

Abstract
AI-driven approaches are widely used in drug discovery, where candidate molecules are generated and tested on a target protein for binding affinity prediction. However, generating new compounds with desirable molecular properties such as Quantitative Estimate of Drug-likeness (QED) and Dopamine Receptor D2 activity (DRD2) while adhering to distinct chemical laws is challenging. To address these challenges, we proposed a graph-based deep learning framework to generate potential therapeutic drugs targeting the SARS-CoV-2 protein. Our proposed framework consists of two modules: a novel reinforcement learning (RL)-based graph generative module with knowledge graph (KG) and a graph early fusion approach (GEFA) for binding affinity prediction. The first module uses a gated graph neural network (GGNN) model under the RL environment for generating novel molecular compounds with desired properties and a custom-made KG for molecule screening. The second module uses GEFA to predict binding affinity scores between the generated compounds and target proteins. Experiments show how fine-tuning the GGNN model under the RL environment enhances the molecules with desired properties to generate valid and unique compounds using different scoring functions. Additionally, KG-based screening reduces the search space of generated candidate molecules by while retaining of promising binding molecules against SARS-CoV-2 protein, i.e., 3C-like protease (3CLpro). We achieved a binding affinity score of 8.185 from the top rank of generated compound. In addition, we compared top-ranked generated compounds to Indinavir on different parameters, including drug-likeness and medicinal chemistry, for qualitative analysis from a drug development perspective.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13721-023-00409-2.
Keywords: Binding affinity prediction, Graph neural network, Knowledge graph, Molecule generation, Reinforcement learning
Introduction
Artificial Intelligence (AI) models are being used in a growing number of disciplines and have effectively been used for sequential processes (like text or speech) (Vaswani et al. 2017), and in different image-related tasks (like object detection, classification, etc.) (Ren et al. 2015). These models are also finding applications in the chemistry domain. They are being utilized to improve drug design by narrowing the initial search space in the early phases of drug discovery. Drug discovery (Drews 2000) includes the process of generating new molecular compounds with a particular set of targeted therapeutic potential from scratch. These processes can be accomplished using an AI-based algorithm trained on the required data to create novel compounds with the desired properties.
In contributing to the rise of the COVID-19 global epidemic, researchers have worked hard to discover the existing medications that could be adapted for SARS-CoV-2 diagnosis (Lu 2020; Zhou et al. 2020). However, generating molecules with specific desirable properties for SARS-CoV-2 remains a challenging task for the scientific community. The process of producing new drugs has to go through the distinct aspect of polymer structure space, and also the enormous size of chemical space (Arús-Pous et al. 2019) which makes it rather complicated, time-consuming, and costly. These limitations arise from the fact that the possible chemical space contains over compounds that should ideally be investigated for therapeutic design before discovering a lead for a target protein, and the candidates with appropriate activity against specific proteins only decrease the search space to – structures.
Additionally, a slight change in molecule design can result in a significant difference in the molecular attribute like binding strength. This process can take several years to complete and is relatively inefficient. Therefore, AI-driven workflows should speed up the design process for appropriate drug development that serves as a search-and-screen process of the reduced chemical space as a simplified molecular-input line-entry system
The candidate molecules are first generated by deep generative models and then further verified via drug–protein-binding prediction techniques (Ranjan et al. 2022). Further research has been carried out for molecule generation, where the molecules are recognized as simplified molecular-input line-entry system (SMILES) sequences. When using SMILES, specific crucial chemical properties of a molecule, such as structural information, are lost, affecting the prediction accuracy of a model and the functional validity of the learned latent space. To overcome these limitations, researchers developed models based on heterogeneous graph representations of compounds (Li et al. 2018a, b), which provide a realistic depiction of chemical compounds, where the node represents the atom, and an edge represents the bond between atoms. Different varieties of Graph Neural Networks (GNNs) (Scarselli et al. 2008a) have since been documented in the literature, the majority of which have only recently been applied to molecular compounds (Gilmer et al. 2017a; Kipf and Welling 2016). Another technique relies on reinforcement learning (RL) (Mnih et al. 2015), a kind of artificial intelligence in which models learn how to make decisions to maximize their reward. A wide range of work uses RL algorithms for fine-tuning the generative model with desired properties to produce SMILES string of molecules (Guimaraes et al. 2017; Putin et al. 2018), but a majority of these models use SMILES strings for learning. They were able to produce molecules with the desired properties, but they ran into problems with chemical validity. Although RL-based models provide direct learning on graph structures, still these models are less researched on compound generation tasks. Recently, (Zhou et al. 2019; You et al. 2018) introduced deep RL-based models for generating graph representations of molecules and obtained 100% validity.
Similarly, various graph-based approaches have been proposed for estimating the binding affinity score of medical compounds with a particular protein (Nguyen et al. 2021a; Öztürk et al. 2018; Tsubaki et al. 2019; Torng and Altman 2019; Jiang et al. 2020). To estimate binding affinity, most of these models employ a late fusion technique, in which compounds and protein features are collected separately and then fused. These models leave out the fact that binding happens inside a protein residue rather than throughout the complete protein. When a medicine attaches to a protein, the protein’s function gets altered, resulting in the expected pharmacological effects. These change the protein’s structure and, as a result, its appearance. Similarly, the late fusion approach fails to detect these structural alterations when a compound binds to the protein. It also ignores the concept of site-specific interaction, making it challenging for the model to focus on affiliated sites, resulting in slower learning and less interpretable results. One approach to addressing this problem is presented in (Nguyen et al. 2021b), wherein the description features for a particular drug from its molecular structure are identified. In this approach, the molecular graph representation is included in the protein network graph before the actual protein embedding process begins. It is indeed a graph architecture that is stacked within another graph architecture. Because of the graph-in-graph technique, the model can reflect changes in the protein network graph caused by drug molecule-bond formation.
Continuing with the above concepts, in this work, we proposed a graph-based framework for high-throughput generation of anti-SARS therapeutic candidates. We utilized and extended two ideas into a single unified framework. The first idea incorporates the concept of RL for fine-tuning a pre-trained graph generative model (i.e., GGNN) to generate candidate molecules and later perform molecular screening using a custom-made knowledge graph. The second idea uses the concept of a GEFA-based approach for binding affinity prediction of screened compounds and the target 3C-Like protease.
Methodology
This section presents the technical specifications of our proposed graph-based framework that contains two major modules. The first module of our framework utilizes the RL-based fine-tuning of Gated Graph Neural Network model, which is responsible for generating unique and valid molecules with desired properties using different scoring functions while preserving the chemistry laws. And later, the generated molecules are screened using a custom-made knowledge graph to reduce the search space. The second module is used for the validation of top-ranking candidates against a Graph Early Fusion-based approach (Nguyen et al. 2021b) to assess their potential activity against SARS-CoV-2 viral protein. The flowchart of our proposed framework is shown in Fig. 1.
Fig. 1.

An overview of the proposed framework containing the two modules: RL-based graph generative model for molecule generation followed by knowledge graph-based molecular screening and graph early fusion approach for binding affinity prediction
Gated graph neural network
Graph Neural Networks (GNNs) (Scarselli et al. 2008b) are a type of graph network that has lately gained popularity as a powerful technique for graph representation learning (Kipf and Welling 2016). The graph representation learning feature of GNNs is used to generate a graphical structure of molecules. GNNs can be seen as a generic neural network architecture that takes input in the form of graph-structured data and generates a latent representation of the input data as output, with nodes taking unique values from 1, ..., |V| and edges being paired . The output of graph representation is produced by aggregating the hidden node states received in the various propagation blocks of the graph network.
GNNs work in two stages to map the input graphs to outputs. The first stage is a propagation step in which node representations for each node are computed. The propagation step of GNNs can be seen as a convolution layer in an artificial neural network. Mathematically, a GNN architecture with an L propagation block that uses a non-linear propagation rule can be represented as
| 1 |
where E represents the adjacency matrix, and represents the state of a hidden node.
The second stage is a graph readout function that maps the node representations (or node embedding or node vector) and appropriate labels to each node’s output Y to find a node embedding for the molecular graph. Inspired from (Mercado et al. 2021; Ranjan et al. 2022), a graph-based generative model used in this work consists of two segments:
GNN segment
Global readout segment.
GNN segment
For the GNN segment, we used gated graph neural network (GGNN) (Li et al. 2015) as message-passing neural network (MPNN) (Gilmer et al. 2017b). Mathematical representation of GNN segment for propagation stage and graph readout stage is given below:
- Message-Passing Stage: Messages are sent over the graph network’s various nodes, and are mathematically described as
2
In the above equation, and denote the messages received from neighboring nodes and the current state of the node , respectively. The current neighboring nodes are given by . The edge feature between two adjacent nodes and is given by . and represent message passing and update operations, respectively. The graph readout phase follows the message-passing phase.3 - Graph Readout Stage: Mathematically, the graph readout stage is described as
where g represents resulted graph feature extraction and R stands for readout function, which takes input and output of various node, transforms it, and later generates the graph embedding.4
Global readout segment
A global readout segment comes after the GNN segment. To predict the APD, global readout segment employs node-level data and graph-level data. The global readout segment is organized in a layered multi-layered perceptron (MLP) architecture, with the starting two MLPs generating initial and , which are subsequently concatenated with the graph embedding g. Later, this concatenated matrix is fed into the next MLP block, which returns the final APD after performing concatenation and normalization operations. It is worth mentioning that fterm relies on graph embedding g
| 5 |
| 6 |
| 7 |
| 8 |
| 9 |
| 10 |
where represents the probability that a new node is being added to the existing graph. The likelihood of connecting the graph’s latest inserted node to another current node is stored in . The probability of the graph termination is given by .
The Kullback–Leibler (KL) divergence loss between actual and estimated APDs is used to train the model. At each epoch, the best model that minimized the validation loss was chosen and utilized as that of the prior model for RL framework.
Reinforcement learning framework
Several RL tasks are formulated as Markov decision processes, indicating that the current state has all of the information needed to make a decision. The RL-based models work in a manner that let us say we have given a specific state , and the model must decide which action to take, where S denotes the set of possible states and A(s) represents the set of potential actions for that state. An agent’s policy links a state to the likelihood of each action that has to be taken throughout.
Inspired from the previous work (Olivecrona et al. 2017) for fine-tuning, by updating the agent policy, our goal is to improve the predicted score for the action sequences utilized to generate a molecular graph by updating the agent policy from the pre-trained graph generative model. A visual representation of the RL framework is depicted in Fig. 2. The initial agent policy is retrieved from the prior model (i.e., the GGNN model). Our GGNN model, which estimates an APD based on a given input graph, also parameterizes the policy. For training the agent, policy-based RL is used with the given states and actions. Also, we used a reward-shaping loss function that records the best agent and updates after a few iterations. Mathematically, the best agent reminder (BAR) loss function is given as
| 11 |
where is the hyper-parameter, and denotes the prior model. indicates the action set chosen to generate a molecular graph by the latest agent, . denotes the action set chosen to generate a molecular graph by the best agent, . M represents the set of molecule m that the latest agent generates. represents the set of molecule that the best agent generates. N indicates the total no. of generated molecules collected from every model. Similarly, for every molecule
| 12 |
where is the hyper-parameter. If the given input graph is and the action probability distribution of sampling action is , then . Also is the probability given by the reference model for similar sequence of actions. S(Seq) denotes the generated score for molecule with sequence of actions Seq. The detailed description for training the graph-based RL framework is mentioned in Algorithm 1.
Fig. 2.

Illustration of the RL framework where the agent is updated using the best agent reminder loss function

Scoring function for desired properties
We fine-tuned the prior model and applied two separate scoring functions to improve the potential of our RL framework. The main goal of these scoring functions is to guide the prior model toward molecule generation with specific desired properties.
We first start by calculating a scoring function based on the generated molecules’ QED values. This scoring function leads the prior model toward molecule generation, which is more drug-like. A higher QED value indicates a more drug-like generated compound. This scoring function has a range of 0–1 as its value. Mathematically, it is shown as
| 13 |
where Mol(A) represents the molecules generated via actions A. PVU stands for Properly terminated, Valid, and Unique molecules.
The second scoring function guides the prior model to generate molecules that are likely to be active for DRD2 activity. Again, the range of this scoring function also lies between 0 and 1. For active or inactive molecules, we set a threshold of 0.5, and more specifically
| 14 |
Knowledge graph for molecule screening
To uncover potential therapeutic candidates against SARS-CoV-2 protein, we build a custom knowledge graph to perform screening of the molecules. Our goal is to reduce the number of calculations while improving the accuracy of the drug–target-binding affinity prediction. We begin with collecting the SARS-CoV-2 protein, i.e., 3C-like protease (3CLpro), which is responsible for propagation of the virus inside human cells (Chen et al. 2020) and adding them to our KG. We concentrate on the SARS-CoV-2 protein node and choose the molecules node and protein structure nodes with the least path distance from it. Our KG is defined as an undirected graph , where denotes the set of nodes and denotes the set of edges; m denotes the number of nodes and n the number of edges and . The KG used in this study is built using two entities and their relationships, i.e., drug and protein, and their interactions. These details are saved in a triplet, with each triplet representing the interaction of the two entities. The entities and relations are represented in a continuous vector. The two entities are represented in the form of nodes, where the target protein node’s first-hop neighbor is a protein with a similar structure, the second-hop neighbor is a set of molecules that can bind to similar proteins, and the third hop is a set of generated molecules. The association among drug–protein interaction, drug–drug interaction, and protein–protein interaction is represented in the form of edges. The edges among the two nodes are assigned using two homogeneous similarity matrices, drug–drug and protein–protein similarity matrices, as well as the DTI (Drug–Target Interaction). A visual depiction of our constructed KG network is shown in Fig. 4. The two similarity measures used to construct the KG are described below.
Fig. 4.
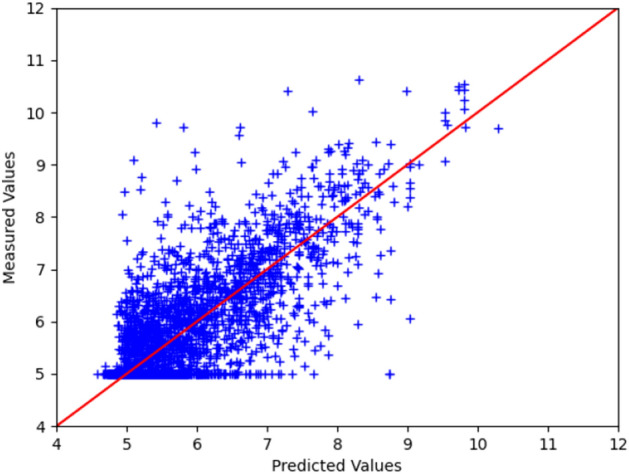
Predicted and measured binding affinity values for the test sets
- Drug–drug similarity measure There are two primary components to drug–drug similarity:
- Molecular descriptors: A metric used to compare the molecular structure.
- Similarity coefficient: A metric for calculating a quantitative score that uses weighted values of structural descriptors.
Protein–protein similarity measure The protein–protein similarity was calculated using the Smith–Waterman algorithm (Yamanishi et al. 2008). By doing local sequencing, the Smith–Waterman method detects similar regions among two strings of amino acids or protein sequence sequences. To build the KG, it is now necessary to define an affinity score threshold as well as a similarity score threshold, both of which will be used to construct a bond among the drug–target and protein–protein graphs. We picked a value of 7.0 ( value of 100 nM) for binding affinity as it is a commonly used DAVIS dataset threshold. The global clustering coefficient metric, which was computed from KG, was utilized to determine the threshold value of protein–protein similarity score.
Fig. 3.
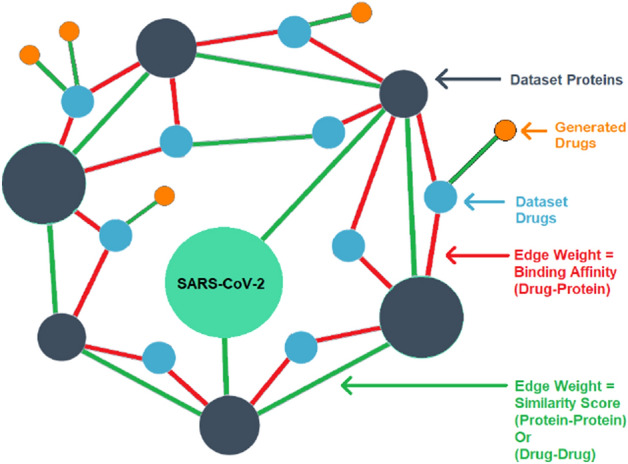
The custom-made Knowledge Graph utilizes two different similarity measures and the Drug–Target interaction dataset. First neighbor hopped of the SARS-CoV-2 protein contains a protein with a 3CLpro structure. The second neighbor hop contains the compound from the dataset that can bind with the first neighbor protein. Finally, the third-hop neighbor contains compounds that are similar to the second-hop neighbor compounds
Drug–target-binding affinity prediction
The potency of both the protein and the drug’s binding force (Ma et al. 2018) is referred to as drug–target-binding affinity. The equilibrium dissociation constant () determines the binding strength. A lower value suggests that the protein and ligand have a stronger binding force (Ma et al. 2018). Estimating the binding affinity score is a regression problem and is mathematically represented as
| 15 |
where F represents the prediction function and represents model parameters, and A represents the affinity score among the target–protein P and given drug D.
In this work, we used the GEFA model (Nguyen et al. 2021b), which takes the drug-graph structure and the target protein-graph structure as input and predicts the binding affinity score. For learning over the graphical representation of drug and target protein, GEFA employs the Graph Convolutional Network (GCN) (Kipf and Welling 2016). The justification for adopting GEFA as a DTA prediction model is that it considers changes in the structural representation of protein graphs caused by drug–protein interactions, which other deep learning models often do not.
First, the drug graph’s feature for a particular drug is extracted. Before the protein-graph learning phase, the extracted drug features are merged into the protein graph. This process can be thought of as a graph-in-graph architecture that enables the training of the deep learning model to learn the changes in protein graphs introduced by the drug–protein interactions. The tertiary structure is represented by 2D contact map information, which is used to represent the protein graph. In the protein network, 2D contact maps show the connections among the two nodes of residue. The secondary structure is essential, since it determines the target protein’s backbone structure, which influences the shape of the binding site. The protein contact map and protein sequence embedding features are then merged to create a graph representation of the target protein. The contextual residue embedding representation is generated using TAPE (Rao et al. 2019) in which an embedded representation is learned from an unlabeled sequence of protein datasets. In particular, TAPE is a protein representation model that uses a sequential language approach. Given a protein residue of size L, the protein’s graph node feature is represented by
| 16 |
where h represents size of TAPE-provided embedding tensor . Every is contextual, that means the residue exists in relation to other residues. As a result, the structural information of protein is implicitly contained in the embedding. Finally, the residue node features within a target protein network are represented using a combination of the two vectors: the embedding vector retrieved by TAPE and the secondary structure feature vector. To represent the drug molecule graph, atoms in the drug molecule are represented as nodes, whereas bonds show the edges among the atoms. The node of a drug molecule contains five features: the symbol of an atom, degree of an atom, connected hydrogen atoms, implicit value of an atom, and aromaticity.
To learn about the changes in protein representation, a graph in graph fusion technique is used to integrate the drug graph and protein graph through a self-attention process. The weights of attention indicate those residues which are highly inclined toward participating in binding with drug nodes. The weights of attention are calculated using the following formula:
| 17 |
where ; shows the feature of ith residue; and represent the trainable parameters.
A cross-domain graph containing the nodes of drugs and their linkage to the target protein residues is constructed. Before graph feature extraction, the drug nodes are separated from the cross-domain graph to assure that the graph only comprises the residue nodes. Finally, the drug and protein embedding are merged and given to a layered FCN (Fully Connected Layer) network for estimating the affinity score.
Experiments and results
We performed a simulation of our framework on the Microsoft Azure platform utilizing the pipeline assets of Azure with Standard_NC64as_T4_v3 as compute instance. With a configuration of 64 cores, 440 GB RAM, and 2816 GB disk, it took around 214 h to complete our simulation successfully.
Dataset details
We used two different datasets for training our framework. For training the RL-based graph generative model, the MOSES dataset was utilized, taken from the MOSES GitHub repository (Polykovskiy et al. 2020), and for training GEFA, DAVIS dataset (Davis et al. 2011) was used. The description of both the datasets is mentioned in Tables 1 and 2.
Table 1.
MOSES dataset description
| Measures | Values |
|---|---|
| Number of training graphs | 3 M |
| Number of testing graphs | 2 M |
| Number of validation graphs | 500 K |
| Atom types | (C, N, O, F, S, Cl, Br) |
| Max. number of nodes in a graph | 27 |
| Formal charges | [0] |
Table 2.
DAVIS dataset description
| Measures | Values |
|---|---|
| Number of compounds | 68 |
| Number of proteins | 442 |
| Max. length of compound | 103 |
| Max. length of protein | 2549 |
| Formal charges | [0] |
Molecule generation setup
We trained an RL-based framework for molecule generation tasks consisting of two major components: GGNN (prior) and RL-based fine-tuning. For training the GGNN, the Adam optimizer is used with no weight decay. We trained the model for a total of 30 epochs. The initial learning rate is set to , and after several iterations, the model achieves a final learning rate of . Since the MOSES dataset is quite large, we fixed the batch size with 1000 sub-graphs. We used SELUs as the activation function. For MLP of GGNN, we fixed the hidden node features to 100, a depth of 4, and hidden features to 250. We have used a total of three message-pass for the message-passing segment. The weights are initialized from Xavier’s normal distribution. To provide the reason behind opting for the GGNN model (as prior) for the molecule generation task, the results in (Ranjan et al. 2022) support the fact that how GGNN performs better than different state-of-the-art models on the MOSES dataset.
For training the agent in RL framework, we used the Adam optimizer and set the initial learning rate of . After several iterations, we found the final learning rate that works best for the agent during training is . For both scoring functions, we employed a batch size of 64 molecules and trained the model for 100 epochs. After a comprehensive examination of the hyper-parameter values, we discovered that the optimal value for is 0.5. For the activity scoring function, the value is set to 20, while for the QED scoring function, the value is set to 10.
Desired properties guided molecule generation via scoring function
The agent was initially trained to optimize molecules with specific properties as a proof of principle, including the scoring functions mentioned in Eqs. 13 and 14. Result obtained from the QED scoring function from both the prior model and the fine-tuned model is shown in Table 3. Later, to explore the agent toward DRD2 activity, we used the second scoring function that maps the agent toward the generation of molecules with predicted biological activity. The obtained results for the DRD2 scoring function are also shown in Table 3.
Table 3.
Comparison of the results obtained from the prior model and that of the fine-tuned model with different evaluation metrics
| Metric | Prior | Fine-tune with QED as scoring function | Fine-tune with DRD2 as scoring function |
|---|---|---|---|
| Valid | 1 | 1 | 1 |
| Unique | 1 | 1 | 0.99 |
| QED | 0.19 | 0.65 | 0.21 |
| DRD2-active | 0.03 | 0.02 | 0.89 |
| Avg. no. of nodes in graph | 21.38 | 21.61 | 21.61 |
Binding affinity prediction
We used the GEFA model trained on the standard benchmark DAVIS dataset for binding affinity prediction. GEFA extracts protein embedding features using TAPE, which uses the BERT model and outputs a protein embedding vector of size 768. The model is trained on mini-batches of size 128 with a learning rate of 0.0005 for 150 epochs. We used Adam as the optimizer. Further, we evaluated GEFA using the DAVIS dataset and compared the model performance with different state-of-the-art methods that use late fusion approaches like DeepDTA (Öztürk et al. 2018), GCNConvNet (Nguyen et al. 2021a), and DGraphDTA (Jiang et al. 2020). We used three other evaluation metrics, namely Concordance Index (CI), Root Mean Squared Error (RMSE), and Mean Squared Error (MSE), to evaluate model performance. The results are shown in Table 4 clearly show that the GEFA outperforms different state-of-the-art methods. The predicted and measured binding affinity values for drug-target pairs for the test sets of the DAVIS dataset are given in Fig. 4. Also the train, test, and validation curves for GEFA model are shown in Fig. 5.
Table 4.
A comparison of the findings received from various state-of-the-art methods with GEFA using DAVIS dataset
Fig. 5.

a GEFA model’s RMSE curves for train, test, and validation, b train, test, and validation curve for MSE metric, and c train, test, and validation curves for CI metric
Use case considering SARS-CoV-2 protein structure
Once we successfully trained our framework, we tested with a viral protein of SARS-CoV-2, i.e., 3CLpro. We used two scoring functions in our RL-based graph framework to fine-tune the prior model. One with the QED scoring function and the second with the DRD2-active scoring function. For each scoring function, we generated 11,647 molecules each, and by combining these two, we have a total list of 23,494 SMILES sequences. We filtered out duplicate molecules and those with a smaller number of elements from the list of generated molecules. We then choose the top 21,000 fruitful SMILE sequences for further processing. The generated candidate SMILES are given input to the custom build KG for screening, and then, the screened molecules are fed into GEFA model along with 3CLpro to predict the binding affinity score.
Only of the 21,000 generated compounds were expected to have a binding affinity with the 3CLpro. After screening the compounds with KG, 705 candidate molecules were chosen, with having a binding affinity greater than 7.0. As shown in Fig. 6, out of 671 binding molecules, 640 were managed to retained after screening, whereas more than 20,000 non-binding molecules were removed throughout the screening process.
Fig. 6.
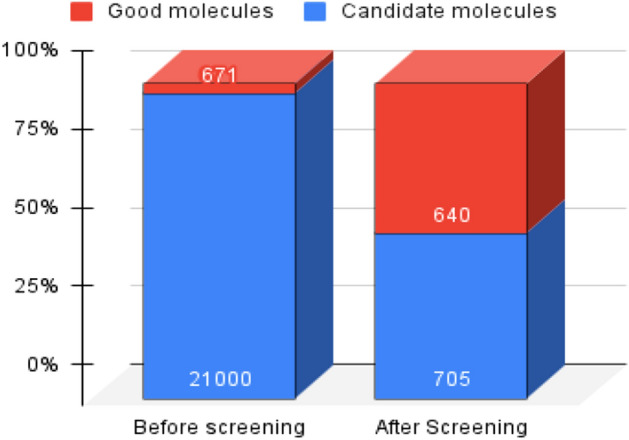
Effect of molecular screening using knowledge graph. Before screening, only 3.19% of candidate molecules possess good binding affinity, while after screening, it becomes 90.78%
Those SMILE which results in an affinity score greater than 7 can be seen as a potential drug to cure the viral protein. The obtained results of SMILE having a binding affinity are shown in Table 5, and the molecular structures of those compounds are shown in Fig. 7.
Table 5.
List of top 20 predicted binding affinity scores of generated compounds considering 3CLPro as the target protein
| S. no. | Generated SMILES | Target name | Affinity score |
|---|---|---|---|
| 1 | CC1NNSC1C(=O)N1CCCC2(COOC2)C1 | 8.185683 | |
| 2 | CC1C(-N2CNNN2)C(=O)[NH]N1C1(C)CCCC1 | 8.044197 | |
| 3 | COC1CC(OC)CC(-N2NNNC2SCC(=O)N2CCCC2)C1 | 8.034753 | |
| 4 | COCCCN1CCC2C(CNC3NC(C)NN32)C1=O | 8.018201 | |
| 5 | CCOC1CC(-C2NNN(CC(=O)N3CCCCC3)N2)CCC1OC | 7.9934325 | |
| 6 | CCN(CC)C(=O)CSC1NNC(C)N1-C1CCC(OC)CC1 | 7.9907875 | |
| 7 | CCOC(=O)C1SC2NC(C)NC(N(CC)CC)C2C1C | 7.9905686 | |
| 8 | CCOC(=O)C1SC2NC(C)NC(N)C2C1C | 7.988122 | |
| 9 | CCN1C(SCC(=O)N2CCCC2(C)C)NNC1-C1CCSC1 | 7.985457 | |
| 10 | CN1NNNC1C(C#N)=C1C2CCC1C(C)(C)C2 | 7.9777865 | |
| 11 | CC1NNC(N(C)C(=O)CC(C)(C)C)S1 | 7.975503 | |
| 12 | COC1CCCC(-C2NNN(CC(=O)N3CCCC3)N2)C1 | 7.9716773 | |
| 13 | CCN(CC)C(=O)CSC1NNNN1-C1CCCC(OC)C1 | 7.966434 | |
| 14 | O=C(CSC1NNC2C(N1)[NH]C1CCCCC12)N1CCOCC1 | 7.9602075 | |
| 15 | CC1NOC(C)C1C(=O)OCC1NC(N)NC(N(C)C)N1 | 7.9530053 | |
| 16 | COC(=O)N1C(SC)NC2SC3C(C2C1=O)CC(C)(C)OC3 | 7.952138 | |
| 17 | CCOC(=O)C1SC2NCNC(NC(C)C)C2C1C | 7.949462 | |
| 18 | COCC(C)NC(=O)CSC1NC(C)NC2SC(C)C(C)C12 | 7.946905 | |
| 19 | CC1CCCCC1-N1C(C)NNC1SCC(=O)N1CCC(C)CC1 | 7.9461155 | |
| 20 | CCCNC(=O)CC1CSC2NC(NC)C(C(N)=O)N12 | 7.9441066 |
Fig. 7.

The molecular graphs of generated compounds that possess a good binding affinity score with 3C-like protease protein
Qualitative analysis of generated compounds
From a qualitative perspective, we analyzed top-ranking generated compounds with higher binding affinity scores with the anti-HIV drug Indinavir, proposed as a potential drug to cure the SARS-CoV-2 protein virus (Harrison 2020). To support drug discovery, we compared the generated compounds and the Indinavir drug on different parameters using (Daina et al. 2014). Table 6 shows the obtained values on various parameters of drug-likeliness and medicinal chemistry of the top generated compounds along with Indinavir.
Table 6.
Comparison of Indinavir and top-ranked generated compounds with different properties of drug-likeness and medicinal chemistry
| Drug-likeness | Medicinal Chemistry | |||||
|---|---|---|---|---|---|---|
| Compound | Vebera | Egana | Bio- availability | PAINS | Lead- likeness | Synthetic accessibility |
| Indinavir | No | No | 0.55 | 0 alert | No | 5.60 |
| Yes | No | 0.55 | 0 alert | Yes | 4.39 | |
| Yes | No | 0.55 | 0 alert | Yes | 4.10 | |
| Yes | No | 0.55 | 0 alert | No | 4.86 | |
| Yes | No | 0.55 | 0 alert | Yes | 4.30 | |
| Yes | No | 0.55 | 0 alert | No | 4.82 | |
| Yes | No | 0.55 | 0 alert | No | 5.08 | |
| Yes | No | 0.55 | 0 alert | Yes | 5.12 | |
| Yes | No | 0.55 | 0 alert | Yes | 4.64 | |
| Yes | No | 0.55 | 0 alert | No | 4.94 | |
| Yes | No | 0.55 | 0 alert | No | 4.86 | |
| Yes | No | 0.55 | 0 alert | No | 4.06 | |
| Yes | No | 0.55 | 0 alert | Yes | 5.03 | |
| Yes | No | 0.55 | 0 alert | Yes | 4.85 | |
| Yes | No | 0.55 | 0 alert | Yes | 5.05 | |
| Yes | No | 0.55 | 0 alert | No | 5.38 | |
| Yes | No | 0.55 | 0 alert | Yes | 4.47 | |
| Yes | No | 0.55 | 0 alert | Yes | 5.28 | |
| Yes | No | 0.55 | 0 alert | No | 6.93 | |
| Yes | No | 0.55 | 0 alert | Yes | 5.45 | |
| Yes | No | 0.55 | 0 alert | No | 5.38 | |
aRefers to the occurrence of violations
Conclusion
In this work, we proposed a graph-based deep learning framework to generate novel drug compounds and later use them to predict the binding affinity score for a target protein structure. We use molecular graph-based deep learning models for both modules, i.e., molecule generation and binding affinity prediction, as the graph-based methods have a specific advantage over string-based models. Since using a 1D representation of the molecule, many of the molecules’ valuable structural characteristics may be lost by the model during training, which can be retrieved from their tertiary structures. Also, string-based models only manage to extract the local features of the compound; on the other hand, graph-based methods can extract the local and global features of a molecular graph. The two modules in our framework are assigned to perform different tasks. This first module uses a GGNN on the molecular graph of drugs to generate drug compounds. Later, reinforcement learning is used to fine-tune the GGNN model with desired properties using different scoring functions to get active, valid, and unique molecules. Then, the generated candidate molecules are screened using KG. The KG is used to reduce the search space using different similarity matrices. The second module uses GEFA, a graph-based deep learning model that predicts the screened compound’s affinity score for a target 3C-like protease (3CLpro) protein structure. The GEFA can efficiently handle the changes inside the protein structure by incorporating the drug embedding into the protein embedding. This allows the model to identify the active binding sites in the protein structure after forming new chemical bonds. Our result supports this claim, which shows we generated valid and unique molecules by following different chemistry rules. Also, using reinforcement learning techniques toward QED and DRD2-active property optimization with different scoring functions enhances the quality of generated molecules. This concept can further be utilized to add additional substituent optimization at the desired target. Additionally, the use of KG, constructed using different similarity matrices for screening the generated molecules, provides efficient optimization in the identification of highly effective pharmaceuticals against a target protein. Even though only 671 of the 21,0000 novel compounds were anticipated to bind, all of the generated novel molecules were required to be tested against the target protein without using the screening technique. However, after the screening process, 705 molecules (out of 21, 000) were chosen as candidate molecules, with of the binding molecules turning out to be good molecules. Before feeding the binding molecules into the Early Fusion model for affinity prediction, the screening procedure eliminated of them. The obtained affinity scores of top rank generated compounds against 3CLpro show that they possess the potential to cure the virus. One major limitation of our work includes the time consumption during simulation. It takes a lot of time to train the model in a reinforcement learning environment.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
Financial support by Microsoft Corporation and its affiliates, Redmond USA, for providing us Microsoft Azure service under Grant ID: 00011000011 is gratefully acknowledged.
Availability of data and materials
All the data sets used in this study are open source and are publicly available on GitHub repositories.
Code availability
On demand, code is made available.
Declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethics approval
None of the authors have conducted any research on human subjects for this article.
Contributor Information
Amit Ranjan, Email: amit_1921cs18@iitp.ac.in.
Hritik Kumar, Email: hritik_1901ee26@iitp.ac.in.
Deepshikha Kumari, Email: deepshikha_1901cs19@iitp.ac.in.
Archit Anand, Email: archit_1901mm09@iitp.ac.in.
Rajiv Misra, Email: rajivm@iitp.ac.in.
References
- Arús-Pous J, Blaschke T, Ulander S, Reymond JL, Chen H, Engkvist O. Exploring the gdb-13 chemical space using deep generative models. J Cheminf. 2019;11(1):1–14. doi: 10.1186/s13321-019-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YW, Yiu C-PB, Wong K-Y (2020) Prediction of the sars-cov-2 (2019-ncov) 3c-like protease (3cl pro) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research 9 [DOI] [PMC free article] [PubMed]
- Daina A, Michielin O, Zoete V. ilogp: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the gb/sa approach. J Chem Inf Model. 2014;54(12):3284–3301. doi: 10.1021/ci500467k. [DOI] [PubMed] [Google Scholar]
- Dalke A (2014) Maccs key 44. figshare http://www.dalkescientific.com/writings/diary/archive/2014/10/17/maccs_key_44.html
- Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29(11):1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Drews J. Drug discovery: a historical perspective. Science. 2000;287(5460):1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- Gilmer J, Schoenholz SS, Riley PF, Vinyals O, Dahl GE (2017a) Neural message passing for quantum chemistry. In: International conference on machine learning, pp 1263–1272
- Gilmer J, Schoenholz SS, Riley PF, Vinyals O, Dahl GE (2017b) Neural message passing for quantum chemistry. In: International conference on machine learning, pp 1263–1272
- Guimaraes GL, Sanchez-Lengeling B, Outeiral C, Farias PLC, Aspuru-Guzik A (2017) Objective-reinforced generative adversarial networks (organ) for sequence generation models. Preprint at arXiv:1705.10843
- Harrison C. Coronavirus puts drug repurposing on the fast track. Nat Biotechnol. 2020;38(4):379–381. doi: 10.1038/d41587-020-00003-1. [DOI] [PubMed] [Google Scholar]
- Jiang M, Li Z, Zhang S, Wang S, Wang X, Yuan Q, Wei Z. Drug-target affinity prediction using graph neural network and contact maps. RSC Adv. 2020;10(35):20701–20712. doi: 10.1039/D0RA02297G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipf TN, Welling M (2016) Semi-supervised classification with graph convolutional networks. Preprint at arXiv:1609.02907
- Li Y, Tarlow D, Brockschmidt M, Zemel R (2015) Gated graph sequence neural networks. Preprint at arXiv:1511.05493
- Li Y, Vinyals O, Dyer C, Pascanu R, Battaglia P (2018a) Learning deep generative models of graphs. Preprint at arXiv:1803.03324
- Li Y, Zhang L, Liu Z. Multi-objective de novo drug design with conditional graph generative model. J Cheminf. 2018;10(1):1–24. doi: 10.1186/s13321-018-0287-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H. Drug treatment options for the 2019-new coronavirus (2019-ncov) Biosci Trends. 2020;14(1):69–71. doi: 10.5582/bst.2020.01020. [DOI] [PubMed] [Google Scholar]
- Ma W, Yang L, He L. Overview of the detection methods for equilibrium dissociation constant kd of drug-receptor interaction. J Pharmac Anal. 2018;8(3):147–152. doi: 10.1016/j.jpha.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado R, Rastemo T, Lindelöf E, Klambauer G, Engkvist O, Chen H, Bjerrum EJ. Graph networks for molecular design. Mach Learn: Sci Technol. 2021;2(2):025023. [Google Scholar]
- Mnih V, Kavukcuoglu K, Silver D, Rusu AA, Veness J, Bellemare MG, et al. Human-level control through deep reinforcement learning. Nature. 2015;518(7540):529–533. doi: 10.1038/nature14236. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Le H, Quinn TP, Nguyen T, Le TD, Venkatesh S. Graphdta: predicting drug-target binding affinity with graph neural networks. Bioinformatics. 2021;37(8):1140–1147. doi: 10.1093/bioinformatics/btaa921. [DOI] [PubMed] [Google Scholar]
- Nguyen TM, Nguyen T, Le TM, Tran T (2021b) Gefa: early fusion approach in drug-target affinity prediction. In: IEEE/ACM transactions on computational biology and bioinformatics [DOI] [PubMed]
- Olivecrona M, Blaschke T, Engkvist O, Chen H. Molecular de-novo design through deep reinforcement learning. J Cheminf. 2017;9(1):1–14. doi: 10.1186/s13321-017-0235-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Öztürk H, Özgür A, Ozkirimli E. Deepdta: deep drug-target binding affinity prediction. Bioinformatics. 2018;34:i821–i829. doi: 10.1093/bioinformatics/bty593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polykovskiy D, Zhebrak A, Sanchez-Lengeling B, Golovanov S, Tatanov O, Belyaev S, et al. Molecular sets (moses): a benchmarking platform for molecular generation models. Front Pharmacol. 2020;11:1931. doi: 10.3389/fphar.2020.565644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putin E, Asadulaev A, Ivanenkov Y, Aladinskiy V, Sanchez-Lengeling B, Aspuru-Guzik A, Zhavoronkov A. Reinforced adversarial neural computer for de novo molecular design. J Chem Inf Model. 2018;58(6):1194–1204. doi: 10.1021/acs.jcim.7b00690. [DOI] [PubMed] [Google Scholar]
- Ranjan A, Shukla S, Datta D, Misra R. Generating novel molecule for target protein (sars-cov-2) using drug-target interaction based on graph neural network. Netw Model Anal Health Inf Bioinform. 2022;11(1):1–11. doi: 10.1007/s13721-021-00351-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao R, Bhattacharya N, Thomas N, Duan Y, Chen P, Canny J et al (2019) Evaluating protein transfer learning with tape. In: Advances in neural information processing systems, 32 [PMC free article] [PubMed]
- Ren S, He K, Girshick R, Sun J (2015) Faster r-cnn: towards real-time object detection with region proposal networks. In: Advances in neural information processing systems 28 [DOI] [PubMed]
- Scarselli F, Gori M, Tsoi AC, Hagenbuchner M, Monfardini G. The graph neural network model. IEEE Trans Neural Netw. 2008;20(1):61–80. doi: 10.1109/TNN.2008.2005605. [DOI] [PubMed] [Google Scholar]
- Scarselli F, Gori M, Tsoi AC, Hagenbuchner M, Monfardini G. The graph neural network model. IEEE Trans Neural Netw. 2008;20(1):61–80. doi: 10.1109/TNN.2008.2005605. [DOI] [PubMed] [Google Scholar]
- Torng W, Altman RB. Graph convolutional neural networks for predicting drug-target interactions. J Chem Inf Model. 2019;59(10):4131–4149. doi: 10.1021/acs.jcim.9b00628. [DOI] [PubMed] [Google Scholar]
- Tsubaki M, Tomii K, Sese J. Compound-protein interaction prediction with end-to-end learning of neural networks for graphs and sequences. Bioinformatics. 2019;35(2):309–318. doi: 10.1093/bioinformatics/bty535. [DOI] [PubMed] [Google Scholar]
- Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez AN et al (2017) Attention is all you need. In: Advances in neural information processing systems 30
- Yamanishi Y, Araki M, Gutteridge A, Honda W, Kanehisa M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics. 2008;24(13):i232–i240. doi: 10.1093/bioinformatics/btn162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You J, Liu B, Ying Z, Pande V, Leskovec J (2018) Graph convolutional policy network for goal-directed molecular graph generation. In: Advances in neural information processing systems 31
- Zhou Z, Kearnes S, Li L, Zare RN, Riley P. Optimization of molecules via deep reinforcement learning. Sci Reports. 2019;9(1):1–10. doi: 10.1038/s41598-019-47148-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Hou Y, Shen J, Huang Y, Martin W, Cheng F. Network-based drug repurposing for novel coronavirus 2019-ncov/sars-cov-2. Cell Discov. 2020;6(1):1–18. doi: 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data sets used in this study are open source and are publicly available on GitHub repositories.
On demand, code is made available.