-field gradients and preservation of coherence pathways. J. Magn. Reson. Ser. A. 1994;111:121–126. doi: 10.1006/jmra.1994.1236. [DOI] [Google Scholar]
- 33.Farrow N.A., Muhandiram R., Singer A.U., Pascal S.M., Kay C.M., Gish G., Shoelson S.E., Pawson T., Forman-Kay J.D., Kay L.E. Backbone dynamics of a free and a phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 34.Lipari G., Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 1982;104:4546–4559. doi: 10.1021/ja00381a009. [DOI] [Google Scholar]
- 35.Lipari G., Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J. Am. Chem. Soc. 1982;104:4559–4570. doi: 10.1021/ja00381a010. [DOI] [Google Scholar]
- 36.Graziadei A., Schildhauer F., Spahn C., Kraushar M., Rappsilber J. SARS-CoV-2 Nsp1 N-terminal and linker regions as a platform for host translational shutoff. bioRxiv. 2022 doi: 10.1101/2022.02.10.479924. Preprint at. [DOI] [Google Scholar]
- 37.Wacker A., Weigand J.E., Akabayov S.R., Altincekic N., Bains J.K., Banijamali E., Binas O., Castillo-Martinez J., Cetiner E., Ceylan B., et al. Secondary structure determination of conserved SARS-CoV-2 RNA elements by NMR spectroscopy. Nucleic Acids Res. 2020;48:12415–12435. doi: 10.1093/nar/gkaa1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 39.Vranken W.F., Boucher W., Stevens T.J., Fogh R.H., Pajon A., Llinas M., Ulrich E.L., Markley J.L., Ionides J., Laue E.D. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 40.Rieping W., Habeck M., Bardiaux B., Bernard A., Malliavin T.E., Nilges M. ARIA2: automated NOE assignment and data integration in NMR structure calculation. Bioinformatics. 2007;23:381–382. doi: 10.1093/bioinformatics/btl589. [DOI] [PubMed] [Google Scholar]
- 41.Bodenhausen G., Ruben D.J. Natural abundance nitrogen-15 NMR by enhanced heteronuclear spectroscopy. Chem. Phys. Lett. 1980;69:185–189. doi: 10.1016/0009-2614(80)80041-8. [DOI] [Google Scholar]
- 42.Piotto M., Saudek V., Sklenár V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 43.Santoro J., King G.C. A constant-time 2D overbodenhausen experiment for inverse correlation of isotopically enriched species. J. Magn. Reson. 1992;97:202–207. doi: 10.1016/0022-2364(92)90250-B. [DOI] [Google Scholar]
- 44.Vuister G.W., Bax A. Resolution enhancement and spectral editing of uniformly 13C-enriched proteins by homonuclear broadband 13C decoupling. J. Magn. Reson. 1992;98:428–435. doi: 10.1016/0022-2364(92)90144-V. [DOI] [Google Scholar]
- 45.Bax A., Clore G., Gronenborn A.M. 1H correlation via isotropic mixing of 13C magnetization, a new three-dimensional approach for assigning 1H and 13C spectra of 13C-enriched proteins. J. Magn. Reson. 1990;88:425–431. doi: 10.1016/0022-2364(90)90202-K. [DOI] [Google Scholar]
- 46.Kay L.E., Xu G.-Y., Singer A.U., Muhandiram D.R., FORMAN-KAY J.D. A gradient-enhanced HCCH-TOCSY experiment for recording side-chain 1H and 13C correlations in H2O samples for proteins. J. Magn. Reson. Ser. A. 1993;101:333–337. doi: 10.1006/jmrb.1993.1053. [DOI] [Google Scholar]
- 47.Logan T.M., Olejniczak E.T., Xu R.X., Fesik S.W. Side chain and backbone assignments in isotopically labeled proteins from two heteronuclear triple resonance experiments. FEBS Lett. 1992;314:413–418. doi: 10.1016/0014-5793(92)81517-. [DOI] [PubMed] [Google Scholar]
- 48.Montelione G.T., Lyons B.A., Emerson S.D., Tashiro M. An efficient triple resonance experiment using carbon-13 isotropic mixing for determining sequence-specific resonance assignments of isotopically-enriched proteins. J. Am. Chem. Soc. 1992;114:10974–10975. doi: 10.1021/ja00053a051. [DOI] [Google Scholar]
- 49.Yamazaki T., Forman-Kay J.D., Kay L.E. Two-Dimensional NMR experiments for Correlatingˆ 1ˆ 3C andˆ 1H/chemical shifts of aromatic residues inˆ 1ˆ 3C-labeled proteins via scalar couplings. J. Am. Chem. Soc. 1993;115:11054–11055. doi: 10.1021/ja00076a099. [DOI] [Google Scholar]
- 50.Neri D., Szyperski T., Otting G., Senn H., Wüthrich K. Stereospecific nuclear magnetic resonance assignments of the methyl groups of valine and leucine in the DNA-binding domain of the 434 repressor by biosynthetically directed fractional carbon-13 labeling. Biochemistry. 1989;28:7510–7516. doi: 10.1021/bi00445a003. [DOI] [PubMed] [Google Scholar]
- 51.Fesik S.W., Zuiderweg E.R. Heteronuclear three-dimensional NMR spectroscopy. A strategy for the simplification of homonuclear two-dimensional NMR spectra. J. Magn. Reson. 1988;78:588–593. doi: 10.1016/0022-2364(88)90144-8. [DOI] [Google Scholar]
- 52.Marion D., Kay L.E., Sparks S.W., Torchia D.A., Bax A. Three-dimensional heteronuclear NMR of nitrogen-15 labeled proteins. J. Am. Chem. Soc. 1989;111:1515–1517. doi: 10.1021/ja00186a066. [DOI] [Google Scholar]
- 53.Ikura M., Kay L.E., Tschudin R., Bax A. Three-dimensional NOESY-HMQC spectroscopy of a 13C-labeled protein. J. Magn. Reson. 1990;86:204–209. doi: 10.1016/0022-2364(90)90227-Z. [DOI] [Google Scholar]
- 54.Muhandiram D., Farrow N., XU G.-Y., Smallcombe S., Kay L. A gradient 13C NOESY-HSQC experiment for recording NOESY spectra of 13C-labeled proteins dissolved in H2O. J. Magn. Reson. Ser. A. 1993;102:317–321. doi: 10.1006/jmrb.1993.1102. [DOI] [Google Scholar]
- 55.Hwang T.-L., Mori S., Shaka A.J., van Zijl P.C.M. Application of phase-modulated CLEAN chemical EXchange spectroscopy (CLEANEX-PM) to detect water− protein proton exchange and intermolecular NOEs. J. Am. Chem. Soc. 1997;119:6203–6204. doi: 10.1021/ja970160j. [DOI] [Google Scholar]
- 56.Ottiger M., Delaglio F., Bax A. Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J. Magn. Reson. 1998;131:373–378. doi: 10.1006/jmre.1998.1361. [DOI] [PubMed] [Google Scholar]
- 57.Ottiger M., Bax A. Determination of relative N− HN, N− C ‘, Cα− C ‘, and Cα− Hα effective bond lengths in a protein by NMR in a dilute liquid crystalline phase. J. Am. Chem. Soc. 1998;120:12334–12341. doi: 10.1021/ja9826791. [DOI] [Google Scholar]
- 58.Yao L., Ying J., Bax A. Improved accuracy of 15N–1H scalar and residual dipolar couplings from gradient-enhanced IPAP-HSQC experiments on protonated proteins. J. Biomol. NMR. 2009;43:161–170. doi: 10.1007/s10858-009-9299-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Geen H., Freeman R. Band-selective radiofrequency pulses. J. Magn. Reson. 1991;93:93–141. doi: 10.1016/0022-2364(91)90034-Q. [DOI] [Google Scholar]
- 60.Korzhnev D.M., Skrynnikov N.R., Millet O., Torchia D.A., Kay L.E. An NMR experiment for the accurate measurement of heteronuclear spin-lock relaxation rates. J. Am. Chem. Soc. 2002;124:10743–10753. doi: 10.1021/ja0204776. [DOI] [PubMed] [Google Scholar]
- 61.Hansen D.F., Kay L.E. Improved magnetization alignment schemes for spin-lock relaxation experiments. J. Biomol. NMR. 2007;37:245–255. doi: 10.1007/s10858-006-9126-6. [DOI] [PubMed] [Google Scholar]
- 62.LI Y.-C., Montelione G. Solvent saturation-transfer effects in pulsed-field-gradient heteronuclear single-quantum-coherence (PFG-HSQC) spectra of polypeptides and proteins. J. Magn. Reson. Ser. A. 1993;101:315–319. doi: 10.1006/jmrb.1993.1049. [DOI] [Google Scholar]
- 63.Ferrage F., Cowburn D., Ghose R. Accurate sampling of high-frequency motions in proteins by steady-state 15N−{1H} nuclear overhauser effect measurements in the presence of cross-correlated relaxation. J. Am. Chem. Soc. 2009;131:6048–6049. doi: 10.1021/ja809526q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Losonczi J.A., Andrec M., Fischer M.W., Prestegard J.H. Order matrix analysis of residual dipolar couplings using singular value decomposition. J. Magn. Reson. 1999;138:334–342. doi: 10.1006/jmre.1999.1754. [DOI] [PubMed] [Google Scholar]
- 65.Zweckstetter M., Bax A. Prediction of sterically induced alignment in a dilute liquid crystalline phase: aid to protein structure determination by NMR. J. Am. Chem. Soc. 2000;122:3791–3792. doi: 10.1021/ja0000908. [DOI] [Google Scholar]
- 66.Dosset P., Hus J.-C., Blackledge M., Marion D. Efficient analysis of macromolecular rotational diffusion from heteronuclear relaxation data. J. Biomol. NMR. 2000;16:23–28. doi: 10.1023/A:1008305808620. [DOI] [PubMed] [Google Scholar]
- 67.Clore G.M., Szabo A., Bax A., Kay L.E., Driscoll P.C., Gronenborn A.M. Deviations from the simple two-parameter model-free approach to the interpretation of nitrogen-15 nuclear magnetic relaxation of proteins. J. Am. Chem. Soc. 1990;112:4989–4991. doi: 10.1021/ja00168a070. [DOI] [Google Scholar]
- 68.Brunger A.T. Version 1.2 of the crystallography and NMR system. Nat. Protoc. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 69.Shen Y., Bax A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR. 2013;56:227–241. doi: 10.1007/s10858-013-9741-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
R1, R1ρ and R2 relaxation rate-constants, and {1H}15N heteronuclear NOEs (hNOE). The columns headed “σ[P]” list the associated uncertainties in the values of parameter P. For the R1, R1ρ and R2 rate-constants, the associated uncertainties are derived from the least-squares fitting of the experimental intensity-decay curves to the respective exponential-decay functions as implemented by FuDA (D. Flemming Hansen; https://www.ucl.ac.uk/hansen-lab/fuda/). For the {1H}15N heteronuclear NOEs, the associated uncertainties are calculated from the uncertainties of the peak-intensities in the saturated and reference sub-spectra (according to standard error-propagation formulae); the uncertainties in the peak-intensities themselves are derived from the least-square fitting of the spectral peak-shapes, as implemented by FuDA.
For EMSAs, samples of 5'-UTR and Nsp1 were prepared by diluting the appropriate volumes of their concentrated stock solutions.
Data Availability Statement
The NMR data of SARS-CoV-2 Nsp1 has been deposited at the BioMagResBank (https://bmrb.io) under accession number 34748 (BMRB: 34748). The atomic coordinates reported in this paper have been deposited in the Protein Data Bank (https://www.wwpdb.org) under accession code 8AOU (PDB: 8AOU). Accession numbers are also listed in the key resources table. Any data reported in this paper that is not included in the above depositions will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.