

Abstract
The mechanisms by which excessive systemic activation of adaptive T lymphocytes, as in cytokine release syndrome (CRS), leads to innate immune cell-mediated acute lung injury (ALI) or acute respiratory distress syndrome, often in the absence of any infection, remains unknown. Here, we investigated the roles of IFN-γ and IL-17A, key T-cell cytokines significantly elevated in patients with CRS, in the immunopathogenesis of CRS-induced extrapulmonary ALI. CRS was induced in wild-type (WT), IL-17A- and IFN-γ knockout (KO) human leukocyte antigen-DR3 transgenic mice with 10 μg of the superantigen, staphylococcal enterotoxin B, given intraperitoneally. Several ALI parameters, including gene expression profiling in the lungs, were studied 4, 24, or 48 hours later. Systemic T-cell activation with staphylococcal enterotoxin B resulted in robust upregulation of several chemokines, S100A8/A9, matrix metalloproteases, and other molecules implicated in tissue damage, granulocyte as well as agranulocyte adhesion, and diapedesis in the lungs as early as 4 hours, which was accompanied by subsequent neutrophil/eosinophil lung infiltration and severe ALI in IFN-γ KO mice. These pathways were significantly underexpressed in IL-17A KO mice, which manifested mildest ALI and intermediate in WT mice. Neutralization of IFN-γ worsened ALI in WT and IL-17A KO mice, whereas neutralizing IL-17A did not mitigate lung injury in IFN-γ KO mice, suggesting a dominant protective role for IFN-γ in ALI and that IL-17A is dispensable. Ruxolitinib, a Janus kinase inhibitor, increased ALI severity in WT mice. Thus, our study identified novel mechanisms of ALI in CRS and its differential modulation by IFN-γ and IL-17A.
Keywords: acute lung injury, acute respiratory distress syndrome, cytokine release syndrome, IFN-γ, IL-17A
Acute lung injury (ALI), or its more severe form, acute respiratory distress syndrome (ARDS), are common presentations of cytokine storm syndrome or cytokine release syndrome (CRS), the umbrella terms used to describe the systemic illnesses resulting from generalized hyperactivation of adaptive T cells accompanied by excessive production of Th1, Th2, and Th17 cytokines among others (1–4). Although cancer immunotherapies are the primary causes of CRS, lung injuries after certain viral (influenza and coronavirus, including severe acute respiratory syndrome coronavirus 2 [SARS-CoV-2]), as well as bacterial infections caused by Staphylococcus aureus and Streptococcus pyogenes producing superantigen exotoxins may also result from CRS, and not by the pathogens per se (1, 2, 5, 6). However, the mechanisms by which systemic hyperactivation of adaptive T cells leads to innate immune cell (neutrophil) mediated ALI/ARDS are not known. Delineating the mechanisms may lead to novel ARDS treatments (7). This mandates a novel mouse model.
Most ALI models are limited in this regard because lung injuries in these models typically result from direct activation of the innate immune system through pattern recognition receptors. Most importantly, ALI in these models can occur even in the absence of T cells, thus failing to mimic human CRS (8–10). Even most pathogen-induced ALI models do not recapitulate human CRS because manipulation of adaptive T cells or cytokines interferes with pathogen clearance resulting in direct pathogen-mediated rather than immune-mediated lung injury (10, 11). Therefore, extrapulmonary ALI (eALI) induced with purified bacterial superantigen is ideal for modeling human CRS because of its unique biological properties (12, 13).
Bacterial superantigens (BSAg), a family of exotoxins produced by S. aureus and S. pyogenes, bind directly to major histocompatibility complex class II molecules outside of the peptide binding groove without undergoing any processing (14). Subsequently, BSAgs activate 30–50% of CD4+ and CD8+ T cells bearing certain T-cell receptor (TCR) α or β chain variable gene families, resulting in a profound systemic cytokine storm and multiorgan dysfunction with ALI that is analogous to human CRS. Because of the more efficient binding of BSAg to human major histocompatibility complex (called human leukocyte antigen or HLA) class II molecules compared with their murine counterparts, we and others have shown that transgenic mice expressing HLA-DR and HLA-DQ molecules respond far more robustly to BSAg than regular laboratory mice, produce significantly higher concentrations of T-cell–derived cytokines, including IFN-γ and IL-17A, and readily develop severe eALI after systemic BSAg challenge (15–19). Thus, whereas direct intratracheal administration of purified lipopolysaccharide, a gram-negative bacterial derivative, is often used to study the immunopathogenesis of innate immune cell-driven pulmonary or direct ALI, we used purified BSAg produced by the gram-positive bacterial pathogen S. aureus to investigate how systemic activation of adaptive T cells causes innate immune cell-mediated eALI and how IFN-γ and IL-17A significantly elevated in CRS differentially modulate the eALI accompanying CRS.
Methods
Experimental Animals
HLA-DR3 transgenic mice (wild type [WT]) and HLA-DR3 transgenic mice lacking IL-17A (IL-17 knockout [KO]) or IFN-γ (IFN-γ KO) are described elsewhere (15, 19).
Induction of eALI
eALI was induced in 8- to 16-week-old male and female mice by intraperitoneal injection of 10 micrograms of endotoxin-reduced staphylococcal enterotoxin B (SEB) (Toxin Technologies) in PBS.
Measurement of ALI Parameters
At indicated time points, BAL fluids were collected, and BAL cells were separated and counted. BAL cell differential counts were determined using Hema 3 stained cytospin preparation (20). Total protein, lactate dehydrogenase (LDH), neutrophil elastase, and neutrophil extracellular traps in BAL fluids were determined as described in detail elsewhere (21). BAL Ang-2 (angiopoietin-2) and S100A8/S100A9 concentrations were determined using ELISA kits (R&D Systems). Concentrations of various cytokines and chemokines were determined using multiplex kits (EMD Millipore).
In vivo Neutralization of IFN-γ and IL-17A
Mice received 0.5 mg of anti–IFN-γ, anti–IL-17A, or isotype control antibodies (Bioxcell) by intraperitoneal injection, and eALI was induced 2 hours later as above.
Administration of Ruxolitinib
The Janus kinase (JAK)1/2 inhibitor, ruxolitinib, was given twice daily (50 mg/kg per mouse, Selleckchem) by gavage on Days −1, 0, and 1. eALI was induced with SEB on Day 0, and the experiment was terminated 48 hours after SEB challenge.
Histopathology
After transcardial perfusion of mice with 10 ml of PBS, lungs were removed, fixed in buffered formalin, sectioned, and stained with hematoxylin and eosin at the Yale Histology Core Facility. Sections were reviewed by Dr. Homer in a blinded fashion. Arbitrary scores of 0, 1, or 2 were given on the basis of the extent of inflammatory infiltrate. Presence of inflammation in less than 5% of the tissue = 0, 5–20% of tissue = 1, and more than 25% of tissue = 2.
Flow Cytometry
Flow cytometry was performed on BAL cells to determine the distribution of various leukocyte subsets per standard procedure (Figure E1 in the data supplement). The following antibodies were used. Anti-mouse CD45 (clone 3C-F11, Invitrogen), anti-mouse Siglec F (clone E50–2440; BD Pharmingen), anti-mouse CD11c (clone N418, eBioscience), anti-mouse F4/80, (clone BMB; Invitrogen), anti-mouse CD11b (clone M1/70; BD Biosciences), and anti-Ly6G (clone RB6–8C5).
Gene Expression Profiling Using Microarrays
Total RNA was extracted from perfused lungs at 4 and 24 hours after SEB challenge. Gene expression profiling was determined using mouse Clariom S arrays at ThermoFisher Corporation. CEL data files were exported into Transcriptome Analysis Console (TAC; Applied Biosystems) software and analyzed as per the recommended procedure. Both unadjusted P values and adjusted P values (false discovery rate) were determined. Genes passing the filtering criteria (fold change greater than 2 or less than −2 and P value < 0.05 compared with naive mice) were included in further analyses. Normalized expression data were also analyzed using Ingenuity Pathway Analysis (IPA) program through institutional licensing, and comparative analysis was performed to identify pathways differentially expressed between different groups. Because several genes passed the initial selection criteria (fold change > 2 or < −2 and P < 0.05 compared with naive mice), only genes with a false discovery rate of P < 0.05 were included in subsequent pathway analysis using IPA.
Statistics
All statistical analysis was performed using GraphPad Prism software (Prism 9). Where the effects of genotype (WT, IFN-γ KO, and IL-17 KO) or treatments (test antibody vs. isotype control subjects) in relation to different time points (4, 24, 48, or 72 h) were studied, two-way ANOVA with multiple comparisons was used to determine the statistical significance. Otherwise, the Mann-Whitney test was used for two-group comparisons. Statistical comparisons and P values within the figures were inserted using the built-in function in the software. Serum and BAL cytokine/chemokine data were also reanalyzed using SAS PROC GLM (by the coauthor B.H., who is a programmer/analyst) for conformity with Prism results. The GLM procedure uses the method of least squares to fit general linear models. The class variables are time points and genotypes.
Results
Induction of Extrapulmonary ALI with Systemically Administered BSAg and Its Modulation by IFN-γ and IL-17A
Consistent with our prior studies, HLA-DR3 WT mice systemically challenged with low-dose SEB developed lung injury, predominantly perivascular inflammation, which peaked at 48 hours, the last time point studied (Figures 1A and 1B, histopathology data not shown for additional time points) (15, 19). Paradoxically, unlike in other models of ALI wherein IFN-γ KO mice are protected (11, 12, 22–25), HLA-DR3 IFN-γ KO mice developed the most severe lung inflammation at all time points (Figures 1A and 1B, histopathology data not shown for additional time points), whereas the lung injury was lowest in HLA-DR3 IL-17 KO mice (Figures 1A and 1B). Consistent with ALI severity, several markers of lung injury such as total protein (Figure 1C), LDH (Figure 1D), neutrophil elastase (Figure 1E), neutrophil extracellular traps (Figure 1F), Ang-2 (Figure 1G), and S100A8/A9 (Figure 1H) were significantly elevated in BAL from IFN-γ KO mice, intermediate in WT mice, and lowest in IL-17 KO mice.
Figure 1.
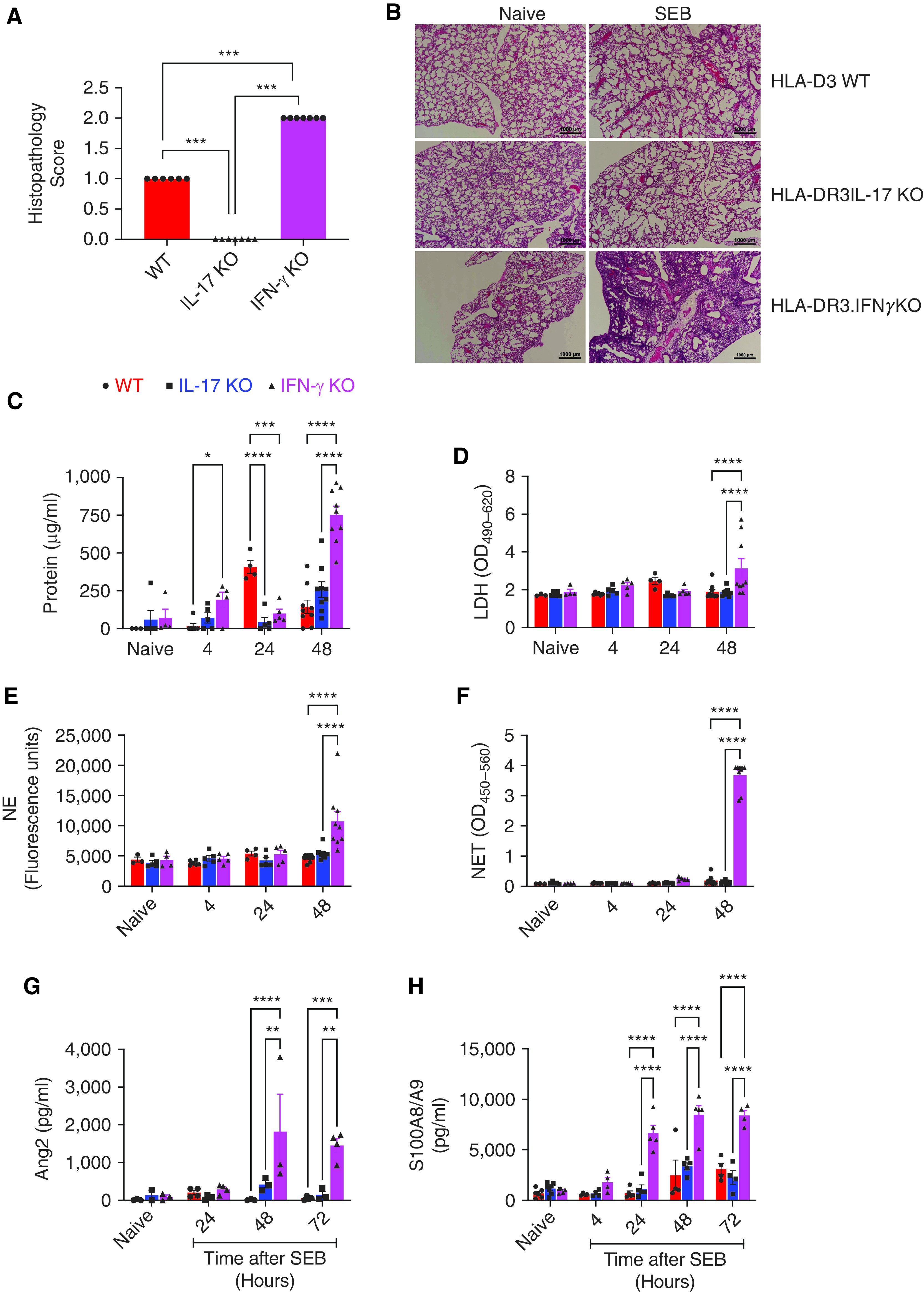
Systemic administration of the staphylococcal superantigen, staphylococcal enterotoxin B (SEB), induces acute lung injury in HLA-DR3 mice and is differentially modulated by IFN-γ and IL-17: HLA-DR3.WT, HLA-DR3.IL-17 KO and HLA-DR3.IFN-γ KO mice were challenged intraperitoneally with 10 μg of SEB. (A and B) Naive as well as mice challenged with SEB were killed at 48 hours. Lungs were collected in buffered formalin, paraffin-embedded, sectioned, and stained with HandE. Slides were evaluated in a blinded fashion and scored as described in the Methods section. Data represent mean ± SE from at least six mice per group. Representative images are shown in panel B. (C–H). BAL fluids from naive as well as mice challenged with SEB were collected at indicated time points. Concentrations of (C) protein, (D) LDH, (E) NE, (F) NET, (G) Ang-2, and (H) S100A8/S100A9 were determined as described in the Methods section. Data represent mean ± SE from at least 4–9 mice per group. *P < 0.5, **P < 0.005, ***P < 0.0005, and ****P < 0.00005. Ang2 = angiopoietin-2; HandE = hematoxylin and eosin; HLA = human leukocyte antigen; KO = knockout; LDH = lactate dehydrogenase; NE = neutrophil elastase; NET = neutrophil extracellular trap; WT = wild-type.
Next, total BAL cell counts were made using an automated cell counter, and differential counts were determined by light microscopy at different time points. Total BAL cells were significantly higher in IFN-γ KO mice compared with the other two groups at 48 hours (Figure 2A). By light microscopy on Hema 3 stained cytospin preparation of BAL cells, multinucleated cells morphologically resembling polymorphonuclear cells (PMNs), as well as eosinophils (Eos), were significantly elevated in IFN-γ KO mice (data not shown). As identifying and distinguishing PMNs and Eos by light microscopy could be challenging, flow cytometry was performed in subsequent experiments to precisely identify BAL cell types. As shown in Figure 2B, at 48 hours, the percentage of neutrophils, aka PMNs, within the CD45+ cell gate was significantly elevated in BAL from SEB-challenged IFN-γ KO mice compared with SEB-challenged WT or IL-17 KO mice (Figure 2B). Although Eos were also higher in SEB-challenged IFN-γ KO mice compared with SEB-challenged WT mice, it did not reach statistical significance (Figure 2B). Interestingly, whereas IL-17 KO mice tended to have higher Eos compared with WT mice, PMNs were comparable to WT mice (Figure 2B). Overall, 1) systemic activation of adaptive T cells with BSAg induced ALI in WT mice; and 2) IFN-γ and IL-17A differentially modulated the outcome of ALI with IFN-γ deficiency being pathogenic and IL-17A deficiency being protective.
Figure 2.

BAL differential cell counts in mice with systemic SEB-induced lung injury. HLA-DR3.WT, HLA-DR3.IL-17 KO, and HLA-DR3.IFN-γ KO mice were challenged intraperitoneally with 10 μg of SEB. BAL fluids from naive as well as mice challenged with SEB were collected at indicated time points. (A) Total cells, (B) BAL differentials by flow cytometry at 48 hours. Data represent mean ± SE from at least 4–8 mice per group. ****P < 0.00005.
BAL Cytokine Profiles Reflect Differential PMN and Eos Recruitment by IFN-γ and IL-17A
We next determined the concentrations of various cytokines and chemokines in BAL fluid from these mice. As anticipated, IFN-γ and IL-17 were below the detectable range in respective KO mice (Figure 3). However, IFN-γ and IL-17 concentrations were higher in IL-17 KO and IFN-γ KO mice, respectively, compared with WT mice, which indicated reciprocal regulation. In addition, the prototypical Th2 cytokines, IL-4 and IL-5, were significantly higher in BAL of IFN-γ KO, which was consistent with elevated BAL Eos in IFN-γ KO mice. Many CC chemokine ligand (CCL) chemokines with demonstrated chemotactic activity toward PMNs (such as MCP-1 [CCL2] [26, 27], MIP1α [CCL3] (27), MIP-1β [CCL4], and RANTES (regulated upon activation, normal T-cell expressed and presumably secreted) [CCL5] [28]) were significantly elevated in BAL fluids of IFN-γ KO mice, which correlated with the extent of neutrophilic lung inflammation, BAL cellularity, and lung injury. Interestingly, these chemokines also show chemotactic activity to Eos (29–31), thus explaining higher Eos in IFN-γ KO mice. Conversely, IL-17 KO mice had significantly elevated concentrations of IFN-γ and the IFN-dependent chemokines MIP-2 (CXCL2) and IP-10 (CXCL10) in BAL at 48 hours compared with the other two groups. Also, compared with WT mice, BAL from IL-17 KO mice had consistently higher (though not significant) concentrations of IL-4, IL-5, MIP-2 (CXCL2), and KC (CXCL1) (Figure 3), all of which have been associated with eosinophilic inflammation (32). This might explain higher Eos in BAL of IL-17 KO compared with WT mice (Figure 2B). Overall, the BAL cytokine/chemokine profiles correlated with the presence of PMNs/Eos in the BAL and the extent of lung pathology.
Figure 3.

BAL cytokine/chemokine profiles in mice with eALI caused by systemically delivered SEB. HLA-DR3.WT, HLA-DR3.IL-17 KO, and HLA-DR3.IFN-γ KO mice were challenged intraperitoneally with 10 μg of SEB. BAL fluids from naive as well as mice challenged with SEB were collected at indicated time points. Concentrations of indicated cytokines and chemokines were determined using multiplex assays. Data represent mean ± SE from at least 4–6 mice per group. *P < 0.5, **P < 0.005, ***P < 0.0005, and ****P < 0.00005. eALI = extrapulmonary acute lung injury.
Serum Cytokine Surge Precedes Lung Cytokine Elevation and Injury
We next correlated the serum cytokine concentrations with BAL cytokine concentrations. As shown in Figure 4, consistent with CRS, many cytokines were significantly higher in serum of WT mice even at 4 hours compared with naive mice. As expected, IL-17A and IFN-γ were below the detection limit in respective KO mice. IL-2, TNF-α, and Th1-promoting cytokines IL-12p40 and IL-12p70 were significantly lower in IFN-γ KO mice, whereas some chemokines and cytokines, including IL-10, were significantly elevated in IL-17 KO mice. Another important observation is that not only were the concentrations of various cytokines/chemokines much higher in serum compared with BAL, the concentrations of cytokines/chemokines in serum tended to fall by 48 hours, whereas they continued to rise in the BAL with time correlating with lung injury at later time points.
Figure 4.

Systemic cytokine storm precedes lung injury. HLA-DR3.WT, HLA-DR3.IL-17 KO, and HLA-DR3.IFN-γ KO mice were challenged intraperitoneally with 10 μg of SEB. Sera from naive as well as mice challenged with SEB were collected at indicated time points. Concentrations of indicated cytokines and chemokines were determined using multiplex assays. Data represent mean ± SE from at least 4–6 mice per group. *P < 0.5, **P < 0.005, ***P < 0.0005, and ****P < 0.00005. Hrs = hours.
Gene Expression Profiling by Microarray Identifies Pathways That Lead to PMN/Eos-mediated Lung Injury in eALI Associated with CRS and its Modulation by IL-17A and IFN-γ
To delineate the molecular pathways underlying the development of eALI after systemic activation of T cells in WT mice and to understand why the extent of lung injury is distinct in IL-17 KO and IFN-γ KO mice, we profiled the gene expression patterns in the whole lungs at 4 and 24 hours after SEB challenge using microarrays. In HLA-DR3 WT mice, at 4 hours, out of 22,206 total genes, 2,464 genes passed the filtering criteria (fold change greater than 2 or less than −2 and P < 0.05 compared with naive mice). From this, 1,427 genes were upregulated and 1,037 downregulated (Figure 5A). The number of differentially regulated genes was significantly fewer at 24 hours, with only 409 upregulated genes and 382 downregulated genes. Several genes that were up- or downregulated at 4 hours were also still highly induced/suppressed, respectively, at 24 hours. However, the extent of up- or downregulation was considerably lower at 24 hours.
Figure 5.

Gene expression profiling in the lungs of mice with eALI induced with systemic SEB. HLA-DR3.WT, HLA-DR3.IL-17 KO, and HLA-DR3.IFN-γ KO mice were challenged intraperitoneally with 10 μg of SEB. Total RNA extracted from naive and SEB-challenged mice were collected at 4 and 24 hours after the SEB challenge, and gene expression patterns were analyzed using Clariom S microarrays. (A) Number of genes up- or downregulated at 4 and 24 hours. (B) Venn diagram depicting gene expression pattern at 4 and 24 hours. (C) Heat map of selected canonical pathways on the basis of the Benjamini-Hochberg method corrected P value in the three groups of mice identified using the IPA tool. (D) Heat map of genes involved in agranulocyte diapedesis and degranulation by expression ratio. hr = hours; IPA = Ingenuity Pathway Analysis; Pathw. = Pathway.
In IFN-γ KO mice, at 4 hours, 1,214 genes were upregulated, and 792 genes downregulated (Figure 5A), whereas in IL-17 KO mice, 1,739 genes were upregulated, and 919 genes downregulated. In IFN-γ KO mice, the number of upregulated genes slightly increased at 24 hours compared with 4 hours, whereas the number of downregulated genes was far fewer (Figure 5A). However, the number of genes that were differentially regulated at 24 hours was significantly lower in IL-17 KO mice compared with the 4-hour time point similar to WT mice. Figure 5A summarizes the numbers of up and downregulated genes in the three strains of mice at 4 and 24 hours. Details regarding the individual genes and changes in their expression are given in Tables E1 and E2.
We next performed a comparison analysis of all the genes differentially expressed in the three strains of mice using Transcriptome Analysis Console (TAC) software and plotted them in a Venn diagram (Figure 5B). A comparison analysis of all the genes differentially expressed in the three mice strains was performed first using the TAC software. Unique, as well as shared genes, were identified. Among the unique genes, Granzyme A and several matrix metalloproteinases (MMPs) were highly induced in IFN-γ KO mice with ALI, whereas Sprr2A (small proline-rich protein 2A), 1, and 3 were upregulated by more than 740-fold in the lungs of IL-17 KO mice with ALI (Table E3). Subsequently, we sorted the genes on the basis of the highest fold induction in IFN-γ KO mice at 4 hours, followed by WT mice and then IL-17 KO mice, in the decreasing order of ALI severity (Table 1). This analysis identified S100A8 and S100A9 as the top two genes upregulated in IFN-γ KO mice, followed by several MMPs (such as Mmp8, Mmp3, Mmp9, and Mmp12), a member of the resistin-like molecules gene family, RetnIg, and Cxcl5. The expression of Il1b also followed the pattern of ALI severity (Table 1).
Table 1.
Top 20 Genes Upregulated in the Lungs of IFN-γ Knockout Mice Followed by Wild-type and IL-17 Knockout Mice Identified Using Microarrays
| Gene | IFN-γ KO | WT | IL-17 KO |
|---|---|---|---|
| S100a8 | 37.1 | 16.5 | 4.3 |
| Mmp8 | 19.7 | 10.8 | 7.3 |
| S100a9 | 19.5 | 11.5 | 4.2 |
| Retnlg | 19.1 | 12.0 | 5.9 |
| Slc26a4 | 18.5 | 18.3 | 11.7 |
| Cxcl5 | 10.6 | 7.5 | 5.8 |
| Lcn2 | 8.8 | 8.7 | 6.4 |
| Ccr1 | 6.3 | 5.2 | 2.0 |
| Gzma | 6.2 | 2.2 | 2.1 |
| Il1b | 6.0 | 3.1 | 1.3 |
| Il1r2 | 5.7 | 4.6 | 3.9 |
| Cxcr2 | 5.4 | 4.0 | 2.2 |
| Cd177 | 5.0 | 4.8 | 3.3 |
| Ccl12 | 4.7 | 4.5 | 1.2 |
| Mmp3 | 4.4 | 1.3 | 1.1 |
| Mmp9 | 4.2 | 2.2 | 1.0 |
| Ifitm6 | 4.1 | 3.8 | 1.8 |
| Nfil3 | 3.8 | 3.0 | 2.2 |
| Arg1 | 3.8 | 1.5 | 1.2 |
| Arg2 | 3.8 | 2.4 | 1.6 |
Definition of abbreviations: SEB = staphylococcal enterotoxin B; WT = wild-type.
Table shows fold-increase in gene expression at 4 hours in SEB-challenged mice compared to respective naïve mice in each genotype.
We then tabulated the genes that are highly induced in IL-17 KO mice at 4 hours, followed by WT and then in IFN-γ KO mice (i.e., in decreasing order of ALI severity) (Table 2). Several IFN-γ–inducible chemokines, such as CXCL9, CXCL10, and CXCL11, and inflammatory markers, such as SAA3, were identified by this sorting. Many other IFN-γ–inducible genes (GTPase, Ifgga3 protein, and Ido1) were also strongly upregulated in IL-17 KO mice followed by WT mice, correlating with higher BAL IFN-γ.
Table 2.
Top 20 Genes Upregulated in the Lungs of IL-17 Knockout Mice Followed by Wild-type and IFN-γ Knockout Mice Identified Using Microarrays
| Gene | IL-17 KO | WT | IFN-γ KO |
|---|---|---|---|
| Cxcl9 | 444.4 | 156.2 | 16.2 |
| Cxcl10 | 153.7 | 116.7 | 12.4 |
| Cxcl11 | 107.5 | 58.7 | 4.5 |
| Gm4841 | 93.6 | 17.4 | 6.5 |
| Ido1 | 66.7 | 25.4 | 7.6 |
| Gbp2b | 61.1 | 32.1 | 9.2 |
| Saa3 | 55.3 | 30.0 | 40.7 |
| Sprr2a3 | 53.1 | 5.7 | 1.3 |
| Iigp1 | 41.9 | 18.5 | 8.2 |
| Gbp2 | 38.9 | 25.1 | 6.4 |
| Serpina3f | 37.6 | 23.8 | 24.3 |
| Igtp | 33.0 | 13.9 | 11.0 |
| Sprr2a2 | 28.8 | 7.9 | 1.1 |
| Ngp | 28.2 | 57.6 | 38.5 |
| Ccl11 | 26.2 | 16.2 | 7.6 |
| Mnda | 25.7 | 15.4 | 6.2 |
| Selp | 24.6 | 27.1 | 19.3 |
| Gbp11 | 24.4 | 15.4 | 5.5 |
| F830016B08Rik | 23.0 | 14.8 | 4.0 |
| Clca1 | 22.9 | 0.4 | 26.9 |
For definition of abbreviations, see Table 1.
Table shows fold-increase in gene expression at 4 hours in SEB-challenged mice compared to respective naïve mice in each genotype.
We next performed a comparison analysis using IPA to identify the major pathways and cellular processes that were upregulated at 4 hours in all three strains and sorted on the basis of the Benjamini-Hochberg (B-H) corrected P value. As shown in Figures 5C and 5D, granulocyte adhesion and diapedesis and agranulocyte adhesion and diapedesis were the two top canonical pathways identified in the comparison analysis. These two pathways were highly altered in IFN-γ KO mice (B-H scores of 17 and 11), followed by DR3.WT (B-H scores of 6 and 5) and lowest in IL-17 KO mice (B-H scores of 2 and 2), which correlated with the severity of lung injury. When the canonical pathways were sorted on the basis of z-score, IL-17A signaling (3.4, 1.8, and 1.3), the role of hyper cytokinemia/hyper chemokinemia in the pathogenesis of influenza (3.2, 2.5, and NA) were the two top pathways with highest z-scores in IFN-γ KO, WT, and IL-17 KO mice, respectively (Table E4).
Given the higher BAL PMNs and Eos counts in IFN-γ KO mice, we then interrogated how many chemokine genes are uniquely expressed in IFN-γ KO mice at 4 hours but not in IL-17 KO mice. We could identify a high expression of chemokines that are chemotactic to PMNs or Eos in IFN-γ KO mice (Table 3). We also examined whether any tissue protective genes are enriched in IL-17 KO mice that could explain less severe ALI in this strain. Interestingly, this process identified CD274 (PD-L1 [programmed death ligand-1]) and several members of the suppressor of cytokine signaling (SOCS) family (Table 3).
Table 3.
Expression of Unique Chemokine Genes and Tissue Protective Factors in the Lungs During Cytokine Release Syndrome-induced Acute Lung Injury
| Gene | IFN-γ KO | WT | IL-17 KO |
|---|---|---|---|
| Ccl12 | 11.6 | 8.3 | ND |
| Ccl8 | 5.5 | ND | ND |
| Cxcl15 | 3.5 | ND | ND |
| Ccl1 | 3 | ND | ND |
| Cd274 (pd l-1) | 31 | 151 | 291 |
| Socs3 | 12 | 71 | 60 |
| Socs1 | 6 | 15 | 61 |
| Socs2 | 2 | 3 | 4 |
Definition of abbreviations: ND = not detected.
Table shows fold-increase in expression of indicated genes at 4 hours in SEB-challenged mice compared to respective naïve mice in each genotype.
A biomarker identification tool in IPA was used to identify novel biomarkers that could be identifiable in BAL, sputum, or lung from mice. Although there was no unique biomarker(s) in any single strain, 24 shared biomarkers were identified. Of these, MMP8 and MMP9 were the top biomarkers at 4 hours, and MMP9 was the top biomarker at 24 hours. To summarize, the expression of IFN-γ–inducible chemokines/pathways in the lungs negatively correlated with the extent of eALI in our CRS model, whereas the expression of genes/pathways involved in PMN as well as Eos chemotaxis, activation, and degranulation, and extracellular matrix damage likely induced by Th17 and Th2 cytokines, were highly expressed in IFN-γ KO lungs and positively correlated with ALI severity.
In vivo Neutralization of IFN-γ Enhances ALI in WT HLA-DR3 Mice
Genetic deletion of IFN-γ or IL-17A may have resulted in an inherent skewing of cytokine patterns (33, 34), which could alter the outcome of T-cell–mediated lung injury. To disprove this possibility, neutralizing antibodies were administered before inducing CRS and the extent of lung injury was determined using BAL cellularity, BAL protein content, and BALS100A8/A9 as representative indicators. Significant elevation in PMN as well as Eos in the BAL of HLA-DR3.WT mice treated with anti–IFN-γ antibody compared with those treated with isotype control confirmed that IFN-γ protects from ALI, reproducing the findings from IFN-γ KO mice (Figures 6A and 6B). Although BAL protein content was not significantly elevated at 48 hours, at 72 hours, anti–IFN-γ antibody-treated mice had significantly higher BAL protein content compared with those treated with isotype control (Figure 6C). However, BAL S100A8/A9 concentrations were significantly higher in anti–IFN-γ antibody-treated mice at 48 and 72 hours (Figure 6D).
Figure 6.

Modulation of systemically delivered SEB-induced lung injury by anti–IFN-γ antibodies in WT and IL-17 KO mice. Neutralizing anti–IFN-γ antibodies or isotype control antibodies (500 microgram/per mouse) were delivered by intraperitoneal injection to (A–D) WT and (E–I) HLA-DR3.IL-17 KO mice. Two hours later, all mice were challenged with 10 micrograms of SEB by intraperitoneal route. BAL fluids were collected at 48 or 72 hours, distribution of various cell types was determined by flow cytometry. BAL protein content and the concentrations of S100A8/A9 were determined as described in the Methods section. Data represent mean ± SE from at least 5–8 mice per group. (I) Representative HandE images at low and high power of lungs from IL-17 KO mice. *P < 0.5, **P < 0.005, ***P < 0.0005, and ****P < 0.00005. Ab = antibody.
PMN Recruitment Occurs in the Absence of IL-17A
Next, when IFN-γ was neutralized in IL-17 KO mice with antibodies, this resulted in a significant increase in total BAL cellularity (Figure 6E) as well as an increase in BAL Eos, and particularly PMNs, in the BAL (Figure 6F). BAL protein and S100A8/A9 concentrations were also high in the anti–IFN-γ antibody group (Figures 6G and 6H), as was the severity of lung immunopathology (Figure 6I). On the contrary, neutralization of IL-17A did not significantly alter BAL differential in IFN-γ KO mice (Figure E2). These experiments conveyed that 1) IFN-γ plays a dominant protective role in CRS-induced ALI; and 2) whereas IL-17A is required to induce ALI when the dominantly protective IFN-γ is neutralized, then IL-17A is no longer required to induce ALI.
Janus Kinase Inhibitor (JAKi) Promotes eALI Caused by SEB
We recently demonstrated that in the SEB-induced CRS model, the JAK1/2 inhibitor, ruxolitinib, significantly suppresses IFN-γ production and its functions (19). Therefore, we next investigated whether the administration of ruxolitinib would promote ALI. Interestingly, as shown in Figure 7, even though ruxolitinib significantly decreased total BAL cell counts (0.3 ± 0.02 and 0.185 ± 0.018 × 103 for corn oil and ruxolitinib group, respectively), PMN count, as well as S100A8/A9 concentrations, were significantly increased in the BAL of ruxolitinib-treated mice. However, the Eos percentage did not change significantly, suggesting distinct effects of ruxolitinib on PMN- and Eos-mediated pulmonary inflammatory responses in CRS.
Figure 7.

Treatment with the JAKi ruxolitinib promotes ALI. HLA-DR3. WT mice were gavaged with ruxolitinib or corn oil on Days −1, 0, and 1. Mice were challenged with 10 micrograms of SEB by the intraperitoneal route and killed on Day 2. BAL fluids were collected and analyzed by flow cytometry, and the concentration of S100A8/A9 was determined by ELISA using a kit as described in the Methods section. Data represent mean ± SE from at least 8–10 mice per group. **P < 0.005, ***P < 0.0005. JAKi = Janus Kinase inhibitor.
Discussion
ALI and ARDS are common complications of CRS. However, the mechanisms by which excessive systemic activation of adaptive T cells in CRS leads to neutrophil-mediated lung injury are poorly understood because of the paucity of robust animal models. Using three different strains of HLA-DR3 transgenic mice, WT, IL-17 KO, and IFN-γ KO, all of which develop CRS when challenged with BSAg, we teased out the immunopathogenesis of ALI associated with CRS and delineated the distinct roles of IL-17 and IFN-γ in this process.
First, in WT mice that developed a certain degree of ALI after systemic BSAg challenge, together with IFN-γ, several IFN-γ-inducible chemokines (such as CXCL9, CXCL10, and CXCL11), as well as other molecules (e.g., members of the guanylate binding proteins and interferon-inducible GTPases) (35), were highly upregulated either systemically and/or locally in the lungs compared with naive WT mice. Therefore, data from WT mice alone would have implied that IFN-γ is pathogenic even in CRS-associated eALI as in most other models of ALI (11, 12, 22–25, 36–38). However, compared with WT mice, these mediators were more robustly upregulated in IL-17 KO mice that manifested the mildest form of ALI and least upregulated in IFN-γ KO mice that developed the most severe ALI. These observations not only implied that IL-17 is pathogenic whereas IFN-γ is protective in ALI associated with CRS, but elevated CXCL9, CXCL10, CXCL11, and upregulation of many IFN-γ–inducible mediators merely reflected heightened IFN-γ activity, and they may not be pathogenic in CRS-induced ALI unlike in other models of lung injury (28, 39, 40). Hence, the role of these chemokines in the immunopathogenesis of ALI/ARDS and the relevance of these mediators as biomarkers of ALI/ARDS needs careful validation.
Second, even though IL-17 was pathogenic when present, it was dispensable for the development of ALI associated with CRS because IL-17 KO mice still developed ALI when IFN-γ was neutralized (Figure 6), and anti–IL-17 antibodies did not protect IFN-γ KO mice from developing ALI (Figure E2). It remains to be elucidated which other cytokines compensate for the absence of IL-17 in our model. Third, IFN-γ is dominantly protective in ALI associated with CRS. More severe ALI in WT mice after in vivo neutralization of IFN-γ pronounced ALI in IL-17 KO mice after in vivo neutralization of IFN-γ and failure of neutralizing anti–IL-17 antibodies to protect IFN-γ KO mice from developing ALI support these conclusions. Finally, this study provided a possible explanation as to why the severity of lung injury is highly variable in human CRS and how it could be dictated by the relative concentrations of Th1, Th2, and Th17 cytokines.
T-cell–derived cytokines can be broadly classified into type 1 (Th1), type 2 (Th2), or type 3 (Th17) on the basis of their biological functions. Generally, IFN-γ, the major Th1 cytokine, activates monocyte/macrophages and promotes monocyte/macrophage-mediated inflammatory responses. The Th2 cytokines, IL-4, IL-5, and IL-13, promote eosinophilic inflammation, whereas the Th17 cytokines, IL-17, IL-21, and IL-22, drive neutrophil-mediated inflammatory responses. Given the central role of PMNs in causing lung injury in ALI/ARDS, Th17 cytokines are thought to be pathogenic. As Eos have also been implicated in the immunopathogenesis of ALI/ARDS (41–45), Th2 cytokines can also promote ALI/ARDS. IFN-γ is a potent inhibitor of Th2 and Th17 cytokines, both their production as well as biological functions (33, 34, 46–49). Hence, it is conceivable that IFN-γ may be protective in ALI/ARDS by inhibiting Th17 and Th2 cytokines. However, in CRS, when IFN-γ is low and other Th17 and Th2 cytokines are elevated, this may result in more severe ALI/ARDS. Overall, the relative concentrations of Th1, Th2, and Th17 cytokines may determine the nature and severity of the inflammatory lung injury.
In our model, SEB primarily activates T cells in a T-cell receptor-dependent but antigen nonspecific manner (although toll-like receptor-2 may be involved in SEB-mediated immune activation [50]), resulting in a rapid significant systemic elevation in Th1, Th2, as well as Th17 cytokines, analogous to human CRS (Figure 4) (1–4, 51). However, IFN-γ production is much more robust compared with Th2 and Th17 cytokines (Figure 4) (20, 52–55). Therefore, higher concentrations of IFN-γ likely suppressed the production as well as the functions of Th2 and Th17 cytokines, resulting in moderate lung injury in HLA-DR3.WT mice. However, antibody-mediated neutralization of IFN-γ in WT mice removed the inhibitory effects of IFN-γ, leading to higher production as well as uninhibited functioning of Th17 and Th2 cytokines, resulting in more severe PMN and Eos-mediated lung injury, respectively (Figure 6). The same is applicable to IFN-γ KO mice. Because genetic inactivation results in complete loss of IFN-γ compared with antibody-mediated neutralization, more severe ALI in IFN-γ KO mice is not unexpected.
The protection of IL-17 KO mice from ALI could be attributed to the lack of IL-17, the key driver of neutrophilic inflammation. However, significantly elevated IFN-γ concentrations in IL-17 KO mice and more severe ALI in IL-17 KO mice after in vivo neutralization of IFN-γ suggested that milder ALI in IL-17 KO mice is because of elevated IFN-γ rather than lack of IL-17. Thus, when IFN-γ was neutralized in IL-17 KO mice, other Th17 cytokines (possibly IL-21 and/or IL-22) and Th2 cytokines were produced at higher concentrations leading to severe ALI.
A similar mechanism has been proposed to operate in Mycobacterium tuberculosis (Mtb)-induced lung inflammation. Mtb, similar to SEB, is a potent inducer of IFN-γ. Thus, in WT mice, after Mtb infection, elevated IFN-γ suppressed Th17 as well as Th2 cytokines, thereby limiting neutrophil- and Eos-mediated lung injury. However, blocking IFN-γ signaling results in excessive production of Th17 as well as Th2 cytokines by adaptive T cells leading to more severe PMN-mediated as well as Eos-mediated lung injury, comparable to our findings (56–60).
Mechanistically, in the lungs of IFN-γ KO mice that developed severe ALI, many members of the multifunctional tissue-destructive enzyme family MMPs were highly induced. MMPs play a very important role in basement membrane degradation that leads to vascular leakage and loss of alveolar integrity, which results in edema and hypoxemia (61). Further, many chemokines, such as CXCL5, CXCL15, CCL8, CCL12, and CCL1, some chemotactic for neutrophils and others for Eos, are readily induced in the lungs of IFN-γ KO mice compared with WT and IL-17 KO mice, which explains greater recruitment of PMNs and Eos in IFN-γ KO mice (62–70).
In addition to these chemokines, other factors that promote PMN/Eos activation/migration were also upregulated in the lungs of IFN-γ KO mice. Prominent among them were S100A8 and S100A9. S100A8 (calgranulin A or MRP-8 [migration inhibitory factor-related protein 8]) and its binding partner S100A9 (calgranulin B or MRP-14), both members of the S100 calcium-binding family of proteins, are alarmins that stimulate the innate immune responses (reviewed in Reference [71]). Although constitutively expressed by cells of myeloid origin, S100A8/A9 genes can be readily induced in a variety of other cell types, including alveolar epithelial cells. S100A8/A9 have been shown to be chemotactic to PMNs (72, 73) and have been implicated in neutrophilic inflammation affecting a variety of tissues/organs, including the lungs, heart and vasculature, joints, and even in some malignancies (74–77). High expression of these genes very early on in the lungs at 4 hours, when the inflammatory infiltrates were minimal, followed by a decrease in expression at 24 hours, when PMNs start appearing in the BAL, suggest that these genes are likely induced in the lung cells and possibly act in concert with other chemokines in recruiting PMNs to the lungs in ALI (72, 73). Because S100A8/A9 proteins are abundantly present in PMNs, when PMNs infiltrate the lungs and start dying at later time points, they release S100A8/A9 proteins into the BAL, which was detectable by ELISA. S100A8/A9 are known ligands for the receptor for advanced glycation end products (RAGE), and blockade of RAGE has been shown to reduce the severity of ALI in mice (78).
Interestingly, many tissue-protective, antiinflammatory pathways were highly upregulated in the lungs of WT and more so in IL-17 KO mice compared with IFN-γ KO mice suggesting additional mechanisms that may determine the severity/outcome of ALI and their modulation by IL-17 and IFN-γ (Table 3). For example, CD274 (PD-L1) is the ligand for PD-1. PDL-1 is expressed on epithelial cells and other nonimmune cells, whereas PD-1 is expressed on T cells. Engagement of PD-1 on activated T cells by PDL-1 on tissues delivers inhibitory signals to T cells and protects the target cells from T-cell–mediated lysis (79). Thus, overexpression of PDL-1 in the lungs of IL-17 KO mice followed by WT mice compared with IFN-γ KO mice may have protected lung epithelial cells in IL-17 KO and WT from damage caused by T-cell–mediated lung injury in CRS. Overexpression of SOCS genes in IL-17 KO and WT lungs compared with IFN-γ KO may also explain the disparity in lung injuries in these strains. In the lungs, it is known that low expression of SOCS is associated with exaggerated inflammatory responses as well as allergy (80, 81). It remains to be elucidated whether all these genes/molecules that are differentially expressed in IFN-γ KO mice are directly regulated by IFN-γ or these are regulated by Th2/Th17 cytokines that are elevated in IFN-γ KO mice.
The gene expression was studied using bulk RNA but not single cells. Hence, the precise cell type(s) expressing these genes could not be pinpointed. As there were minimal inflammatory changes in the lungs by histological, biochemical (LDH, protein, neutrophil extracellular trap, or neutrophil elastase), or BAL cellular analysis at 4 hours, the genes identified were likely induced in the lung resident cells (such as alveolar epithelial cells, pulmonary endothelial cells, or alveolar macrophages) but not in other infiltrating inflammatory cells.
Clinically, our findings suggest that the extent of lung injury in CRS could be determined by relative concentrations of protective IFN-γ compared with pathogenic cytokines such as IL-4, IL-5, IL-13, IL-17, IL-21, or IL-22 that promote ALI/ARDS. Patients with lower IFN-γ may develop severe ARDS and vice versa. In support of our hypothesis, IFN-γ has been shown to stabilize lung function and help in the recovery of patients with community-acquired pneumonia and viral infections, hence being tested in a clinical trial (82). Even in coronavirus disease (COVID-19) caused by SARS-CoV-2, IFN-γ concentrations were significantly lower in patients with severe COVID-19 (83, 84). Also, an inverse relationship between IFN-γ concentrations and lung fibrosis at discharge in patients with COVID-19 has been reported (85). Further, administration of IFN-γ significantly improved ventilator-acquired pneumonia in patients with COVID-19 (82, 86, 87). Although IFN-γ is believed to improve the outcome by increasing monocyte functions and increasing HLA-DR expression, the potential inhibitory effects of IFN-γ on PMNs/Eos, either directly or indirectly via mechanisms discussed earlier, cannot be ruled out (82, 86, 87).
Interestingly, in a cohort of patients with COVID-19 with certain parasitic infections (such as Toxoplasma gondii and Cryptosporidium, among others, that are known to induce a robust IFN-γ response, similar to SEB and Mtb), patients who produced higher amounts of IFN-γ had milder COVID-19 compared with those with severe COVID-19 who produced only low concentrations of IFN-γ or were parasite-free, further supporting a protective role for IFN-γ in ALI/ARDS (88). On the basis of the idea that IFN-γ is pathogenic in ALI/ARDS, antibodies to neutralize IFN-γ and JAKi are proposed as treatments for ALI/ARDS (89, 90). However, our data suggest that JAKi and anti–IFN-γ antibodies may promote ALI/ARDS in certain conditions. Hence, the pros and cons of IFN-γ antagonism in ALI associated with CRS need to be further explored.
As with any other experimental model of ALI, our study also has some limitations. The translatability of our model to human ALI/ARDS accompanying CRS needs further validation. Also, the effects of IFN-γ and IL-17A, as well as the effects of blocking these cytokines, were investigated only at specific time points during the progression of ALI. The role of other cytokines and different effector cell types at distinct stages of ALI/ARDS need to be investigated. Understanding the long-term effects of these cytokines on lung functions and the therapeutic use of blocking/enhancing the functions of these cytokines in ALI/ARDS needs further investigation. In addition, these experiments were conducted using healthy inbred mice of either sex belonging to a certain age group raised under a controlled specific pathogen-free environment. However, sex, age, genetic heterogeneity, and other comorbidities may have a major impact on the outcome of ALI/ARDS in humans. Nonetheless, our model could still be valuable in studying the role of adaptive T cells in inducing ALI/ARDS.
Acknowledgments
Acknowledgment
The authors are thankful to Dr. Andrew Wang, Section of Rheumatology, Allergy, and Immunology, Yale School of Medicine, and Dr. Seyedtaghi Takyar, PCCSM, Yale School of Medicine, for helpful discussion. The authors thank Susan Ardito for editing the manuscript.
Footnotes
Supported by the National Institutes of Health (NIH) 1R01HL125897 and NIH RFA-A1-17-040 U19 (J.L.K.) and NIH R21 AI142243 (G.R.).
Author Contributions: Y.S.: conducting experiments and acquiring data. B.H.: analyzing data and writing the manuscript. G.S. and Z.M.H.: design of the work, analyzing data, and writing the manuscript. S.G.: design of the work providing reagents and writing the manuscript. R.H.: evaluating pathology slides and writing the manuscript. J.L.K.: designing research studies, analyzing data, and writing the manuscript. G.R.: designing research studies, conducting experiments, acquiring data, analyzing data, and writing the manuscript.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2022-0117OC on September 20, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med . 2020;383:2255–2273. doi: 10.1056/NEJMra2026131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shimabukuro-Vornhagen A, Gödel P, Subklewe M, Stemmler HJ, Schlößer HA, Schlaak M, et al. Cytokine release syndrome. J Immunother Cancer . 2018;6:56. doi: 10.1186/s40425-018-0343-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood . 2014;124:188–195. doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xhangolli I, Dura B, Lee G, Kim D, Xiao Y, Fan R. Single-cell analysis of CAR-T cell activation reveals a mixed TH1/TH2 response independent of differentiation. Genomics Proteomics Bioinformatics . 2019;17:129–139. doi: 10.1016/j.gpb.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ryabkova VA, Churilov LP, Shoenfeld Y. Influenza infection, SARS, MERS and COVID-19: cytokine storm – the common denominator and the lessons to be learned. Clin Immunol . 2021;223:108652. doi: 10.1016/j.clim.2020.108652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Faulkner L, Cooper A, Fantino C, Altmann DM, Sriskandan S. The mechanism of superantigen-mediated toxic shock: not a simple Th1 cytokine storm. J Immunol . 2005;175:6870–6877. doi: 10.4049/jimmunol.175.10.6870. [DOI] [PubMed] [Google Scholar]
- 7. Beitler JR, Thompson BT, Baron RM, Bastarache JA, Denlinger LC, Esserman L, et al. Advancing precision medicine for acute respiratory distress syndrome. Lancet Respir Med . 2021;10:107–120. doi: 10.1016/S2213-2600(21)00157-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol . 2008;295:L379–L399. doi: 10.1152/ajplung.00010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Verjans E, Kanzler S, Ohl K, Rieg AD, Ruske N, Schippers A, et al. Initiation of LPS-induced pulmonary dysfunction and its recovery occur independent of T cells. BMC Pulm Med . 2018;18:174. doi: 10.1186/s12890-018-0741-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aeffner F, Bolon B, Davis IC. Mouse models of acute respiratory distress syndrome: a review of analytical approaches, pathologic features, and common measurements. Toxicol Pathol . 2015;43:1074–1092. doi: 10.1177/0192623315598399. [DOI] [PubMed] [Google Scholar]
- 11. Verma AK, Bauer C, Palani S, Metzger DW, Sun K. IFN-γ drives TNF-α hyperproduction and lethal lung inflammation during antibiotic treatment of postinfluenza Staphylococcus aureus pneumonia. J Immunol . 2021;207:1371–1376. doi: 10.4049/jimmunol.2100328. [DOI] [PubMed] [Google Scholar]
- 12. Svedova J, Ménoret A, Mittal P, Ryan JM, Buturla JA, Vella AT. Therapeutic blockade of CD54 attenuates pulmonary barrier damage in T cell-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol . 2017;313:L177–L191. doi: 10.1152/ajplung.00050.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alghetaa H, Mohammed A, Zhou J, Singh N, Nagarkatti M, Nagarkatti P. Resveratrol-mediated attenuation of superantigen-driven acute respiratory distress syndrome is mediated by microbiota in the lungs and gut. Pharmacol Res . 2021;167:105548. doi: 10.1016/j.phrs.2021.105548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tuffs SW, Haeryfar SMM, McCormick JK. Manipulation of innate and adaptive immunity by staphylococcal superantigens. Pathogens . 2018;7:53. doi: 10.3390/pathogens7020053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tilahun AY, Marietta EV, Wu TT, Patel R, David CS, Rajagopalan G. Human leukocyte antigen class II transgenic mouse model unmasks the significant extrahepatic pathology in toxic shock syndrome. Am J Pathol . 2011;178:2760–2773. doi: 10.1016/j.ajpath.2011.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sriskandan S, Unnikrishnan M, Krausz T, Dewchand H, Van Noorden S, Cohen J, et al. Enhanced susceptibility to superantigen-associated streptococcal sepsis in human leukocyte antigen-DQ transgenic mice. J Infect Dis . 2001;184:166–173. doi: 10.1086/322018. [DOI] [PubMed] [Google Scholar]
- 17. Yeung RS, Penninger JM, Kündig T, Khoo W, Ohashi PS, Kroemer G, et al. Human CD4 and human major histocompatibility complex class II (DQ6) transgenic mice: supersensitivity to superantigen-induced septic shock. Eur J Immunol . 1996;26:1074–1082. doi: 10.1002/eji.1830260518. [DOI] [PubMed] [Google Scholar]
- 18. Roy CJ, Warfield KL, Welcher BC, Gonzales RF, Larsen T, Hanson J, et al. Human leukocyte antigen-DQ8 transgenic mice: a model to examine the toxicity of aerosolized staphylococcal enterotoxin B. Infect Immun . 2005;73:2452–2460. doi: 10.1128/IAI.73.4.2452-2460.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kale SD, Mehrkens BN, Stegman MM, Kastelberg B, Carnes H, McNeill RJ, et al. “Small” intestinal immunopathology plays a “big” role in lethal cytokine release syndrome, and its modulation by interferon-gamma, IL-17A and a Janus kinase inhibitor. Front Immunol . 2020;11:1311. doi: 10.3389/fimmu.2020.01311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rajagopalan G, Iijima K, Singh M, Kita H, Patel R, David CS. Intranasal exposure to bacterial superantigens induces airway inflammation in HLA class II transgenic mice. Infect Immun . 2006;74:1284–1296. doi: 10.1128/IAI.74.2.1284-1296.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gautam S, Stahl Y, Young GM, Howell R, Cohen AJ, Tsang DA, et al. Quantification of bronchoalveolar neutrophil extracellular traps and phagocytosis in murine pneumonia. Am J Physiol Lung Cell Mol Physiol . 2020;319:L661–L669. doi: 10.1152/ajplung.00316.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mock JR, Tune MK, Dial CF, Torres-Castillo J, Hagan RS, Doerschuk CM. Effects of IFN-γ on immune cell kinetics during the resolution of acute lung injury. Physiol Rep . 2020;8:e14368. doi: 10.14814/phy2.14368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu B, Bao L, Wang L, Li F, Wen M, Li H, et al. Anti-IFN-γ therapy alleviates acute lung injury induced by severe influenza A (H1N1) pdm09 infection in mice. J Microbiol Immunol Infect . 2019;54:396–403. doi: 10.1016/j.jmii.2019.07.009. [DOI] [PubMed] [Google Scholar]
- 24. Chen ES, Greenlee BM, Wills-Karp M, Moller DR. Attenuation of lung inflammation and fibrosis in interferon-γ-deficient mice after intratracheal bleomycin. Am J Respir Cell Mol Biol . 2001;24:545–555. doi: 10.1165/ajrcmb.24.5.4064. [DOI] [PubMed] [Google Scholar]
- 25. Muralimohan G, Rossi RJ, Guernsey LA, Thrall RS, Vella AT. Inhalation of Staphylococcus aureus enterotoxin A induces IFN-γ and CD8 T cell-dependent airway and interstitial lung pathology in mice. J Immunol . 2008;181:3698–3705. doi: 10.4049/jimmunol.181.5.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maus UA, Waelsch K, Kuziel WA, Delbeck T, Mack M, Blackwell TS, et al. Monocytes are potent facilitators of alveolar neutrophil emigration during lung inflammation: role of the CCL2-CCR2 axis. J Immunol . 2003;170:3273–3278. doi: 10.4049/jimmunol.170.6.3273. [DOI] [PubMed] [Google Scholar]
- 27. Reichel CA, Rehberg M, Lerchenberger M, Berberich N, Bihari P, Khandoga AG, et al. Ccl2 and Ccl3 mediate neutrophil recruitment via induction of protein synthesis and generation of lipid mediators. Arterioscler Thromb Vasc Biol . 2009;29:1787–1793. doi: 10.1161/ATVBAHA.109.193268. [DOI] [PubMed] [Google Scholar]
- 28. Metzemaekers M, Vanheule V, Janssens R, Struyf S, Proost P. Overview of the mechanisms that may contribute to the non-redundant activities of interferon-inducible CXC chemokine receptor 3 ligands. Front Immunol . 2018;8:1970. doi: 10.3389/fimmu.2017.01970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kobayashi Y, Konno Y, Kanda A, Yamada Y, Yasuba H, Sakata Y, et al. Critical role of CCL4 in eosinophil recruitment into the airway. Clin Exp Allergy . 2019;49:853–860. doi: 10.1111/cea.13382. [DOI] [PubMed] [Google Scholar]
- 30. Isgrò M, Bianchetti L, Marini MA, Bellini A, Schmidt M, Mattoli S. The C-C motif chemokine ligands CCL5, CCL11, and CCL24 induce the migration of circulating fibrocytes from patients with severe asthma. Mucosal Immunol . 2013;6:718–727. doi: 10.1038/mi.2012.109. [DOI] [PubMed] [Google Scholar]
- 31. Rose CE, Jr, Lannigan JA, Kim P, Lee JJ, Fu SM, Sung SS. Murine lung eosinophil activation and chemokine production in allergic airway inflammation. Cell Mol Immunol . 2010;7:361–374. doi: 10.1038/cmi.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blanchard C, Stucke EM, Rodriguez-Jimenez B, Burwinkel K, Collins MH, Ahrens A, et al. A striking local esophageal cytokine expression profile in eosinophilic esophagitis. J Allergy Clin Immunol . 2011;127:208–217.e7. doi: 10.1016/j.jaci.2010.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang ZE, Reiner SL, Zheng S, Dalton DK, Locksley RM. CD4+ effector cells default to the Th2 pathway in interferon gamma-deficient mice infected with Leishmania major. J Exp Med . 1994;179:1367–1371. doi: 10.1084/jem.179.4.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ajendra J, Chenery AL, Parkinson JE, Chan BHK, Pearson S, Colombo SAP, et al. IL-17A both initiates, via IFNγ suppression, and limits the pulmonary type-2 immune response to nematode infection. Mucosal Immunol . 2020;13:958–968. doi: 10.1038/s41385-020-0318-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pilla-Moffett D, Barber MF, Taylor GA, Coers J. Interferon-inducible GTPases in host resistance, inflammation and disease. J Mol Biol . 2016;428:3495–3513. doi: 10.1016/j.jmb.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mohammed A, Alghetaa H, Sultan M, Singh NP, Nagarkatti P, Nagarkatti M. Administration of Δ9-tetrahydrocannabinol (THC) post-staphylococcal enterotoxin B exposure protects mice from acute respiratory distress syndrome and toxicity. Front Pharmacol . 2020;11:893. doi: 10.3389/fphar.2020.00893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rao R, Rieder SA, Nagarkatti P, Nagarkatti M. Staphylococcal enterotoxin B-induced microRNA-155 targets SOCS1 to promote acute inflammatory lung injury. Infect Immun . 2014;82:2971–2979. doi: 10.1128/IAI.01666-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Faggioni R, Gatti S, Demitri MT, Delgado R, Echtenacher B, Gnocchi P, et al. Role of xanthine oxidase and reactive oxygen intermediates in LPS- and TNF-induced pulmonary edema. J Lab Clin Med . 1994;123:394–399. [PubMed] [Google Scholar]
- 39. Callahan V, Hawks S, Crawford MA, Lehman CW, Morrison HA, Ivester HM, et al. The pro-inflammatory chemokines CXCL9, CXCL10 and CXCL11 are upregulated following SARS-CoV-2 infection in an AKT-dependent manner. Viruses . 2021;13:1062. doi: 10.3390/v13061062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lang S, Li L, Wang X, Sun J, Xue X, Xiao Y, et al. CXCL10/IP-10 neutralization can ameliorate lipopolysaccharide-induced acute respiratory distress syndrome in rats. PLoS One . 2017;12:e0169100. doi: 10.1371/journal.pone.0169100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Al Duhailib Z, Farooqi M, Piticaru J, Alhazzani W, Nair P. The role of eosinophils in sepsis and acute respiratory distress syndrome: a scoping review. Can J Anaesth . 2021;68:715–726. doi: 10.1007/s12630-021-01920-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hällgren R, Borg T, Venge P, Modig J. Signs of neutrophil and eosinophil activation in adult respiratory distress syndrome. Crit Care Med . 1984;12:14–18. doi: 10.1097/00003246-198401000-00004. [DOI] [PubMed] [Google Scholar]
- 43. Hällgren R, Samuelsson T, Venge P, Modig J. Eosinophil activation in the lung is related to lung damage in adult respiratory distress syndrome. Am Rev Respir Dis . 1987;135:639–642. doi: 10.1164/arrd.1987.135.3.639. [DOI] [PubMed] [Google Scholar]
- 44. Giacoppe GN, Degler DA. Rapidly evolving adult respiratory distress syndrome with eosinophilia of unknown cause in previously healthy active duty soldiers at an Army training center: report of two cases. Mil Med . 1999;164:911–916. [PubMed] [Google Scholar]
- 45. Ashitani J, Yanagi S, Arimura Y, Sano A, Mukae H. Acute respiratory distress syndrome induced by rifampicin with high levels of neutrophil and eosinophil products in bronchoalveolar lavage fluid. Respiration . 2003;70:541–543. doi: 10.1159/000074216. [DOI] [PubMed] [Google Scholar]
- 46. Dow SW, Schwarze J, Heath TD, Potter TA, Gelfand EW. Systemic and local interferon gamma gene delivery to the lungs for treatment of allergen-induced airway hyperresponsiveness in mice. Hum Gene Ther . 1999;10:1905–1914. doi: 10.1089/10430349950017266. [DOI] [PubMed] [Google Scholar]
- 47. Tanaka T, Hu-Li J, Seder RA, Fazekas de St Groth B, Paul WE. Interleukin 4 suppresses interleukin 2 and interferon gamma production by naive T cells stimulated by accessory cell-dependent receptor engagement. Proc Natl Acad Sci USA . 1993;90:5914–5918. doi: 10.1073/pnas.90.13.5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cohn L, Herrick C, Niu N, Homer R, Bottomly K. IL-4 promotes airway eosinophilia by suppressing IFN-γ production: defining a novel role for IFN-γ in the regulation of allergic airway inflammation. J Immunol . 2001;166:2760–2767. doi: 10.4049/jimmunol.166.4.2760. [DOI] [PubMed] [Google Scholar]
- 49. Fulkerson PC, Zimmermann N, Brandt EB, Muntel EE, Doepker MP, Kavanaugh JL, et al. Negative regulation of eosinophil recruitment to the lung by the chemokine monokine induced by IFN-γ (Mig, CXCL9) Proc Natl Acad Sci USA . 2004;101:1987–1992. doi: 10.1073/pnas.0308544100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mandron M, Ariès MF, Brehm RD, Tranter HS, Acharya KR, Charveron M, et al. Human dendritic cells conditioned with Staphylococcus aureus enterotoxin B promote TH2 cell polarization. J Allergy Clin Immunol . 2006;117:1141–1147. doi: 10.1016/j.jaci.2005.12.1360. [DOI] [PubMed] [Google Scholar]
- 51. Li X, Shao M, Zeng X, Qian P, Huang H. Signaling pathways in the regulation of cytokine release syndrome in human diseases and intervention therapy. Signal Transduct Target Ther . 2021;6:367. doi: 10.1038/s41392-021-00764-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nooh MM, El-Gengehi N, Kansal R, David CS, Kotb M. HLA transgenic mice provide evidence for a direct and dominant role of HLA class II variation in modulating the severity of streptococcal sepsis. J Immunol . 2007;178:3076–3083. doi: 10.4049/jimmunol.178.5.3076. [DOI] [PubMed] [Google Scholar]
- 53. DaSilva L, Welcher BC, Ulrich RG, Aman MJ, David CS, Bavari S. Humanlike immune response of human leukocyte antigen-DR3 transgenic mice to staphylococcal enterotoxins: a novel model for superantigen vaccines. J Infect Dis . 2002;185:1754–1760. doi: 10.1086/340828. [DOI] [PubMed] [Google Scholar]
- 54. Tilahun AY, Chowdhary VR, David CS, Rajagopalan G. Systemic inflammatory response elicited by superantigen destabilizes T regulatory cells, rendering them ineffective during toxic shock syndrome. J Immunol . 2014;193:2919–2930. doi: 10.4049/jimmunol.1400980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tilahun AY, Karau M, Ballard A, Gunaratna MP, Thapa A, David CS, et al. The impact of Staphylococcus aureus-associated molecular patterns on staphylococcal superantigen-induced toxic shock syndrome and pneumonia. Mediators Inflamm . 2014;2014:468285. doi: 10.1155/2014/468285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nandi B, Behar SM. Regulation of neutrophils by interferon-γ limits lung inflammation during tuberculosis infection. J Exp Med. 2011;208:2251–2262. doi: 10.1084/jem.20110919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cruz A, Khader SA, Torrado E, Fraga A, Pearl JE, Pedrosa J, et al. Cutting edge: IFN-γ regulates the induction and expansion of IL-17-producing CD4 T cells during mycobacterial infection. J Immunol . 2006;177:1416–1420. doi: 10.4049/jimmunol.177.3.1416. [DOI] [PubMed] [Google Scholar]
- 58. Desvignes L, Ernst JD. Interferon-gamma-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity . 2009;31:974–985. doi: 10.1016/j.immuni.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kirman J, Zakaria Z, McCoy K, Delahunt B, Le Gros G. Role of eosinophils in the pathogenesis of Mycobacterium bovis BCG infection in gamma interferon receptor-deficient mice. Infect Immun . 2000;68:2976–2978. doi: 10.1128/iai.68.5.2976-2978.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bohrer AC, Castro E, Hu Z, Queiroz ATL, Tocheny CE, Assmann M, et al. Tuberculosis Imaging Program Eosinophils are part of the granulocyte response in tuberculosis and promote host resistance in mice. J Exp Med . 2021;218:e20210469. doi: 10.1084/jem.20210469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Davey A, McAuley DF, O’Kane CM. Matrix metalloproteinases in acute lung injury: mediators of injury and drivers of repair. Eur Respir J . 2011;38:959–970. doi: 10.1183/09031936.00032111. [DOI] [PubMed] [Google Scholar]
- 62. Puneet P, Moochhala S, Bhatia M. Chemokines in acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol . 2005;288:L3–L15. doi: 10.1152/ajplung.00405.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zemans RL, Matthay MA. What drives neutrophils to the alveoli in ARDS? Thorax . 2017;72:1–3. doi: 10.1136/thoraxjnl-2016-209170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Islam SA, Chang DS, Colvin RA, Byrne MH, McCully ML, Moser B, et al. Mouse CCL8, a CCR8 agonist, promotes atopic dermatitis by recruiting IL-5+ T(H)2 cells. Nat Immunol . 2011;12:167–177. doi: 10.1038/ni.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Metzemaekers M, Gouwy M, Proost P. Neutrophil chemoattractant receptors in health and disease: double-edged swords. Cell Mol Immunol . 2020;17:433–450. doi: 10.1038/s41423-020-0412-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rossi DL, Hurst SD, Xu Y, Wang W, Menon S, Coffman RL, et al. Lungkine, a novel CXC chemokine, specifically expressed by lung bronchoepithelial cells. J Immunol . 1999;162:5490–5497. [PubMed] [Google Scholar]
- 67. Bishop B, Lloyd CM. CC chemokine ligand 1 promotes recruitment of eosinophils but not Th2 cells during the development of allergic airways disease. J Immunol . 2003;170:4810–4817. doi: 10.4049/jimmunol.170.9.4810. [DOI] [PubMed] [Google Scholar]
- 68. Nouailles G, Dorhoi A, Koch M, Zerrahn J, Weiner J, III, Faé KC, et al. CXCL5-secreting pulmonary epithelial cells drive destructive neutrophilic inflammation in tuberculosis. J Clin Invest . 2014;124:1268–1282. doi: 10.1172/JCI72030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Berger S, Wienhold S-M, Goekeri C, Behrendt U, Müller-Redetzky H, Dietert K, et al. CXCL5-dependent neutrophil recruitment harms lung barrier function in acute lung injury. Eur Respir J . 2018;52:PA4295. [Google Scholar]
- 70. Jia GQ, Gonzalo JA, Lloyd C, Kremer L, Lu L, Martinez-A C, et al. Distinct expression and function of the novel mouse chemokine monocyte chemotactic protein-5 in lung allergic inflammation. J Exp Med . 1996;184:1939–1951. doi: 10.1084/jem.184.5.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. S100A8/A9 in inflammation. Front Immunol . 2018;9:1298. doi: 10.3389/fimmu.2018.01298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ryckman C, Vandal K, Rouleau P, Talbot M, Tessier PA. Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J Immunol . 2003;170:3233–3242. doi: 10.4049/jimmunol.170.6.3233. [DOI] [PubMed] [Google Scholar]
- 73. Ryckman C, McColl SR, Vandal K, de Médicis R, Lussier A, Poubelle PE, et al. Role of S100A8 and S100A9 in neutrophil recruitment in response to monosodium urate monohydrate crystals in the air-pouch model of acute gouty arthritis. Arthritis Rheum . 2003;48:2310–2320. doi: 10.1002/art.11079. [DOI] [PubMed] [Google Scholar]
- 74. Crowe LAN, McLean M, Kitson SM, Melchor EG, Patommel K, Cao HM, et al. S100A8 & S100A9: alarmin mediated inflammation in tendinopathy. Sci Rep . 2019;9:1463. doi: 10.1038/s41598-018-37684-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tirkos S, Newbigging S, Nguyen V, Keet M, Ackerley C, Kent G, et al. Expression of S100A8 correlates with inflammatory lung disease in congenic mice deficient of the cystic fibrosis transmembrane conductance regulator. Respir Res . 2006;7:51. doi: 10.1186/1465-9921-7-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chakraborty D, Zenker S, Rossaint J, Hölscher A, Pohlen M, Zarbock A, et al. Alarmin S100A8 activates alveolar epithelial cells in the context of acute lung injury in a TLR4-dependent manner. Front Immunol . 2017;8:1493. doi: 10.3389/fimmu.2017.01493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Deguchi A, Yamamoto T, Shibata N, Maru Y. S100A8 may govern hyper-inflammation in severe COVID-19. FASEB J . 2021;35:e21798. doi: 10.1096/fj.202101013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Blondonnet R, Audard J, Belville C, Clairefond G, Lutz J, Bouvier D, et al. RAGE inhibition reduces acute lung injury in mice. Sci Rep . 2017;7:7208. doi: 10.1038/s41598-017-07638-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, et al. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci USA . 2004;101:10691–10696. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhang L, Xu C, Chen X, Shi Q, Su W, Zhao H. SOCS-1 suppresses inflammation through inhibition of NALP3 inflammasome formation in smoke inhalation-induced acute lung injury. Inflammation . 2018;41:1557–1567. doi: 10.1007/s10753-018-0802-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Draijer C, Speth JM, Penke LRK, Zaslona Z, Bazzill JD, Lugogo N, et al. Resident alveolar macrophage-derived vesicular SOCS3 dampens allergic airway inflammation. FASEB J . 2020;34:4718–4731. doi: 10.1096/fj.201903089R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Nguyen LS, Ait Hamou Z, Gastli N, Chapuis N, Pène F. Potential role for interferon gamma in the treatment of recurrent ventilator-acquired pneumonia in patients with COVID-19: a hypothesis. Intensive Care Med . 2021;47:619–621. doi: 10.1007/s00134-021-06377-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mudd PA, Crawford JC, Turner JS, Souquette A, Reynolds D, Bender D, et al. Distinct inflammatory profiles distinguish COVID-19 from influenza with limited contributions from cytokine storm. Sci Adv . 2020;6:eabe3024. doi: 10.1126/sciadv.abe3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Palacios-Gutiérrez J-J, Rodríguez-Guardado A, Arias-Guillén M, Alonso-Arias R, Palacios-Penedo S, García-García J-M, et al. Clinical and epidemiological correlates of low IFN-gamma responses in mitogen tube of QuantiFERON assay in tuberculosis infection screening during the COVID-19 pandemic: a population-based marker of COVID-19 mortality? Arch Bronconeumol . 2022;58:649–659. doi: 10.1016/j.arbres.2022.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hu Z-J, Xu J, Yin J-M, Li L, Hou W, Zhang L-L, et al. Lower circulating interferon-gamma is a risk factor for lung fibrosis in COVID-19 patients. Front Immunol . 2020;11:585647. doi: 10.3389/fimmu.2020.585647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Myasnikov AL, Berns SA, Talyzin PA, Ershov FI. [Interferon gamma in the treatment of patients with moderate COVID-19] Vopr Virusol . 2021;66:47–54. doi: 10.36233/0507-4088-24. [DOI] [PubMed] [Google Scholar]
- 87. Grimm C, Dickel S, Grundmann J, Payen D, Schanz J, Zautner AE, et al. Case report: interferon-γ rescues monocytic human leukocyte antigen receptor (mHLA-DR) function in a COVID-19 patient with ARDS and superinfection with multiple MDR 4MRGN bacterial strains. Front Immunol . 2021;12:753849. doi: 10.3389/fimmu.2021.753849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Abdel-Hamed EF, Ibrahim MN, Mostafa NE, Moawad HSF, Elgammal NE, Darwiesh EM, et al. Role of interferon gamma in SARS-CoV-2-positive patients with parasitic infections. Gut Pathog . 2021;13:29. doi: 10.1186/s13099-021-00427-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mehta P, Ciurtin C, Scully M, Levi M, Chambers RC. JAK inhibitors in COVID-19: the need for vigilance regarding increased inherent thrombotic risk. Eur Respir J . 2020;56:2001919. doi: 10.1183/13993003.01919-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Luo W, Li Y-X, Jiang L-J, Chen Q, Wang T, Ye D-W. Targeting JAK-STAT signaling to control cytokine release syndrome in COVID-19. Trends Pharmacol Sci . 2020;41:531–543. doi: 10.1016/j.tips.2020.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]