

Abstract
Chronic obstructive pulmonary disease (COPD) is associated with airway inflammation, increased infiltration by CD8+ T lymphocytes, and infection-driven exacerbations. Although cigarette smoke is the leading risk factor for COPD, the mechanisms driving the development of COPD in only a subset of smokers are incompletely understood. Lung-resident mucosal-associated invariant T (MAIT) cells play a role in microbial infections and inflammatory diseases. The role of MAIT cells in COPD pathology is unknown. Here, we examined MAIT cell activation in response to cigarette smoke-exposed primary human bronchial epithelial cells (BECs) from healthy, COPD, or smoker donors. We observed significantly higher baseline MAIT cell responses to COPD BECs than healthy BECs. However, infected COPD BECs stimulated a smaller fold increase in MAIT cell response despite increased microbial infection. For all donor groups, cigarette smoke-exposed BECs elicited reduced MAIT cell responses; conversely, cigarette smoke exposure increased ligand-mediated MR1 surface translocation in healthy and COPD BECs. Our data demonstrate that MAIT cell activation is dysregulated in the context of cigarette smoke and COPD. MAIT cells could contribute to cigarette smoke- and COPD-associated inflammation through inappropriate activation and reduced early recognition of bacterial infection, contributing to microbial persistence and COPD exacerbations.
Keywords: chronic obstructive pulmonary disease, cigarette smoke, MAIT cells, MR1, Streptococcus pneumoniae
Despite continued smoking cessation programs, smoking remains a major health concern, with 8 million deaths in 2017 attributed to tobacco usage (1). Cigarette smoking is associated with a variety of immunological impacts, such as higher susceptibility to microbial infections (2–4). The components of cigarette smoke act as both proinflammatory and immunosuppressive factors that modulate innate and adaptive immunity (3, 5). For example, cigarette smoke activates caspase-1 to secrete interleukin (IL-1β) and IL-18 in-vivo (6–9), resulting in emphysema and small airway remodeling (10, 11) and accumulation of CD8+ T cells through IFN-γ signaling (12–14). In the context of infection, cigarette smoke inhibits the production of proinflammatory cytokines in response to microbial infection or LPS stimulation (15), increases adhesion of Streptococcus pneumoniae to bronchial epithelial cells (16), and delays clearance of Pseudomonas aeruginosa (17). Others have observed that repeated cigarette smoke exposure in mice with persistent S. pneumoniae airway infection resulted in increased release of proinflammatory cytokines including IL-12 and IL-1β, greater bacterial load, and reduced lung function (18), suggesting the interplay between cigarette smoke, the airway, and microbial infections is complex.
Cigarette smoking also results in long-term airway changes, evidenced by its role as the primary risk factor for the development of chronic obstructive pulmonary disease (COPD) (19, 20), the third leading cause of death worldwide (21). COPD manifests as a number of clinical phenotypes, including small airway disease (e.g., bronchitis) and emphysema, all of which are characterized by chronic inflammation and airflow limitation in the lung and airway (19). Further complicating COPD pathology are exacerbations triggered by bacterial or viral colonization and infection, which can increase inflammation and play an important role in the morbidity and mortality associated with COPD (19, 20).
The immune mechanisms underlying the development of airway damage and inflammation leading to COPD in some smokers but not others are poorly defined (22). CD8+ T cells, which are often increased in the lungs of patients with bacterial infections, are the main inflammatory cell subset increased in the lungs of smokers with COPD compared with asymptomatic smokers (13). Increased frequencies of CD8+ T cells were also observed at the onset of acute exacerbations (23). CD8+ T cells were specifically correlated with airflow limitation and COPD pathology (13). Central to this, CD8+ T lymphocytes have increased expression of chemokine receptors, cytotoxic effector molecules, and proinflammatory cytokines in human COPD lung tissue (reviewed in Reference [23]), and chronic cigarette smoke exposure alone resulted in persistent clonal expansion of CD8+ T cells in mice (24). Mucosal-associated invariant T (MAIT) cells are an innate-like subset of T lymphocytes that make up a relatively large proportion of the total CD8+ T-cell population in the blood and lungs in healthy individuals (25). MAIT cells are critical to the early control of respiratory infections, including S. pneumoniae, Haemophilus influenzae, and Legionella longbeachae (26, 27). Despite the overall increase in CD8+ T cells in COPD, the frequency of both peripheral blood and lung-resident MAIT cells in individuals with COPD is decreased (28–31), and lower MAIT cell counts are associated with increased hospitalization (32). This observation is different from many other infectious and inflammatory lung conditions, and the mechanisms underlying MAIT cell loss in COPD lungs are not yet defined. In fact, little is known about the role of MAIT cells in cigarette smoke- and COPD-associated inflammatory processes.
The antigens presented to MAIT cells by the MHC class I related molecule, MR1, are primarily small molecule metabolites generated during riboflavin biosynthesis by many microbial organisms (33–35), including those implicated in COPD-associated exacerbations (28, 36, 37). MAIT cells can also be activated through antigen-independent, cytokine-mediated mechanisms (38). IL-12 and IL-18, the cytokines which elicit this type of antigen-independent response, are among those produced by airway epithelial cells and other inflammatory cells in the context of cigarette smoke and COPD (6, 8, 39). We hypothesized that exposure of bronchial epithelial cells (BECs) to cigarette smoke and the inflammatory COPD airway environment would result in dysregulated MAIT cell responses through altered MR1 function, contributing to inflammation and exacerbation. We found that BEC from COPD lungs induced greater overall MAIT cell responses compared with healthy control subjects. Furthermore, cigarette smoke exposure to BECs decreased both microbe-independent and microbe-dependent MAIT cell responses. Exposure to cigarette smoke did not affect the transcriptional expression of MR1 but did result in increased MR1 surface expression, suggesting that smoking may interfere with the ability of MR1 to encounter microbial ligands. Our data demonstrate that impaired interactions between airway epithelial cells and MAIT cells, resulting in the dysregulated release of proinflammatory cytokines and other molecules, may play a role in COPD-associated inflammation in the context of both cigarette smoke as well as bacterial colonization and infection. Some of the results of these studies have been previously reported in the form of conference abstracts (40–43).
Methods
Human Subjects
This study was conducted according to the principles expressed in the Declaration of Helsinki. Additional information on study participants, protocols, and consent are described in the data supplement.
Cells and Reagents
Primary BECs (Table 1) from Lonza Bioscience or isolated from lung tissue obtained from the Pacific Northwest Transplant Bank were cultured as previously described (44). BEAS-2B cells (ATCC CRL-9609), BEAS-2B:doxMR1-GFP cells, and the MR1-restricted T-cell clone D426G11 were used as described previously (45–48).
Table 1.
Description of Bronchial Epithelial Cell Donors
| Donor Information |
Medical History |
Assays Performed |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ID | Source | Age, yr | Sex | Race | COPD Diagnosis | Smoking | Smoking Notes | ELISPOT | RT-PCR | Flow |
| Healthy | ||||||||||
| H276 | Lonza | 68 | M | W | — | — | — | Y | Y | Y |
| H527 | Lonza | 47 | M | W | — | — | — | Y | Y | Y |
| H608 | Lonza | 67 | M | W | — | — | — | Y | Y | Y |
| H619 | Lonza | 53 | M | W | — | — | — | Y | Y | Y |
| H628 | Lonza | 42 | M | B | — | — | — | Y | N* | N* |
| H544 | Lonza | 48 | M | W | — | — | — | Y | N† | Y |
| H063 | PNTB | 57 | M | W | — | — | — | Y | Y | N‡ |
| COPD | ||||||||||
| C141 | Lonza | 73 | M | W | 12 yr; emphysema |
1–2 ppd; 20 yr |
— | Y | Y | Y |
| C179 | Lonza | 69 | M | W | Unknown; inhaler |
2–3 ppd; 40 yr |
Decreased smoking (recently); smoked marijuana |
Y | Y | Y |
| C409 | Lonza | 53 | M | W | Unknown; inhaler, oxygen |
2 ppd; 27 yr |
— | Y | Y | Y |
| C415 | Lonza | 53 | M | B | Unknown; emphysema |
1 ppd; 20 yr |
Quit smoking (5 yr) | Y | Y | Y |
| C436 | Lonza | 59 | M | W | 20 yr; steroid inhalers |
2–3 ppd; 35 yr |
— | Y | Y | Y |
| C147 | PNTB | 66 | M | W | 2 yr | 1.5 ppd; 40 yr |
Quit smoking (10 yr); smoked marijuana (49 yr) |
Y | N§ | N‡ |
| Smoker | ||||||||||
| S118 | Lonza | 56 | Fǁ | B | — | 0.5 ppd; 26 yr |
— | Y | Y | Y |
| S123 | PNTB | 39 | M | W | — | Occasional; unknown |
— | Y | Y | Y |
| S149 | PNTB | 57 | M | W | — | 1 ppd; 12 yr |
Quit smoking (23 yr) | Y | Y | Y |
| S150 | PNTB | 55 | M | Am Ind | — | Unknown; 25 yr |
— | Y | Y | Y |
| S151 | PNTB | 41 | M | W | — | Unknown; 25 yr |
— | Y | Y | Y |
| S011 | PNTB | 50 | M | W | — | 0.5 ppd; >20 yr |
— | Y | N§ | N‡ |
Definition of abbreviations: Am Ind = American Indian; B = Black; COPD = chronic obstructive pulmonary disease; ELISPOT = enzyme-linked immunospot; ppd = packs per day; PNTB = Pacific Northwest Transplant Bank (now Cascade Alliance); RT-PCR = real-time quantitative PCR; W = White.
Loss of cell viability after first expansion.
Failed to isolate RNA of sufficient quality and quantity.
Positive and negative flow cytometry controls failed.
Irregular amplification of internal reference gene.
Sole female donor.
S. pneumoniae and Mycobacterium smegmatis Mc2155 (ATCC) were cultured as previously described (36, 49). Antibodies are described in the data supplement. Phytohemagglutinin PHA-L (Sigma), NucBlue Cell Stain ReadyProbes (ThermoFisher), and AlexaFluor 488 succinimidyl ester (ThermoFisher) were used per manufacturers’ protocols. Doxycycline (Sigma) was used at 2 μg/ml. 6-FP (6-formylpterin; Schirck’s Laboratories) was suspended in 0.01 M NaOH and used at a final concentration of 100 μM.
Cigarette Smoke Extract Preparation
Cigarette smoke extract (CSE) was prepared using research-grade cigarettes (1R6F, University of Kentucky Tobacco and Health Research Foundation) (50). Sterile filtered CSE (pH = 7.4) was stored at −20°C. Thawed aliquots of RPMI (“0% CSE”) or CSE (“30% CSE”) were diluted to 30% vol/vol final concentration in the culture medium.
ELISPOT Assay
IFN-γ ELISPOT (enzyme-linked immunospot) assays were performed as previously described (51) with modifications where indicated: BECs were incubated with 0% or 30% CSE medium for 3 hours prior to other indicated treatments in ELISPOT plate. For antibody blocking experiments, plated cells were incubated with isotype control subjects, α-MR1, or α-IL-12 and α-IL-18 antibodies for 4 hours. Antigens were added for 1 hour before the addition of MAIT cell clones and overnight incubation.
Fluorescence Microscopy
Primary BECs were seeded in glass-bottom chamber slides (Nunc). Where indicated, cells were incubated with CSE for 3 hours, washed, then infected with fluorescently labeled S. pneumoniae as previously described (36). Fixed slides were stained with α-HLA-A,B,C antibody and NucBlue nuclear stain and imaged with a high-resolution widefield CoreDV microscope (Applied Precision). Approximately 20 fields per condition were selected by unbiased nuclear stain and analyzed as described in the data supplement.
Real-Time Quantitative PCR
Qiagen RNeasy Plus RNA isolation and Life Technologies High Capacity cDNA synthesis kits were used per manufacturers’ protocols. Real-time PCR was performed using Taqman gene expression assays for MR1 (Hs01042278_m1) and HPRT1 (Hs02800695_m1). Relative expression levels for each target gene were determined using the 2−ΔΔCt method (52).
Surface MR1 and MHC-Ia Flow Cytometry
Primary BECs, wild-type BEAS-2B cells, and BEAS-2B:doxMR1-GFP cells were incubated with CSE and 6-FP as indicated, then stained with APC-conjugated 26.5 α-MR1 antibody and analyzed by flow cytometry. All analyses were performed using FlowJo10 (TreeStar).
IL-18 Expression
IL-18 immunoassay was performed using the ProQuantum Human IL-18 Immunoassay Kit (A35613; Invitrogen) per manufacturer’s protocols with a 1:3 dilution of supernatants.
Data Analysis
ELISPOT statistical analysis was performed as described in the data supplement. All other data were analyzed using Prism 8 (GraphPad) or R 4.0, as described in the data supplement.
Results
BEC from COPD Lungs Induce Increased Microbe-independent, MR1-dependent Activation of MAIT Cells
Inappropriate MR1 antigen presentation and activation of lung-resident MAIT cells could contribute to the inflammatory airway environment present in COPD airways and after cigarette smoking. As such, we tested the ability of MAIT cells to respond to primary human BECs from the lungs of COPD or smoker donors compared with healthy control subjects and in the context of cigarette smoke exposure. BECs were isolated from the lungs of healthy (n = 7), COPD (n = 6), or smoker (n = 6) donors between the ages of 41 and 73 (Table 1). BECs from these donors were incubated with a previously described MAIT cell clone (D426 G11) (45, 53) after treatment with CSE and infection with M. smegmatis or S. pneumoniae in an ELISPOT assay with IFN-γ production by the MAIT cell clone as the readout. A linear mixed effects model with square root transformation of the IFN-γ spot forming units (SFUs) was used to analyze the data for significant effects of donor BEC groups on MAIT cell responses.
We first analyzed the response of the MAIT cell clone to uninfected BECs. We observed significantly greater microbe-independent IFN-γ SFUs in response to BECs from COPD donors than from healthy or smoker donors (P = 0.0416) (Figure 1A and Table 2). These microbe-independent MAIT cell responses to COPD donors were greater than responses observed in the uninfected bronchial epithelial cell line (BEAS-2B) control (Figure E1 in the data supplement). There were no differences in the MAIT cell response to smoker donors compared with healthy control subjects (P = 0.5173) (Figure 1A and Table 2). We hypothesized that an increase in proinflammatory cytokines capable of mediating MAIT cell responses, such as IL-18 (38), which is produced by primary BECs from the lungs of subjects with COPD (6, 7, 39), could induce increased MR1-independent MAIT cell responses absent microbial antigens. To determine whether stimulation of IFN-γ production by the MAIT cells occurred through MR1- or cytokine-dependent pathways, we used antibodies to block MR1 or IL-12 and IL-18 in BECs from a representative healthy and COPD donor. There was an almost complete blockade of the IFN-γ SFU response for the healthy and COPD donors in the presence of the 26.5 α-MR1 antibody, with very little impact on blocking IL-12 and IL-18 (Figures 1B and 1C). This suggests that despite the lack of antigen from microbial infection, there are nonetheless MR1-dependent MAIT cell responses to primary BECs from all donors. We did observe diffuse IFN-γ staining haze in all ELISPOT wells containing both BEC and MAIT cells (Figure 1C). This haze was completely abrogated in the context of IL-12 and IL-18 blocking for both donors, demonstrating that there are likely cytokine-mediated MAIT cell responses to the primary BECs in addition to the MR1-dependent responses. Quantification of non-spot forming IFN-γ is not possible in the context of an ELISPOT assay. Therefore, we were unable to determine whether there was also a meaningful difference in this cytokine-dependent response to the healthy or COPD donor BECs. We did perform an assay to detect IL-18 secretion by a representative healthy, COPD, and smoker donor. All donors produced less than 2 pg/ml of IL-18, with no difference between the donors (Figure E2). Taken together with the abrogation of IFN-γ spots in the presence of the α-MR1 antibody, our data suggest that microbe-independent MAIT cell activation is largely mediated through MR1-dependent mechanisms and is increased in response to COPD BECs.
Figure 1.
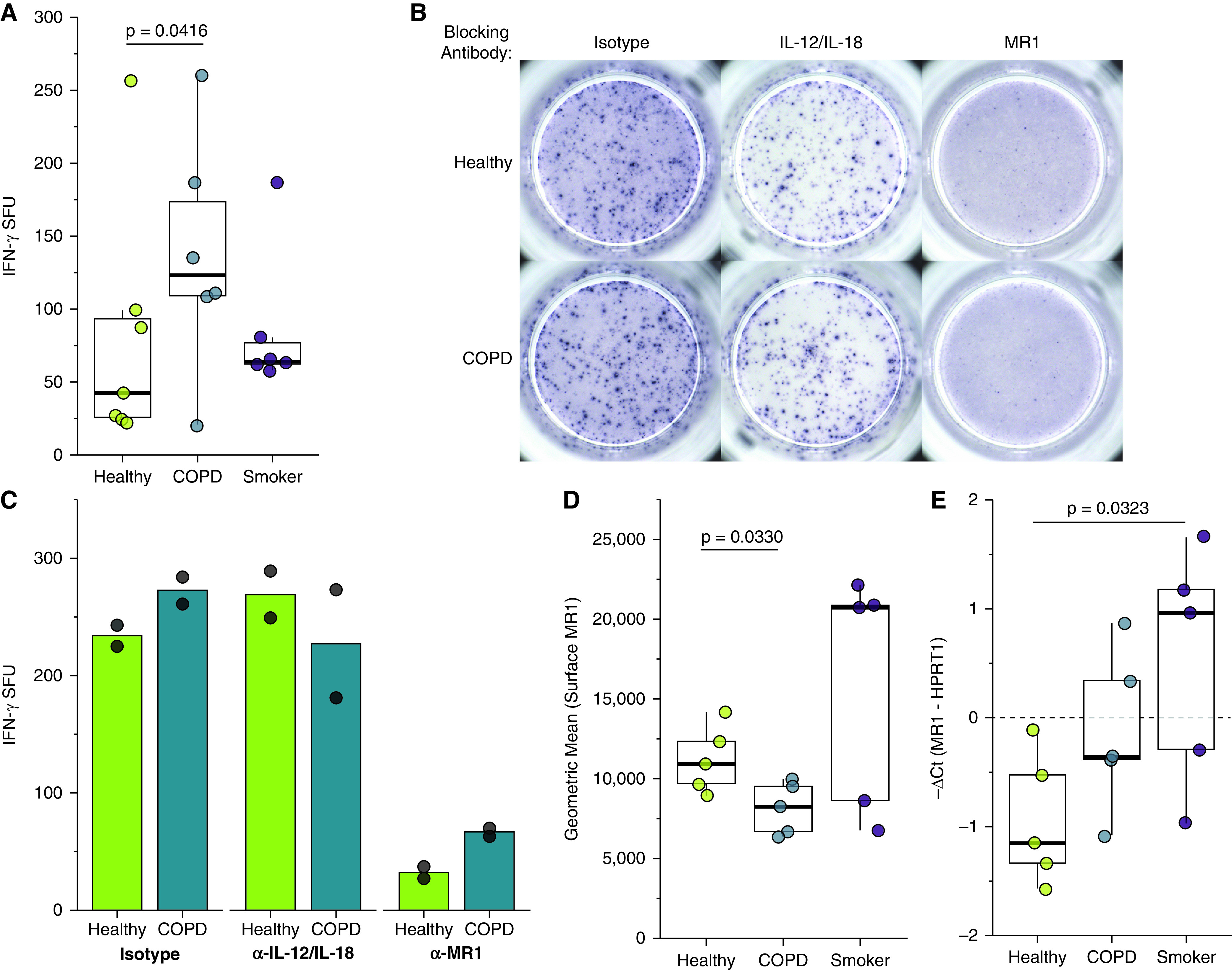
Primary bronchial epithelial cells (BECs) elicit microbe-independent, MR1-dependent responses by mucosal‐associated invariant T (MAIT) cells. (A) Primary BECs from healthy (n = 7), chronic obstructive pulmonary disease (COPD) (n = 6), or smoker (n = 6) donors were incubated with the D426 G11 MAIT cell clone in an enzyme-linked immunospot (ELISPOT) assay with IFN-γ production as the readout. Data points are the mean IFN-γ spot-forming units (SFUs) of two technical replicates per donor. Statistical analysis was performed as described in the data supplement and is summarized in Table 2. (B and C) BECs from the healthy and COPD donors that induced the greatest IFN-γ SFUs in A were incubated with blocking antibodies to IL-12/IL-18 or MR1 5 hours before the addition of the MAIT cells in an IFN-γ ELISPOT assay. Results are presented as (B) ELISPOT well images from one representative experiment and (C) the mean of two experimental replicates. Control IgG1 and IgG2a isotype antibodies are pooled from one representative experiment each. (D) Primary BECs from healthy, COPD, or smoker donors (n = 5) were stained for surface expression of MR1 by flow cytometry. (E) RNA was isolated from healthy, COPD, or smoker donor BECs (n = 5), and real-time quantitative PCR (RT-PCR) was performed to detect amplification of MR1 and the internal control, HPRT1. Data points are the mean of three technical replicates per donor. Two-tailed unpaired t tests were performed to determine statistical significance for D and E. MR1 = MHC class I related molecule.
Table 2.
Statistical Analysis of Enzyme-linked Immunospot Data: Fixed Effects Results from Linear Mixed Model
| Variable | P Value |
|---|---|
| Healthy | (Intercept) |
| COPD | 0.0416 |
| Smoker | 0.5173 |
| CSE | 0.5279 |
| Msm | <0.0001 |
| Sp | <0.0001 |
| COPD:CSE | 0.0003 |
| Smoker:CSE | <0.0001 |
| COPD:Msm | <0.0001 |
| Smoker:Msm | 0.8718 |
| COPD:Sp | 0.4193 |
| Smoker:Sp | 0.2432 |
| CSE:Msm | <0.0001 |
| CSE:Sp | 0.0482 |
Definition of abbreviations: COPD = chronic obstructive pulmonary disease; CSE = cigarette smoke extract; Msm = Mycobacterium smegmatis; Sp = Streptococcus pneumoniae.
We considered the possibility that altered MR1 expression in these cells could explain these changes. We quantified surface MR1 molecules by staining cells with 26.5 α-MR1 antibody for flow cytometry (Figure 1D). Consistent with our previous studies, the degree of endogenous MR1 surface expression in ex-vivo primary BECs is relatively low compared with cell lines (46), particularly those that overexpress MR1 (49). As such, we included BEAS-2B cells overexpressing MR1 in each assay as a control to confirm the detection and surface translocation of MR1 (Figure E3). At baseline, COPD donor BECs express less MR1 than healthy donor BECs (P = 0.0330) (Figure 1D), demonstrating that the increased MAIT cell responses to COPD donor BECs are not because of increased MR1 molecules. Although MR1 expression has been confirmed in all cell types studied to date (54), nearly all analyses of MR1 expression and regulation have focused on the surface expression of MR1 protein. There are a limited number of studies examining MR1 gene expression in bulk cells from the lung parenchyma or peripheral blood of COPD donors (30, 31); however, we are unaware of any analysis of the impact of COPD or smoking on MR1 expression in primary BECs. Therefore, we collected mRNA from the BECs and quantified MR1 expression relative to the internal control gene, hypoxanthine phosphoribosyltransferase 1 (HPRT1). Baseline Ct values and ΔCt analysis of MR1 mRNA across all donors revealed significantly higher expression in smoker donors compared with healthy control at baseline (Figure 1E). The smaller ΔCt value for COPD BECs compared with healthy donors suggested higher MR1 expression in contrast to the significantly reduced surface MR1 expression in COPD donor BECs (Figures 1D and 1E). Together these results suggest existing differences in MR1 expression at baseline are not enough to explain altered MAIT cell responses.
Exposure to Cigarette Smoke Decreases MAIT Cell Activation in Response to Primary BECs
We next examined whether treating primary BECs with cigarette smoke impacts microbe-independent MAIT cell responses. BECs were treated with 30% CSE for 3 hours, then washed and replated for incubation with MAIT cell clones in an IFN-γ ELISPOT assay. CSE treatment did not significantly affect overall MAIT cell IFN-γ responses for healthy and COPD donor BECs (Figure 2A and Table 3). We compared the fold change in IFN-γ SFUs from 0% to 30% CSE treatment to account for the increased baseline MAIT cell responses to COPD donor BECs. Although healthy donor BECs were inconsistently impacted by CSE treatment, MAIT cell responses to BECs from COPD and smoker donors were significantly more reduced after incubation with CSE (P = 0.0003 and P < 0.0001) (Figure 2B and Table 2).
Figure 2.

Decreased MAIT cell responses to primary BECs after treatment with cigarette smoke extract (CSE). (A and B) Primary BECs from healthy (n = 7), COPD (n = 6), or smoker (n = 6) donors were infected with media containing 0% or 30% CSE for 3 hours before the addition of D426 G11 MAIT cells in an IFN-γ ELISPOT assay. Statistical analysis was performed as described in the data supplement and is summarized in Tables 2 and 3. (A) Data points are the mean IFN-γ SFUs of two experimental replicates paired by an individual donor. (B) Fold change IFN-γ SFUs between 0% and 30% CSE-treated primary BECs from healthy, COPD, or smoker donors, calculated pairwise by donor. (C and D) Primary BECs from healthy, COPD, or smoker donors (n = 5) were incubated with 0% or 30% CSE for 3 hours. (C) RNA was isolated from BECs, and real-time quantitative PCR (RT-PCR) was performed to detect amplification of MR1 and the internal control, HPRT1. MR1 expression was calculated by 2−ΔΔCt method, relative to 0% CSE pairwise control and HPRT1 expression. (D) Cells were washed, incubated overnight, then stained for surface expression of MR1 by flow cytometry. Data points are mean fluorescence intensities paired by individual donor. Statistical significance was determined by two-tailed paired t tests for same-donor 0% and 30% CSE treatment in C and D and unpaired t tests for donor group comparison in D.
Table 3.
Statistical Analysis of Enzyme-linked Immunospot Data: Multiple Comparisons of Means
| Donor | Infection | CSE Treatment | P Value |
|---|---|---|---|
| Healthy | UI vs. Msm | No CSE | <0.0001 |
| UI vs. Sp | No CSE | <0.0001 | |
| Msm vs. Sp | No CSE | <0.0001 | |
| UI vs. Msm | +CSE | <0.0001 | |
| UI vs. Sp | +CSE | <0.0001 | |
| Msm vs. Sp | +CSE | <0.0001 | |
| UI | No CSE vs. +CSE | 1 | |
| Msm | No CSE vs. +CSE | <0.0001 | |
| Sp | No CSE vs. +CSE | 1 | |
| COPD | UI vs. Msm | No CSE | <0.0001 |
| UI vs. Sp | No CSE | <0.0001 | |
| Msm vs. Sp | No CSE | <0.0001 | |
| UI vs. Msm | +CSE | <0.0001 | |
| UI vs. Sp | +CSE | <0.0001 | |
| Msm vs. Sp | +CSE | <0.0001 | |
| UI | No CSE vs. +CSE | 0.9964 | |
| Msm | No CSE vs. +CSE | <0.0001 | |
| Sp | No CSE vs. +CSE | 0.004 | |
| Smoker | UI vs. Msm | No CSE | <0.0001 |
| UI vs. Sp | No CSE | <0.0001 | |
| Msm vs. Sp | No CSE | <0.0001 | |
| UI vs. Msm | +CSE | <0.0001 | |
| UI vs. Sp | +CSE | <0.0001 | |
| Msm vs. Sp | +CSE | <0.0001 | |
| UI | No CSE vs. +CSE | 0.0006 | |
| Msm | No CSE vs. +CSE | <0.0001 | |
| Sp | No CSE vs. +CSE | <0.0001 | |
| Healthy vs. COPD | UI | No CSE | 0.9964 |
| Msm | No CSE | 1 | |
| Sp | No CSE | 0.999 | |
| UI | +CSE | 1 | |
| Msm | +CSE | 1 | |
| Sp | +CSE | 1 | |
| Healthy vs. Smoker | UI | No CSE | 1 |
| Msm | No CSE | 1 | |
| Sp | No CSE | 1 | |
| UI | +CSE | 1 | |
| Msm | +CSE | 1 | |
| Sp | +CSE | 1 | |
| COPD vs. Smoker | UI | No CSE | 1 |
| Msm | No CSE | 1 | |
| Sp | No CSE | 1 | |
| UI | +CSE | 0.9998 | |
| Msm | +CSE | 1 | |
| Sp | +CSE | 0.9995 |
Definition of abbreviations: UI = uninfected.
Our ELISPOT results suggested that MR1-dependent MAIT cell activation was impacted in cells after acute treatment with CSE. We looked at MR1 gene expression in BECs from COPD and smoker lungs compared with healthy control subjects and sought to determine if exposure to CSE had any impact on MR1 mRNA expression in BECs from all donors. We isolated mRNA from BECs after CSE treatment and corresponding control conditions and measured the expression of MR1 and the internal control HPRT1. For all BEC donor groups, paired comparisons demonstrated no statistically significant impacts of acute CSE exposure on MR1 expression (Figure 2C). These results do not demonstrate a consistent role for CSE-mediated transcriptional regulation of MR1 in the observed ability of MAIT cells to respond to BECs.
We then quantified surface MR1 expression by flow cytometry. Similar to the baseline comparison, CSE-treated COPD donor BECs expressed lower overall degrees of cell surface MR1 protein than healthy donor BECs (P = 0.0475) (Figure 2D). Although COPD and smoker donor BECs were relatively unaffected by CSE treatment, surface MR1 expression was significantly increased in CSE-treated healthy donor BECs (P = 0.0408) (Figure 2D). This increase in MR1 surface expression may compensate for the CSE-mediated reduction in MAIT cell responses to healthy donor BECs. Together, these data suggest that MAIT cell responses to CSE-treated COPD and smoker donor BECs are impaired.
Increased Infection of Primary BECs from COPD Donors or after CSE Treatment
Our data demonstrate that microbe-independent, MR1-dependent MAIT cell responses to primary BECs are increased for COPD donor BECs but decreased after CSE treatment. We next explored the MAIT cell activation in the context of bacterial infection by the pneumonia-causing pathogen S. pneumoniae. Cigarette smoke and COPD are associated with increased bacterial adhesion and colonization of airway epithelial cells in both in vitro assays and directly ex vivo samples (16, 55–57). To measure this, we infected a representative healthy, COPD, and smoker donor BEC with fluorescent S. pneumoniae, then quantified the number of bacteria per cell using fluorescence microscopy. We observed significantly more S. pneumoniae cocci associated with BECs from the COPD donor than healthy or smoker donors (P = 0.0001 and P = 0.0271) (Figures 3A and 3C). To assess the impact of cigarette smoke, representative BECs were incubated with media containing 0% or 30% CSE before infection. As expected, CSE-treated healthy and COPD donor BECs had increased bacterial burdens compared with the 0% control subjects (P = 0.0027 and P = 0.0227) (Figures 3B and 3C). We again observed significantly greater infection in CSE-treated COPD donor BECs than in CSE-treated healthy or smoker donor BECs (P = 0.0404 and P = 0.0067) (Figure 3C). These data demonstrate that bacterial burden is increased in COPD donor BECs and in the context of acute cigarette smoke exposure.
Figure 3.

Increased infection of primary BECs from COPD donors or after CSE treatment. (A and B) BECs from representative healthy, COPD, or smoker donors were incubated with media containing 0% or 30% CSE for 3 hours where indicated and infected with fluorescently labeled Streptococcus pneumoniae for 3 hours. Fixed cells were stained with α-HLA-A,B,C antibody to label the cell surface and NucBlue nuclear stain. Approximately 20 fields per donor were selected without bias on the basis of nuclear stain, and whole cells within these fields were then analyzed by Imaris to enumerate the number of bacteria associated with individual cells. Shown are representative images of S. pneumoniae-infected primary BECs for each condition. White = surface stain and red = fluorescent S. pneumoniae pseudocolor. Arrows indicate adherent bacteria (yellow) enumerated for analysis or extracellular bacteria (red) excluded from the analysis. Scale bar, 10μM. (C) Quantification of S. pneumoniae per cell. Data points indicate individual cells, analyzed by one-way ANOVA.
BECs from COPD Lungs Induce a Decreased Fold-change in Microbe-dependent, MR1-dependent Activation of MAIT Cells
We next used IFN-γ ELISPOT assays to measure MAIT cell responses to BECd infected with S. pneumoniae, or with M. smegmatis as a positive control. As expected, MAIT cell responses to the S. pneumoniae- or M. smegmatis-infected donor BECs were significantly greater than responses to uninfected BEC for all donors (P < 0.0001) (Figure 4A and Table 3). Similar to the microbe-independent ELISPOT assays, the MAIT cell IFN-γ SFU responses to infected BECs required MR1, as demonstrated by nearly complete blocking in the presence of the 26.5 α-MR1 antibody (Figure 4B). Overall, M. smegmatis- or S. pneumoniae-infected BECs from COPD donors induced higher, but not statistically significant, MAIT cell responses than infected BECs from healthy or smoker donors (Figure 4A and Table 2).
Figure 4.
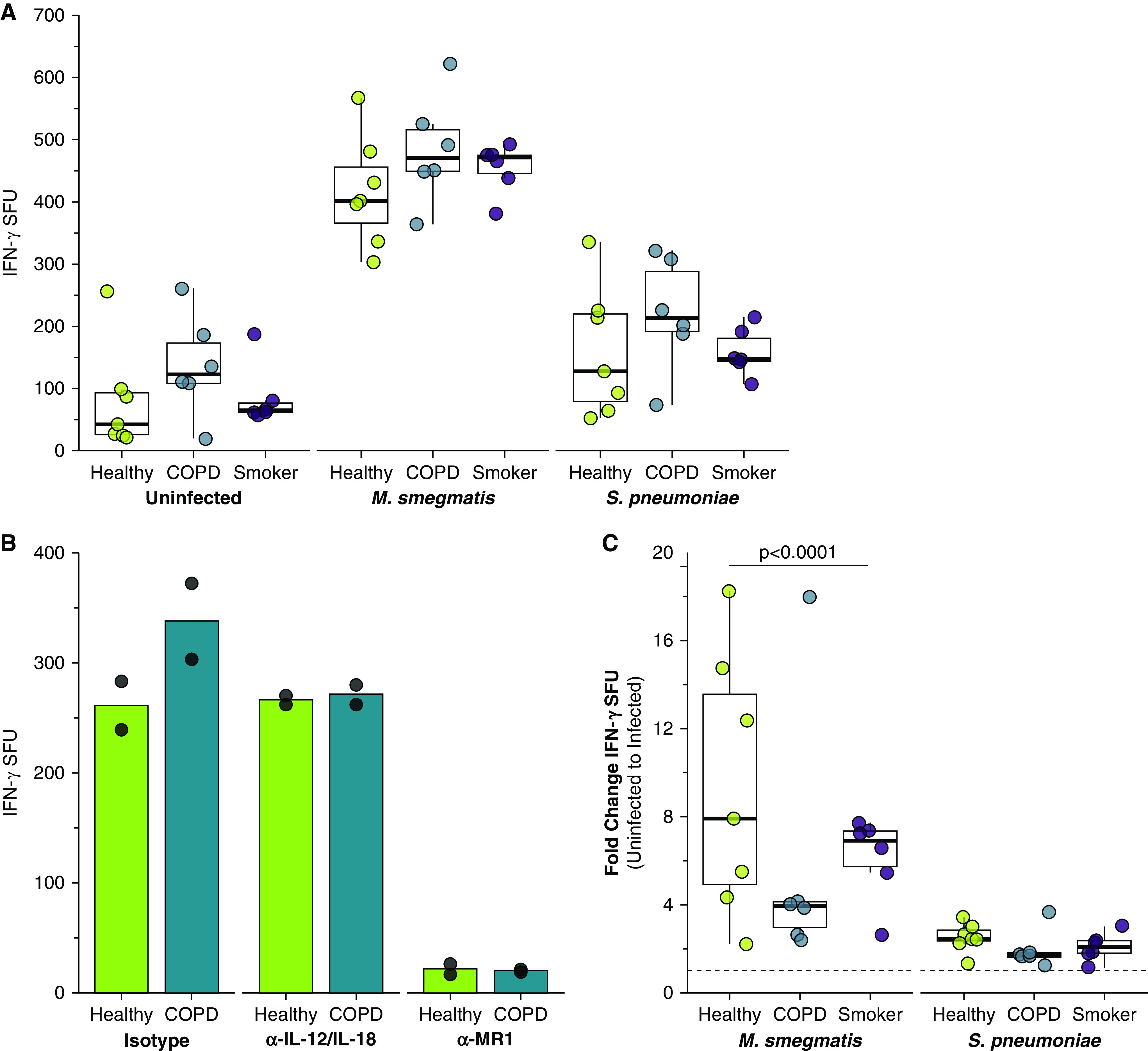
Increased microbe-dependent, MR1-dependent MAIT cell responses to infected primary BECs from COPD donors. (A) Primary BECs from healthy (n = 7), COPD (n = 6), or smoker (n = 6) donors were infected with media control, Mycobacterium smegmatis (0.1 μl/well), or S. pneumoniae (multiplicity of infection [MOI] = 20) for 1 hour before the addition of D426 G11 MAIT cells in an IFN-γ ELISPOT assay. Data points are the mean IFN-γ SFUs of two technical replicates per donor. Statistical analysis was performed as described in the data supplement and is summarized in Tables 2 and 3. (B) BECs from the healthy and COPD donors that induced the greatest IFN-γ SFUs in Figure 1A were incubated with blocking antibodies to IL-12/IL-18 or MR1 for 4 hours and infected with S. pneumoniae (MOI = 20) for 1 hour before the addition of the MAIT cells in an IFN-γ ELISPOT assay. Results are presented as the mean of two experimental replicates. (C) Fold change IFN-γ SFUs between uninfected and microbial-infected BECs from healthy, COPD, or smoker donors, calculated pairwise by donor from raw data in A. Statistical analysis was performed as described in the data supplement and is summarized in Table 2.
To quantify the infection-mediated increase in MAIT cell IFN-γ production and take into account the differences in bacterial burden between the donor groups, we compared the pairwise fold change in IFN-γ SFU responses between uninfected and infected donor BECs. Surprisingly, the infection-mediated increase in MAIT cell responses to infected COPD donor BECs was significantly reduced in comparison with fold-change responses to healthy and smoker donor BECs (P < 0.0001) (Figure 4C). Taken together with the observations of significantly greater bacterial infection per cell and overall higher induction of MAIT cell IFN-γ production, COPD donor BECs stimulated a weaker MAIT cell response on infection. These results suggest that MR1 antigen presentation is impaired in infected BECs from COPD lungs.
Exposure to Cigarette Smoke Decreases MAIT Cell Activation in Response to Infected Primary BECs from Healthy, COPD, and Smoker Lungs
We next explored the impact of CSE treatment in combination with bacterial infection. MAIT cell responses to infected BECs from all donor groups were significantly reduced by CSE treatment (Figures 5A and 5B and Tables 2 and 3). The decreased response with CSE treatment was unexpected, given the increased bacterial infection of CSE-treated cells (Figures 3B and 3C). In the context of this increased infection, our observation of decreased IFN-γ SFU response to CSE-treated cells suggested cigarette smoke may downregulate MR1 antigen presentation to MAIT cells. There were no significant donor group differences in the fold change IFN-γ response to CSE-treated, infected BECs (Figure 5C); these results suggest that the combination of infection and CSE treatment may affect healthy BECs similarly to COPD BECs. CSE treatment did not significantly affect S. pneumoniae infection of BECs from smoker donors (Figure 3C) despite reduced MAIT cell responses, suggesting that cigarette smoke alteration of S. pneumoniae infection and downstream MAIT cell responses may occur through different mechanisms in the context of acute versus chronic smoke exposure. Together, our data suggest a complex role for cigarette smoke in modulating MR1 antigen presentation to MAIT cells.
Figure 5.
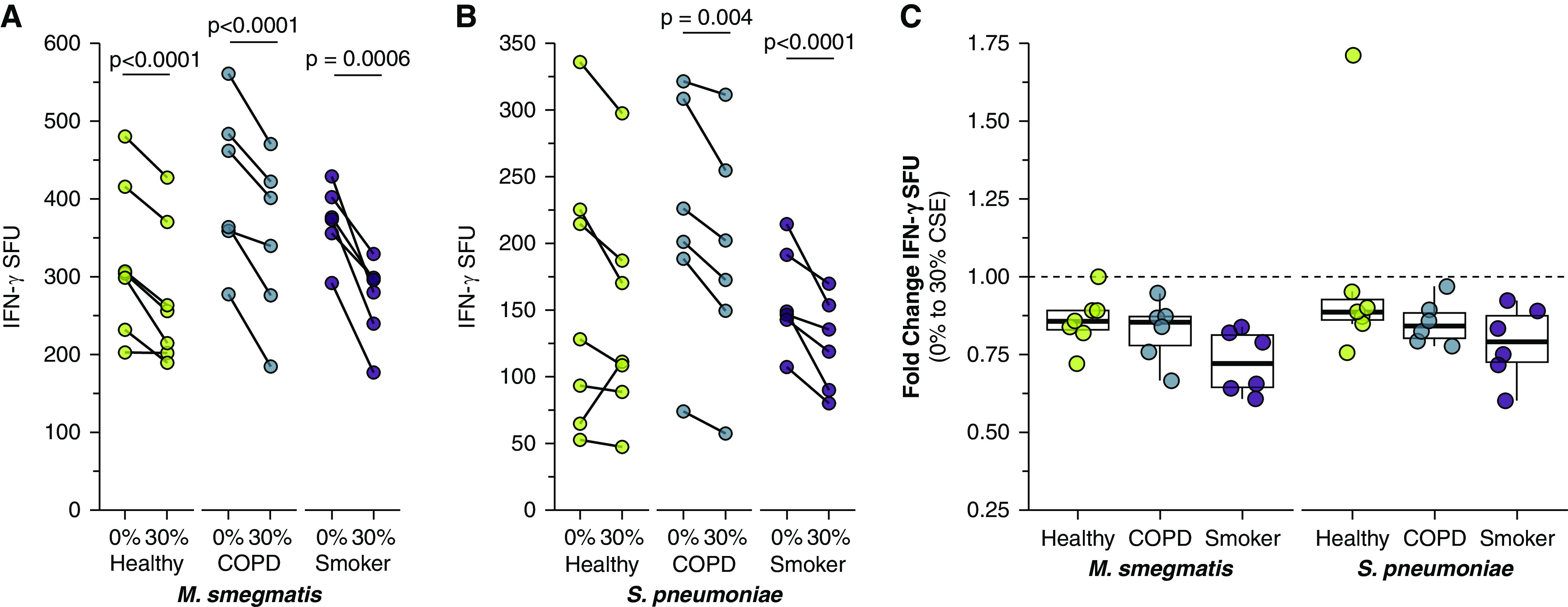
Reduced MAIT cell responses to infected BECs after treatment with CSE. (A and B) Primary BECs from healthy (n = 7), COPD (n = 6), or smoker (n = 6) donors were incubated with media containing 0% or 30% CSE for 3 hours. BECs were infected with (A) M. smegmatis (0.05 μl/well) or (B) S. pneumoniae (20 MOI) for 1 hour before the addition of D426 G11 MAIT cells in an IFN-γ ELISPOT assay. (C) Fold change IFN-γ SFUs between 0% and 30% CSE-treated primary BECs infected with M. smegmatis or S. pneumoniae, calculated pairwise by donor from data in A and B. Statistical analysis was performed as described in the data supplement and is summarized in Tables 2 and 3.
Acute Cigarette Smoke Exposure Increases MR1 Surface Translocation in Ligand-stimulated BECs
We had observed that CSE alone increases surface MR1 on healthy donor BECs but did not significantly affect MR1 expression in COPD or smoker donor BECs. To examine how the presence of MR1 ligand may impact MR1 expression, we used flow cytometry to assess surface expression at basal levels and after induction of MR1 surface translocation through treatment with the ligand 6-FP. 6-FP treatment induced significantly increased surface MR1 expression in healthy donors (P = 0.0149) (Figure 6A). Although not significant, we also observed a modest increase for 6-FP–treated COPD donor BECs (P = 0.0634). We then assessed the role of acute exposure to CSE in modulating these processes. The 6-FP–mediated increase in surface MR1 was increased in the context of CSE treatment for healthy donor BECs (P = 0.0029) (Figure 6B). For all conditions tested, healthy donor BECs expressed significantly higher degrees of surface MR1 than COPD donor BECs (P = 0.0447 and P = 0.0468) (Figures 6A and 6B). To explore whether this CSE- and ligand-mediated increase is because of greater overall expression of MR1, we incubated primary BECs with 0% or 30% CSE before infection with Sp, then harvested RNA to quantify relative MR1 mRNA expression. In the presence of S. pneumoniae infection, CSE treatment significantly reduced MR1 expression in healthy donor BECs (P = 0.0491) (Figure 6C), suggesting that the increase in surface MR1 expression is likely because of posttranslational impacts. We observed no significant pairwise changes from CSE treatment in combination with 6-FP or S. pneumoniae infection for smoker donor BECs (Figures 6B and 6C), further indicating that MR1 expression is differentially affected by acute CSE exposure, long-term cigarette smoking, and intracellular changes induced in BEC during the development of COPD. Together, our results demonstrate that acute exposure to cigarette smoke may impact ligand-dependent surface translocation of MR1.
Figure 6.
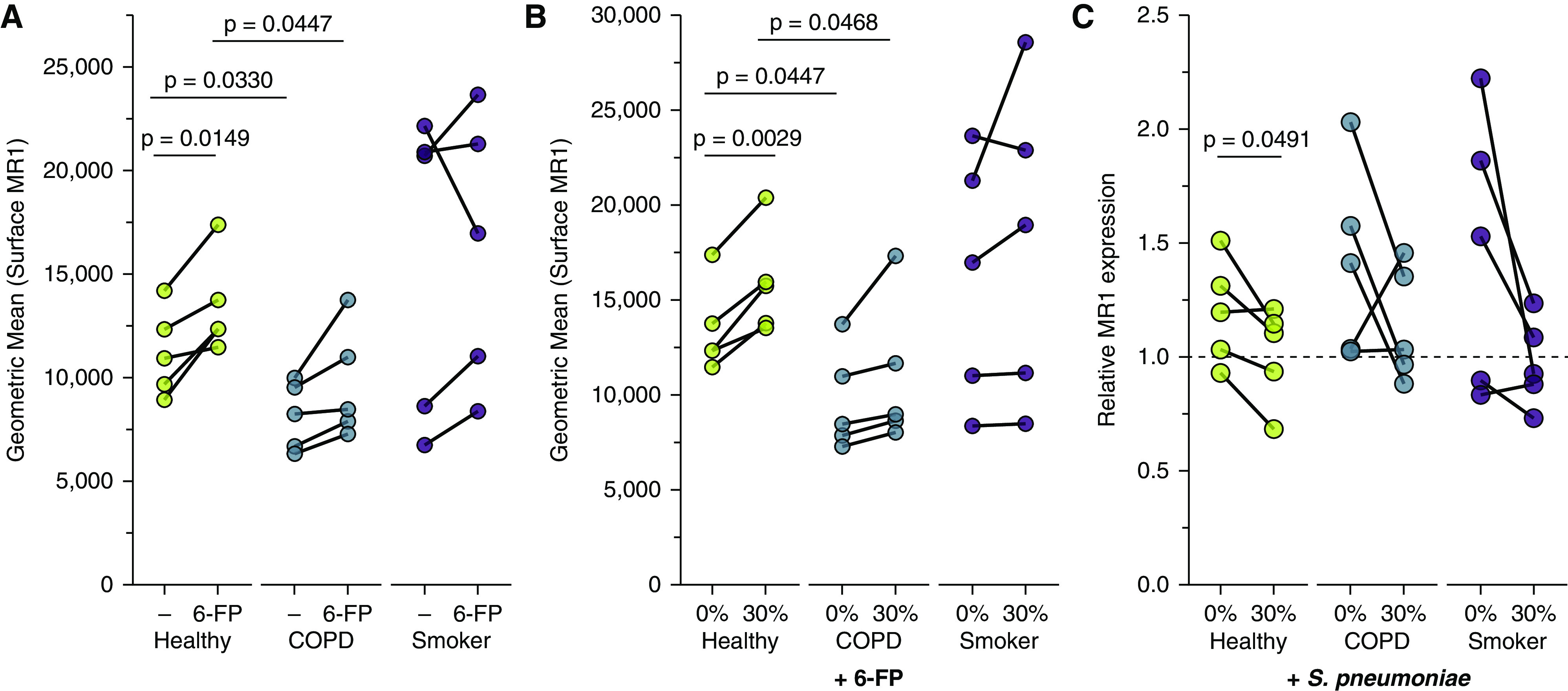
Increased MR1 expression in primary BECs exposed to cigarette smoke. (A and B) Primary BECs from healthy, COPD, or smoker donors (n = 5) were incubated with media containing 0% or 30% CSE for 3 hours where indicated, then incubated overnight with the MAIT cell ligand 6-FP (6-formylpterin) before harvest and staining for surface expression of MR1 by flow cytometry. Data points are mean fluorescence intensities paired by individual donor. (C) Primary BECs from healthy, COPD, or smoker donors were incubated with media containing 0% or 30% CSE for 3 hours, washed, then infected with S. pneumoniae for 3 hours. RNA was isolated from BECs, and real-time quantitative PCR (RT-PCR) was performed to detect amplification of MR1 and the internal control, HPRT1. MR1 expression was calculated by 2−ΔΔCt method, relative to no-treatment pairwise control and HPRT1 expression. Statistical significance was determined by two-tailed paired t tests for same-donor treatment analyses or unpaired t tests for donor group comparison.
Discussion
MAIT cells are an evolutionarily conserved subset of T cells present in high proportions in human blood and peripheral mucosal sites. Although MAIT cells were first described for their role in recognizing and responding to microbial infection (45, 58), evidence continues to grow for their role in inflammatory noninfectious diseases (26). Furthermore, MAIT cells have now been implicated in the homeostasis and repair of various mucosal barrier tissues, including the lung (59). MAIT cell functions may be relevant to the cigarette smoke-mediated development of airway inflammation resulting in COPD pathologies and to airway exacerbations common in COPD. Of note, numerous groups have observed decreased MAIT cell frequencies in both the peripheral blood and lungs of individuals with COPD (28, 29, 31), which is contrary to the increase in MAIT cell frequency observed in many inflammatory conditions. It is tempting to speculate that persistent inflammation and microbial colonization in COPD lungs could result in aberrant activation of MAIT cells leading to exhaustion and loss, as well as inappropriate recruitment of the adaptive lung immune response. Loss of MAIT cells could subsequently be an important factor in the inability to reverse tissue pathology observed in COPD lungs because of the loss of their function in tissue repair. In this way, MAIT cells could be important early immune contributors supporting the Goldilocks hypothesis of COPD pathogenesis proposed by Curtis and colleagues, in which too strong or too weak adaptive immune response can lead to worsened symptoms of COPD (22). Here, we considered how changes to large airway epithelial cells, the first line of defense against external assaults important to the development of COPD pathology, including cigarette smoke and microbial infection, alter MAIT cell activation.
We found that acute exposure BECs to CS generally resulted in decreased MAIT cell responses. This finding was particularly striking in the context of microbially infected BECs, in which despite significantly increased infection of BECs exposed to cigarette smoke, we observed significantly decreased MAIT cell response. We and others have repeatedly demonstrated in vitro and directly ex vivo that increased microbial antigen or infection of healthy, untreated cells results in increased MAIT cell responses (46). During microbial infection, MAIT cells are thought to play an important early role in immune response, for example, through the recruitment of cells like inflammatory monocytes to the site of infection (60, 61). Delayed recruitment of adaptive immune responses in the lungs of otherwise healthy smokers and the context of COPD exacerbations could allow for microbial persistence, inappropriately amplifying and prolonging lung inflammation.
We also observed greater microbe-independent MAIT cell responses to BECs than those observed in response to airway epithelial cell lines. These responses were also significantly higher in response to BECs from COPD lungs. We initially hypothesized this would be the result of cytokine-mediated MAIT cell activation because of reports of increased expression of cytokines like IL-18 in COPD lungs (62–64). To our surprise, these MAIT cell responses did not require IL-12 and IL-18 but were, in fact, dependent on MR1. Therefore, we examined MR1 expression. Little is known about the regulation of MR1 gene expression, although it is known that overexpression of MR1 increases MR1-dependent MAIT cell responses (e.g., Huber and colleagues [49]). In addition, genome-wide studies have identified MR1 as a gene with altered expression or methylation status in the context of e-cigarette smoking (65) and COPD lungs (66). Although our sample size was not sufficiently powered for statistical significance in this area, our real-time PCR data suggest the possibility for increased MR1 mRNA expression in BECs from COPD donors, with significantly decreased surface MR1 expression. Although BECs from smoker donors did express significantly more MR1 mRNA, we did not observe a corresponding increase in MAIT cell response or any significant changes to MR1 surface expression. There was also no impact of acute exposure to cigarette smoke on baseline MR1 expression in donors from any group, complicating the argument for the role of altered transcriptional regulation of MR1 in dysregulated induction of MAIT cell IFN-γ production by uninfected BECs.
We considered other possible explanations for the increased microbe-independent, MR1-dependent responses observed in BECs from COPD lungs. One group has posited the possibility that long-term tissue damage caused by cigarette smoke could lead to the production of T-cell neoantigens that contribute a potential autoimmune component to COPD-associated inflammation (67). There has not yet been an endogenous MR1 ligand identified; however, increasing evidence from cancer MAIT cell biology suggests the existence of self-ligands that can be modified in disease states (68). Because neoantigens are already known to be important MR1 ligands (69), the role of potential novel MR1 neoantigens produced in the context of damage from long-term cigarette smoke and COPD inflammation should be an avenue of interest. Given the small molecule nature of MR1 ligands, we initially hypothesized that cigarette smoke itself could contain novel ligands. However, absent other antigens, we did not observe any significant increase in MR1 expression of CSE-treated BECs. Furthermore, our functional data demonstrate that if cigarette smoke did contain MR1 ligands, they would not be MAIT−T cell receptor stimulatory. If anything, exposure to cigarette smoke decreased the microbe-dependent MAIT cell responses, suggesting that any putative ligands in cigarette smoke would be antagonistic. Alternately, acute exposure to cigarette smoke resulted in an increase in 6-FP–mediated MR1 surface translocation. This increase could be mediated by cigarette smoke through altered MR1 trafficking influencing ligand availability or access to putative chaperones for MR1. Together, these results demonstrate that short-term and long-term exposure to cigarette smoke could influence MR1 antigen presentation and MAIT cell responses in different ways through distinct mechanisms.
The mechanisms underlying COPD onset in some chronic smokers, but not others, remain unclear (22, 70). Dysfunctional MAIT cell activation could play a role in the early development of COPD-associated inflammation. Absent microbial stimulus, the greater overall MAIT cell response to COPD BECs suggests that hyperactive MAIT cells could facilitate inappropriate airway inflammation, possibly through the recruitment of inflammatory monocytes and neutrophils. Conversely, the hypoactivation of MAIT cells in response to infected and CS-exposed COPD BECs could permit microbial colonization and promote chronic stimulation of innate inflammation. In the broader pulmonary context, altered immune signaling from diverse innate and adaptive cell populations (such as alveolar macrophages and neutrophils) may contribute to MAIT cell dysregulation. Our study was limited to exploring MR1 antigen presentation by primary BEC to a healthy MAIT cell clone. Future exploration of inflammatory signaling between primary MAIT and other immune cells from COPD and smoker donors may reveal further insight into COPD development.
Conclusions
We demonstrate that MR1-dependent MAIT cell responses to BECs are altered in the context of COPD and cigarette smoke exposure. Understanding these impacts on MAIT cell activation may inform future therapies to treat these critically important pulmonary diseases.
Acknowledgments
Acknowledgment
The authors thank Dr. Suil Kim at VA Portland Health Care System for technical advice on cigarette smoke extract preparation and use. The authors thank Dr. Yalda Zarnegarnia and Jack Wiedrick of the Oregon Health & Science University Biostatistics & Design Program for their assistance in statistical analysis. The authors also thank the staff at Pacific Northwest Transplant Bank (now Cascade Alliance) for procuring the human lung and airway tissue samples used in this study. The D426G11 MAIT cell clone was a kind gift from Dr. David Lewinsohn.
Footnotes
Supported by the National Institutes of Health (NIH) (R01 AI129976 [M.J.H.] and T32 2T32HL083808 [M.E.H.]) and the U.S. Department of Veterans Affairs (Merit Award I01 CX001562 [M.J.H.]). The DeltaVision Core DV microscope is supported in part by NIH S10 RR023432. The contents of this article do not represent the views of the U.S. Department of Veterans Affairs or the U.S. government.
Author Contributions: M.E.H., E.L., and M.J.H. conceptualized the research design, interpreted the results of experiments, prepared figures, and drafted the manuscript. M.E.H., E.L., T.N.L., and C.M.H. performed experiments and analyzed data. All authors reviewed and approved the final version of the manuscript.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2022-0131OC on September 29, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.World Health Organization. 2019.
- 2. Sopori M. Effects of cigarette smoke on the immune system. Nat Rev Immunol . 2002;2:372–377. doi: 10.1038/nri803. [DOI] [PubMed] [Google Scholar]
- 3. Stämpfli MR, Anderson GP. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat Rev Immunol . 2009;9:377–384. doi: 10.1038/nri2530. [DOI] [PubMed] [Google Scholar]
- 4. Nuorti JP, Butler JC, Farley MM, Harrison LH, McGeer A, Kolczak MS, et al. Active Bacterial Core Surveillance Team Cigarette smoking and invasive pneumococcal disease. N Engl J Med . 2000;342:681–689. doi: 10.1056/NEJM200003093421002. [DOI] [PubMed] [Google Scholar]
- 5. Aghapour M, Raee P, Moghaddam SJ, Hiemstra PS, Heijink IH. Airway epithelial barrier dysfunction in chronic obstructive pulmonary disease: role of cigarette smoke exposure. Am J Respir Cell Mol Biol . 2018;58:157–169. doi: 10.1165/rcmb.2017-0200TR. [DOI] [PubMed] [Google Scholar]
- 6. Eltom S, Stevenson CS, Rastrick J, Dale N, Raemdonck K, Wong S, et al. P2X7 receptor and caspase 1 activation are central to airway inflammation observed after exposure to tobacco smoke. PLoS One . 2011;6:e24097. doi: 10.1371/journal.pone.0024097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kang MJ, Homer RJ, Gallo A, Lee CG, Crothers KA, Cho SJ, et al. IL-18 is induced and IL-18 receptor alpha plays a critical role in the pathogenesis of cigarette smoke-induced pulmonary emphysema and inflammation. J Immunol . 2007;178:1948–1959. doi: 10.4049/jimmunol.178.3.1948. [DOI] [PubMed] [Google Scholar]
- 8. Yi G, Liang M, Li M, Fang X, Liu J, Lai Y, et al. A large lung gene expression study identifying IL1B as a novel player in airway inflammation in COPD airway epithelial cells. Inflamm Res . 2018;67:539–551. doi: 10.1007/s00011-018-1145-8. [DOI] [PubMed] [Google Scholar]
- 9. Li C, Zhihong H, Wenlong L, Xiaoyan L, Qing C, Wenzhi L, et al. The nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 inflammasome regulates bronchial epithelial cell injury and proapoptosis after exposure to biomass fuel smoke. Am J Respir Cell Mol Biol . 2016;55:815–824. doi: 10.1165/rcmb.2016-0051OC. [DOI] [PubMed] [Google Scholar]
- 10. Churg A, Cosio M, Wright JL. Mechanisms of cigarette smoke-induced COPD: insights from animal models. Am J Physiol Lung Cell Mol Physiol . 2008;294:L612–L631. doi: 10.1152/ajplung.00390.2007. [DOI] [PubMed] [Google Scholar]
- 11. Kang XZ, Chen KN. [Chronic obstructive pulmonary disease and lung cancer: advances in the study of epigenetic modification enzymes] Zhonghua Jie He He Hu Xi Za Zhi . 2012;35:286–288. [PubMed] [Google Scholar]
- 12. Freeman CM, Curtis JL, Chensue SW. CC chemokine receptor 5 and CXC chemokine receptor 6 expression by lung CD8+ cells correlates with chronic obstructive pulmonary disease severity. Am J Pathol . 2007;171:767–776. doi: 10.2353/ajpath.2007.061177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saetta M, Di Stefano A, Turato G, Facchini FM, Corbino L, Mapp CE, et al. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 1998;157:822–826. doi: 10.1164/ajrccm.157.3.9709027. [DOI] [PubMed] [Google Scholar]
- 14. Saetta M, Mariani M, Panina-Bordignon P, Turato G, Buonsanti C, Baraldo S, et al. Increased expression of the chemokine receptor CXCR3 and its ligand CXCL10 in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2002;165:1404–1409. doi: 10.1164/rccm.2107139. [DOI] [PubMed] [Google Scholar]
- 15. Strzelak A, Ratajczak A, Adamiec A, Feleszko W. Tobacco smoke induces and alters immune responses in the lung triggering inflammation, allergy, asthma and other lung diseases: a mechanistic review. Int J Environ Res Public Health . 2018;15:1033. doi: 10.3390/ijerph15051033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grigg J, Walters H, Sohal SS, Wood-Baker R, Reid DW, Xu CB, et al. Cigarette smoke and platelet-activating factor receptor dependent adhesion of Streptococcus pneumoniae to lower airway cells. Thorax . 2012;67:908–913. doi: 10.1136/thoraxjnl-2011-200835. [DOI] [PubMed] [Google Scholar]
- 17. Drannik AG, Pouladi MA, Robbins CS, Goncharova SI, Kianpour S, Stämpfli MR. Impact of cigarette smoke on clearance and inflammation after Pseudomonas aeruginosa infection. Am J Respir Crit Care Med . 2004;170:1164–1171. doi: 10.1164/rccm.200311-1521OC. [DOI] [PubMed] [Google Scholar]
- 18.Gou X, Zhang Q, More S, Bamunuarachchi G, Liang Y, Haider Khan F, et al. Repeated exposure to Streptococcus pneumoniae exacerbates chronic obstructive pulmonary disease. Am J Pathol. 2019;189:1711–1720. doi: 10.1016/j.ajpath.2019.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barnes PJ, Burney PG, Silverman EK, Celli BR, Vestbo J, Wedzicha JA, et al. Chronic obstructive pulmonary disease. Nat Rev Dis Primers . 2015;1:15076. doi: 10.1038/nrdp.2015.76. [DOI] [PubMed] [Google Scholar]
- 20. Vestbo J, Hurd SS, Agustí AG, Jones PW, Vogelmeier C, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med . 2013;187:347–365. doi: 10.1164/rccm.201204-0596PP. [DOI] [PubMed] [Google Scholar]
- 21.Organization WH.2020.
- 22. Curtis JL, Freeman CM, Hogg JC. The immunopathogenesis of chronic obstructive pulmonary disease: insights from recent research. Proc Am Thorac Soc . 2007;4:512–521. doi: 10.1513/pats.200701-002FM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Williams M, Todd I, Fairclough LC. The role of CD8 + T lymphocytes in chronic obstructive pulmonary disease: a systematic review. Inflamm Res . 2021;70:11–18. doi: 10.1007/s00011-020-01408-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Motz GT, Eppert BL, Sun G, Wesselkamper SC, Linke MJ, Deka R, et al. Persistence of lung CD8 T cell oligoclonal expansions upon smoking cessation in a mouse model of cigarette smoke-induced emphysema. J Immunol . 2008;181:8036–8043. doi: 10.4049/jimmunol.181.11.8036. [DOI] [PubMed] [Google Scholar]
- 25. Treiner E, Duban L, Bahram S, Radosavljevic M, Wanner V, Tilloy F, et al. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature . 2003;422:164–169. doi: 10.1038/nature01433. [DOI] [PubMed] [Google Scholar]
- 26. Godfrey DI, Koay HF, McCluskey J, Gherardin NA. The biology and functional importance of MAIT cells. Nat Immunol . 2019;20:1110–1128. doi: 10.1038/s41590-019-0444-8. [DOI] [PubMed] [Google Scholar]
- 27. Wang H, D’Souza C, Lim XY, Kostenko L, Pediongco TJ, Eckle SBG, et al. MAIT cells protect against pulmonary Legionella longbeachae infection. Nat Commun . 2018;9:3350. doi: 10.1038/s41467-018-05202-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hinks TS, Wallington JC, Williams AP, Djukanović R, Staples KJ, Wilkinson TM. Steroid-induced deficiency of mucosal-associated invariant T cells in the chronic obstructive pulmonary disease lung. Implications for nontypeable Haemophilus influenzae infection. Am J Respir Crit Care Med . 2016;194:1208–1218. doi: 10.1164/rccm.201601-0002OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kwon YS, Jin HM, Cho YN, Kim MJ, Kang JH, Jung HJ, et al. Mucosal-associated invariant T cell deficiency in chronic obstructive pulmonary disease. COPD . 2016;13:196–202. doi: 10.3109/15412555.2015.1069806. [DOI] [PubMed] [Google Scholar]
- 30. Qiu W, Kang N, Wu Y, Cai Y, Xiao L, Ge H, et al. Mucosal associated invariant T cells were activated and polarized toward Th17 in chronic obstructive pulmonary disease. Front Immunol . 2021;12:640455. doi: 10.3389/fimmu.2021.640455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Szabó M, Sárosi V, Balikó Z, Bodó K, Farkas N, Berki T, et al. Deficiency of innate-like T lymphocytes in chronic obstructive pulmonary disease. Respir Res . 2017;18:197. doi: 10.1186/s12931-017-0671-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pincikova T, Parrot T, Hjelte L, Högman M, Lisspers K, Ställberg B, et al. MAIT cell counts are associated with the risk of hospitalization in COPD. Respir Res . 2022;23:127. doi: 10.1186/s12931-022-02045-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eckle SB, Birkinshaw RW, Kostenko L, Corbett AJ, McWilliam HE, Reantragoon R, et al. A molecular basis underpinning the T cell receptor heterogeneity of mucosal-associated invariant T cells. J Exp Med . 2014;211:1585–1600. doi: 10.1084/jem.20140484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kjer-Nielsen L, Patel O, Corbett AJ, Le Nours J, Meehan B, Liu L, et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature . 2012;491:717–723. doi: 10.1038/nature11605. [DOI] [PubMed] [Google Scholar]
- 35. Hartmann N, Harriff MJ, McMurtrey CP, Hildebrand WH, Lewinsohn DM, Kronenberg M. Role of MAIT cells in pulmonary bacterial infection. Mol Immunol . 2018;101:155–159. doi: 10.1016/j.molimm.2018.06.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hartmann N, McMurtrey C, Sorensen ML, Huber ME, Kurapova R, Coleman FT, et al. Riboflavin metabolism variation among clinical isolates of Streptococcus pneumoniae results in differential activation of mucosal-associated invariant T cells. Am J Respir Cell Mol Biol . 2018;58:767–776. doi: 10.1165/rcmb.2017-0290OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kurioka A, van Wilgenburg B, Javan RR, Hoyle R, van Tonder AJ, Harrold CL, et al. Diverse Streptococcus pneumoniae strains drive a mucosal-associated invariant T-cell response through major histocompatibility complex class I-related molecule-dependent and cytokine-driven pathways. J Infect Dis . 2018;217:988–999. doi: 10.1093/infdis/jix647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ussher JE, Bilton M, Attwod E, Shadwell J, Richardson R, de Lara C, et al. CD161++ CD8+ T cells, including the MAIT cell subset, are specifically activated by IL-12+IL-18 in a TCR-independent manner. Eur J Immunol . 2014;44:195–203. doi: 10.1002/eji.201343509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kang MJ, Choi JM, Kim BH, Lee CM, Cho WK, Choe G, et al. IL-18 induces emphysema and airway and vascular remodeling via IFN-γ, IL-17A, and IL-13. Am J Respir Crit Care Med . 2012;185:1205–1217. doi: 10.1164/rccm.201108-1545OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harriff MJ, Larson E, Leiser MR.2020. [Google Scholar]
- 41. Huber ME, Larson E, Lust TN, Heisler CM, Harriff MJ. MR1 expression and surface translocation in airway epithelial cells from COPD or smoker lungs is altered following exposure to IFNγ or MAIT cells. J immunol . 2021;206:93.08. [Google Scholar]
- 42.Huber ME, Larson E, Lust TN, Heisler CM, Harriff MJ.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huber ME, Larson E, Lust TN, Heisler CM, Harriff MJ.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wong J, Korcheva V, Jacoby DB, Magun BE. Proinflammatory responses of human airway cells to ricin involve stress-activated protein kinases and NF-kappaB. Am J Physiol Lung Cell Mol Physiol . 2007;293:L1385–L1394. doi: 10.1152/ajplung.00207.2007. [DOI] [PubMed] [Google Scholar]
- 45. Gold MC, Cerri S, Smyk-Pearson S, Cansler ME, Vogt TM, Delepine J, et al. Human mucosal associated invariant T cells detect bacterially infected cells. PLoS Biol . 2010;8:e1000407. doi: 10.1371/journal.pbio.1000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Harriff MJ, Karamooz E, Burr A, Grant WF, Canfield ET, Sorensen ML, et al. Endosomal MR1 trafficking plays a key role in presentation of Mycobacterium tuberculosis ligands to MAIT cells. PLoS Pathog . 2016;12:e1005524. doi: 10.1371/journal.ppat.1005524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lewinsohn DM, Briden AL, Reed SG, Grabstein KH, Alderson MR. Mycobacterium tuberculosis-reactive CD8+ T lymphocytes: the relative contribution of classical versus nonclassical HLA restriction. J Immunol . 2000;165:925–930. doi: 10.4049/jimmunol.165.2.925. [DOI] [PubMed] [Google Scholar]
- 48. Karamooz E, Harriff MJ, Narayanan GA, Worley A, Lewinsohn DM. MR1 recycling and blockade of endosomal trafficking reveal distinguishable antigen presentation pathways between Mycobacterium tuberculosis infection and exogenously delivered antigens. Sci Rep . 2019;9:4797. doi: 10.1038/s41598-019-41402-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Huber ME, Kurapova R, Heisler CM, Karamooz E, Tafesse FG, Harriff MJ. Rab6 regulates recycling and retrograde trafficking of MR1 molecules. Sci Rep . 2020;10:20778. doi: 10.1038/s41598-020-77563-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Higashi T, Mai Y, Noya Y, Horinouchi T, Terada K, Hoshi A, et al. A simple and rapid method for standard preparation of gas phase extract of cigarette smoke. PLoS One . 2014;9:e107856. doi: 10.1371/journal.pone.0107856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Heinzel AS, Grotzke JE, Lines RA, Lewinsohn DA, McNabb AL, Streblow DN, et al. HLA-E-dependent presentation of Mtb-derived antigen to human CD8+ T cells. J Exp Med . 2002;196:1473–1481. doi: 10.1084/jem.20020609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods . 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 53. Gold MC, McLaren JE, Reistetter JA, Smyk-Pearson S, Ladell K, Swarbrick GM, et al. MR1-restricted MAIT cells display ligand discrimination and pathogen selectivity through distinct T cell receptor usage. J Exp Med . 2014;211:1601–1610. doi: 10.1084/jem.20140507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McWilliam HEG, Villadangos JA. How MR1 presents a pathogen metabolic signature to mucosal-associated invariant T (MAIT) cells. Trends Immunol . 2017;38:679–689. doi: 10.1016/j.it.2017.06.005. [DOI] [PubMed] [Google Scholar]
- 55. El Ahmer OR, Essery SD, Saadi AT, Raza MW, Ogilvie MM, Weir DM, et al. The effect of cigarette smoke on adherence of respiratory pathogens to buccal epithelial cells. FEMS Immunol Med Microbiol . 1999;23:27–36. doi: 10.1111/j.1574-695X.1999.tb01713.x. [DOI] [PubMed] [Google Scholar]
- 56. Mahajan B, Panhotra BR. Adherence of Streptococcus pneumoniae to buccal epithelial cells of smokers & non-smokers. Indian J Med Res . 1989;89:381–383. [PubMed] [Google Scholar]
- 57. Murakami D, Kono M, Nanushaj D, Kaneko F, Zangari T, Muragaki Y, et al. Exposure to cigarette smoke enhances pneumococcal transmission among littermates in an infant mouse model. Front Cell Infect Microbiol . 2021;11:651495. doi: 10.3389/fcimb.2021.651495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Le Bourhis L, Martin E, Péguillet I, Guihot A, Froux N, Coré M, et al. Antimicrobial activity of mucosal-associated invariant T cells. Nat Immunol . 2010;11:701–708. doi: 10.1038/ni.1890. [DOI] [PubMed] [Google Scholar]
- 59. Nel I, Bertrand L, Toubal A, Lehuen A. MAIT cells, guardians of skin and mucosa? Mucosal Immunol . 2021;14:803–814. doi: 10.1038/s41385-021-00391-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Meierovics A, Yankelevich WJ, Cowley SC. MAIT cells are critical for optimal mucosal immune responses during in vivo pulmonary bacterial infection. Proc Natl Acad Sci USA . 2013;110:E3119–E3128. doi: 10.1073/pnas.1302799110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Meierovics AI, Cowley SC. MAIT cells promote inflammatory monocyte differentiation into dendritic cells during pulmonary intracellular infection. J Exp Med . 2016;213:2793–2809. doi: 10.1084/jem.20160637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Christenson SA, van den Berge M, Faiz A, Inkamp K, Bhakta N, Bonser LR, et al. An airway epithelial IL-17A response signature identifies a steroid-unresponsive COPD patient subgroup. J Clin Invest . 2019;129:169–181. doi: 10.1172/JCI121087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Imaoka H, Hoshino T, Takei S, Kinoshita T, Okamoto M, Kawayama T, et al. Interleukin-18 production and pulmonary function in COPD. Eur Respir J . 2008;31:287–297. doi: 10.1183/09031936.00019207. [DOI] [PubMed] [Google Scholar]
- 64. Petersen AM, Penkowa M, Iversen M, Frydelund-Larsen L, Andersen JL, Mortensen J, et al. Elevated levels of IL-18 in plasma and skeletal muscle in chronic obstructive pulmonary disease. Lung . 2007;185:161–171. doi: 10.1007/s00408-007-9000-7. [DOI] [PubMed] [Google Scholar]
- 65. Rebuli ME, Glista-Baker E, Hoffman JR, Duffney PF, Robinette C, Speen AM, et al. Electronic-cigarette use alters nasal mucosal immune response to live-attenuated influenza virus. A clinical trial. Am J Respir Cell Mol Biol . 2021;64:126–137. doi: 10.1165/rcmb.2020-0164OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sundar IK, Yin Q, Baier BS, Yan L, Mazur W, Li D, et al. DNA methylation profiling in peripheral lung tissues of smokers and patients with COPD. Clin Epigenetics . 2017;9:38. doi: 10.1186/s13148-017-0335-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Taraseviciene-Stewart L, Douglas IS, Nana-Sinkam PS, Lee JD, Tuder RM, Nicolls MR, et al. Is alveolar destruction and emphysema in chronic obstructive pulmonary disease an immune disease? Proc Am Thorac Soc . 2006;3:687–690. doi: 10.1513/pats.200605-105SF. [DOI] [PubMed] [Google Scholar]
- 68. Crowther MD, Dolton G, Legut M, Caillaud ME, Lloyd A, Attaf M, et al. Genome-wide CRISPR-Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nat Immunol . 2020;21:178–185. doi: 10.1038/s41590-019-0578-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Corbett AJ, Eckle SB, Birkinshaw RW, Liu L, Patel O, Mahony J, et al. T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature . 2014;509:361–365. doi: 10.1038/nature13160. [DOI] [PubMed] [Google Scholar]
- 70. Nguyen JMK, Robinson DN, Sidhaye VK. Why new biology must be uncovered to advance therapeutic strategies for chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol . 2021;320:L1–L11. doi: 10.1152/ajplung.00367.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]