

Abstract
Purpose of Review
The right ventricle (RV) and left ventricle (LV) have different developmental origins, which likely plays a role in their chamber-specific response to physiological and pathological stress. RV dysfunction is encountered frequently in patients with congenital heart disease (CHD) and right heart abnormalities emerge from different causes than increased afterload alone as is observed in RV dysfunction due to pulmonary hypertension (PH). In this review, we describe the developmental, structural, and functional differences between ventricles while highlighting emerging therapies for RV dysfunction.
Recent Findings
There are new insights into the role of fibrosis, inflammation, myocyte contraction, and mitochondrial dynamics in the pathogenesis of RV dysfunction. We discuss the current state of therapies that may potentially improve RV function in both experimental and clinical trials.
Summary
A clearer understanding of the differences in molecular alterations in the RV compared to the LV may allow for the development of better therapies that treat RV dysfunction.
Keywords: Right ventricle, Left ventricle, Adult congenital heart disease, Congenital heart disease, Pulmonary hypertension, Right ventricular failure
Introduction
The right ventricle (RV) and left ventricle (LV) have different embryologic origins, and each responds differently to stress. Specifically, pressure and volume overload contribute to RV dysfunction, which are encountered commonly in congenital heart disease (CHD). RV failure is an important predictor of morbidity and mortality in CHD; therefore, understanding the pathophysiology of pressure and volume overload in this disease spectrum is imperative.
The pathogenesis of RV failure includes myocardial stress, cytokine and neurohormonal activation, fibrosis, inflammation, and reduced contractility. Treatment of RV failure is largely extrapolated from treatment of LV failure; however, there are gaps with this management strategy because each ventricle exerts variable response patterns to stress. Emerging evidence shows that targeting molecular parameters implicated in the pathogenesis of RV dysfunction may become a novel therapeutic approach in the treatment of right heart failure.
Embryological, Anatomical, and Physiological Differences Between the RV and LV
This section contrasts the developmental biology, structure, and function of the RV and LV.
Embryological Differences Between Ventricles
Embryonic development of the cardiovascular system in humans occurs between the third and eighth weeks of gestation [1•]. The LV myocardium derives from the primary heart field and the RV myocardium derives from the anterior (secondary) heart field [1•]. These events develop successively and are driven by preprogrammed genetic signals. Basic helix-loop-helix transcription factors are important regulators of embryonic development [2]. Specifically, after cardiac looping, cardiomyocyte differentiation is dependent on basic helix-loop-helix transcription factors expressed in the RV (driven by dHAND) and the LV (driven by eHAND) [2, 3]. It has been demonstrated that deletion of dHAND in a murine model resulted in hypoplasia of the RV [2]. Identification of dHAND and eHAND was important as it provides a pathway through which molecular signaling controlling cardiogenesis may be further elucidated [3].
Anatomical and Physiological Differences Between Ventricles
In utero, the RV contributes to 60% of overall cardiac output with both ventricles having equal wall thickness [4, 5•]. Functionally, poorly oxygenated blood from the vena cava travels across the tricuspid valve to the RV into the pulmonary artery (PA) with minimal amounts of blood entering the lung due to elevated pulmonary vascular resistance (PVR) [4]. The low PA saturation maintains a state of high PVR and blood shunts through the foramen ovale and ductus arteriosus, largely bypassing the lungs [6]. There are many physiological transitions that occur at birth with important structural and functional changes.
The placental circulation in utero is under high PVR and there is a rapid decrease in PVR once the umbilical cord is clamped at birth [7]. As a result, RV wall thickness decreases and LV mass increases [6]. In the face of a lower impedance pulmonary circuit, the normal postnatal RV maintains a cardiac output equal to the LV (in the absence of intracardiac shunting) at one-sixth the energy expenditure of the LV [8].
The RV can be divided into three anatomical sections: inlet (including tricuspid valve), trabeculated apex, and outflow tract (infundibulum, a muscular structure that supports pulmonary valve leaflets) (Fig. 1A). The crista supraventricularis separates the tricuspid valve and pulmonary valve, which is different from the aortomitral continuity of the LV. Structurally, the normal adult RV is thin-walled and crescent-shaped (Fig. 1B), whereas the LV is thick-walled and bullet-shaped. As previously mentioned, myocyte differentiation is directed by chamber-specific expression of basic transcription factors that likely alters the plane of myocyte contractility for the respective chamber. Although the LV has three distinct myocardial fibers, the RV consists of superficial (circumferential) fibers and deep (longitudinal) fibers with contraction from inlet to outlet and from free wall to septum; the RV relies more on longitudinal shortening compared to the LV and is heavily influenced by loading conditions [6]. Some embryological, physiological, and anatomical differences between ventricles are summarized below (Table 1).
Fig. 1.

Pathologic specimens. A Pathologic specimen of the right ventricle with the free wall removed to demonstrate the 3 anatomic regions [9]. B Pathologic specimen of the heart cut transversely demonstrating the crescent shape of the right ventricle [9]. Image from Warnes [9] reproduced with permission. Please remove Photo courtesy of Dr. W. D. Edwards, consultant in pathology, Mayo Clinic
Table 1.
Embryological, physiological, and anatomical differences between ventricles
| Embryological, anatomical, and functional comparisons of LV and RV |
Left ventricle | Right ventricle |
|---|---|---|
| Embryologic origins | Primary heart field | Anterior heart field |
| Helix-loop helix transcription factors | eHAND | dHAND |
| Myocardium | Thick walls; fine trabeculations | Thin walls; coarse trabeculations |
| Wall thickness (mm) | 7–11 | 2–3 |
| Morphological features | Bullet shape; ellipsoid | Crescentic shape |
| Papillary muscles | Two large | Several small |
| Contraction properties | Concentric | Peristaltic |
| Response to pathologic load | Responds better to pressure overload | Responds better to volume overload |
| Coronary supply | Two coronary arteries | One coronary artery |
RV-PA Coupling
The RV is coupled with the compliant PA, leading to a pressure–volume relationship distinct from the LV. Specifically, the RV is trapezoidal-shaped with few isovolumic periods, whereas the LV is rectangular-shaped with distinct isovolumic periods [10]. Functionally, the LV continues to generate pressure until the aortic valve closes, whereas the RV pressure falls prior to pulmonary valve closure, but RV output continues in the face of low pulmonary resistance [10]. The RV takes advantage by producing a similar output to the LV with reduced stroke work but remains sensitive to afterload. RV-PA coupling is a measure of RV performance and refers to the relationship between RV afterload and contractility [11, 12]. More specifically, both RV and PA are “coupled” where the RV contractility should “match” the afterload [12]. If RV afterload decreases, RV contractility should decrease; if RV afterload increases, RV contractility should increase to maintain RV performance and preserve RV-PA coupling [12].
Difference between RV dysfunction and RV Failure
Difference Between RV Dysfunction and RV Failure
RV dysfunction is any abnormality of filling or contraction without clinical heart failure (HF), whereas RV failure results in clinical HF from a structural or functional impairment of the RV [13].
Causes of RV Failure
RV failure can be categorized by its mechanism of injury and chronicity. The RV may fail from pressure or volume overload, inflow obstruction, myocardial disease, pericardial disease, or myocardial ischemia.
This review focuses on pressure and volume overload, specifically as it relates to CHD; however, it is worth highlighting the spectrum of diseases that impact RV function (Table 2).
Table 2.
The spectrum of diseases that impact RV function
| Causes of RV failure |
Pressure overload | Volume overload |
|---|---|---|
| Acute | Pulmonary embolism Acute respiratory distress syndrome Myocarditis Hypoxia |
Sepsis Excessive blood or saline transfusion |
| Chronic | Congenital pulmonary valve stenosis Pulmonary hypertension* Double-chambered RV Systemic RV |
Transposition of the great arteries Pulmonary regurgitation Tricuspid regurgitation Ebstein anomaly Atrial septal defect Partial anomalous pulmonary venous drainage |
ASD indicates atrial septal defect; ARDS, acute respiratory distress syndrome; DCRV.
Includes pulmonary arterial hypertension (PAH)
RV Pressure Overload Overview
There are many causes of RV failure from non-physiologic pressure overload, such as PH, pulmonary embolism (PE), congenital pulmonary valve stenosis, systemic RV dysfunction, double-chambered RV, and peripheral pulmonary stenosis [13]. The RV fails from both acute and chronic pressure overload but in different ways, and it is important to understand how.
Acute RV Pressure Overload
Acute PE causes pulmonary obstruction resulting in increased RV afterload and pulmonary arterial vasoconstriction. The rapid rise in afterload increases RV wall tension leading to RV dilatation and systolic dysfunction [14]. As the RV pressure increases, the septum shifts into the LV thereby reducing LV filling and compromising LV output [14]. Furthermore, coronary perfusion is restricted by elevated RV wall stress. The final event in this death spiral is worsening systemic hypotension and sudden cardiac arrest. The RV does not respond well to acute changes in afterload but responds better to long-term pressure overload through a hypertrophic adaptive response and expansion of extracellular matrix [15].
Chronic RV Pressure Overload
The adaptation to chronic pressure overload becomes maladaptive for the RV, resulting in dilation, decreased systolic performance, and reduced output [16]. RV pressure overload leads to concentric hypertrophy (sarcomeres are in parallel) thereby increasing myocardial thickness and reducing chamber diameter [17].
Concentric hypertrophy is a remodeling process that helps defend cardiac output, but remodeling progresses from adaptive to maladaptive leading to cardiac failure. RV pressure overload generates oxidant stress and capillary rarefaction, leading to fibrosis, cardiomyocyte dysfunction, and cardiomyocyte loss [18]. It is unclear what triggers this transition from adaptive to maladaptive, but genetics, neurohormonal over-activation, and ischemia play roles [19].
Congenital PS and systemic RV are pressure overload conditions seen in CHD. Congenital PS is one of the most common congenital heart defects and the degree of RV hypertrophy varies with the severity of obstruction [20]. The RV is the systemic ventricle in D-loop transposition of the great arteries (D-TGA) post atrial switch (Fig. 2A-C) and L-loop transposition of the great arteries (L-TGA) or congenitally corrected TGA. In these defects, the aorta arises from the RV and PA from the LV. The chronic increase in afterload causes the systemic RV to assume a pressure–volume loop like the LV where ejection of blood during RV pressure decline no longer occurs [21]. This results in compensatory RV dilation to maintain stroke volume; these series of events lead to increase in myocardial wall stress and oxygen demand [21]. Lack of contractile reserve is another concern in systemic RVs [22]. Because of this exposure to systemic pressures, RV failure is the most clinically important problem we see in patients with TGA. In D-loop TGA post atrial switch, systemic AV valve (AVV) (tricuspid valve) regurgitation contributes to progressive decline in RV function [23]. In L-loop TGA, systemic AVV (tricuspid valve) regurgitation and RV failure are associated with increased mortality [20]. Therefore, the tricuspid valve function should be closely monitored in patients with TGA and a decline in RV systemic function should prompt a search for worsening AV valve regurgitation. The pathophysiological mechanisms of systemic RV failure are multifactorial involving issues with preload and afterload (Fig. 3).
Fig. 2.

Four-chamber view on a transthoracic echocardiogram shows the intra-atrial baffles seen in a patient with D-loop TGA post atrial switch where baffles are used to restore physiological circulation. A An atrial baffle diverts blood from both vena cava across to the mitral valve and LV (blue arrow is in the systemic venous baffle), which ejects blood to the PA. B The oxygenated pulmonary venous blood returns to the tricuspid valve and systemic RV (red arrow is in the pulmonary venous baffle), which ejects blood to the aorta. C Apical short-axis view on a transthoracic echocardiogram shows a dilated and hypertrophied systemic RV where the interventricular septum bulges into the “banana” shaped—a finding expected in a patient with systemic RV post atrial switch repair
Fig. 3.
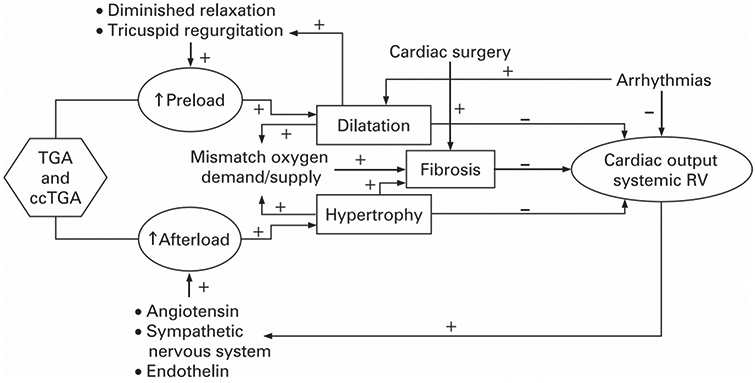
Pathophysiological pathways of systemic right ventricular (RV) dysfunction from Winter [24]. The pathophysiology of systemic RV dysfunction is multifactorial and includes arrhythmias, tricuspid valve regurgitation, myocar-dial fibrosis, and myocardial ischemia. Image from Winter [24] reproduced with permission
Ebstein anomaly (EA) is a congenital heart defect that imposes a significant RV volume load. EA has an accompanying RV myopathy and involves failure of delamination of the septal leaflet (sometimes posterior) of the tricuspid valve where the valve is apically displaced with an “atrialized” RV and a true RV below (Fig. 4).
Fig. 4.

Pathologic specimen cut in the 4-chamber plane from a patient with Ebstein anomaly from Warnes [9]. The tricuspid valve is displaced markedly inferiorly, and the right ventricular wall is extremely thin. Image from Warnes [9] reproduced with permission. Please remove Photo courtesy of Dr. W. D. Edwards, consultant in pathology, Mayo Clinic
Pathogenesis of Chronic RV Pressure Overload: Summary
The key pathophysiological principle in RV failure from chronic pressure overload is the prolonged exposure to increased afterload. RV pressure overload is associated with myocardial ischemia caused by reduced right coronary perfusion that promotes cardiomyocyte injury [25, 26]. Due to these changes, there is an increase in mitochondrial reactive oxygen species (ROS) accumulation, resulting in hypoxia-induced factor-1α (HIF-1α) inhibition and p53 activation [27]. These events lead to further reduction in angiogenesis. Furthermore, vascular endothelial growth factor (VEGF) and apelin are downregulated, contributing to impaired capillary growth [28].
Chronic RV Volume Overload
The RV adapts more favorably to volume overload compared to pressure overload. The thin RV wall permits it to accommodate changes in preload without significant changes in pressure. States of chronic volume overload, such as an ASD or PR after repair of tetralogy of Fallot (TOF), can persist many years prior to the development of RV failure.
ASDs are commonly diagnosed initially in adulthood and result in a net left-to-right shunt with direction and magnitude of blood flow determined largely by the size of the defect and ventricular compliance [29]. The shunt poses a volume load on the RV and pulmonary vessels. RV volume overload is associated with LV dysfunction due to altered ventricular geometry and reduced myofiber preload [30]. In long-standing ASDs (in the absence of Eisenmenger physiology), an increased rate of morbidity is driven by the increased net left-to-right shunt because of progressive LV stiffness from systemic hypertension or aging [20].
TOF is the most common cyanotic CHD and obstruction along the RV outflow tract is a key element of its pathophysiology, but surgical mitigation of obstruction frequently results in PR, which leads to RV dilatation [31].
Pathogenesis of Chronic RV Volume Overload: Summary
The key pathophysiological principle in RV failure from chronic volume overload is a dilated tricuspid annulus that permits TR, exacerbating the volume load on the RV and septal shift. There is significant septal shift because the pericardium is unable to distend and, thus, cannot geometrically accommodate changes in end-diastolic volume manifest by RV dilation. The septal shift impairs LV filling that impairs LV end-diastolic filling, increases left atrial pressure, and often promotes pulmonary hypertension [31]. In volume overload, the RV is more prone than the LV to developing fibrosis, as demonstrated in an experimental high-flow porcine model [32]. Furthermore, patients with post-surgical repair of TOF and PR can develop RV fibrosis, even at areas remote from surgical incision sites, which is clinically relevant owing to the effect of replacement and interstitial collagen deposition on electromechanical stability and susceptibility to RV failure [33, 34].
The molecular mechanisms underlying RV volume overload in humans remain elusive, but some recent animal studies have shown the detrimental effects of volume overload on the RV, such as hypertrophy and angiogenesis [35]. Volume overload was shown recently to induce an immune response in the RV during the neonatal period in vivo [36]. Moreover, immune responses may be an initiating factor for RV remodeling and, therefore, immune modulating therapies have been proposed as one potential path forward to prevent potential deleterious effects of volume overload in neonatal right heart failure syndromes [36]. More data are needed before immunosuppressants should be considered for use under clinical circumstances, however, owing to the pathogenic effects reported for these therapies on myocardial tone and structural integrity.
Diagnosis and Assessment of RV Failure
A thorough history and physical examination is required. The symptoms of RV failure may be extremity swelling, early satiety, shortness of breath, and exercise intolerance [13]. The physical examination may reveal elevated jugular venous pressure with prominent V wave, RV heave, right-sided S3 gallop, ascites, and peripheral edema. A prominent pulmonic component of the second heart sound (P2) indicates the presence of PH. The pulmonic component is defined as loud if it is greater than the aortic component in the second left intercostal space or if audible at the cardiac apex [37].
The electrocardiogram may show right axis deviation and right atrial enlargement (p-wave amplitude > 2.5 mm in leads II, III, and aVF) [8]. RV hypertrophy may be identified as a dominant R wave in V1 (>7 mm tall or R/S ratio >1) [38].
Ideally, using serum biomarkers to help guide therapy is prudent. The serum N-terminal pro-brain natriuretic peptide (NT-proBNP) is a biomarker that may be useful in management of patients with HF due to RV dysfunction, but is not specific to RV heart failure per se [39].
The chest radiograph may demonstrate RV enlargement as manifest by a globular appearance of the cardiac silhouette and loss of the retrosternal airspace on the lateral projection.
Two-dimensional echocardiography aids in the diagnosis of RV dysfunction; however, there are limitations to the quantification of RV function. Tricuspid annular systolic velocity, tricuspid annular plane systolic excursion (TAPSE), and functional area change (FAC) are standard parameters used for the quantitative assessment of RV function but are load dependent [40]. RV strain is less load dependent and has high predictive value in patients with CHD and PH. Importantly, it enables detection of subclinical RV dysfunction even when TAPSE, FAC, or annular velocities are in the normal range but have not been used routinely in clinical practice [41].
Cardiac MRI provides a full unimpeded examination of the heart’s structure and function. It is the gold standard for quantitative measurement of mass, EF, and volumes. It provides delayed gadolinium enhancement aiding in identifying fibrosis and velocity-encoded methods to measure blood flow. Multidetector computed tomography provides information about RV size and function. A hemodynamic catheterization is informative if the volume status is uncertain, worsening renal function in response to therapy, or hemodynamic instability [8].
Management of RV Failure
Management should focus on identifying the underlying cause while focusing on afterload reduction, preload optimization, and myocardial contractility support with pharmacotherapy. Advanced mechanical circulatory support may be utilized in select cases, if needed.
Despite the increase in mortality from RV failure in patients with CHD, there are no adequately powered trials to assess the role of pharmacotherapy in this group. Moreover, patients with CHD have historically been excluded from left-sided HF clinical trials. Therefore, it is important to understand that the guideline-directed medical therapies (GDMT) in HF should be regarded as LV-centric. Small series suggest a potential benefit of β-blockade in patients with systemic RV, including improvement in symptoms; however, in a large clinical trial, carvedilol did not improve HF outcomes [42]. Some reports suggest mixed results but overall, there have been no demonstrable benefits of angiotensin-converting enzyme inhibitor or angiotensin-2 receptor blocker use in systemic RV dysfunction [43].
Patients with RV failure and CHD should be referred to a center specializing in the care of CHD to potentially correct any reversible anatomical or physiological lesions contributing to RV failure. Patients with a systemic RV routinely have systemic ventricular dysfunction, which is commonly associated with systemic AVV (tricuspid) regurgitation. AVV repair or replacement can improve the course of disease, if performed before a reduction in systemic ventricular function [44]. Cardiac resynchronization therapy may be considered in patients with CHD and reduced systemic RV function [45].
The current data do not recommend GDMT used for left-sided HF to patients with a systemic RV. A more comprehensive management strategy for RV dysfunction in CHD can be found in the newest iteration of the adult congenital heart disease guidelines [46]. Some patients may require consideration for heart only or heart–lung transplantation [46].
Novel Clinical and Experimental Therapeutic Approaches to RV Dysfunction
Clinical trials have been constructed to better understand PAH treatment, but most assessed cardiac improvement secondary to reduced PA pressure instead of assessing it as a primary objective. In other words, most clinical trials focused on improving RV function as a consequence of reduction in PA pressures; however, there are recent studies aimed at assessing RV function by measuring various structural and functional parameters.
Some clinical trials report improvements in RV ejection fraction at varying degrees: ~3.9% after 3 months of trimetazidine (NCT03273387 [47]), 10.1% after 6 months of macitentan (NCT02310672 [48]), and 10.4% after 6 months of carvedilol (NCT00964678 at clinical trials.gov [49]) suggesting an overall improvement in RV function. Changes in RV volumes have also been demonstrated. For instance, 6 months of carvedilol therapy caused a change of 22.6 mL in RVESV (NCT00964678 [49]) and 6 months of macitentan therapy caused changes in RVEDV of −6.22 mL, and RVESV of 16.39 mL (NCT02310672 [48]). Reductions in RV mass have been observed. Macitentan therapy for 6 months caused a reduction of 10.10 g in RV mass (NCT02310672 [48]), suggesting an improvement in RV remodeling. These clinical trials reported improvement in RV function using imaging-related metrics; however, they did not focus on molecular parameters such as fibrosis, myocyte contraction, inflammation, and mitochondrial content, which could be possible novel therapeutic targets to address RV dysfunction. For example, current antifibrotic therapies effective in LV do not reverse RV fibrosis, which may be explained by the differences in extracellular matrix composition [50]. RV has more dendritic and macrophage cells, suggesting that inflammation plays a more important role [51]. Protein kinases A activators have been shown to improve sarcomere function as RV myofilaments have lower calcium sensitivity [52]. The RV has less mitochondrial content and lower rate of oxidation; therefore, preservation of mitochondrial integrity improves RV performance [53, 54]. These are potential molecular pathways to target in RV dysfunction, but they require further investigation.
Summary
Both ventricles are different in respect to their development, structure, and function. Acknowledging the differences in molecular alterations in both ventricles may facilitate the development of novel therapies. This is especially important for the RV because the mechanisms related to its dysfunction remain unclear. Advances in therapeutics for RV dysfunction are needed to improve morbidity and mortality, and though there are promising new pathways to target, they require further investigation.
Funding
B.A.M.: NIH 1R01HL139613-01, R01HL153502, R01HL155096-01, 2021A007243 BWH/MIT-Broad Institute; Cardiovascular Medical Research Foundation.
Footnotes
Conflict of Interest B.A.M., Deerfield Corporation (beyond the scope of this work); Actelion Sciences (beyond the scope of this work), Tenax Company (beyond the scope of this work), and Regeneron (beyond the scope of this work); patent or patent pending (beyond the scope of this work): patent 9,605,047; PCT/US2019/059890; PCT/US2020/066886.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
- 1.•. Sanz J, Sánchez-Quintana D, Bossone E, Bogaard HJ, Naeije R. Anatomy, function, and dysfunction of the right ventricle: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73(12):1463–82. In this article, the authors provide a contemporaneous review of right ventricular anatomy, physiology, and hemodynamics while highlighting how the right ventricle plays an essential role in determining prognosis in nearly all cardiovascular disorders.
- 2.Srivastava D, Olson EN. A genetic blueprint for cardiac development. Nature. 2000;407(6801):221–6. 10.1038/35025190. [DOI] [PubMed] [Google Scholar]
- 3.Thomas T, Yamagishi H, Overbeek PA, Olson EN, Srivastava D. The bHLH factors, dHAND and eHAND, specify pulmonary and systemic cardiac ventricles independent of left-right sidedness. Dev Biol. 1998;196(2):228–36. 10.1006/dbio.1998.8849. [DOI] [PubMed] [Google Scholar]
- 4.Rudolph AM. Congenital cardiovascular malformations and the fetal circulation. Arch Dis Child Fetal Neonatal Ed. 2010;95(2):F132–6. 10.1136/adc.2007.128777. [DOI] [PubMed] [Google Scholar]
- 5.•. Friedberg MK, Redington AN. Right versus left ventricular failure: differences, similarities, and interactions. Circulation. 2014;129(9):1033–44. The authors emphasize the differences between the left and right ventricle but also appropriately acknowledge that both ventricles share similar features in adaptation when faced with pressure or volume overloading conditions.
- 6.Walker LA, Buttrick PM. The right ventricle: biologic insights and response to disease: updated. Curr Cardiol Rev. 2013;9(1):73–81. 10.2174/157340313805076296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guimaron S, Guihaire J, Amsallem M, Haddad F, Fadel E, Mercier O. Current knowledge and recent advances of right ventricular molecular biology and metabolism from congenital heart disease to chronic pulmonary hypertension. Biomed Res Int. 2018;17(2018):1981568. 10.1155/2018/1981568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konstam MA, Kiernan MS, Bernstein D, Bozkurt B, Jacob M, Kapur NK, et al. American Heart Association Council on Clinical Cardiology; Council on Cardiovascular Disease in the Young; and Council on Cardiovascular Surgery and Anesthesia. Evaluation and Management of Right-Sided Heart Failure: a scientific statement from the American Heart Association. Circulation. 2018;137(20):e578–e622. 10.1161/CIR.0000000000000560. [DOI] [PubMed] [Google Scholar]
- 9.Warnes CA. Adult congenital heart disease importance of the right ventricle. J Am Coll Cardiol. 2009;17;54(21):1903–10. 10.1016/j.jacc.2009.06.048. [DOI] [PubMed] [Google Scholar]
- 10.Redington AN. Right ventricular function. Cardiol Clin. 2002;20(3):341–9. 10.1016/s0733-8651(02)00005-x. [DOI] [PubMed] [Google Scholar]
- 11.Sathananthan G, Grewal J. The complex relationship that is RV-PA coupling and its relevance to managing congenital heart disease. Can J Cardiol. 2019;35(7):816–8. 10.1016/j.cjca.2019.04.027. [DOI] [PubMed] [Google Scholar]
- 12.Egbe AC, Kothapalli S, Miranda WR, Pislaru S, Ammash NM, Borlaug BA, et al. Assessment of right ventricular-pulmonary arterial coupling in chronic pulmonary regurgitation. Can J Cardiol. 2019;35(7):914–22. 10.1016/j.cjca.2019.03.009. [DOI] [PubMed] [Google Scholar]
- 13.Haddad F, Doyle R, Murphy DJ, Hunt SA. Right ventricular function in cardiovascular disease, part II: pathophysiology, clinical importance, and management of right ventricular failure. Circulation. 2008;117(13):1717–31. 10.1161/CIRCULATIONAHA.107.653584. [DOI] [PubMed] [Google Scholar]
- 14.Kürkciyan I, Meron G, Sterz F, Janata K, Domanovits H, Holzer M, et al. Pulmonary embolism as a cause of cardiac arrest: presentation and outcome. Arch Intern Med. 2000;160(10):1529–35. 10.1001/archinte.160.10.1529. [DOI] [PubMed] [Google Scholar]
- 15.Bogaard HJ, Abe K, VonkNoordegraaf A, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest. 2009;135(3):794–804. 10.1378/chest.08-0492. [DOI] [PubMed] [Google Scholar]
- 16.Simon MA, Pinsky MR. Right ventricular dysfunction and failure in chronic pressure overload. Cardiol Res Pract. 2011;23(2011):568095. 10.4061/2011/568095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anaruma CP, Pereira RM, Cristina da Cruz Rodrigues K, Ramos da Silva AS, Cintra DE, Ropelle ER, et al. Rock protein as cardiac hypertrophy modulator in obesity and physical exercise. Life Sci. 2020;254:116955. 10.1016/j.lfs.2019.116955. [DOI] [PubMed] [Google Scholar]
- 18.Reddy S, Bernstein D. Molecular mechanisms of right ventricular failure. Circulation. 2015;132(18):1734–42. 10.1161/CIRCULATIONAHA.114.012975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van der Bruggen CEE, Tedford RJ, Handoko ML, van der Velden J, de Man FS. RV pressure overload: from hypertrophy to failure. Cardiovasc Res. 2017;113(12):1423–32. 10.1093/cvr/cvx145. [DOI] [PubMed] [Google Scholar]
- 20.Davlouros PA, Niwa K, Webb G, Gatzoulis MA. The right ventricle in congenital heart disease. Heart. 2006;92 Suppl 1(Suppl 1):i27–38. 10.1136/hrt.2005.077438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Redington AN, Gray HH, Hodson ME, Rigby ML, Oldershaw PJ. Characterisation of the normal right ventricular pressure-volume relation by biplane angiography and simultaneous micromanometer pressure measurements. Br Heart J. 1988;59(1):23–30. 10.1136/hrt.59.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Connelly MS, Liu PP, Williams WG, Webb GD, Robertson P, McLaughlin PR. Congenitally corrected transposition of the great arteries in the adult: functional status and complications. J Am Coll Cardiol. 1996;27(5):1238–43. 10.1016/0735-1097(95)00567-6. [DOI] [PubMed] [Google Scholar]
- 23.Warnes CA. Transposition of the great arteries. Circulation. 2006;114(24):2699–709. 10.1161/CIRCULATIONAHA.105.592352. [DOI] [PubMed] [Google Scholar]
- 24.Winter MM, Bouma BJ, Groenink M, Konings TC, Tijssen JG, van Veldhuisen DJ, et al. Latest insights in therapeutic options for systemic right ventricular failure: a comparison with left ventricular failure. Heart. 2009;95(12):960–3. 10.1136/hrt.2008.156265. [DOI] [PubMed] [Google Scholar]
- 25.Ryan JJ, Archer SL. The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ Res. 2014;115(1):176–88. 10.1161/CIRCRESAHA.113.301129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andersen S, Nielsen-Kudsk JE, Vonk Noordegraaf A, de Man FS. Right ventricular fibrosis. Circulation. 2019;139(2):269–85. 10.1161/CIRCULATIONAHA. [DOI] [PubMed] [Google Scholar]
- 27.Sutendra G, Dromparis P, Paulin R, Zervopoulos S, Haromy A, Nagendran J, Michelakis ED. A metabolic remodeling in right ventricular hypertrophy is associated with decreased angiogenesis and a transition from a compensated to a decompensated state in pulmonary hypertension. J Mol Med (Berl). 2013;91(11):1315–27. 10.1007/s00109-013-1059-4. [DOI] [PubMed] [Google Scholar]
- 28.Drake JI, Bogaard HJ, Mizuno S, Clifton B, Xie B, Gao Y, et al. Molecular signature of a right heart failure program in chronic severe pulmonary hypertension. Am J Respir Cell Mol Biol. 2011;45(6):1239–47. 10.1165/rcmb.2010-0412OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brida M, Chessa M, Celermajer D, Li W, Geva T, Khairy P, Griselli M, et al. Atrial septal defect in adulthood: a new paradigm for congenital heart disease. Eur Heart J. 2021:ehab646. 10.1093/eurheartj/ehab646. [DOI] [PubMed] [Google Scholar]
- 30.Walker RE, Moran AM, Gauvreau K, Colan SD. Evidence of adverse ventricular interdependence in patients with atrial septal defects. Am J Cardiol. 2004;93(11):1374–7, A6. 10.1016/j.amjcard.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 31.Valente AM, Geva T. How to image repaired tetralogy of Fallot. Circ Cardiovasc Imaging. 2017;10(5):e004270. 10.1161/CIRCIMAGING.116.004270. [DOI] [PubMed] [Google Scholar]
- 32.Modesti PA, Vanni S, Bertolozzi I, Cecioni I, Lumachi C, Perna AM, et al. Different growth factor activation in the right and left ventricles in experimental volume overload. Hypertension. 2004;43(1):101–8. 10.1161/01.HYP.0000104720.76179.18. [DOI] [PubMed] [Google Scholar]
- 33.Babu-Narayan SV, Kilner PJ, Li W, Moon JC, Goktekin O, Davlouros PA, et al. Ventricular fibrosis suggested by cardiovascular magnetic resonance in adults with repaired tetralogy of Fallot and its relationship to adverse markers of clinical outcome. Circulation. 2006;113(3):405–13. 10.1161/CIRCULATIONAHA. [DOI] [PubMed] [Google Scholar]
- 34.Wald RM, Haber I, Wald R, Valente AM, Powell AJ, Geva T. Effects of regional dysfunction and late gadolinium enhancement on global right ventricular function and exercise capacity in patients with repaired tetralogy of Fallot. Circulation. 2009;119(10):1370–7. 10.1161/CIRCULATIONAHA.108.816546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bossers GPL, Hagdorn QAJ, Ploegstra MJ, Borgdorff MAJ, Silljé HHW, Berger RMF, et al. Volume load-induced right ventricular dysfunction in animal models: insights in a translational gap in congenital heart disease. Eur J Heart Fail. 2018;20(4):808–12. 10.1002/ejhf.931. [DOI] [PubMed] [Google Scholar]
- 36.Cui Q, Sun S, Zhu H, Xiao Y, Jiang C, Zhang H, et al. Volume overload initiates an immune response in the right ventricle at the neonatal stage. Front Cardiovasc Med. 2021;16(8):772336. 10.3389/fcvm.2021.772336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Felner JM. The Second Heart Sound. In: Walker HK, Hall WD, Hurst JW, editors. Clinical methods: the history, physical, and laboratory examinations. 3rd ed. Boston: Butterworths; 1990. Chapter 23. [PubMed] [Google Scholar]
- 38.Surawicz B, Knilans T (2008) Chou’s electrocardiography in clinical practice: adult and pediatric, 6th ed. W.B. Saunders, Philadelphia. [Google Scholar]
- 39.Cepkova M, Kapur V, Ren X, Quinn T, Zhuo H, Foster E, et al. Clinical significance of elevated B-type natriuretic peptide in patients with acute lung injury with or without right ventricular dilatation: an observational cohort study. Ann Intensive Care. 2011;1(1):18. 10.1186/2110-5820-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hayek S, Sims DB, Markham DW, Butler J, Kalogeropoulos AP. Assessment of right ventricular function in left ventricular assist device candidates. Circ Cardiovasc Imaging. 2014;7(2):379–89. 10.1161/CIRCIMAGING.113.001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tadic M, Nita N, Schneider L, Kersten J, Buckert D, Gonska B, et al. The predictive value of right ventricular longitudinal strain in pulmonary hypertension, heart failure, and valvular diseases. Front Cardiovasc Med. 2021;17(8):698158. 10.3389/fcvm.2021.698158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shaddy RE, Boucek MM, Hsu DT, Boucek RJ, Canter CE, Mahony L, Ross RD, et al. Pediatric Carvedilol Study Group. Carvedilol for children and adolescents with heart failure: a randomized controlled trial. JAMA. 2007;298(10):1171–9. 10.1001/jama.298.10.1171. [DOI] [PubMed] [Google Scholar]
- 43.Book WM, Shaddy RE. Medical therapy in adults with congenital heart disease. Heart Fail Clin. 2014;10(1):167–78. 10.1016/j.hfc.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 44.Graham TP Jr, Bernard YD, Mellen BG, Celermajer D, Baumgartner H, Cetta F, et al. Long-term outcome in congenitally corrected transposition of the great arteries: a multi-institutional study. J Am Coll Cardiol. 2000;36(1):255–61. 10.1016/s0735-1097(00)00682-3. [DOI] [PubMed] [Google Scholar]
- 45.Dubin AM, Janousek J, Rhee E, Strieper MJ, Cecchin F, Law IH, et al. Resynchronization therapy in pediatric and congenital heart disease patients: an international multicenter study. J Am Coll Cardiol. 2005;46(12):2277–83. 10.1016/j.jacc.2005.05.096. [DOI] [PubMed] [Google Scholar]
- 46.Stout KK, Daniels CJ, Aboulhosn JA, Bozkurt B, Broberg CS, Colman JM, et al. 2018 AHA/ACC Guideline for the Management of Adults With Congenital Heart Disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2019;73(12):e81–192. 10.1016/j.jacc.2018.08.1029. [DOI] [PubMed] [Google Scholar]
- 47.Muliawan HS, Widyantoro B, Soerarso R, Hersunarti N, Sahara E, Atmadikoesoemah CA, et al. P194 Trimetazidine preserves right ventricular function on pulmonary arterial hypertension patients in National Cardiovascular Center Harapan Kita Hospital Indonesia. Eur Heart J. 2020. 10.1093/ehjci/ehz872.068. [DOI] [Google Scholar]
- 48.Vonk Noordegraaf A, Channick R, Cottreel E, Kiely DG, Marcus JT, Martin N, et al. The REPAIR study: effects of macitentan on RV structure and function in pulmonary arterial hypertension. JACC Cardiovasc Imaging. 2022;15(2):240–53. 10.1016/j.jcmg.2021.07.027. [DOI] [PubMed] [Google Scholar]
- 49.Grinnan D, Bogaard HJ, Grizzard J, Van Tassell B, Abbate A, DeWilde C, et al. Treatment of group I pulmonary arterial hypertension with carvedilol is safe. Am J Respir Crit Care Med. 2014;189(12):1562–4. 10.1164/rccm.201311-2025LE. [DOI] [PubMed] [Google Scholar]
- 50.Andersen S, Birkmose Axelsen J, Ringgaard S, Randel Nyengaard J, Holm Nielsen S, Genovese F, et al. Pressure overload induced right ventricular remodeling is not attenuated by the anti-fibrotic agent pirfenidone. Pulm Circ. 2019;9(2):2045894019848659. 10.1177/2045894019848659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gorr MW, Sriram K, Chinn AM, Muthusamy A, Insel PA. Transcriptomic profiles reveal differences between the right and left ventricle in normoxia and hypoxia. Physiol Rep. 2020;8(2):e14344. 10.14814/phy2.14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rain S, Andersen S, Najafi A, Gammelgaard Schultz J, da Silva Gonçalves Bós D, Handoko ML, et al. Right ventricular myocardial stiffness in experimental pulmonary arterial hypertension: relative contribution of fibrosis and myofibril stiffness. Circ Heart Fail. 2016;9(7):e002636. 10.1161/CIRCHEARTFAILURE.115.002636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bernal-Ramírez J, Silva-Platas C, Jerjes-Sánchez C, Ramos-González MR, Vázquez-Garza E, Chapoy-Villanueva H, et al. Resveratrol prevents right ventricle dysfunction, calcium mishandling, and energetic failure via SIRT3 stimulation in pulmonary arterial hypertension. Oxid Med Cell Longev. 2021;20(2021):9912434. 10.1155/2021/9912434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nagendran J, Gurtu V, Fu DZ, Dyck JR, Haromy A, Ross DB, Rebeyka IM, Michelakis ED. A dynamic and chamber-specific mitochondrial remodeling in right ventricular hypertrophy can be therapeutically targeted. J Thorac Cardiovasc Surg. 2008;136(1):168–78, 178.e1-3. 10.1016/j.jtcvs.2008.01.040. [DOI] [PubMed] [Google Scholar]