

Abstract
Caveolin-1, the structural and signaling protein of caveolae, is an important negative regulator of endothelial nitric oxide synthase (eNOS). We observed that mice lacking caveolin-1 (Cav1−/−) had twofold increased plasma NO levels but developed pulmonary hypertension. We measured pulmonary vascular resistance (PVR) and assessed alterations in small pulmonary arteries to determine the basis of the hypertension. PVR was 46% greater in Cav1−/− mice than wild-type (WT), and increased PVR in Cav1−/− mice was attributed to precapillary sites. Treatment with NG-nitro-l-arginine methyl ester (l-NAME) to inhibit NOS activity raised PVR by 42% in WT but 82% in Cav1−/− mice, indicating greater NO-mediated pulmonary vasodilation in Cav1−/− mice compared with WT. Pulmonary vasculature of Cav1−/− mice was also less reactive to the vasoconstrictor thromboxane A2 mimetic (U-46619) compared with WT. We observed redistribution of type I collagen and expression of smooth muscle α-actin in lung parenchyma of Cav1−/− mice compared with WT suggestive of vascular remodeling. Fluorescent agarose casting also showed markedly decreased density of pulmonary arteries and artery filling defects in Cav1−/− mice. Scanning electron microscopy showed severely distorted and tortuous pulmonary precapillary vessels. Thus caveolin-1 null mice have elevated PVR that is attributed to remodeling of pulmonary precapillary vessels. The elevated basal plasma NO level in Cav1−/− mice compensates partly for the vascular structural abnormalities by promoting pulmonary vasodilation.
Keywords: nitric oxide, pulmonary hypertension, fluorescent angiography, corrosion casting
caveolae are 50–100 nm diameter plasma membrane invaginations and cytoplasmic vesicles that are prominent in endothelial cells, vascular smooth muscle cells, and adipocytes (5, 32). Molecular complexes consisting of GTP-binding proteins, kinases, endothelial nitric oxide synthase (eNOS), matrix metalloproteases, and others are bound to caveolin-1, the primary structural protein of caveolae (3, 16, 27). Interaction of these proteins with caveolin-1 often results in inhibition of their activities (19, 29). Caveolin-1 regulates vasomotor tone through its ability to control nitric oxide (NO) production (6, 20, 29). eNOS binding to caveolin-1 leads to eNOS inhibition and reduced NO production (10, 13, 20, 31). However, NO levels were increased in smooth muscle cell cultures and plasma derived from caveolin-1 knockout mice (Cav1−/−; Refs. 6, 40). Little is known about the control of pulmonary vasomotor tone in Cav1−/− mice in the face of the high basal NO production. Phenylephrine preconstricted aortic rings of Cav1−/− mice showed enhanced vasodilation in response to acetylcholine (6, 26). These mice also had poor exercise performance, pulmonary hypertension with right ventricular hypertrophy, and decreased survival (6, 25, 26, 40). Additionally, lungs of Cav1−/− mice were notable in having abnormal connective tissue staining, alveolar septal thickening, and hypercellularity (6, 26). Given that NO is a pulmonary vasodilator (4, 20), the finding of pulmonary hypertension in Cav1−/− mice is unexpected. To resolve these questions, we assessed pulmonary vascular resistance (PVR) by determining the pressure-flow relationship of the mouse lung vasculature and the role of NO in regulating pulmonary vasomotor tone. We also related vascular structural alterations to the PVR changes. Our results in Cav1−/− mice show a critical requirement for caveolin-1 in normal lung vascular homeostasis. We showed that increased NO production in Cav1−/− mice helps to compensate for the severe pulmonary vascular remodeling by modulating the increase in PVR.
METHODS
Animals.
Animal studies were approved by the University of Illinois Animal Care and Use Committee. Caveolin-1 knockout (Cav1tm1Mls) mice were purchased from The Jackson Laboratories (Bar Harbor, ME). Black Swiss (Taconic, Hudson, NY) and B6:129SF2/J mice were used as the strain-matched controls (B6:129SF2/J mice were the appropriate control strain after the Cav1−/− strain had been backcrossed). In all cases, adult, age-matched mice were used; no differences were noted between the early and current Cav1−/− strains or between the two control strains.
Lung and ventricle weights.
The lungs were removed from ketamine/xylazine-anesthetized animals, and wet weights were determined. The tissue was dried in an oven at 65°C for at least 48 h and weighed. Wet-to-dry lung weight ratio was determined. The right ventricle was separated from the left ventricle and septum and weighed (11).
Antibodies and reagents.
Rabbit anti-caveolin-1 polyclonal anti-body (PAb) was purchased from Transduction Laboratories, mouse anti-α-tubulin MAb was purchased from Sigma Chemical, mouse MAb to native type I collagen was from Sigma, rabbit MAb to smooth muscleα-actin (α-SMA) was from Epitomics, rat MAb to platelet endothelial cell adhesion molecule-1 (PECAM-1; CD31) was from BD Biosciences, TO-PRO3 and 4,6-diamidino-2-phenylindole (DAPI) were from Molecular Probes, and rabbit anti-von Willebrand factor PAb was from Chemicon. All other reagents were obtained from Sigma or as specified.
Western blotting.
Lungs were homogenized for 30 s using a Polytron homogenizer. Lysates were run on 10% SDS-polyacrylamide gels, transferred to nitrocellulose membranes, and probed with the appropriate primary and secondary antibodies as previously described (22).
Light microscopy.
Airways of excised lungs were perfusion-fixed with formaldehyde to a pressure of 25 cmH2O to distend the lungs to approximately total lung capacity. The tissue was then dehydrated and embedded in paraffin. Sections were stained using hematoxylin and eosin, elastin van Gieson, and Masson’s trichrome. Cryostat sectioning (15 μm) of lungs was carried out using standard methodology.
Fluorescent arteriography.
GenePure LE agarose was dissolved in double-distilled water by heating in a microwave. Fluorescent latex beads were added to the hot solution to a final concentration of 0.2% solids and kept at 50°C for 2–5 h. The agarose/fluorescent microbead solution was buffered with 10× Media 199, and pH was adjusted to neutral with 1 N NaOH and further buffered with HEPES (0.2 M final) and BSA (0.2% final). For differential arteriography, we used 1% agarose to visualize the muscular small pulmonary arteries and 0.3% agarose to visualize capillaries (Ref. 7; see the data supplement online at the AJP-Lung Cellular and Molecular Physiology web site).
Wild-type (WT) and Cav1−/− mice (5 mo old) were anesthetized and intubated by tracheotomy, placed on a heating pad (~40°C), and continuously ventilated. A median sternolaparotomy was performed, and 50 units of heparin (Baxter) were injected directly into the right atrium. After the abdominal aorta was transected, the pulmonary artery was cannulated and perfused while the pressure was monitored. The lungs were then perfused via a left atrial cannula with HEPES-buffered RPMI 1640 medium (40°C) containing 0.5 U/ml of heparin and 0.5% BSA at a flow rate of 1 ml/min until blood free. To visualize small pulmonary arteries, 300-μl polystyrene fluorescent beads in 1% agarose were injected via a side arm syringe into the perfusion system. Lungs were dissected en block and placed in cold 4% paraformaldehyde solution (PFA; 50 ml) and stored overnight at 4°C.
Whole mount immunohistochemistry.
Fixed lungs were dissected and washed with fresh 4% PFA fixative at 4°C. Each lobe was cut into 4 × 4-mm2 fragments, and comparable regions were selected for analysis from WT and Cav1−/− mice. Fragments were washed with cold PBS and permeabilized with 0.5% Triton X-100 for 1 day under constant agitation at 4°C. After several washes with cold PBS, the fragments were blocked with 5% horse serum in PBS containing 0.05% Tween 20 and sodium azide (NaN3) at room temperature (RT) on a rocking stage. The fragments were then placed into 1.5-ml Eppendorf tubes with antibodies in blocking solution and incubated for 48 h on a rocking stage at 4°C. Primary antibodies were washed 3–5 times with PBS + NaN3 for 2 h at RT and incubated with secondary antibodies diluted 1:600 in PBS + 0.5% BSA solution overnight. If nuclear counterstain was desired, TO-PRO3 was added at 1:3,000 dilution together with secondary antibodies. After extensive washings, stained fragments were mounted on glass-bottom dishes (MatTek) in PBS +70% glycerol NaN3. Serial optical sections were acquired in 1-μm step increments with a ×10 objective and Zeiss 510 LSM confocal microscope. Three-dimensional reconstructions were generated using Zeiss AxioVision v.4.5 software.
Vascular casting.
Mice were anesthetized and heparinized. The thoracic aorta was cannulated with an 18-gauge catheter; 2 ml of partially polymerized methyl methacrylate (Mercox; Ladd Research Industries, Burlington, VT) mixed with the accelerator benzoyl peroxide was injected over 1 min. The lung microvasculature was filled in a retrograde fashion. The tissue was dissolved in a dilute sodium hydroxide solution. The casts were mounted onto aluminum studs, coated with palladium-gold, and examined with a Hitachi S-3000N scanning electron microscope as previously described (2).
Plasma NO levels.
Plasma nitrite levels were measured using a NO sensor (Apollo 4000; World Precision Instruments, Sarasota, FL). Clarke-type electrode (39) was used to measure the NO generated by the reduction of total plasma nitrite in deproteinized venous plasma by catalytic conversion of nitrate to nitrite as described by the manufacturer’s protocol.
Arterial blood gases.
Arterial blood was collected from the aorta of anesthetized mice. Partial pressures of oxygen and carbon dioxide and pH were measured using a blood gas analyzer (GEM Premier 3000, Instrumentation Laboratories).
Pulmonary artery pressure-flow relationship.
Mice were anesthetized and prepared for perfusion as previously described (35). Lungs were ventilated with room air at a peak inspiratory pressure of 12 cmH2O, and HEPES-buffered RPMI (pH = 7.4) was perfused into the pulmonary artery at a flow of 2.0 ml/min through PE-10 tubing. Pulmonary arterial (Ppa) and venous (Ppv) pressures were monitored continuously through the pulmonary arterial and left atrial cannula, which were connected to a pressure transducer. We determined pulmonary capillary pressure (Ppc) using a previously described method (14) on 5-s double occlusion of the pulmonary and atrial catheters by electronic valves. After a period of 20 min during which isogravimetric conditions were attained (defined as stable pressure and lung weight at the baseline flow of 2 ml/min), the flow was stepped cumulatively from 2 to 3.5 and then to 5 ml/min. Ppa was measured at steady state and was plotted against the flow, which was measured by weighing the effluent collected over a 1-min period. PVR (cmH2O·ml−1·min−1) was derived as Ppa (as the left atrial pressure was held constant) divided by the flow. Pre- and postcapillary segmental resistances were calculated as previously described (14). In brief, we simultaneously occluded arterial inflow and venous outflow paths by closing electronic pinch valves for 5 s. We measured double-occlusion pressure Pdo, which is equivalent to Ppc. Precapillary resistance was calculated from Pdo/(Ppa − Ppc), and postcapillary resistance was calculated from Pdo/(Ppc − Ppv), where Ppv is pulmonary venous pressure. In some cases, thromboxane A2 mimetic U-46619 (9,11-dideoxy-9α,11α-methanoepoxy PGF2α) was infused (20 nM for 2 min), and pressure-flow curves were obtained as described above. For this experiment, flow points were 2, 2.5, and 3 ml/min to avoid excessive vascular pressures. For eNOS inhibition, 300 μM NG-nitro-l-arginine methyl ester (l-NAME) was added to the perfusate for 45 min before starting the pressure-flow curve determinations.
Statistical analysis.
Data are presented as means ± SD. Statistical significance of differences between group means was determined using one-way ANOVA or Student’s t-test; P < 0.05 was considered significant.
RESULTS
Lung and ventricle weights in Cav1−/− mice.
Our studies showed the absence of the 22-kDa caveolin-1 protein by Western blot analysis of lung homogenates from Cav1−/− mice (Fig. 1A). Cav1−/− mice showed an increase of 37% in right ventricular weight compared with WT (44.2 ± 3.0 mg, n = 14 vs. 32.3 ± 5.8 mg, n = 8; P < 0.05). Right (RV) and left (LV) ventricular weight-to-total body weight (BW) ratios as well as RV/LV were significantly increased in Cav1−/− mice compared with WT (Fig. 1B). Although both heart chambers were enlarged in Cav1−/− mice, there was relatively greater increase in RV weight compared with LV weight (Fig. 1B). No differences in (100 ± 33 mmHg vs. 106 ± 47 mmHg), (54 ± 20 mmHg vs. 53 ± 12 mmHg), or pH (7.15 ± 0.1 vs. 7.16 ± 0.02) (Fig. 1C) were found between WT and Cav1−/− (P > 0.05; n = 3–4/group). Wet-to-dry lung weight ratio (Fig. 1D) was the same (4.84 ±.04 vs. 4.80 ±.060; P > 0.05; n = 5) in the WT and Cav1−/− lungs.
Fig. 1.

A: absence of caveolin-1 expression in caveolin-1 knockout mice. Western blot analysis of mouse lung homogenate shows absence of caveolin-1 expression in lungs of animals lacking caveolin-1 (Cav1−/−) in contrast to wild-type (WT) lungs. B: right ventricular hypertrophy in caveolin-1 knockout mice. Right ventricles (RV) were removed from the left ventricle + septum (LV), weighed, and normalized to the body weight (BW). RV- and LV-to-total BW ratios as well as RV/LV were significantly increased in Cav1−/− mice compared with WT. *P < 0.05 vs. WT. C: arterial blood gases. Blood samples from the aorta of anesthetized Cav1−/− and WT mice were analyzed for oxygen and carbon dioxide partial pressures and pH. No differences in , , or pH were found between WT and knockout mice (100 ± 33 mmHg vs. 106 ± 47 mmHg, 54 ± 20 mmHg vs. 53 ± 12 mmHg, and 7.15 ± 0.1 vs. 7.16 ± 0.02, respectively; P > 0.05; n = 3–4/group). D: lung weights and wet-to-dry ratio. No difference was found in wet-to-dry weight (wet/dry wt) ratios between WT and Cav1−/− (4.84 ± 0.037 for WT vs. 4.80 ± 0.06 for Cav1−/−; n = 5).
Lung pathology in Cav1−/− mice.
In hematoxylin and eosin-stained sections, the Cav1−/− lungs had increased thickness of the alveolar septa with a marked increase in the number of cells in the interstitium (Fig. 2, B, D, and F) relative to WT (Fig. 2, A, C, and E). ECM deposition was increased and prominent in Cav1−/− lungs (Fig. 2, D and F) in contrast to WT lungs. Excess ECM was present in alveolar septa and in perivascular space in Cav1−/− lungs (Fig. 2F). There were no areas of lung destruction, scarring, honeycombing, or departitioning of alveoli characteristic of severe fibrotic lung disease. There was also no evidence of plexiform or onionskin lesions, small artery obstruction, or vascular necrosis (Fig. 2, C and E).
Fig. 2.

Lung histology and collagen deposition. Histological analysis was performed on formalin-inflated and -fixed lung sections stained with hematoxylin and eosin (A and B, ×20) or Masson’s trichrome stain (C and D, ×20; E and F ×630). Cav1−/− mouse lungs had thickened (B, D, solid arrow, and F) and hypercellular (F, open arrow) alveolar walls as well as increased connective tissue deposition (blue) compared with WT (A, C, and E). The collagen deposits were localized within alveolar septa and around capillaries and other small blood vessels (F). There were no areas of lung destruction, confluent scarring, honeycombing, or departitioning of alveoli (n = 4)
To examine type I collagen expression, sections from WT and Cav1−/− mouse lungs were stained with type I collagen and α-SMA Abs followed by fluorescently labeled secondary Abs and nuclear stain DAPI. As shown in confocal images in Fig. 3A, we observed an increase in both α-SMA and collagen I staining in Cav1−/− lung parenchyma. α-SMA staining of muscle layers of resistance vessels and airways were similar (Fig. 3B). We also observed a decrease in type I collagen staining of postcapillary vessels of Cav1−/− mice compared with WT (Fig. 3B).
Fig. 3.

Smooth muscle α-actin (α-SMA) and type I collagen immunostaining in lungs. Paraformaldehyde-fixed lungs were frozen in optimum cutting temperature compound, and 20-μm sections were cut and labeled with a 1:1,000 dilution of α-SMA MAb (green, top row), type I collagen MAb (1:1,000 dilution; red, 2nd row), and 4,6-diamidino-2-phenylindole (DAPI; 1:1,000 dilution; blue, 3rd row) + Alexa 488 and Alexa Fluor 555-labeled secondary Abs (1:300 dilution). A: confocal image shows increased α-SMA and collagen I staining in the Cav1−/− lung parenchyma. Scale bar = 50 μm. B: confocal image of postcapillary vessels in WT and Cav1−/− lungs showing equivalent α-SMA staining of the vessel wall but reduced type I collagen staining. Scale bar = 50 μm. Data are representative of 3 separate mouse lungs for each genotype.
PVR and pulmonary vasoreactivity.
Lungs of Cav1−/− mice had a 46% increase in PVR compared with WT as indicated by the slope of the pressure-flow curves (Fig. 4A; mean data are shown in Fig. 4B; 0.75 ± 0.18 cmH2O·ml−1·min−1 vs. 1.10 ± 0.02 cmH2O·ml−1·min−1; P < 0.05; n = 3). By partitioning total PVR into precapillary and postcapillary resistances as previously described (23), we determined that the increase in PVR in the Cav1−/− lung was located primarily in the precapillary segment (Fig. 4C).
Fig. 4.
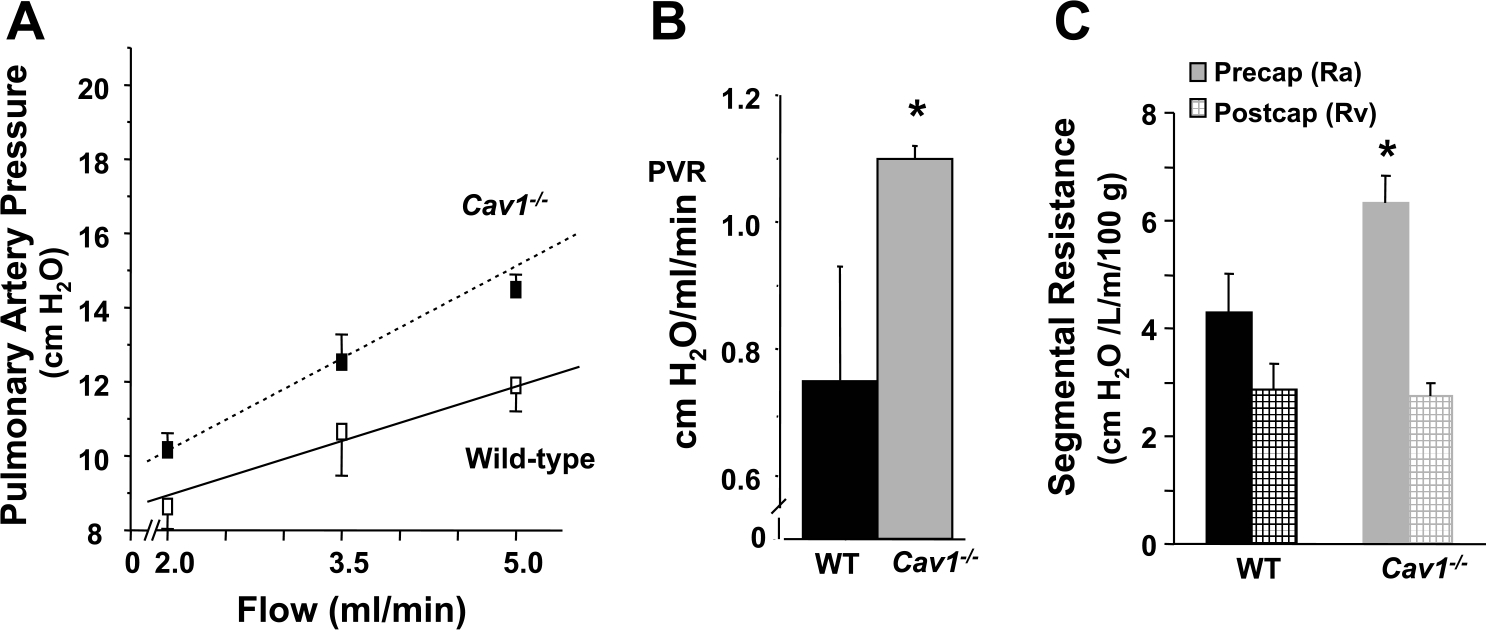
Increased pulmonary vascular resistance (PVR) in Cav1−/− mice. A and B: isolated mouse lung preparations were perfused with HEPES-buffered RPMI using a peristaltic pump. Pulmonary artery pressure was recorded at increasing flows in the range of 2–5 ml/min. The slope of the pressure-flow curve yielded PVR. Lungs from Cav1−/−animals showed an average increase of 46% in PVR compared with WT (0.75 ± 0.2 cmH2O·ml−1·min−1 vs. 1.1 ± 0.02 cmH2O·ml−1·min−1; *P < 0.05 vs. WT; n = 3). C: the increased total PVR in Cav1−/− lungs was localized to the precapillary-capillary segment of the pulmonary vasculature with the aid of the capillary pressure determination. Lungs were perfused at 2 ml/min, and the capillary pressure was measured during a 5-s occlusion of the in- and outflow catheters. Precapillary-capillary (artery resistance, Ra) and postcapillary fractions (vein resistance, Rv) of the PVR were then calculated as described in METHODS. The precapillary resistance was greater in Cav1−/− mouse lungs than in WT (69% vs. 60% of total), and the postcapillary resistance was lower in Cav1−/− mouse lungs than in WT (31% vs. 40% of total).
To investigate pulmonary vasoreactivity in Cav1−/− mice, we determined the response to the thromboxane A2 mimetic, U-46619, by comparing the slope of pressure-flow curves. As shown in Fig. 5A, the pressor response to U-46619 was significantly reduced in lungs of Cav1−/− mice compared with WT (2.50 ± 0.30 vs. 6.67 ± 1.00; P < 0.05; n = 3; Fig. 5, A and B).
Fig. 5.

Blunted rise in PVR following infusion of U-46619 in Cav1−/− mice. A: perfused lung pressure-flow curves were assessed to determine the PVR response to vasoconstrictor by bolus infusion of the thromboxane A2 mimetic U-46619 (20 nM for 2 min). B: there was a markedly reduced mean PVR response to U-46619 in Cav1−/− lungs compared with WT (2.5 ± 0.3 vs. 6.67 ± 1.0; *P < 0.05 vs. WT; n = 3–4).
NO regulation of PVR in Cav1−/− mice.
We next addressed the question whether increased NO production in Cav1−/− mice serves to modulate PVR. Isolated perfused mouse lungs were treated for 45 min with 300 μM l-NAME to inhibit eNOS, and PVR was determined as above. In WT lungs, l-NAME increased PVR from 0.75 ± 0.18 cmH2O·ml−1·min−1 to 1.10 ± 0.38 cmH2O·ml−1·min−1, n = 3–4; P < 0.05; (Fig. 6, A and C). Interestingly, in Cav1−/− mouse lungs, the effect of l-NAME was greater; PVR increased from 1.1 ± 0.02 cmH2O·ml−1·min−1 to 2.0 ± 0.20 cmH2O·ml−1·min−1 (n = 3–4; P < 0.05; Fig. 6, B and C). As shown in Fig. 6C, eNOS inhibition with l-NAME resulted in an 82% increase in PVR in Cav1−/− mouse lungs compared with a 47% increase in WT lungs.
Fig. 6.

PVR alterations following nitric oxide synthase (NOS) inhibition. PVR was calculated in buffer-perfused lungs in response to NOS inhibitor NG-nitro-l-arginine methyl ester (l-NAME). Acute inhibition of NOS with 300 M l-NAME for 45 min led to a greater increase in PVR in Cav1−/− lungs (B) compared with WT (A) (82% vs. 47%, respectively). In the WT group, PVR increased from 0.75 ± 0.18 cmH2O·ml−1·min−1 to 1.1 ± 0.38 cmH2O·ml−1·min−1 (P < 0.05; n = 3–4); in the Cav1−/− group, PVR increased from 1.1 ± 0.02 cmH2O·ml−1·min−1 to 2.0 ± 0.2 cmH2O·ml−1 ·min−1 (*P < 0.05 vs. control) (C). D: increased plasma NO in Cav1−/− mice. Total nitrite in deproteinized plasma samples was measured using an NO electrode following catalytic conversion of nitrate to nitrite. Nitrate levels of the Cav1−/− mice (n = 4) showed an average 2-fold increase compared with WT (*P < 0.05 vs. WT; n = 4).
To investigate NO generation in Cav1−/− mice, we assessed the nitrite concentration in plasma of Cav1−/− mice using an NO-specific electrode (METHODS). As shown in Fig. 6D, plasma NO was twofold greater in Cav1−/− mice compared with WT (3.5 ± 0.49 μM vs. 7.6 ± 1.5 μM; P < 0.05; n = 4).
Pulmonary artery filling defects in Cav1−/− mice.
To determine alterations in the pulmonary circulation of Cav1−/− mice, we examined blood vessels using a modified vessel contrasting method (7). Fluorescent probe of high viscosity was used to determine filling of precapillary vessels and distinguish these from the capillary network (see supplemental data). We filled pulmonary precapillary vessels with 1% agarose solution containing 0.5-μm fluorescently labeled microbeads (Fig. 7A). Fluorescence was seen in small arteries of WT lungs, but it was considerably less in lungs of Cav1−/− mice in which we observed a severe filling defect (Fig. 7B). To assess collagen deposition in the pulmonary artery wall and lung parenchyma, we determined the colocalization of arteries with type I collagen immunostaining using a specific antibody. We observed type I collagen in association with pulmonary arteries in WT but altered collagen I deposition coupled to impaired filling of small pulmonary arteries of Cav1−/− mice (Fig. 7C).
Fig. 7.

Arterial agarose casting and lung tissue collagen immunostaining. A: representative epifluorescent arteriograms from regions of the right anterior border of the left upper lobe of WT and Cav1−/− lungs showing alterations of arterial vessel structure and density of the surface precapillaries filled with fluorescent polystyrene beads in 1% agarose solution (scale bar = 200 μm). B: quantification of surface arterial vessel density/region of interest (ROI) from WT and Cav1−/− lungs (*P < 0.05 vs. WT; n = 3). C: 3-dimensional (3-D) reconstructions of arterial casts (green), cellular nuclei (TO-PRO3 whole mount stain; blue), and whole mount collagen I immunostaining (1:1,000 dilution; red) of WT and Cav1−/− lungs showing alterations in distribution of native collagen I, hypercellularity (increased number of nuclei), and perfusion defects in lungs of Cav1−/− mice. Reconstructions are from 300 confocal serial sections collected at 1-μm step increments. Scale bar = 150 μm.
In other studies, we assessed the relationship between pulmonary artery filling and endothelial cell marker PECAM-1 (CD31). As shown in Fig. 8A, the density of PECAM-1-positive capillaries in Cav1−/− lungs was greater than WT (capillary network is shown in red). The greater capillarity was coupled to decreased filling of the pulmonary arteries in Cav1−/− mice relative to WT (Fig. 8A). In line with a reduction in “functional” small lung arteries, we observed a significant decrease in the uniformity of filling of microvasculature networks in Cav1−/− mice relative to WT (Fig. 8B).
Fig. 8.

Pulmonary vessel casting and platelet endothelial cell adhesion molecule-1 (PECAM-1) immunostaining. A: 3-D reconstruction image of arterial cast (green; perfused with green-fluorescent microspheres and 1% agarose solution) and anti-PECAM-1 immunostaining (1:500 dilution; shown in red) of the lung. Combined 3-D views of both reconstructions show perfusion defects on the arterial side and increased microvessel density in Cav1−/− mice. Optical serial sections (n = 300; 1-μm step increment) from the edge of right inferior lobe were acquired by laser scanning confocal microscopy. B: pulmonary capillaries (perfused with green-fluorescent microspheres and 0.3% agarose solution) showing nonhomogenous filling of surface capillaries in Cav1−/− mice. Scale bar = 200 μm for A and B. C: vascular corrosion casts obtained by injection of the pulmonary microvasculature with the sclerosing agent methyl methacrylate followed by tissue digestion were sectioned and studied by scanning electron microscopy. Microvessels from WT mouse lungs showed smooth surfaces and nearly constant diameter. Cav1−/− lung vascular casts showed abnormal surfaces of the endothelium, which appeared rough and wrinkled (solid thick arrow).There were also segments of flattening of the capillary lumen, focal stenosis, and torsion of the capillaries (open thick arrow). Miniature vessels (1–3 μm, too small to admit erythrocytes) were seen (thin arrows), but more extensive signs of sprouting, corkscrew, and intussusception patterns were not identified.
Scanning electron microscopy analysis.
To visualize pulmonary microvascular changes at the electron microscopic level, retrograde methacrylate casts of the lung were examined by scanning electron microscopy. We found striking abnormalities in the corrosion casts (Fig. 8C) including abnormal luminal surface of pulmonary endothelia of Cav1−/− mice that appeared rough and wrinkled, flattening of capillary lumen (Fig. 8C, solid arrow), focal stenosis, and torsion of capillaries (Fig. 8C, open arrow) as contrasted with WT lung casts. There was no evidence of angiogenesis such as sprouting, corkscrew, and intussusception patterns in the Cav1−/− lungs. We observed abrupt termination of pulmonary precapillary vessels in Cav1−/− mice (Fig. 8C, thin arrow).
DISCUSSION
Here, we determined the relationship between alterations in PVR in Cav1−/− mice and the structural changes in pulmonary vessels. Although previous studies have described pulmonary hypertension in these mice (6, 26, 40), its basis is unclear. A key observation has been the finding of pulmonary hypertension despite increased plasma levels of NO in Cav1−/− mice (6, 26, 40). We observed marked remodeling of the pulmonary vascular bed of Cav1−/− mice that contributed to the increased PVR. Moreover, the high NO production in these mice was shown to be an important compensatory mechanism that modulates the increased PVR in the face of vascular disease.
Studies previously showed thickening and hypercellularity of alveolar septa as well as increased matrix deposition in the pericapillary space of Cav1−/− mice (6, 26). Other studies showed carotid artery thickening coupled to cellular hyperproliferation in the vessel wall 14 days after artery ligation in Cav1−/− mice (37), suggesting a role of caveolin-1 in maintaining vascular morphology and function. In the present study, besides the pericapillary remodeling, we observed severe defects in precapillary lung vessels of Cav1−/− mice. Using fluorescence arteriography, we showed markedly reduced fluorescence in lungs of Cav1−/− mice compared with WT indicating severe vascular filling defects as the result of disruption of caveolin-1 gene. There were 40% fewer filled arteries in Cav1−/− mice compared with WT. The marked filling defect was consistent with the abrupt termination of precapillary vessel as seen by scanning electron microscopy as well as fewer arteries. In addition, type I collagen, seen to be associated with postcapillary vessels of WT mice, was redistributed in Cav1−/− mice. These structural alterations primarily at the level of small pulmonary arteries are the likely explanation for the observed increase of PVR in Cav1−/− mice. On the basis of the double-occlusion method of partitioning PVR (14), we identified that the increased PVR was largely ascribed to the precapillary vessels, the site of the structural vascular alterations.
We also assessed the relationship between pulmonary artery filling and the endothelial cell marker PECAM-1 (CD31). The density of PECAM-1-positive capillaries in Cav1−/− lungs (reflecting overall capillary density) was surprisingly greater than in WT, indicating that greater capillarity in Cav1−/− mice was coupled to decreased filling of the pulmonary arteries. The basis of increased capillarity in the absence of caveolin-1 is not clear, but the finding raises the interesting possibility that caveolin-1 may negatively regulate capillary formation. Similar findings were made in mice overexpressing VEGF (38); thus it is possible that increased VEGF signaling may be responsible for increased capillarity formation in Cav1−/− mice. The increase in PECAM-1 staining and capillarity is also consistent with the data of Drab et al. (6) who showed an increase in VEGF receptor 2 (Flk1) immunostaining in the lung suggesting the hypercellularity may be due to increased numbers of endothelial cells.
The changes in PVR were assessed in the isolated perfused mouse lung preparation by determining the pressure-flow relationships. We observed that basal PVR was increased in Cav1−/− mice in association with right ventricular hypertrophy, suggesting that increased right ventricular afterload was a determinant of the hypertrophy. Cav1−/− and WT mice had no differences in or indicating that the increase in PVR was not the result of hypoxemia or hypercarbia per se. Lungs from Cav1−/− mice were also not edematous, thus ruling out vasoconstriction secondary to perivascular water accumulation as the basis of the rise in PVR. However, we observed that l-NAME produced a markedly greater increase in PVR in Cav1−/− mice than in WT. This finding indicates the importance of the greater NO production in Cav1−/− mice in dampening the rise in PVR in the face of severe structural defects of precapillary lung vessels. We also observed that the pulmonary vasoconstrictor response to the stable thromboxane A2 analog U-46619 was significantly reduced in Cav1−/− mice compared with WT, consistent with higher NO production in these mice and the lung vascular abnormalities.
The source of excess NO in the Cav1−/− mice is most likely eNOS (NOS 3) rather than one of the other two isoforms, neuronal NOS (NOS 2) or inducible NOS (iNOS; NOS 1). eNOS is activated in Cav1−/− mice, and this can be reversed by endothelial-specific reconstitution of caveolin-1 expression (23). Even though caveolin-1 can interact with all three known NOS isoforms (12), their expression is not upregulated in the absence of caveolin-1 (8, 28, 40). Given that iNOS is not expressed in unstimulated lung endothelial cells (9), the observed effects in our study are likely related to eNOS disinhibition by lack of caveolin-1.
Because Cav1−/− mice have higher levels of eNOS-derived NO, it is important to discuss its possible relevance to the development of lung vascular pathology. Using fluorescence vascular imaging, Han et al. (15) described severe pulmonary vasculopathy in eNOS−/− mice, which share some similarities with the Cav1−/− phenotype described in our study, including reduced pulmonary artery filling and hypertension (15). It is therefore striking that both abundance as well as lack of eNOS-derived NO are associated with similar pulmonary vascular pathology. However, important differences in vascular phenotypes exist between the eNOS−/− and Cav1−/− mutant mouse strains. In contrast to eNOS−/−, neonatal viability was not reduced in Cav1−/− (26, 40). Also endothelial cell hyperproliferation has been noted in Cav1−/− (6, 26) but not in eNOS−/− eNOS−/− mice that survived into adulthood appeared to have normal lung vascular anatomy (33) in contrast to Cav1−/− mice in our study.
The basis for pericapillary alterations seen in Cav1−/− mice is not clear. Caveolae are known to contain a variety of signal transduction molecules (3, 5, 27, 32), which are held in an inactive state when bound to caveolin-1 (3, 18, 19). One possibility therefore is that, in the absence of caveolin-1, bone morphogenetic protein (BMP) receptors may be activated (24), leading to increased production of collagen and other matrix proteins. Studies showed that BMP receptors, BRIa and BRII, colocalized with caveolin-1, and stimulation of cells with BMP-2 induced redistribution of BRII into domains enriched in caveolin-1 (24). As caveolin-1 normally inhibits BMP signaling (24), it is possible that BMP signaling is activated in the absence of caveolin-1 expression, contributing to the mechanism of fibrosis. Studies have also shown PDGF receptors in the caveolae membrane of cultured mesangial cells (34) and that decreased caveolin-1 protein resulted in increased PDGF-mediated signaling (36). In addition, caveolin-1 downregulation in models of monocrotaline toxicity has been associated with mitogenesis through activation of ERK and transcription factor STAT3 (21, 30). Caveolin-1 scaffold domain peptide blocked the development of pulmonary hypertension (17), indicating that the binding of one or more signaling molecules by the scaffold peptide prevented monocrotaline-induced pulmonary hypertension. Thus caveolin-1 expression may normally serve to dampen the responses of growth factors by inhibiting growth factor receptors or their signaling downstream. In the absence of caveolin-1, this regulatory mechanism fails, resulting in unchecked fibrosis in alveolar septa and pericapillary areas (1). However, the absence of caveolin-1 in pulmonary arteries was associated with decreased type I collagen deposition in the vessel wall, reiterating the cell-specific functions of caveolin-1 in the lung.
In summary, we show here extensive pulmonary vascular pathology at the level of precapillary vessels of Cav-1−/− mice, suggesting that Cav-1 is required for maintenance and homeostasis of the normal pulmonary vascular bed in the adult. PVR at the precapillary sites is elevated in Cav-1−/− mice as the result of the vascular structural defects. The elevated level of NO in Cav-1−/− mice is an important compensatory mechanism that serves to dampen the rise in PVR.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the technical assistance of David Visintine and Maricela Castellon.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants R01-HL-71626 (R. D. Minshall) and P01-HL-60678 (R. D. Minshall and A. B. Malik).
REFERENCES
- 1.Achcar RO, Demura Y, Rai PR, Taraseviciene-Stewart L, Kasper M, Voelkel NF, Cool CD. Loss of caveolin and heme oxygenase expression in severe pulmonary hypertension. Chest 129: 696–705, 2006. [DOI] [PubMed] [Google Scholar]
- 2.Agha-Majzoub R, Becker RP, Schraufnagel DE, Chan LS. Angiogenesis: the major abnormality of the keratin-14 IL-4 transgenic mouse model of atopic dermatitis. Microcirculation 12: 455–476, 2005. [DOI] [PubMed] [Google Scholar]
- 3.Anderson RG. The caveolae membrane system. Annu Rev Biochem 67: 199–225, 1998. [DOI] [PubMed] [Google Scholar]
- 4.Cherry PD, Gillis CN. Evidence for the role of endothelium-derived relaxing factor in acetylcholine-induced vasodilatation in the intact lung. J Pharmacol Exp Ther 241: 516–520, 1987. [PubMed] [Google Scholar]
- 5.Couet J, Belanger M, Roussel E, Drolet MC. Cell biology of caveolae and caveolin. Adv Drug Deliv Rev 49: 223–235, 2001. [DOI] [PubMed] [Google Scholar]
- 6.Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 293: 2449–2452, 2001. [DOI] [PubMed] [Google Scholar]
- 7.Dutly AE, Kugathasan L, Trogadis JE, Keshavjee SH, Stewart DJ, Courtman DW. Fluorescent microangiography (FMA): an improved tool to visualize the pulmonary microvasculature. Lab Invest 86: 409–416, 2006. [DOI] [PubMed] [Google Scholar]
- 8.El-Yazbi AF, Cho WJ, Boddy G, Daniel EE. Caveolin-1 gene knockout impairs nitrergic function in mouse small intestine. Br J Pharmacol 145: 1017–1026, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ermert M, Ruppert C, Gunther A, Duncker HR, Seeger W, Ermert L. Cell-specific nitric oxide synthase-isoenzyme expression and regulation in response to endotoxin in intact rat lungs. Lab Invest 82: 425–441, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feron O, Belhassen L, Kobzik L, Smith TW, Kelly RA, Michel T. Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells. J Biol Chem 271: 22810–22814, 1996. [DOI] [PubMed] [Google Scholar]
- 11.Fulton RM, Hutchinson EC, Jones AM. Ventricular weight in cardiac hypertrophy. Br Heart J 14: 413–420, 1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia-Cardena G, Martasek P, Masters BS, Skidd PM, Couet J, Li S, Lisanti MP, Sessa WC. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the NOS caveolin binding domain in vivo. J Biol Chem 272: 25437–25440, 1997. [DOI] [PubMed] [Google Scholar]
- 13.Garcia-Cardena G, Oh P, Liu J, Schnitzer JE, Sessa WC. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: implications for nitric oxide signaling. Proc Natl Acad Sci USA 93: 6448–6453, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hakim TS, Malik AB. Hypoxic vasoconstriction in blood and plasma perfused lungs. Respir Physiol 72: 109–121, 1988. [DOI] [PubMed] [Google Scholar]
- 15.Han RN, Babaei S, Robb M, Lee T, Ridsdale R, Ackerley C, Post M, Stewart DJ. Defective lung vascular development and fatal respiratory distress in endothelial NO synthase-deficient mice: a model of alveolar capillary dysplasia? Circ Res 94: 1115–1123, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Hnasko R, Lisanti MP. The biology of caveolae: lessons from caveolin knockout mice and implications for human disease. Mol Interv 3: 445–464, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Jasmin JF, Mercier I, Dupuis J, Tanowitz HB, Lisanti MP. Short-term administration of a cell-permeable caveolin-1 peptide prevents the development of monocrotaline-induced pulmonary hypertension and right ventricular hypertrophy. Circulation 114: 912–920, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Lee CG, Link H, Baluk P, Homer RJ, Chapoval S, Bhandari V, Kang MJ, Cohn L, Kim YK, McDonald DM, Elias JA. Vascular endothelial growth factor (VEGF) induces remodeling and enhances TH2-mediated sensitization and inflammation in the lung. Nat Med 10: 1095–1103, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li S, Couet J, Lisanti MP. Src tyrosine kinases, Galpha subunits, and H-Ras share a common membrane-anchored scaffolding protein, caveolin. Caveolin binding negatively regulates the auto-activation of Src tyrosine kinases. J Biol Chem 271: 29182–29190, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maniatis NA, Brovkovych V, Allen SE, John TA, Shajahan AN, Tiruppathi C, Vogel SM, Skidgel RA, Malik AB, Minshall RD. Novel mechanism of endothelial nitric oxide synthase activation mediated by caveolae internalization in endothelial cells. Circ Res 99: 870–877, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Mathew R, Huang J, Shah M, Patel K, Gewitz M, Sehgal PB. Disruption of endothelial-cell caveolin-1alpha/raft scaffolding during development of monocrotaline-induced pulmonary hypertension. Circulation 110: 1499–1506, 2004. [DOI] [PubMed] [Google Scholar]
- 22.Minshall RD, Tiruppathi C, Vogel SM, Niles WD, Gilchrist A, Hamm HE, Malik AB. Endothelial cell-surface gp60 activates vesicle formation and trafficking via G(i)-coupled Src kinase signaling pathway. J Cell Biol 150: 1057–1070, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murata T, Lin MI, Huang Y, Yu J, Bauer PM, Giordano FJ, Sessa WC. Reexpression of caveolin-1 in endothelium rescues the vascular, cardiac, and pulmonary defects in global caveolin-1 knockout mice. J Exp Med 204: 2373–2382, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nohe A, Keating E, Underhill TM, Knaus P, Petersen NO. Dynamics and interaction of caveolin-1 isoforms with BMP-receptors. J Cell Sci 118: 643–650, 2005. [DOI] [PubMed] [Google Scholar]
- 25.Park DS, Cohen AW, Frank PG, Razani B, Lee H, Williams TM, Chandra M, Shirani J, De Souza AP, Tang B, Jelicks LA, Factor SM, Weiss LM, Tanowitz HB, Lisanti MP. Caveolin-1 null (−/−) mice show dramatic reductions in life span. Biochemistry 42: 15124–15131, 2003. [DOI] [PubMed] [Google Scholar]
- 26.Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou H Jr, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 276: 38121–38138, 2001. [DOI] [PubMed] [Google Scholar]
- 27.Sargiacomo M, Sudol M, Tang Z, Lisanti MP. Signal transducing molecules and glycosyl-phosphatidylinositol-linked proteins form a caveolin-rich insoluble complex in MDCK cells. J Cell Biol 122: 789–807, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schubert W, Frank PG, Woodman SE, Hyogo H, Cohen DE, Chow CW, Lisanti MP. Microvascular hyperpermeability in caveolin-1 (−/−) knock-out mice. Treatment with a specific nitric-oxide synthase inhibitor, L-NAME, restores normal microvascular permeability in Cav-1 null mice. J Biol Chem 277: 40091–40098, 2002. [DOI] [PubMed] [Google Scholar]
- 29.Sessa WC. Regulation of endothelial derived nitric oxide in health and disease. Mem Inst Oswaldo Cruz 100: 15–18, 2005. [DOI] [PubMed] [Google Scholar]
- 30.Shah M, Patel K, Sehgal PB. Monocrotaline pyrrole-induced endothelial megalocytosis involves a Golgi blockade mechanism. Am J Physiol Cell Physiol 288: C850–C862, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Shaul PW, Smart EJ, Robinson LJ, German Z, Yuhanna IS, Ying Y, Anderson RG, Michel T. Acylation targets emdothelial nitric-oxide synthase to plasmalemmal caveolae. J Biol Chem 271: 6518–6522, 1996. [DOI] [PubMed] [Google Scholar]
- 32.Stan RV, Roberts WG, Predescu D, Ihida K, Saucan L, Ghitescu L, Palade GE. Immunoisolation and partial characterization of endothelial plasmalemmal vesicles (caveolae). Mol Biol Cell 8: 595–605, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steudel W, Ichinose F, Huang PL, Hurford WE, Jones RC, Bevan JA, Fishman MC, Zapol WM. Pulmonary vasoconstriction and hypertension in mice with targeted disruption of the endothelial nitric oxide synthase (NOS 3) gene. Circ Res 81: 34–41, 1997. [DOI] [PubMed] [Google Scholar]
- 34.Tamai O, Oka N, Kikuchi T, Koda Y, Soejima M, Wada Y, Fujisawa M, Tamaki K, Kawachi H, Shimizu F, Kimura H, Imaizumi T, Okuda S. Caveolae in mesangial cells and caveolin expression in mesangial proliferative glomerulonephritis. Kidney Int 59: 471–480, 2001. [DOI] [PubMed] [Google Scholar]
- 35.Tiruppathi C, Freichel M, Vogel SM, Paria BC, Mehta D, Flockerzi V, Malik AB. Impairment of store-operated Ca2+ entry in TRPC4(−/−) mice interferes with increase in lung microvascular permeability. Circ Res 91: 70–76, 2002. [DOI] [PubMed] [Google Scholar]
- 36.Yamamoto M, Toya Y, Jensen RA, Ishikawa Y. Caveolin is an inhibitor of platelet-derived growth factor receptor signaling. Exp Cell Res 24: 380–388, 1999. [DOI] [PubMed] [Google Scholar]
- 37.Yu J, Bergaya S, Murata T, Alp IF, Bauer MP, Lin MI, Drab M, Kurzchalia TV, Stan RV, Sessa WC. Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels. J Clin Invest 116: 1284–1291, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeng X, Wert SE, Federici R, Peters KG, Whitsett JA. VEGF enhances pulmonary vasculogenesis and disrupts lung morphogenesis in vivo. Dev Dyn 211: 215–227, 1998. [DOI] [PubMed] [Google Scholar]
- 39.Zhang X Real time and in vivo monitoring of nitric oxide by electrochemical sensors–from dream to reality. Front Biosci 9: 3434–3446, 2004. [DOI] [PubMed] [Google Scholar]
- 40.Zhao YY, Liu Y, Stan RV, Fan L, Gu Y, Dalton N, Chu PH, Peterson K, Ross J Jr, Chien KR. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci USA 99: 11375–11380, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.