

SUMMARY
Metabolic dysfunction mutations can impair energy sensing and cause cancer. Loss of function of the mitochondrial tricarboxylic acid (TCA) cycle enzyme subunit succinate dehydrogenase B (SDHB) results in various forms of cancer typified by pheochromocytoma (PC). Here we delineate a signaling cascade where the loss of SDHB induces the Warburg effect, triggers dysregulation of [Ca2+]i, and aberrantly activates calpain and protein kinase Cdk5, through conversion of its cofactor from p35 to p25. Consequently, aberrant Cdk5 initiates a phospho-signaling cascade where GSK3 inhibition inactivates energy sensing by AMP kinase through dephosphorylation of the AMP kinase γ subunit, PRKAG2. Overexpression of p25-GFP in mouse adrenal chromaffin cells also elicits this phosphorylation signaling and causes PC. A potent Cdk5 inhibitor, MRT3-007, reverses this phospho-cascade, invoking a senescence-like phenotype. This therapeutic approach halted tumor progression in vivo. Thus, we reveal an important mechanistic feature of metabolic sensing and demonstrate that its dysregulation underlies tumor progression in PC and likely other cancers.
In brief
Gupta et al. describe a signaling cascade by which TCA cycle deficiency inactivates bioenergetic sensing to cause adrenal gland cancer. Animal models and drugs targeting the cascade support the discovery, which suggests an additional explanation for Warburg’s description of cancers as uncontrolled cell proliferation despite metabolic impairment.
Graphical Abstract
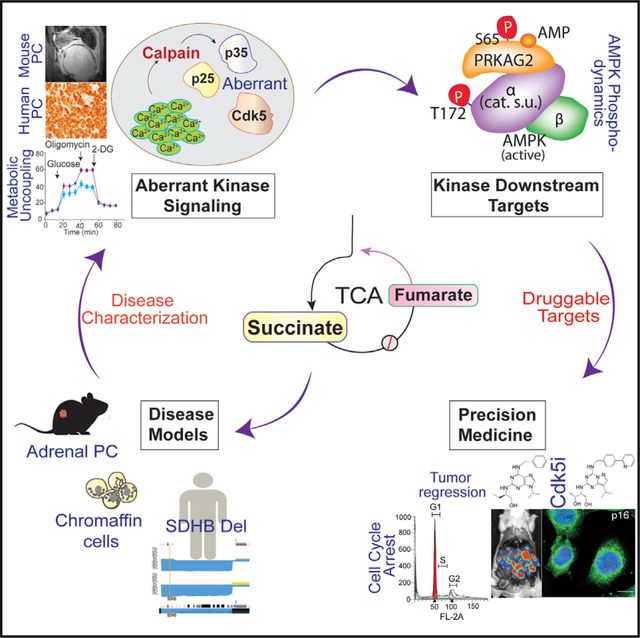
INTRODUCTION
The tricarboxylic acid (TCA) cycle is the critical energy-yielding component of cellular respiration and the centerpiece of cell metabolism. Impairments in TCA cycle components cause a spectrum of metabolic disorders (Krebs and Johnson, 1980). These alterations may still allow embryonic and postnatal development but result in organ-system dysfunction or context-specific pathology (Jeanmonod et al., 2020; Stockholm et al., 2005). Multiple enzymes of the TCA cycle, such as fumarate hydratase (FH), isocitrate dehydrogenase (IDH), and succinate dehydrogenase (SDH) are altered in numerous sporadic or hereditary forms of cancer, causing the production of onco-metabolites. Pheochromocytoma (PC) and paraganglioma (PG) are archetypical metabolic tumors in which TCA cycle malfunction results in tumors arising from chromaffin cells of the adrenal medulla or sympathetic and parasympathetic ganglia (Dahia, 2014; Erez et al., 2011). High rates of SDHx-related abnormalities correlate with an increased risk of metastatic PCPG in patients who often developed primary tumors in childhood or adolescence (King et al., 2011).
The SDHx complex is a hetero-tetrameric protein (composed of four subunits: SDHA, SDHB, SDHC, and SDHD) that functions in both the TCA cycle and electron transport chain (ETC), catalyzing the oxidation of succinate to fumarate and converting ubiquinone to ubiquinol, respectively (Bardella et al., 2011). Patients harboring SDHB mutations have a lifetime cancer risk of 76%, with 50% penetrance by the age of 35 years (Neumann et al., 2004). A wide array of intragenic mutations occur in SDHx complex genes, including frameshifts, splicing defects, and single/multiple exon deletions. However, genetic analysis has clearly shown that heterozygous mutations of SDHx genes predominate in PCPG patients, and very few patients harbor tumors completely lacking SDH function. Approximately 60% of SDHB-related adrenal or extra-adrenal PCs present allelic imbalances (Gimenez-Roqueplo et al., 2002, 2003). A significant focal deletion in the SDHB locus of chromosome 1 in 91 out of 93 patients was identified in a human PC/PG dataset (NCIC3326) (https://progenetix.org/progenetix-cohorts/TCGA/). These exhibit complicated phenotypes that affect gene expression and enzymatic function to varying degrees (Huang et al., 2018; Solis et al., 2009). SDHB mutant PC typically exhibited intact mRNA expression but significantly reduced protein compared with non-SDHB PC/PG (Yang et al., 2012). Retention of a haploinsufficient wild-type allele results in reduced SDHB expression and function. For example, impaired SDH maturation and compartmentalization arising from germline mutation p.Tyr147Cys in SDHB manifested as reduced enzymatic function (Maignan et al., 2017). Germline SDHB mutations such as c.343C>T and c.541-2A>G still exhibit some level of protein expression in patients (Huang et al., 2018). Thus, there is a strong rationale to model heterozygous SDHB genotype that elicits reduced but not complete loss of SDH function.
Malfunction due to loss of SDHx genes causes abnormal accumulation of succinate, thereby activating angiogenic/hypoxia-responsive genes, which further promote metabolic reprogramming and tumor progression (Anderson et al., 2018). Other malignancies affected by SDHB loss of function include renal carcinoma, gastrointestinal stromal tumors, pancreatic neuroendocrine tumors, pituitary adenoma, and pulmonary chondroma (Eijkelenkamp et al., 2020). For all these, attenuating SDHB impairs mitochondrial respiration and causes a metabolic shift in favor of aerobic glycolysis to meet the high energetic and biosynthetic demands of tumor cells, a process known as the Warburg effect (Favier et al., 2009; Kim and Dang, 2006). Impaired TCA cycle function due to SDHB loss not only perturbs cellular bioenergetics but also elicits tumor-promoting signaling mediated by Ca2+ or reactive oxygen species (ROS) released by mitochondria (Hadrava Vanova et al., 2020; Jana et al., 2019). Moreover, TCA cycle dysfunction can also override cell-cycle checkpoint regulators whereby cells may become poorly differentiated, and malignant cell-cycle progression can ensue.
While SDHB loss characterizes highly aggressive forms of PC and other diseases (Jochmanova and Pacak, 2016), the underlying mechanisms by which metabolism is reprogrammed and connected to the loss of cell cycle control is not yet fully understood. As a result, it has been challenging to model these diseases or identify therapeutic targets. Consequently, outcome improvement for patients with these metabolic errors has been limited. Here we studied the impact of metabolic dysfunction caused by loss of SDHB on bioenergetics, Ca2+ dynamics, intracellular signaling, and tumor progression. Our findings provide additional understanding of the mechanisms that control proliferation and senescence-like features and reveal drug targets for the treatment of PC and other SDHB mutation-derived disorders.
RESULTS
SDHB loss alters cellular metabolism and dysregulates Cdk5 signaling
Heterozygous SDHB mutations often associate with reduced mRNA expression, or truncated protein (Cascon et al., 2006; Huang et al., 2018; Yang et al., 2012), which may result in impaired but not necessarily complete loss of SDH activity. Analysis of a PCPG dataset revealed genetic alterations within SDHx complex in 54% of patients (n = 161). The highest portion of 39% occurs in SDHB/SDHD where a majority of samples exhibit reduced SDHB expression (Figures 1A and 1B). Also, ~60% of patients with shallow SDHB deletions have corresponding reductions in SDHB mRNA relative to individuals with an unaltered copy of genes (Figure 1C). A significant decrease in SDHB transcripts in PC compared with normal adrenals indicates SDHB haploinsufficiency and thus sensitivity to copy number alterations (Figure 1D). Haploinsufficiency in SDHB develops clinically aggressive metastatic tumors manifesting stem-like properties (Baysal and Maher, 2015; Buffet et al., 2012). This also indicates that SDHB null is relevant to only a small subset of patients and not the absolute presentation of SDHx-associated disease to study downstream signaling mechanisms. Therefore, we conducted CRISPR-Cas9 targeted gene deletion of SHDB from progenitor human PC tumor-derived cell line, hPheo1 (Ghayee et al., 2013) and selected a partial knockout (KO) clone expressing 20% of basal SDHB levels as an accurate cellular model of the human disease (Figure 1E).
Figure 1. Alterations of SDHx genes in pheochromocytoma.

(A) Oncoprint depicts alterations in SDHx complex genes in patients. Genetic alterations are indicated by color codes.
(B) Expression heatmap of SDHx complex genes.
(C) Bar plots presenting copy number alteration and mRNA expression of SDHB in patients with PC tumors (del., deletions; alter., alterations).
(D) Boxplots of SDHB gene expression in PCPG tumors (n = 179) versus normal adrenal (n = 3).
(E) Exemplary immunoblot showing SDHB protein expression in WT versus SDHB KO hPheo1.
(F) Glycolytic profile of WT versus SDHB KO hPheo1 cells indicated as extracellular acidification rate (ECAR). Quantitative ECAR analysis indicated by glycolytic capacity, glycolysis, and glycolytic reserve.
(G) Oxygen consumption rate (OCR) measured in WT and SDHB KO cells. Bar plots show OCR analysis indicated as basal and ATP-linked respiration, n = 3, 10 replicates per group. Values are means ± SEM, ***p < 0.001, ****p < 0.0001 compared by Student’s t test.
Altered metabolism in cancer cells is a common feature where TCA cycle impairment in tumors harboring mutations in SDHB or other key components of aerobic respiration contributes to the Warburg effect (Vander Heiden et al., 2009). In agreement, SDHB KO dramatically altered the cellular bioenergetics profile. Sequential metabolic perturbation with glucose, oligomycin, and 2-deoxy-D-glucose (2-DG) enhanced the extracellular acidification rate (ECAR), which corresponded to metabolic shifts toward increased glycolysis, glycolytic capacity, and glycolytic reserve in SDHB KO cells (Figure 1F). These effects in the KO hPheo1 recapitulate the metabolic phenotype that characterizes SDHB mutant human PC (Favier et al., 2009; Jochmanova and Pacak, 2016). Concomitant comparison of mitochondrial function indicated an elevated oxygen consumption rate (OCR) and higher basal and ATP-linked respiration in wild type (WT) hPheo1, suggesting decreased efficiency of mitochondrial function due to loss of SDHB (Figure 1G). Nevertheless, when challenged with high energy demand via disruption of mitochondrial membrane potential with carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP), a significant spike in maximum respiration was induced in both cell types, suggesting glycolytic preference but not complete shut-off of mitochondrial function in SDHB KO PC cells. In accordance, it has been shown that chromaffin cells retain mitochondrial fitness despite SDHx loss (Kluckova et al., 2020).
Loss of SDHx function not only alters metabolism but also may dysregulate Ca2+ homeostasis (Nasr et al., 2003; Ranganayaki et al., 2021). To evaluate the effect of SDHB loss on Ca2+, single-cell confocal imaging was performed to compare intracellular [Ca2+]i in WT versus SDHB KO hPheo1 cells. In WT cells, a rapid time-dependent recovery to the ionomycin-induced spike in [Ca2+]i occurred, where [Ca2+]i surge returned to basal levels within 5 min. This response was absent in SDHB KO cells where increased [Ca2+]i levels were sustained without significant reduction over the same period of analysis (Figure 2A).
Figure 2. SDHB loss activates a succinate-Ca2+-calpain-Cdk5 cascade.

(A) [Ca2+]i activity reported by time-lapse live-cell imaging of cells loaded with Fluo-4 AM, images acquired pre or post stimulation with ionomycin (10 μM). Representative pseudo-colored images are shown with time course of fluorescence intensity quantification as %[Ca2+]i, scale bar, 80 μm.
(B) Liquid chromatography-mass spectrometry (LC-MS) quantitation of succinate in WT and KO cell extracts.
(C) Quantitation of fluorescence intensity and photomicrographs showing effects of ionomycin on %[Ca2+]i in WT hPheo1 cells treated with dimethyl succinate (DMS, 2 mM) or controls.
(D) Time-lapse measurement of [Ca2+]i in SDHB KO cells pretreated with SUCNR1 antagonist, NF-56-EJ40, with representative images, scale bar, 50 μm
(E) Schematic workflow of calpain sensor, and representative pseudocolor FRET map and normalized FRET index of WT and SDHB KO hPheo1.
(F) Immunoblots of Cdk5, p25/p35 in WT and SDHB KO cells with quantitation.
(G) 3D surface plot of immunostained cells comparing relative levels of p35/25 between WT and KO hPheo1, scale bar, 80 μm.
(H and I) Representative confocal 3D photomicrographs and quantitation showing p35/25 expression in WT hPheo1 treated with increasing concentrations of succinate as indicated.
(J) Quantitative immunoblotting of Cdk5/p35/p25 levels in tumor lysates derived from WT (n = 4) and KO (n = 6) xenografts. Values are means ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001; n.s., non-significant compared by t test and/or one-way ANOVA.
(K) Schematic illustrating the mechanism of action mediated via succinate accumulation in chromaffin cells.
In addition to causing an imbalance in [Ca2+]i, SDHB loss can trigger buildup in the TCA metabolite, succinate. Indeed, succinate concentrations were significantly higher in SDHB KO cells compared with controls (Figure 2B). We hypothesized that this increase in basal intracellular succinate could contribute to the loss in [Ca2+]i dynamics observed in KO cells. To test this, WT PC cells were treated with cell-permeable dimethyl succinate (DMS). Similar to the effect of SDHB KO, the addition of exogenous succinate disrupted ionomycin-induced [Ca2+]i homeostasis recovery in parent cells (Figure 2C).
Succinate may alter [Ca2+]i homeostasis through autocrine signaling. Specifically, excessive succinate freely shuttles between the mitochondria, cytosol, and across the cell membrane to stimulate the SUCNR1 receptor in SDHx-related PC (Matlac et al., 2021). Interestingly, hPheo1 expressed appreciable levels of SUCNR1 and SDHB KO causing a 1.75-fold increase in SUCNR1 expression (Figure S1A). To test if SDHB loss and consequent succinate accumulation triggers SUCNR1-mediated disruption of [Ca2+]i homeostasis, KO cells were treated with SUCNR1 antagonist (NF-56-EJ40). [Ca2+]i homeostasis was impaired, as was observed earlier in KO cells. However, pretreatment with NF-56-EJ40, rescued the WT phenotype for [Ca2+]i recovery in SDHB KO cells (Figures 2D and S1B). These data indicate that SDHB loss caused an increase in succinate, which then destabilized [Ca2+]i through constitutive activation of SUCNR1 receptors.
The Ca2+-dependent protease calpain is a key downstream effector activated by loss of Ca2+ homeostasis (Crespo-Biel et al., 2007; Nasr et al., 2003; Pang et al., 2003). To determine if SDHB KO activated calpain, a fluorescence resonance energy transfer (FRET) probe harboring a calpain-specific substrate was used (Stockholm et al., 2005). SDHB KO caused an intracellular calpain activity increase, as indicated by a higher FRET index in parent hPheo1 versus SDHB KO cells expressing the calpain reporter (Figures 2E and S2A). Accordingly, an increase in the 145-kDa breakdown product of spectrin confirmed elevated ubiquitous calpain activity in SDHB-deficient cells (Figure S3A) (Rajgopal and Vemuri, 2002).
The Cdk5 activating cofactor p35 is an important substrate of calpain that is cleaved to p25 in response to the loss of [Ca2+]i homeostasis. Increased expression of coactivators p35/25 is pivotal to stimulating aberrant or pathological Cdk5 hyper-activation (Patrick et al., 1999; Pozo et al., 2013), which can cause neuronal death and may drive neuroendocrine cell proliferation (Barros-Minones et al., 2013; Carter et al., 2020; Crespo-Biel et al., 2009). Interestingly, SDHB KO triggered a cumulative increase of both p35 and p25 levels compared with WT cells (Figure 2F). Concomitant immunofluorescent quantitation showed a 22.1 ± 2.4 pixel intensity increase in p35/p25 signals in KO versus parent cell lines (p = 0.002) (Figure 2G). The expression patterns of SDHB, p35/p25, and Cdk5 were recapitulated in a second independent partial knockout clone (KO-8) (Figure S3B), confirming that aberrant Cdk5 signaling is not selective to a particular clone but rather a potential common target linked to SDHB loss. Furthermore, exogenous succinate induced a dose-dependent increase in p35/p25 levels, with a simultaneous increase in spectrin cleavage in parent cells (Figures 2H, 2I, and S3C). The increased expression of p35 and p25 generation in response to SDHB KO was also observed in vivo when WT or SDHB KO hPheo1 cells were used to create xenografts in SCID mice. SDHB KO cells were more likely to produce viable tumors with rapid onset compared with WT cells (Figure S3D). Tumors derived from SDHB KO cells exhibited increased calpain activity (Figure S3E) and exhibited higher levels of p35 and p25, while Cdk5 levels were comparable in both WT and KO tumors (Figure 2J). Tumor sections from both WT and SDHB KO xenografts immunostained for tyrosine hydroxylase (TH) and chromogranin A (CgA), as hallmarks of human neuroendocrine PC (Figure S3F) (Fliedner et al., 2010). There was also a trend toward higher TH and ChrA levels in KO mice (Figure S3G). Together these data show that a shallow deletion of SDHB, similar to what occurs in humans, alters the metabolic profile, induces succinate buildup, and perturbs Ca2+/calpain/Cdk5 signaling in PC cells (Figures 2K and S3H).
SDHB and Cdk5 correlation in human PC
Aberrant Cdk5 has been previously implicated in human neuroendocrine tumors (NETs) (Pozo and Bibb, 2016). However, the link between Cdk5 and SDHx-related NETs has heretofore not been investigated. Given that SDHB loss elicits elevated p35/25 levels in human PC cells, we assessed p35 transcripts and protein levels in human PC tissues. Interestingly, SDHB and Cdk5R1 (i.e., encodes p35) were differentially expressed between PCs and adjacent normal adrenal medulla in The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) datasets. A significant reduction in SDHB mRNA (see Figure 1D) corresponds to increased Cdk5R1 expression in PC compared with normal adrenals (Figure 3A). In addition, co-expression analysis of 161 PCPGs highlighted a significant negative correlation between SDHB and Cdk5R1 (R = −0.41, p = 6.78 × 10−8; Figure 3B). No such significant correlation was observed between Cdk5R1 and other SDH subunit genes (such as SDHA, SDHC, and SDHD) or with other tumor-suppressive genes of the TCA cycle (e.g., FH, IDH1, CS) (Figures S4A–S4F). Also, a PCPG GEO dataset of 84 patients showed an inverse correlation between the expression of Cdk5 activating components (Cdk5, Cdk5R1, Cdk5R2) and SDHB (p < 0.001), while no such relationships were observed with SDHA, SDHC, and SDHD subunits (Figures S5A–S5C). Moreover, treatment of WT hPheo1 cells with cell-permeable succinate increased Cdk5R1 mRNA, suggesting that the inverse relationship between SDHB and Cdk5R1 may be mediated via succinate signaling (Figure S5D).
Figure 3. SDHB and Cdk5 coactivator inversely correlate in human PC.

(A) Boxplots of Cdk5R1 in PCPG tumors (n = 179) versus normal adrenal (n = 3).
(B) Correlation between SDHB and Cdk5R1, data quarried from cBioportal PCPG dataset; (R is Spearman coefficient; p = 6.78e).
(C) Quantitation and representative immunoblots showing protein levels of SDHB, Cdk5, and p35/25 in human sporadic PC (n = 11) compared with normal medulla (n = 5).
(D) Expression analysis of SDHB, Cdk5, p35/25 in human SDHB mutant tumors (n = 7) versus normal medulla (n = 7).
(E) Hematoxylin eosin (HE) and immunostains of Cdk5 and p35/p25 in human PC, scale bar, 50 μm.
(F) Histological assessment of Cdk5 and p35/25 in tissue microarray sections of adrenocortical adenoma (ACA, n = 40) and PC (n = 30); quantification presented as optical density; *p < 0.05, **p < 0.01, ***p < 0.001 compared using t test with Welch’s correction; n.s., non-significant.
Germline mutations in SDHx genes are responsible for 38%–80% of metastatic familial PCPGs, while ~10% of sporadic PC also harbored SDHx mutations (Gottlieb and Tomlinson, 2005; Korpershoek et al., 2011). To determine if the inverse correlation between SDHB and p35 gene expression characterizes PC tumors at the post-translation level, immunoblots were performed with sporadic PC tumors versus human normal adrenal medulla. Consistent with the mRNA levels, SDHB protein levels decreased, whereas p35/p25 cumulative protein band intensities were increased (Figure 3C). This effect was mirrored in PC in which SDHB mutations were observed (Figure 3D).
Both sporadic and SDHx mutated PCs immunostained for Cdk5 and p35/p25 (Figure 3E). To determine cell type selectivity, differential expression of p35/25 was determined between PC tissues and a different form of adrenal tumor derived from cortical cells known as adrenocortical adenoma (ACA). Analysis of a human adrenal tissue microarray (30 cases of PC and 40 cases of ACA) showed significantly higher levels of p35/25 in PC compared with ACA (1.5-fold, p = 0.0002) with no measurable change in Cdk5 (Figure 3F), implicating selective functional significance for aberrant Cdk5 in chromaffin cell-derived tumors. These results illustrate that human PCs manifest a negative correlation between SDHB and p35/p25, supporting the concept that aberrant Cdk5 can function as a driver of tumor cell proliferation in PC.
Aberrant activation of Cdk5 in mouse chromaffin cells causes human-like PC
To determine if aberrant Cdk5 activation recapitulates human PC, a PiggyBac transgenic mouse carrying single-copy PNMT (gene encodes the enzyme catalyzing the final step of catecholamine biosynthesis) transgene promoter was designed to drive chromaffin cell-specific tetracycline transactivator (tTA) expression (Goldstein et al., 1972; Ross et al., 1990) (Figures 4A and S6A). This animal was then crossed with the Tet-Op-p25GFP line (Bujard, 1999) to generate bitransgenic mice in which p25 overexpression (p25OE) could be induced in adrenal medulla chromaffin cells by removal of dietary doxycycline (Figure S6B) (Cruz et al., 2003). Here, P25GFP expression status is denoted as p25-ON (Dox-OFF) or p25-OFF (Dox-ON), and PNMT-tTA littermate controls are referred to as WT. Aberrant Cdk5 induced via activation of p25OE caused bilateral 12- to 50-mm3 PCs to develop in 20–21 weeks with no effect on adrenal glands of WT mice (Figures 4B and 4C).
Figure 4. Aberrant Cdk5 develops PC in bitransgenic mice.

(A) Schematic showing design of PiggyBac PNMT expression system to generate Dox-ON/OFF bitransgenic model.
(B) T2-weighted representative MRI coronal images indicating temporal changes in adrenal gland size in WT versus p25-ON mice.
(C) MRI quantitation of adrenal size in p25-ON mice (n = 11) compared with WT (n = 6).
(D) Immunoblots showing expression of p25-GFP, Cdk5, TH, and ChrA in tissue lysates derived from p25-ON or WT adrenal glands.
(E) Histological assessment of NE markers in WT adrenals or PC tissue sections, scale bar 100 μM.
(F–H) Measurements of plasma nor-metanephrines (F), metanephrines (G), and tail-cuff blood pressure (H); (n = 6–11), *p < 0.05, **p < 0.01 compared by using t test with Welch’s correction.
Higher levels of p25GFP, TH, and ChrA occurred in p25-ON PC tissue compared with WT adrenal glands (Figure 4D). Moreover, p25-ON PC expressed the highest levels of these proteins compared with other organs, including liver, brain, spleen, and lung, demonstrating tissue type specificity. P25-ON tumor tissue showed neuroendocrine pseudo-rosettes surrounding blood vessels immunostained for TH/ChrA that typify human PC. While both WT adrenal medulla and p25-ON PC stained for Cdk5, only p25ON tumors were p25-GFP positive (Figure 4E).
Patients exhibit hypertensive crises due to PC-derived overproduction of catecholamines (Eisenhofer et al., 1999). Similarly, p25-ON mice had elevated plasma levels of nor-metanephrine, metanephrine, and higher systolic blood pressure (Figures 4F–4H). These data show that aberrant Cdk5 activity, such as that resulting from SDHB mutations, can drive the formation and progression of PC and the clinical symptoms with which it is associated in a mouse model.
Screening functional targets of aberrant Cdk5
Loss- or gain-of-function mutations in kinases dysregulate signaling pathways associated with cancer (Hanahan and Weinberg, 2000). To understand how the aberrant Cdk5 invokes PC tumorigenesis, we analyzed our published library of phosphorylation sites derived from p25-ON/OFF NETs (Carter et al., 2020). Of 2,000 proline-directed phosphorylation sites detected, 200 were upregulated, while 122 were downregulated, suggesting that p25OE both positively and negatively regulates protein phosphorylation in growing versus arrested tumors (Figure 5A). To gain a wider mechanistic perspective we focused on phosphorylation sites suppressed in p25-ON tumors. Forty-four phosphoproteins were downregulated by >60% in growing tumors (Figure 5B). Functional analysis of these phosphoproteins revealed a network of enriched pathways involved in metabolic processes, cell-cycle regulation, cell size, kinase activity, protein modification, and RNA processing (Figures 5C, S7, S8A, and S8B).
Figure 5. Characterization of functional Cdk5 targets in human PC.

(A) Schematic showing NE tumors from p25-ON/OFF model analyzed for potential Cdk5 phosphorylation sites; function of phosphosite ratio (log2 fold change) between p25-ON and -OFF tumors is plotted against p values.
(B) Table listing the Cdk5 phospho-targets downregulated in growing p25-ON tumors.
(C) Graphical presentation of network tree highlighting enriched pathways associated with downregulated Cdk5 targets; each node represents enriched pathway where two nodes or pathways are connected if they share 20% or more genes; bigger nodes indicate larger gene sets.
(D) Table summarizing phosphosites selected to evaluate their effects on PC cellular proliferation.
(E) Effects of phospho- and dephospho-mimetics (D/E/A) on cell growth determined by dual fluorescence acridine orange/propidium iodide (AO/PI) viability staining. EV, empty vector.
(F) Schematic of AMP-activated protein kinase (AMPK) heterotrimeric complex composed of α, β, and γ subunits. Phosphorylation site, S65, is located in PRKAG2 subunit of the trimeric complex.
(G) Immunoblot detection of P-T172 AMPK in response to PRKAG2 S65D/E/A phosphomutant overexpression, n = 4. Values are means ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001, n.s. non-significant, one-way ANOVA multiple comparisons with Tukey’s method.
(H) Quantitative immunoblotting of P-S65 PRKAG2 and -T172 AMPKα normalized to total PRKAG2 and -AMPKα in human PC tissues (n = 9) versus normal adrenal medulla (n = 5). Values are means ± SEM, *p < 0.05, ***p < 0.001, Student’s t test.
(I) Correlation plot between P-S65 PRKAG2 and -T172 AMPK in patients with PC; r is Spearman’s correlation coefficient.
To analyze phosphorylation sites of particular interest in PC, five targets were chosen based on the algorithm of having (1) unknown phosphorylation sites; (2) conserved phosphosite in humans; (3) implicated in cancer metabolism, protein trafficking, cell cycle, autophagy, or protein translation. These phospho-targets included phospho-Ser672 AAK-1, phospho-Ser244 DEPTOR, phosphoSer65 PRKAG2, phospho-Thr517 UVRAG, and phospho-Ser288 SDPR (Figure 5D). To assess functional relevance to PC tumorigenesis, each phosphorylation site was mutated to encode phospho-mimetic (D/E) or phospho-null (A) amino acids and transiently overexpressed in hPheo1 cells. Of the five phospho-targets, S65D/E mutants of PRKAG2 (5’-AMP-activated protein kinase subunit gamma-2) significantly suppressed cell proliferation compared with that of WT or the S65A form (Figure 5E).
PRKAG2 is the non-catalytic regulatory γ2 subunit of AMP-activated protein kinase (AMPK), a heterotrimeric metabolic sensor composed of catalytic α subunit and two regulatory (β and γ) subunits (Figure 5F). AMPK regulates cellular energy homeostasis, glucose sensing, and immune response processes that affect cell growth and proliferation (Zadra et al., 2015). PRKAG2 has been chiefly studied in cardiac and skeletal muscles (Pinter et al., 2013; Zhan et al., 2018). However, the isoform-specific function of PRKAG2 is largely unexplored in cancer. Nucleotide binding to PRKAG2 confers phosphorylation/dephosphorylation upon residue Thr172 in the activation loop of the AMPK catalytic α subunit, as a prerequisite to kinase activation (Gowans et al., 2013; Oakhill et al., 2011; Shaw et al., 2004). Interestingly, D/E mutations of S65 PRKAG2 increased phospho-Thr172 AMPKα (Figures 5G and S8C). This suggests that phosphorylation at Ser65 PRKAG2 controls AMPK activity. These data also implicate the regulation of AMPK via phosphoSer65 PRKAG2 as an important downstream effector of aberrant Cdk5 in NE cells.
Aberrant Cdk5/GSK3 deregulates AMPK pathway
AMPK activity is regulated by either allosteric or non-canonical mechanisms (Hardie, 2014; Hawley et al., 2005). To better understand how Ser65 PRKAG2 phosphorylation could regulate AMPK activity, a phosphorylation state-specific antibody was generated to this site (Figures S9A–S9C). Analysis of human patient tissue showed that PC tumors had significant decreases in both phospho-Ser65 PRKAG2 and -Thr172 AMPKα compared with normal control adrenals, with a linear correlation between the two sites (Figures 5H and 5I). In contrast, no alterations in PRKAG2/AMPK gene expression were observed between normal and tumor tissues (TCGA-Gene Expression Profiling Interactive Analysis [GEPIA] dataset; n = 182; Figure S9D). Thus decreased Ser65 phosphorylation and AMPK inactivation characterize both human and mouse NE tumors.
A decrease in phospho-Ser65 PRKAG2 and AMPK activity in response to aberrant activation of Cdk5 suggests an intermediary signaling step is involved. In fact, both Cdk5 and GSK3 are predicted to phosphorylate Ser65 PRKAG2 (Figure S9E). Since Cdk5 activation indirectly results in GSK3 inactivation (Morfini et al., 2004), we asked if Ser65 PRKAG2 phosphorylation by GSK3 is downstream of aberrant Cdk5/p25. Indeed, GSK3 efficiently phosphorylated purified recombinant PRKAG2 (Figure 6A). The site at which GSK3 phosphorylates PRKAG2 was confirmed as Ser65 by immunoblotting, while Cdk5 did not phosphorylate this site in vitro (Figures 6B and 6C). Also, active GSK3 immunoprecipitated from hPheo1 cell lysates efficiently phosphorylated WT PRKAG2 in vitro compared with the S65A mutant (Figures S9F and S9G). Aberrant Cdk5 activation leads to inhibitory phosphorylation of Ser9 GSK3b (Plattner et al., 2006; Wen et al., 2008). In agreement, SDHB KO PC cells exhibited higher phospho-Ser9 GSK3β levels compared with parent cells (Figure S9H). Also, the expression of kinase-dead (KD) versus WT Cdk5 reduced phospho-Ser9 GSK3 by 50% and caused a concomitant increase in phospho-Ser65 PRKAG2 and Thr172 AMPKα (Figure 6D). As evidence that this pathway is linked to upstream dysregulation of succinate signaling, treatment of hPheo1 KO cells with the SUCNR1 antagonist NF-56-EJ40 also disrupted aberrant Cdk5-GSK3-AMPK signaling (Figure S9I).
Figure 6. Targeting Cdk5 to regulate the P-PRKAG2/P-AMPK cascade.

(A) In vitro phosphorylation of recombinant PRKAG2 by GSK3, with time-dependent 32P incorporation and Coomassie-Brilliant Blue (CBB) stained protein shown with stoichiometry.
(B) Immunoblots showing GSK3 but not Cdk5 phosphorylates Ser65 PRKAG2 in vitro.
(C) Immunoblots of recombinant AMPK holoenzyme trimeric complex (α1β1γ2) phosphorylated by GSK3β.
(D) Effects of ectopic expression of WT versus kinase-dead (KD) Cdk5 on the levels of phosphorylation sites as shown.
(E) Dose-dependent effects of GSK3 (SB216763, 24 h) versus CDK5 inhibition (CP681301, 24 h) on SDHB KO hPheo1 cell viability.
(F) Plot showing dose-dependent effect of AMPK activator AICAR on cell viability (24 h).
(G) Time-dependent effects of AMPK activators, metformin (20 mM), 2-deoxy-D-glucose (2-DG, 20 mM), and AMPK inhibitor, compound C (CC; 10 μM) on KO hPheo1 cell viability. n = 3, values are mean ± SEM, *p < 0.05, **p < 0.01 compared by one-way ANOVA.
(H) Schematic of signaling mechanism showing aberrant CDK5-GSK3β crosstalk and deregulation of downstream phospho-dynamics of AMPK pathway.
(I) Chemical structures of Cdk5 inhibitors, as indicated.
(J) Dose-response effects of five different Cdk5 inhibitors on cell viability of hPheo1. n = 4.
(K) Glycolytic profile of SDHB KO cells treated with or without MRT3–007 [Cdk5in], 25 nM for 12 h (left). Plots comparing basal glycolysis rate, glycolytic capacity, and glycolytic reserve between control versus Cdk5in (right).
(L) Bioenergetic phenotype of KO cells in response to Cdk5in. Values are means ± SD, *p < 0.05, Student t test, n = 2 (10 replicates per group).
(M) Immunoblot quantification showing time-dependent effect of Cdk5in on phosphorylation states as indicated, n = 3, values presented as fold change normalized with time = 0.
(N) Immunoblot analysis of the impact of GSK3 inhibitor, SB216763 (5 μM, pretreatment 10 h) on Cdk5in-induced phosphorylations.
(O) hPheo1 pre-incubated with or without CC (10 μM), immunoblot quantitation comparing effects of Cdk5in alone or in combination with CC.
Inhibitors or activators of GSK3 may induce cellular proliferation or suppression dependent on cell type (Li et al., 2014; Pap and Cooper, 1998; Tang et al., 2012). Here, we found that hPheo1 cells were unaffected by treatment with a GSK3 inhibitor (SB216763), while a Cdk5 inhibitor (CP681301) dose-dependently abrogated cell viability (Figure 6E). Moreover, metabolic modulators that activate AMPK, including AICAR, metformin (Vial et al., 2019), and 2-DG (Wang et al., 2011), induced growth-inhibitory effects, whereas an AMPK inhibitor, compound C (CC; dorsomorphin) (Zhou et al., 2001) had a minimal effect on cell growth (Figures 6F and 6G). These results suggest a pivotal signaling mechanism where aberrant Cdk5 activation inhibits GSK3, thereby disrupting phospho-PRKAG2-dependent AMPK activation (Figure 6H).
Characterization of a potent Cdk5 inhibitor
With the emergence of Cdk5 as a promising target in cancers, there is renewed demand for effective drugs that target this kinase. Currently available Cdk5 inhibitors such as roscovitine (CYC202, seliciclib) act as purine analogs that interfere with ATP binding (Bettayeb et al., 2010). However, lack of specificity, short half-life, rapid degradation, and weak potency limit their potential for clinical use (McClue and Stuart, 2008). Therefore, we screened a small library of selective Cdk5 inhibitors including 25–16, MRT3–007, MRT3–124, and CR8 using roscovitine as a positive control. These compounds share the same general chemical structures (Figure 6I) and relative kinase selectivity (Figure S10A). Each of these compounds exhibited dose-dependent effects on hPheo1 cell viability (Figures 6J and S10B). MRT3–007 showed the highest potency, with an half maximal inhibitory concentration (IC50) value approximately 1,000-fold lower than that of roscovitine (25 ± 10 nM for MRT3–007 versus 26 ± 10 μM for roscovitine).
Effective cancer treatments using kinase inhibitors depend on the precise genetic constitution of individual patients so that differences in molecular signatures between tumor and normal cells can be defined (Broekman et al., 2011; McDermott et al., 2007). Furthermore, it is possible for inhibitors to be effective by targeting the kinase driving neoplasia while exhibiting broader activity in vitro. As a typical example, MRT3–007 shares overlapping selectivity for Cdk1, 2 and 9 in vitro. Therefore, we queried whether its growth-inhibitory effects in PC were Cdk5 dependent. Notably, PC/PG patients showed significantly higher gene expression of Cdk5 over Cdk1/2 (Figure S10C), while Cdk9 expression was absent in the experimental models used here. Correspondingly, MRT3–007 was more effective in suppressing in vitro growth of PC compared with a selective Cdk2 inhibitor (CVT-313) (Figure S10D). Also, MRT3–007 elicited limited efficacy on cancer cell lines expressing lower p25 levels, such as those derived from breast cancer (MDAMB231 cells), liver carcinoma (HepG2 cells), and small cell lung cancer (H1184 cells). In contrast, MRT3–007 showed higher potency for higher p25-expressing cervical cancer cells (HeLa cells), comparable with PC-derived cells (Figure S10E). Thus this class of compounds could be effective in treating tumors that are dependent upon hyperactive Cdk5.
Additional support for ex vivo selectivity of MRT3–007 for Cdk5 over Cdk1/2 was derived by immunoprecipitating Cdk5 or Cdk1/2 from the lysates of hPheo1 cells treated with MRT3–007 using histone H1 as a reporter substrate. Cdk5 but not Cdk1/2-dependent phosphorylation of histone H1 was significantly attenuated, suggesting MRT3–007 is more selective for Cdk5 within the context of the intracellular milieu than in vitro (Figure S10F). MRT3–007 also selectively inhibited the proliferation of cells that overexpress p25 but had no effect on cells lacking p25 expression (Dox-ON; Figure S10G), implying aberrant Cdk5-dependent sensitivity. Together these results suggest that MRT3–007 can be exploited as a potent Cdk5 inhibitor (Cdk5in) in cells, which depend upon aberrant Cdk5 for their growth.
Cdk5-GSK3-AMPK cascade controls bioenergetics and induces senescence
Cdk5 plays a critical role in tumor-associated cell-cycle progression, DNA damage response, and mitochondrial dysfunction (Mao and Hinds, 2010; Sun et al., 2008; Zhang et al., 2015). Here, we showed that SDHB loss caused a metabolic shift toward glycolysis and triggered aberrant Cdk5-AMPK signaling. These findings prompted us to explore the impact of Cdk5 inhibitor on AMPK signaling, downstream metabolism, and cell-cycle progression. Treatment of SDHB KO hPheo1 cells with Cdk5in (MRT3–007) decreased basal glycolysis, glycolytic capacity and glycolytic reserve (Figure 6K). In addition, Cdk5in shifted the cell energy profile (ratio of OCR:ECAR) from a glycolytic to quiescence/low-energy state (Figure 6L). Concomitantly, Cdk5 inhibition caused time-dependent increases in phospho-S65 PRKAG2 and -T172 AMPKα (Figure 6M). These effects corresponded to increased phosphorylation of S79 acetyl-CoA carboxylase (ACC), a defined AMPK activity reporter, confirming that Cdk5in activates the AMPK pathway.
To better understand the relationship of Cdk5-GSK3-AMPK signaling pathway, a selective GSK3 inhibitor, SB216763 was used. SB216763 pretreatment suppressed activation of S65-PRKAG2, T172-AMPK, and S79-ACC phosphorylation induced by Cdk5in (Figure 6N). Also, direct inhibition of AMPK via CC reversed the effects of Cdk5in on phospho-T172 AMPK (Figure 6O). These data further substantiate the Cdk5-GSK3-AMPK cascade as a critical signaling mechanism downstream of SDHB deficiency.
On sensing bioenergetics stress, AMPK-dependent phosphorylation of Ser15 p53 stabilizes the protein causing cell-cycle arrest (Jones et al., 2005; Garcia and Shaw, 2017). In agreement, Cdk5in induced a time-dependent increase in phospho-Ser15 p53 (Figure S11A), implicating p53 as a critical downstream effector of AMPK in PC. P53 functions as a transcription factor controlling cell-cycle regulatory gene expression (Chen, 2016; Mijit et al., 2020), while AMPK-p53 activation can also induce cellular senescence in response to bioenergetics stress or chemotherapy (Jones et al., 2005; Lee et al., 2015; Xue et al., 2007). Here, in addition to increased phospho-Ser15 p53, Cdk5in induced time-dependent increases in the expression of senescence markers, p16INK4a and p27Kip, which associate with the primary G1-S checkpoint (Figures S11B and S11C). Cdk5in also caused a sharp rise in phospho-Ser139 histone H2AX, a potential indicator of a transition from senescent to apoptotic cell death (Figure S11D). These effects were negated in the presence of the p53-specific inhibitor, Pifthrin (Pftα) (Figure S11E). Cdk5in also induced senescence-like cell morphology, marked by enlarged flattened cells (Figure 7A), disorganized cytoskeleton, and elevated β-galactosidase, concomitant with overexpression of p16INK4a, p27, and phospho-S139 H2Ax (Figure 7B). Of note, inactivation of AMPK via CC reversed the cell spreading induced by Cdk5in, indicating that AMPK activation mediated the Cdk5in-induced morphological changes (Figure S11F).
Figure 7. Cdk5in induces senescence-like phenotypic characteristics.

(A) Scanning electron microscopic images of hPheo1 cell morphology in control versus Cdk5in (Indolinone A).
(B) Imaging of proliferating cells and those treated with Cdk5in (20 nM, 48 h) for common senescence markers. Representative confocal photomicrographs and quantitation show phalloidin stain of F-actin, p16INK4a (inset is senescence-associated β-gal), p27Kip, and P-H2AX, respectively; scale bar, 136 μm.
(C) Confocal images of Ki67 staining and bar graph show percentage of Ki67-positive cells.
(D and E) SDHB KO xenografts treated with vehicle (Con, n = 8) or Cdk5in (n = 10) (0.5 mg/kg) were analyzed for tumor volume (D), followed by histological (HE) and Ki67 expression analysis in xenograft tissues (E).
(F and G) Efficacy of Cdk5 inhibition tested on a metastatic allograft PC model, examined via in vivo bioluminescent imaging. Representative images of vivisected liver and tumor spread indicated by histological analysis and photon flux quantitation; n = 4.
(H and I) Representative coronal MRI images and quantitation showing changes in adrenal gland size of mice over time under Dox-ON/OFF conditions.
(J) Quantitation of p25GFP expression determined by immunoblotting.
These phenotypic effects induced by Cdk5 inhibition corresponded to a significant shift toward cell-cycle arrest in G1 phase with a drastic expulsion of cells from G2 phase (Figure S11G). At the same time, a decrease in proliferation marker Ki67 was evident (Figure 7C). The G1/S cell-cycle arrest was also observed in cells that overexpressed S65D PRKAG2 compared with those expressing either WT or S65A forms of the AMPK regulatory subunit (Figure S11H). Thus, Cdk5 inhibition not only alters the bioenergetics of cancer cells but also provokes senescence-like characteristics following activation of PRKAG2/AMPK/p53 signaling cascade.
Cdk5 inhibition as a preclinical treatment for SDHB-mediated disease
The above results implicate Cdk5in as a promising targeted therapy for PC and other SDHB-related disorders. To evaluate its anti-tumor potential in vivo, the maximal tolerated dose (MTD) of Cdk5in was first determined in mice. MRT3–007 was well tolerated up to a dose of 1 mg/kg, intraperitoneal (i.p.) (Figure S11I). Subsequently, mice carrying SDHB KO hPheo1 xenografts were treated with 0.5 mg/kg Cdk5in, which induced a significant reduction in tumor volume and mitotic index (Figures 7D and 7E) with a minimal adverse effect on body weight (Figure S11J). Additionally, in vivo therapeutic efficacy of Cdk5in was confirmed using a metastatic allograft model of PC where luciferase-expressing mouse PC cells (MTT) were injected intravenously and metastases were imaged in vivo (Martiniova et al., 2009). Cdk5in dramatically reduced tumor signal compared with both vehicle and its parent compound, roscovitine (Figures 7F and 7G). Consistent with our ex vivo findings, Cdk5 inhibition reduced phospho-Ser21/9 GSK3 and increased phospho-Ser65 PRKAG2, -Thr172 AMPK, and -Ser79 ACC in the lysates of PC xenografts (Figures S12A–S12D). Additionally, Cdk5in treatment increased phospho-Ser15 p53, p16, and p27 levels, consistent with that of senescence-like markers observed in vitro (Figure S12E). These findings indicate that Cdk5in possesses therapeutic potential in the treatment of PC and other SDHB-related disorders.
Finally, to validate that PC tumor progression is dependent upon aberrant Cdk5 activity, we assessed the effects of halting p25OE in the bitransgenic mouse PC model. Under Dox-OFF conditions, tumor size progressed by 2.5-fold over 18 weeks. However, the replacement of dietary Dox (Dox-ON) significantly limited tumor volume with a corresponding decrease in p25-GFP expression (Figures 7H–7J). Tumor arrest due to halt in p25OE (i.e., p25-OFF) resulted in decreased phospho-Ser21/9 GSK3 and increased phospho-Ser65 PRKAG2, -Thr172 AMPK, and -Ser79 ACC (Figures S12F–S12I). Once again, these tumors achieve a senescent-like state as indicated by increased phospho-Ser15 p53 and p16 levels (Figures S12J and S12K).
Together, these data support a signaling cascade (Figure S12L) where SDHB loss causes accumulation of succinate, which, in the context of metabolic impairment, leads to loss of Ca2+ control and calpain activation. As a result, aberrant Cdk5/p25 accumulates and causes GSK3 inactivation. Consequently, AMPK is inactivated through the reduction in Ser65 PRKAG2 phosphorylation. Attenuated AMPK activity leads to reduced phosphorylation of Ser15 p53 promoting cell proliferation. This signaling cascade appears to be an important additional feature of the Warburg effect.
DISCUSSION
TCA-linked mitochondrial malfunction and elevated glucose utilization is considered the root cause of several human diseases, including cancer, diabetes, and neurodegenerative disorders (Blank et al., 2010; Hsu and Sabatini, 2008). Here, we delineated a signaling cascade that highlights critical phosphorylation hotspots on metabolic checkpoints disrupted by loss of the TCA cycle component, SDHB. Several downstream effects of succinate accumulation have been linked to altered metabolism, pseudohypoxia, and SUCNR1 activation (Dahia et al., 2005; Matlac et al., 2021; Pollard et al., 2006). Although these mechanisms have been demonstrated to serve as components in discrete cellular- or disease-specific contexts, their suggested interactions as part of the multistep process of carcinogenesis have not been fully delineated. Hence, the complete picture of how TCA perturbations can lead to cancer has not yet emerged.
Previously, features of neurodegeneration or ischemic injury have been modeled by inhibiting SDH activity using 3-nitropropionic acid, and malonate, which activates calpain/Cdk5 signaling (Barros-Minones et al., 2013; Pang et al., 2003; Ranganayaki et al., 2021). These findings support the importance of Cdk5 in SDHx deficiency diseases. Here we deciphered several distinct aspects of tumorigenic signaling including metabolic shift coupled with dysregulation of [Ca2+]i/calpain/aberrant Cdk5 in response to SDHB loss in PC tumors.
Recent studies have identified ancillary driver mutations in ATRX (Fishbein et al., 2015), KIF1B, and NF1 (Evenepoel et al., 2017) in patients harboring SDHB mutations. This is congruent with recently discovered mutations in KIF1B and NRAS genes in hPheo1 cells (Rossitti et al., 2020). In addition to the germline driver SDHB mutations, ancillary mutations in hPheo1 create the opportunity to study malignant PC behavior. Moreover, the majority of the SDHx mutations are not complete deletions as was previously thought, but human tumors harboring SDHx mutations still express some of the protein (Gimenez-Roqueplo et al., 2003; Neumann et al., 2004; Yang et al., 2012). Thus, the partial SDHB KO hPheo1 cells provided a valuable clinically relevant tool to decipher distinct tumor phenotypes, and signaling mechanisms.
Extracellular succinate activates SUCNR1, which invokes multiple signaling outcomes, dependent on the cell type, while excess intracellular succinate accumulation causes inhibition of 2-oxoglutarate-dependent dioxygenases, histone, and DNA demethylases. Succinate accumulation could cause [Ca2+]i. dysregulation either through activation of SUCNR1 receptors or mitochondrial ROS (Andrienko et al., 2017). SUCNR1 are metabolic stress sensors that modulate intracellular Ca2+ through an inositol phosphate-dependent mechanism via PLCβ activation or pathways downstream of G protein invocation (Bhuniya et al., 2011; Gilissen et al., 2016; Sundstrom et al., 2013; Wu et al., 2020). We showed that succinate accumulation impaired intracellular Ca2+ dynamics, and caused aberrant Cdk5 activity, while inhibition of SUCNR1 rescued these effects. Thus, we demonstrate succinate-SUCNR1 pathway as a primary source of [Ca2+]i. dysregulation in cancer cells.
Cdk5-GSK3β interactions have been reported in neurodegenerative disorders, although how Cdk5 induces GSK3 inhibition remains to be explained. This may involve either activation of ErbB/Akt (Wen et al., 2008) or inhibition of phosphatases PP1/PP2A (Morfini et al., 2004; Plattner et al., 2006). The data presented here strongly support Cdk5 as the arbitrator regulating the phospho-dynamics of a GSK3/PRKAG2/AMPK cascade, which determines the proliferative state of PC cells. It has been suggested that GSK3 interacts with the AMPKβ regulatory subunit and can inhibit AMPKα through phosphorylation at Thr479 (Suzuki et al., 2013). However, this mechanism is somewhat confounded by report of simultaneous activation of AMPKα and reduction of inhibitory Ser9 GSK3 phosphorylation in neuronal models (King et al., 2011). Thus, the relationship between GSK3 and AMPK may be context dependent.
We identified a functional role for phospho-Ser65 PRKAG2 as a prerequisite to Thr172 AMPKα phosphorylation. In agreement, several mutations in PRAKG2, such as K475E and R302Q, lead to increased AMPK activity associated with cardiomyocyte hypertrophy (Xu et al., 2017; Zhan et al., 2018). AMPK activationcoupled PRKAG2 nuclear translocation also promotes cardioprotection against ischemic injury (Cao et al., 2017) and is likely followed by interactions between PRKAG2-AMPK-LKB1 (Xie et al., 2008). Considering these findings, a cardinal observation of our study is reporting an isoform-specific function for PRKAG2 in cancer.
AMPK-mediated p53 activation is known to cause premature senescence in response to energetic stress. Moreover, AMPK-induced metabolic activation of p53 was not affected by the inhibition of ATM kinase, while loss of p53 negated AMPK-induced effects (Jones et al., 2005). Here, Cdk5in-induced p53 activation appears to be regulated by the metabolic state of the cell where a coordinated increase in P-Ser15 p53 and P-Thr172 AMPK was marked by senescence-like cell morphology. Mitochondrial ROS, DNA damage, and histone methylation are the main drivers of cellular senescence. However, modulation of stress-inducible kinases such as p38MAPK (Freund et al., 2011), AMPK, and mTOR/PTEN (Jung et al., 2019) can also mediate cellular senescence signals independent of DNA damage. Furthermore, AMPK-p53 activation incites anti-glycolytic effects (Thoreen and Sabatini, 2005), consistent with the ability of AMPK to negatively regulate glycolysis as a feature of its anti-tumor effects (Faubert et al., 2013). Interestingly, inhibiting Cdk5 attenuates glycolysis, suggesting an active role for this kinase in aerobic glycolysis while maintaining low levels of AMPK-p53 signaling.
In summary, this study reveals a phospho-dynamic mechanism where a Cdk5/GSK3/PRKAG2-AMPK/p53 signaling cascade acts as a critical downstream effector of SDHB loss. We demonstrated key components of this cascade across cell-based and in vivo models as well as in human tumors. We also derived a clinically accurate model of PC by transgenically invoking aberrant Cdk5 activity. These findings serve as a mechanistic rationale to utilize combinations of Cdk5 inhibitors and AMPK agonists in tumors driven by Cdk5/AMPK-dependent metabolic checkpoints.
Limitations of the study
Our study uncovers an important signaling cascade, presenting a molecular hub of potential anti-cancer targets. However, there are a few missing links that will require further exploration. We show activation of aberrant Cdk5 downstream of SDHB-succinate-Ca2+ signaling via SUCNR1 as one crucial tumorigenic signaling mechanism in PC. However, it remains unclear precisely how SUCNR1 activation mediates these effects. Further delineation of the signaling steps between SUCNR1 and Ca2+/calpain/p25 is warranted. Second, TCGA analysis indicates a significant negative correlation between SDHB and Cdk5R1 in contrast to other subunits. This raises the question of why loss of SDH through different subunit genes causes distinct functional outcomes on the tumorigenic potential of PC patients. The causes of this selective negative correlation between SDHB and Cdk5R1 need further study. There are also likely additional functional aspects of PRKAG regulation to understand. The precise molecular mechanism by which GSK3-dependent Ser65 PRKAG2 phosphorylation mediates Thr172 AMPK phosphorylation remains to be understood, as do any additional features of PRKAG2-AMPK regulation. Finally, it remains to be fully explained how Cdk5 inhibition induces premature cellular senescence. Mechanistically, senescence is a complex orchestration and temporal coordination of numerous cell-cycle regulators. The molecular markers for tracking senescence in the context of SDHx tumors may be context, cell type, and species dependent and should be further studied.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, James A. Bibb (jbibb@uabmc.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Human tumor samples
Human Pheochromocytoma (PC) were acquired from tumor bank of University of Alabama Birmingham, and Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) following institutional review board (IRB) regulations, office for human research protections, NIH guidelines for research involving human subjects, and the health insurance portability and accountability act. All samples were de-identified, coded with no patient data, stored at −80°C until required for assay. Normal adrenal medullae were obtained from cadaveric kidney transplant donors from University of Alabama Birmingham (used as controls for which patients have given consent).
Cell lines and cell culture
The human progenitor PC cell line, hPheo1 (Ghayee et al., 2013) and mouse MTT PC cells were used (Korpershoek et al., 2012). SDHB was deleted from parent hPheo1 using CRISPR/Cas9 technology The goal was to achieve 80–90% KO of SDHB where single KO clones were selected and, subcloned multiple times. The resultant cell line confirmed >80% KO efficiency with a strong glycolytic phenotype in comparison to the uncloned cells. Cells were cultured in RPMI 1640 with 10% FBS (Gibco). HEK293, HeLa, MDAMB-231, and H1184 were maintained in the ATCC-recommended culture.
Pheochromocytoma animal model
All animal research was approved by the University of Alabama Birmingham (UAB) Institutional Animal Care and Use Committee (IACUC). Mice were genotyped and maintained within UAB Animal Resource Program Facility (ARP) and the animal research facilities of the Department of Surgery. Bitransgenic mice- Both male and female wild-type C57BL/6 mice, ages eight to twelve weeks were utilized for all the experiments. PiggyBac technology was used to generate single-copy PNMT–tTA transgenic mice (Cyagen Biosciences). Transgene positive offsprings were confirmed by genotyping four weeks old pups (primer sequence given in Key Resource Table; annealing temp– 60°C; Product– 373 bp). All positive offsprings were confirmed by PCR to not contain any integration of the helper plasmid. Primers used in the PCR to test for helper plasmid integration, Forward– CTGGACGAGCAGAACGTGATCG; Reverse- CGAAGAAGGCGTAGATCTCGTCCTC. Bi-transgenic mice were generated by crossing eight weeks old Tet-Op-p25GFP (Meyer et al., 2008) with that of PNMT-tTA. Transgenes were confirmed by genotyping for PNMT and p25-GFP alleles while control littermates were positive only for PNMT-tTA (Figure S6). Mice were treated with water containing 0.1 g/L doxycycline as required and all experimental mice were group-housed 12 h light/dark cycle with access to food and water ad libitum.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit anti-Cdk5 | Rockland | Cat# 200-301-163; RRID: AB_11182476 |
| Rabbit anti-SDHB | Abcam | Cat#154974; RRID: N/A |
| Rabbit anti-ChrA | Abcam | Cat#15160; RRID: AB_368477 |
| Rabbit anti-Tyrosine Hydroxylase | Abcam | Cat#6211; RRID: AB_2240393 |
| Rabbit anti-GFP | Cell Signaling Technology | Cat#2956; RRID: AB_1196615 |
| Rabbit anti-p35/25 | Cell Signaling Technology | Cat#2680; RRID: AB_1078214 |
| Rabbit anti-phospho-Thr172 AMPKα | Cell Signaling Technology | Cat#2535; RRID: AB_331250 |
| Rabbit anti-AMPKα | Cell Signaling Technology | Cat#2532; RRID: AB_330331 |
| Rabbit anti-phospho-Ser79 ACC | Cell Signaling Technology | Cat#11818; RRID: AB_2687505 |
| Rabbit anti-ACC | Cell Signaling Technology | Cat# #3662; RRID: N/A |
| Rabbit anti-phospho-Ser21/9 GSK3 | Cell Signaling Technology | Cat# 9331; RRID: AB_329830 |
| Rabbit anti-GSK3 | Cell Signaling Technology | Cat#9315; RRID: N/A |
| Rabbit anti-phospho-Ser15 p53 | Cell Signaling Technology | Cat#9284; RRID: AB_331464 |
| Mouse anti-p53 | Thermo Fisher Scientific | Cat# MA5-12557; RRID: AB_10989883 |
| Rabbit anti-p27Kip1 | Cell Signaling Technology | Cat#3686; RRID: AB_2077850 |
| Rabbit anti-phospho-Ser139 H2Ax | Cell Signaling Technology | Cat# 9718; RRID:AB_2118009 |
| Rabbit anti-p16INK4A | Droteintech | Cat#10883-1-AP; RRID: AB_2078303 |
| Mouse anti-Spectrin | Millipore Sigma | Cat#MAB1622; RRID: AB_94295 |
| Mouse anti-GAPDH | Thermo Fisher Scientific | Cat# 39-8600, RRID: AB_2533438 |
| Mouse anti-β actin | Thermo Fisher Scientific | Cat#PA5-78715; RRID: AB_2745831 |
| IRDye® 800CW Goat anti-Rabbit or anti-Mouse IgG | LI-COR Biosciences | Cat#926-32211; RRID: AB_621843, Cat# 926-32210; RRID: AB_621842 |
| IRDye® 680RD Goat anti-Rabbit or anti-Mouse IgG | LI-COR Biosciences | Cat# 926-68071; RRID:AB_10956166, Cat# 926-68070; RRID: AB_10956588 |
|
| ||
| Biological samples | ||
|
| ||
| Human adrenal tumor tissue microarray | US Biomax Inc. | Cat#AG801 |
| Human PC tissues | Tumor bank (University of Alabama Birmingham); Dr.Karel Pacak [Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD)] | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Roscovitine | Dr. Laurent Meijer | ManRos Therapeutics |
| MRT3-007 | Dr. Laurent Meijer | ManRos Therapeutics |
| MRT3-124 | Dr. Laurent Meijer | ManRos Therapeutics |
| CR8 | Dr. Laurent Meijer | ManRos Therapeutics |
| 25-16 | Dr. Amarnath Natarajan (University of Nebraska) | N/A |
| CVT-313 | Selleck Chemicals | Cat#S6537 |
| Metformin | Sigma | CAS#1115-70-4 |
| AICAR | Sigma | CAS#2627-69-2 |
| NF-56-EJ40 | Med Chem Express | Cat#HY-130246 |
| 2-DG | Sigma Aldrich | Cat# D6134 |
| Sulfo-MBS | Sigma Aldrich | Cat# 803227 |
| P-S65 PRKAG2 peptide - RKVDS*PFGC | University of Texas Southwestern | N/A |
| HBSS | Gibco | Cat#14175 |
| Gibco™ Fetal Bovine Serum | Fisher Scientific | Cat# 10-082-147 |
| Doxycycline | Sigma Aldrich | Cat# D9891 |
| Fluo-4AM | Thermo Fischer Scientific | Cat# F14201 |
| FuGENE® HD Transfection Reagent | Promega | Cat# E2311 |
|
| ||
| Critical commercial assays | ||
|
| ||
| 2-MET Plasma ELISA Fast Track | Rocky Mountain Diagnostics | Cat#BA E-8300 |
| Cell Counting Kit-8 | Dojindo Molecular Technologies | Cat#CK04-05 |
| Q5-site directed mutagenesis Kit | New England Biolabs | Cat# E0554S |
| DAKO Immunohistochemistry Kit | Agilent Technologies, Inc. | Cat# K064030-2 |
|
| ||
| Deposited data | ||
|
| ||
| Library of Cdk5 Phosphosites | Carter et al. (2020) | https://www.phosphosite.org/Supplemental_Files.action |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| hPheo1 (Wild-type, SDHB knockout) | Dr. Hans Ghayee (University of Florida) | Developed by Ghayee laboratory |
| MTT cells | Courtesy of Dr. Karel Pacak (NICHD) | Martiniova et al. (2009) |
|
| ||
| Experimental models: Organisms/Strains | ||
|
| ||
| Mouse: PiggyBac PNMT-tTA | Cyagen Biosciences | N/A |
| Mouse: TetOp - P25GFP | Meyer et al. (2008) | N/A |
| Mouse: PNMT-tTA x TetOp-P25GFP | Dr. James Bibb (University of Alabama at Birmingham) | N/A |
| Mouse: nu/nu | Jackson laboratory | JAX:002019 |
| Mouse: C.B-Igh-1b/IcrTac-Prkdcscid | Taconic Biosciences | TAC:cb17sc |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primer: PNMT-tTA Forwardl: CAGTAGTAGATAAAGGGATGGGGAG | This paper | N/A |
| Primer: PNMT-tTA Reversel: GGGGCAGAAGTGGGTATGATG | This paper | N/A |
| Primer: PNMT-tTA Forward2: CAGGAGCATCAAGTAGCAAAAGAG | This paper | N/A |
| Primer: PNMT-tTA Reverse2: CACACCAGCCACCACCTTCT | This paper | N/A |
| gRNA sequence: SDHB: ATGGCAAATTTCTTGATACG | Fisher Scientific | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pLV[Exp]-EGFP:T2A:Puro | VectorBuilder, Cyagen Biosciences | Cat#VB160420-1011mqh |
| CMV pCalpain-sensor | Addgene | Cat#36182 |
|
| ||
| Software and algorithms | ||
|
| ||
| FIJI | Schindelin et al., 2012 | http://imagej.net/software/fiji/ |
| cBioportal | Gao et al., 2013 | http://www.cbioportal.org |
| ShinyGO v0.61 | Ge et al., 2020 | http://ge-lab.org/go/ |
| GEPIA | Tang et al., 2017 | http://gepia.cancer-pku.cn/index.html |
| UALCAN | Chandrashekar et al., 2017 | http://ualcan.path.uab.edu/ |
| XF96 Seahorse Wave | Agilent Technologies | https://www.agilent.com/zh-cn/product/cell-analysis/real-time-cellmetabolic-analysis/xf-software/seahorse-wave-desktop-software-740897 |
| ImageStudio | LI-COR Biosciences | https://www.licor.com/bio/image-studio/ |
| GraphPad Prism | GraphPad Software, Inc | http://www.graphpad.com/scientific-software/prism/ |
|
| ||
| Other | ||
|
| ||
| CODA noninvasive BP system | Courtesy of Dr. David Pollock, University of Alabama at Birmingham | N/A |
| Production of P-S56 PRKAG2 antiserum | SouthernBiotech, Birmingham, AL | N/A |
| Magnetic Resonance Imaging | Institutional Research Core, University of Alabama at Birmingham | N/A |
| Agilent Seahorse XFe96 Analyzers | Bio-Analytical Redox Biology (BARB) Core, UAB Department of Nutrition Sciences | https://www.uab.edu/shp/drc/cores/barb-core |
| Bioluminescence imaging (IVIS Lumina III) | Small Animal Imaging Shared Facility, Department of Radiology & Comprehensive Cancer Center, University of Alabama at Birmingham | N/A |
METHOD DETAILS
Database mining
SDHB copy number alteration, mRNA expression and co-expression analysis were analyzed in cBioportal Cancer Genomics Database (http://www.cbioportal.org) (Gao et al., 2013). According to cBioportal definitions, genomic alterations are defined as: −2 or deep deletion equivalent to homozygous deletion; −1 or shallow deletion indicative of heterozygous deletion; and 0 indicative of diploid with no alteration. UALCAN online TCGA transcriptomic database was used to compare expression levels of SDHB/Cdk5R1 between normal human medulla and PCPG patients (http://ualcan.path.uab.edu/) (Chandrashekar et al., 2017). Publicly available Gene expression Omnibus (GEO) dataset GSE19422 was used for data mining in Figure S5. ShinyGO v0.61 gene-set enrichment tool was used to derive Gene Ontology categories, enriched pathways and graphical pathway network trees (Ge et al., 2020). Gene expression analysis of PRKAG2, AMPKα, (Figure S9D), Cdk5, Cdk2 and Cdk1 (Figure S10A) were performed in GEPIA (The Gene Expression Profiling Interactive Analysis) database (Tang et al., 2017).
Seahorse XF96 metabolic flux analysis
Real time extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) in cells were determined using the Seahorse Extracellular Flux (XFe-96) analyzer (Seahorse Bioscience, MA, USA). 1 × 104 cells were seeded per well of XF96 cell culture plates and incubated for 24 h to allow adherence. Bioenergetic profile was determined by mitochondrial and glycolytic stress tests following the manufacturer’s protocol. The following day cells were washed with pre-warmed XF assay base media (note: for OCR measurement, assay media was supplemented with 10 mM glucose, 1 mM Pyruvate, 2 mM L-glutamine, 5 mM HEPES and adjusted at 7.4 pH; for the glycolysis stress test, cells were washed with glucose-free XF base media). No signs of cytotoxicity, cell detachment or variations in the cell density noticed between the wells. Cells were maintained in final volume of 180 μL/well of media at 37°C, in a non-CO2 incubator for 1 h. Meanwhile, we loaded the cartridge ports with effectors. [For glycolysis stress test–12.5 mM Glucose, 1.5 μM Oligomycin, 50 μM 2-DG. For Mito stress test– 1.5 μM Oligomycin, 1 μM FCCP, 0.5 μM Antimycin and 0.5 μM Rotenone]. Linear correlation between protein concentration and viable cell number was confirmed. Values normalized by CyQUANT DNA quantification (#C35014, Thermo Fisher Scientific) and protein content (BCA assay) was comparable. Data were analyzed using XF96 Wave software and GraphPad Prism.
Ca2+ measurement and live cell imaging
Cells were loaded with cell permeant intracellular Ca2+ flux indicator + in HBSS (Gibco) for 30 min following manufacturer’s instruction. Time-lapse live cell imaging was performed using Nikon A1R HD25 inverted confocal microscope equipped with perfect focus system. Cell imaging chambers were maintained in 5% CO2 and 37°C. Images were capture with Plan Apo λ 20× NA 0.8 wd 1000 objective, frame size– 1024 × 1024, scan speed– 2, time loop– 31, imaging zoom– 1.192, resolution– 3.7 pixels per micron, frame interval – 10 sec, bits per pixel–16, ex/em– 494/525. Pinhole and laser power settings were adjusted based on pre-stimulation background levels. Fiji software (Schindelin et al., 2012) was used to select and quantify ROIs and individual object integrated density (IntDen) values were calculated for each cell using the formula (IntDen- (area of selected cell × mean field background intensity). Individual kinetic profile of [Ca2+]i for WT and SDHB KO cells were generated by plotting fluorescence intensity as a function of time in seconds as described previously (Koh et al., 2016).
Fluorescence resonance energy transfer (FRET)
CMV pCalpain-sensor (Addgene #36182) composed of eCFP (donor) and eYFP (acceptor) linked to calpain cleavage site (GSG-QQEVY GAMPRDGSG) where inactive calpains = High FRET and vice versa). pCalpain-sensor transfected in PC cells using Fugene HD according to the manufacturer’s instruction (Promega). Donor and acceptor bleed through were corrected using eCFP and eYFP only fluorophores. FRET measurements were performed using Nikon C2 confocal microscope (Plan Apo 60x Oil λS DIC N2) attached to a stage- top live-cell incubator to maintain cells at 37°C with 5% CO2 (Tokai Hit environmental chamber). FRET imaging based on sensitized emission were performed to acquire images with following filter combinations: donor (eCFP, ex-430±20, em-485±20); acceptor (eYFP, ex- 480, em-535±25); and FRET channel with a 435 nm (ex) and 535 nm (em). Visualization of FRET index and quantitation were performed as described in the FRET analyzer image J plug-in (Hachet-Haas et al., 2006).
Immunostaining and tissue microarray
Each human PC tumor case was thoroughly reviewed, formalin-fixed, and paraffin-embedded blocks were acquired within Department of Surgery, University of Alabama at Birmingham, and Dept. of Pathology, NICHD followed by the protocol described previously using DAKO immunohistochemistry kit (Pozo et al., 2013). Human adrenal tumor tissue microarray (US Biomax Inc.) was de-waxed at 60°C for 2 h followed by standard IHC protocol. Primary antibodies used included those for Cdk5 (Rockland), -p35/25 (Cell Signaling), ChrA (Abcam), anti-tyrosine hydroxylase (Abcam), and GFP (Cell Signaling). Secondary antibody alone was used as negative control. Quantitative analysis of DAB stained images were performed by using optical density (color deconvolution algorithm) within IHC profiler plug-in compatible with ImageJ digital image analysis software (Varghese et al., 2014).
Blood pressure
Mice blood pressure measurements were evaluated using CODA noninvasive BP system (a tail-cuff Method, Kent Scientific Corporation) as described previously (Wang et al., 2017).
Assessment of Cdk5 inhibitors in vivo
Xenografts- 6–7 week-old C.B-Igh-1b/IcrTac-Prkdcscid mice (Taconic Biosciences) were used for xenografts as previously described (Rai et al., 2020). 2 × 106 hPheo1 tumor cells (WT and SDHB KO) were injected subcutaneously in the right flank of the mice. When average tumor volume reached 150 mm3, mice were divided into two groups and injected intraperitoneally with vehicle or MRT3–007 (0.5 mg/kg every alternate day for three weeks). Body weights and tumor diameters were measured 3 times/week and tumor volumes were calculated using the formula V = ab2 × 0.52, where a and b are major and minor axes of the tumor foci, respectively. The experiment was terminated on day 25, and the tumors were harvested for biochemistry and histological assessment. Allograft and in vivo imaging- 6–7 week old nude mice (nu/nu) (Jackson laboratory) were used for allograft assay described previously (Korpershoek et al., 2012). 1 × 106 MTT luciferase expressing cells were injected via tail vein and imaged one-week post-injection via bioluminescence imaging (Xenogen IVIS). Cohorts bearing comparable size of allografts received IP injection of substrate d-Luciferin (250 μL; 3.75 mg, Caliper Life Science, Hopkinton, MA, USA) 12 min before the whole-body imaging. Data was acquired and analyzed using the Live Imaging software version 3.0 (Caliper Life Science). Fourteen days after injecting tumor cells, all mice were divided into three groups, treated with Vehicle, MRT3–007 (0.75 mg/kg) or Roscovitine (50 mg/kg) every other day for 2 weeks. Animals were re-imaged to measure metastatic lesions via bioluminescence imaging.
Plasmids, site directed mutagenesis and transfections
Lentivirus gene expression vector (3rd generation; pLV-EGFP:T2A:Puro-EF1>ORF/FLAG) was used for subcloning ORF’s of PRKAG2 (NM_016203.3), DEPTOR(NM_022783.3), SDPR (NM_004657.5), AAK1(NM_014911.3) and UVRAG (NM_003369.3) driven by EF1A promoter (Vector Builder, Cyagen Biosciences). Cdk5 wild-type and kinase dead plasmids were described previously (Pozo et al., 2013). DNA modifications were performed in lentiviral pLV[Exp]-EGFP:T2A expression plasmids cloned by VectorBuilder, Cyagen Biosciences to generate phospho- and dephospho-mimetics for PRKAG2 (S-65D/E/A), DEPTOR (S-244D/E/A), SDPR(S-288D/E/A), AAK1(S-676D/E/A) and UVRAG (T-518D/E/A) using Q5-site directed mutagenesis Kit (NEB). Manufacturer’s instructions for mutagenic primer design were followed, and mutations were confirmed by DNA sequencing. Transfections were carried out using FuGene-HD (Promega). Transfection solutions were prepared in Opti-MEM using 1:3 ratio of plasmid DNA to Fugene transfection reagent.
Cell growth assay
Cells were seeded in 6-well plates at the density of 1 × 105, transfected with phospho- or dephospho-mimetics and control vectors. Cell growth was determined 48 h post-transfection by dual fluorescence acridine orange/propidium iodide (AO/PI) viability staining. Total cell number was counted using Cellometer Auto 2000 cell viability counter (Nexcelom) and normalized with total number of GFP-expressing cells. Dose-response curves were generated by cell viability tests using Cell Counting Kit-8 (CCK-8) (Dojindo). Assays were performed in five replicates and repeated at least 3 times.
Phosphorylation state specific antibody generation
Production and affinity purification of phosphorylation state specific polyclonal antibodies were performed as described previously (Hemmings, 1997). Phospho-Ser65 PRKAG2 were raised against a synthetic oligopeptide encompassing the amino acid sequence (RKVDS*PFGC). Cysteine containing phosphopeptide was conjugated to carrier protein Limulus hemocyanin using hetero-bifunctional crosslinker m-maleimidobenzoyl-N-hydroxysulfosuccinimide ester (Sigma# 803227). This conjugate was used to immunize New Zealand white rabbits (Charles River Laboratories). Preimmune sera were obtained and booster injections of 150 μg phosphopeptide conjugate were given at 2, 4, 6 and 8 weeks. Blood was collected at weeks– 5, 7, 9,11,13 and 14. The specificity of the antibodies in anti-serum was characterized by dot blot analysis using dephospho- and phospho-PRKAG2 standards. Phosphorylation-specific antibodies were purified using affinity purification method (Hemmings, 1997).
Immunoblotting, cell cycle analysis, and in vitro phosphorylation
Antibodies to the following phosphorylation sites and proteins were used: phospho-Thr172 AMPK, phospho-Ser79 ACC, phosphoSer21/9 GSK3, phospho-Ser16 p53, p27Kip1, phospho-Ser139 H2Ax from Cell Signaling Technology; and anti-p16INK4A, antispectrin and, anti-GAPDH, anti-actin from Thermo Fisher Scientific. SDS-PAGE and immunoblotting were conducted as previously described (Bibb et al., 1999). Following membrane blocking, immunoblots were incubated with primary and fluorescent secondary antibodies IRDye® 800CW, and IRDye® 680 (goat anti-rabbit or goat anti-mouse) from LI-COR and visualized with Odyssey CLx Imaging System (LI-COR Biosciences, NE). Thereafter, immunoblots’ signal intensity was computationally quantified and analyzed using ImageStudio software (LI-COR Biosciences–GmbH, www.licor.com). For cell cycle analysis, MRT3–007 treatment or phosphomimetics transfected cells were stained with 50 μg/mL of propidium iodide and analyzed for cell-cycle distribution as described previously (Erba et al., 1989). In vitro phosphorylation and immunoprecipitation-kinase assays were performed using optimized protocols described previously (Bibb et al., 1999; Pozo et al., 2013).
Magnetic resonance imaging
MRI experiments were performed using a Bruker Biospec 9.4 Tesla scanner with Paravision 5.1 software (Bruker Biospin, Billerica, MA). A Bruker 72 mm volume coil was used for signal excitation, with a 24 mm surface coil for reception (Doty Scientific Inc., Columbia, SC). Mice were anesthetized with isoflurane gas and respiration observed with an MRI-compatible physiological monitoring system (SA Instruments Inc., Stony Brook, NY). Animals were imaged in supine position on an animal bed with integrated circulating heated water to maintain temperature during the experiment. Scout images were collected in the axial, sagittal, and coronal planes to confirm animal positioning and coil placement. A 2D T2-weighted fast spin echo sequence was used for imaging of kidney and adrenal gland areas. Prospective respiratory gating was enabled to minimize motion artifacts. The following imaging parameters were used: TR/TE = 2000/25 ms, echo spacing = 12.5 ms, ETL = 4, 4 averages, 23 contiguous coronal slices with 0.5 mm thickness, FOV = 30 × 30 mm, and matrix = 300 × 300 for an in-plane resolution of 100 μ. All MRI images were obtained in the DICOM (Digital Imaging and Communications in Medicine) format and were imported into the image processing ITK software to obtain tumor volumes and perform 3D reconstructions. Mean tumor volumes were measured by drawing regions of interest (ROI), to circumscribe the entire tumor.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed by Student’s t-test for comparison of two groups or one-way ANOVA combined with Tukey’s post hoc test for multiple comparisons using Prism 8 version 8.4.2 (Graph Pad Software). The number of experimental replications or number of animals are represented as n, and the definitions of center and precision measures i.e, mean ± SD or mean ± SEM are indicated in the figure legends. p values <0.05 were considered statistically significant, reported as *< 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 in the figure legends. p values >0.05 were considered not significant (n.s.).
Supplementary Material
Highlights.
Dysfunctional SDHB subunit causes aberrant activation of Cdk5 in pheochromocytoma (PC)
Aberrantly activated Cdk5 dysregulates a GSK3/PRKAG2/AMPKα signaling cascade
p25 overexpression in chromaffin cells and consequent aberrant Cdk5 activity causes PC
Cdk5 inhibition activates AMPK/p53 axis to rescue senescence and block PC progression
ACKNOWLEDGMENTS
We thank S. Thomas for assistance with MRI, E. Daniel for technical expertise, S. Barnes for LC-MS succinate measurements, D. Pollack for assistance in measuring mouse blood pressure, L. Bibb and SouthernBiotech for phosphorylation state-specific antibodies, and Pfizer for CP681301. This research was supported by the SDHB Para/Pheo Coalition and the Neuroendocrine Tumor Research Foundation (NETRF, J.A.B.). Portions of this work were also facilitated by the NIH (MH116896, MH126948, J.A.B.), the Robert E Reed Gastrointestinal Oncology Research Foundation, an American Cancer Society Institutional Research Grant Junior Faculty Development Award (S.R.), and Postdoctoral Fellowship (A.M.C.). This research was supported, in part, by Intramural Research Program of the Eunice Kennedy Shriver NICHD, NIH (K.P.); the Gatorade Trust through funds by the University of Florida, Department of Medicine (H.K.G.); and by a Eurostars grant (CYST-ARREST, L.M.). Studies were also supported by the National Cancer Institute Cancer Center Support Grant P30 CA013148 (UAB O’Neal Comprehensive Cancer Center). It was also supported by S10 OD028498-01 (UAB Preclinical Imaging Shared Facility). Some chemical synthesis was also supported by NCI SPORE P50 CA127297 (A.N., Eppley Institute for Research in Cancer and Allied Disease). This work was also supported in part by grant 1145265 from the National Health and Medical Research Council (NHMRC, to J.S.O.), St Vincent’s Institute of Medical Research (Australia), and the Victorian Government’s Operational Infrastructure Support Program. We are grateful to the UAB Diabetes Research Center (NIH P30 DK-079626) for providing core services.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111218.
REFERENCES
- Anderson NM, Mucka P, Kern JG, and Feng H (2018). The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 9, 216–237. 10.1007/s13238-017-0451-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrienko TN, Pasdois P, Pereira GC, Ovens MJ, and Halestrap AP (2017). The role of succinate and ROS in reperfusion injury - a critical appraisal. J. Mol. Cell. Cardiol. 110, 1–14. 10.1016/j.yjmcc.2017.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardella C, Pollard PJ, and Tomlinson I (2011). SDH mutations in cancer. Biochim. Biophys. Acta 1807, 1432–1443. 10.1016/j.bbabio.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Barros-Minones L, Martín-de-Saavedra D, Perez-Alvarez S, Orejana L, Suquía V, Goñi-Allo B, Hervias I, López MG, Jordan J, Aguirre N, and Puerta E. (2013). Inhibition of calpain-regulated p35/cdk5 plays a central role in sildenafil-induced protection against chemical hypoxia produced by malonate. Biochim. Biophys. Acta 1832, 705–717. 10.1016/j.bbadis.2013.02.002. [DOI] [PubMed] [Google Scholar]
- Baysal BE, and Maher ER (2015). 15 YEARS OF PARAGANGLIOMA: Genetics and mechanism of pheochromocytoma-paraganglioma syndromes characterized by germline SDHB and SDHD mutations. Endocr Relat Cancer 22, T71–T82. [DOI] [PubMed] [Google Scholar]
- Bettayeb K, Baunbæk D, Delehouze C, Loaëc N, Hole AJ, Baumli S, Endicott JA, Douc-Rasy S, Bénard J, Oumata N, et al. (2010). CDK inhibitors roscovitine and CR8 trigger Mcl-1 down-regulation and apoptotic cell death in neuroblastoma cells. Genes Cancer 1, 369–380. 10.1177/1947601910369817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuniya D, Umrani D, Dave B, Salunke D, Kukreja G, Gundu J, Naykodi M, Shaikh NS, Shitole P, Kurhade S, et al. (2011). Discovery of a potent and selective small molecule hGPR91 antagonist. Bioorg. Med. Chem. Lett. 21, 3596–3602. 10.1016/j.bmcl.2011.04.091. [DOI] [PubMed] [Google Scholar]
- Bibb JA, Snyder GL, Nishi A, Yan Z, Meijer L, Fienberg AA, Tsai LH, Kwon YT, Girault JA, Czernik AJ, et al. (1999). Phosphorylation of DARPP-32 by Cdk5 modulates dopamine signalling in neurons. Nature 402, 669–671. 10.1038/45251. [DOI] [PubMed] [Google Scholar]
- Blank A, Schmitt AM, Korpershoek E, van Nederveen F, Rudolph T, Weber N, Strebel RT, de Krijger R, Komminoth P, and Perren A (2010). SDHB loss predicts malignancy in pheochromocytomas/sympathethic paragangliomas, but not through hypoxia signalling. Endocr. Relat. Cancer 17, 919–928. 10.1677/ERC-09-0316. [DOI] [PubMed] [Google Scholar]
- Broekman F, Giovannetti E, and Peters GJ (2011). Tyrosine kinase inhibitors: Multi-targeted or single-targeted? World J. Clin. Oncol. 2, 80–93. 10.5306/wjco.v2.i2.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffet A, Venisse A, Nau V, Roncellin I, Boccio V, Le Pottier N, Boussion M, Travers C, Simian C, Burnichon N, et al. (2012). A decade (2001–2010) of genetic testing for pheochromocytoma and paraganglioma. Horm Metab Res 44, 359–366. [DOI] [PubMed] [Google Scholar]
- Bujard H (1999). Controlling genes with tetracyclines. J. Gene Med. 1, 372–374. . [DOI] [PubMed] [Google Scholar]
- Cao Y, Bojjireddy N, Kim M, Li T, Zhai P, Nagarajan N, Sadoshima J, Palmiter RD, and Tian R (2017). Activation of gamma2-AMPK suppresses ribosome biogenesis and protects against myocardial ischemia/reperfusion injury. Circ. Res. 121, 1182–1191. 10.1161/CIRCRESAHA.117.311159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AM, Tan C, Pozo K, Telange R, Molinaro R, Guo A, De Rosa E, Martinez JO, Zhang S, Kumar N, et al. (2020). Phosphoprotein-based biomarkers as predictors for cancer therapy. Proc. Natl. Acad. Sci. USA 117, 18401–18411. 10.1073/pnas.2010103117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascon A, Montero-Conde C, Ruiz-Llorente S, Mercadillo F, Letón R, Rodríguez-Antona C, Martínez-Delgado B, Delgado M, Díez A, Rovira A, et al. (2006). Gross SDHB deletions in patients with paraganglioma detected by multiplex PCR: a possible hot spot? Genes Chromosomes Cancer 45, 213–219. 10.1002/gcc.20283. [DOI] [PubMed] [Google Scholar]
- Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK, and Varambally S (2017). UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 19, 649–658. 10.1016/j.neo.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J (2016). The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med. 6, a026104. 10.1101/cshperspect.a026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo-Biel N, Camins A, Pallàs M, and Canudas AM (2009). Evidence of calpain/cdk5 pathway inhibition by lithium in 3-nitropropionic acid toxicity in vivo and in vitro. Neuropharmacology 56, 422–428. 10.1016/j.neuropharm.2008.09.012. [DOI] [PubMed] [Google Scholar]
- Crespo-Biel N, Camins A, Pelegrí C, Vilaplana J, Pallàs M, and Canudas AM. (2007). 3-Nitropropionic acid activates calpain/cdk5 pathway in rat striatum. Neurosci. Lett. 421, 77–81. 10.1016/j.neulet.2007.05.038. [DOI] [PubMed] [Google Scholar]
- Cruz JC, Tseng HC, Goldman JA, Shih H, and Tsai LH (2003). Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 40, 471–483. 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- Dahia PL (2014). Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat. Rev. Cancer 14, 108–119. 10.1038/nrc3648. [DOI] [PubMed] [Google Scholar]
- Dahia PL, Ross KN, Wright ME, Hayashida CY, Santagata S, Barontini M, Kung AL, Sanso G, Powers JF, Tischler AS, et al. (2005). A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 1, 72–80. 10.1371/journal.pgen.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkelenkamp K, Osinga TE, Links TP, and van der Horst-Schrivers ANA (2020). Clinical implications of the oncometabolite succinate in SDHx-mutation carriers. Clin. Genet. 97, 39–53. 10.1111/cge.13553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhofer G, Lenders JW, Linehan WM, Walther MM, Goldstein DS, and Keiser HR (1999). Plasma normetanephrine and metanephrine for detecting pheochromocytoma in von Hippel-Lindau disease and multiple endocrine neoplasia type 2. N. Engl. J. Med. 340, 1872–1879. 10.1056/NEJM199906173402404. [DOI] [PubMed] [Google Scholar]
- Erba E, Ubezio P, Pepe S, Vaghi M, Marsoni S, Torri W, Mangioni C, Landoni F, and D’Incalci M (1989). Flow cytometric analysis of DNA content in human ovarian cancers. Br. J. Cancer 60, 45–50. 10.1038/bjc.1989.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erez A, Shchelochkov OA, Plon SE, Scaglia F, and Lee B (2011). Insights into the pathogenesis and treatment of cancer from inborn errors of metabolism. Am. J. Hum. Genet. 88, 402–421. 10.1016/j.ajhg.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evenepoel L, Helaers R, Vroonen L, Aydin S, Hamoir M, Maiter D, Vikkula M, and Persu A (2017). KIF1B and NF1 are the most frequently mutated genes in paraganglioma and pheochromocytoma tumors. Endocr. Relat. Cancer 24, L57–L61. 10.1530/ERC-17-0061. [DOI] [PubMed] [Google Scholar]
- Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, et al. (2013). AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 17, 113–124. 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favier J, Brière JJ, Burnichon N, Rivière J, Vescovo L, Benit P, GiscosDouriez I, De Reyniès A, Bertherat J, Badoual C, et al. (2009). The Warburg effect is genetically determined in inherited pheochromocytomas. PLoS One 4, e7094. 10.1371/journal.pone.0007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishbein L, Khare S, Wubbenhorst B, DeSloover D, D’Andrea K, Merrill S, Cho NW, Greenberg RA, Else T, Montone K, et al. (2015). Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat. Commun. 6, 6140. 10.1038/ncomms7140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliedner SM, Breza J, Kvetnansky R, Powers JF, Tischler AS, Wesley R, Merino M, Lehnert H, and Pacak K (2010). Tyrosine hydroxylase, chromogranin A, and steroidogenic acute regulator as markers for successful separation of human adrenal medulla. Cell Tissue Res. 340, 607–612. 10.1007/s00441-010-0965-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund A, Patil CK, and Campisi J (2011). p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 30, 1536–1548. 10.1038/emboj.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1. 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia D, and Shaw RJ (2017). AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 66, 789–800. 10.1016/j.molcel.2017.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge SX, Jung D, and Yao R (2020). ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629. 10.1093/bioinformatics/btz931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghayee HK, Bhagwandin VJ, Stastny V, Click A, Ding LH, Mizrachi D, Zou YS, Chari R, Lam WL, Bachoo RM, et al. (2013). Progenitor cell line (hPheo1) derived from a human pheochromocytoma tumor. PLoS One 8, e65624. 10.1371/journal.pone.0065624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilissen J, Jouret F, Pirotte B, and Hanson J (2016). Insight into SUCNR1 (GPR91) structure and function. Pharmacol. Ther. 159, 56–65. 10.1016/j.pharmthera.2016.01.008. [DOI] [PubMed] [Google Scholar]
- Gimenez-Roqueplo AP, Favier J, Rustin P, Rieubland C, Crespin M, Nau V, Khau Van Kien P, Corvol P, Plouin PF, and Jeunemaitre X; COMETE Network (2003). Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 63, 5615–5621. [PubMed] [Google Scholar]
- Gimenez-Roqueplo AP, Favier J, Rustin P, Rieubland C, Kerlan V, Plouin PF, Rötig A, and Jeunemaitre X (2002). Functional consequences of a SDHB gene mutation in an apparently sporadic pheochromocytoma. J. Clin. Endocrinol. Metab. 87, 4771–4774. 10.1210/jc.2002-020525. [DOI] [PubMed] [Google Scholar]
- Goldstein M, Fuxe K, and Hökfelt T (1972). Characterization and tissue localization of catecholamine synthesizing enzymes. Pharmacol. Rev. 24, 293–309. [PubMed] [Google Scholar]
- Gottlieb E, and Tomlinson IPM (2005). Mitochondrial tumour suppressors: a genetic and biochemical update. Nat. Rev. Cancer 5, 857–866. 10.1038/nrc1737. [DOI] [PubMed] [Google Scholar]
- Gowans GJ, Hawley SA, Ross FA, and Hardie DG (2013). AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 18, 556–566. 10.1016/j.cmet.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachet-Haas M, Converset N, Marchal O, Matthes H, Gioria S, Galzi JL, and Lecat S (2006). FRET and colocalization analyzer–a method to validate measurements of sensitized emission FRET acquired by confocal microscopy and available as an ImageJ Plug-in. Microsc. Res. Tech. 69, 941–956. 10.1002/jemt.20376. [DOI] [PubMed] [Google Scholar]
- Hadrava Vanova K, Kraus M, Neuzil J, and Rohlena J (2020). Mitochondrial complex II and reactive oxygen species in disease and therapy. Redox Rep. 25, 26–32. 10.1080/13510002.2020.1752002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2000). The hallmarks of cancer. Cell 100, 57–70. 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hardie DG (2014). AMPK–sensing energy while talking to other signaling pathways. Cell Metab. 20, 939–952. 10.1016/j.cmet.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, and Hardie DG (2005). Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2, 9–19. 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Hemmings HC (1997). Regulatory Protein Modification: Techniques and Protocols (Springer; ). [Google Scholar]
- Hsu PP, and Sabatini DM (2008). Cancer cell metabolism: Warburg and beyond. Cell 134, 703–707. 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- Huang Y, Wang LA, Xie Q, Pang J, Wang L, Yi Y, Zhang J, Zhang Y, Chen R, Lan W, et al. (2018). Germline SDHB and SDHD mutations in pheochromocytoma and paraganglioma patients. Endocr. Connect. 7, 1217–1225. 10.1530/EC-18-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana F, Bustos G, Rivas J, Cruz P, Urra F, Basualto-Alarcón C, Sagredo E, Ríos M, Lovy A, Dong Z, et al. (2019). Complex I and II are required for normal mitochondrial Ca(2+) homeostasis. Mitochondrion 49, 73–82. 10.1016/j.mito.2019.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanmonod R, Asuka E, and Jeanmonod D (2020). Inborn Errors of Metabolism (StatPearls; ). [PubMed] [Google Scholar]
- Jochmanova I, and Pacak K (2016). Pheochromocytoma: the first metabolic endocrine cancer. Clin. Cancer Res. 22, 5001–5011. 10.1158/1078-0432.CCR-16-0606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, and Thompson CB (2005). AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 18, 283–293. 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Jung SH, Hwang HJ, Kang D, Park HA, Lee HC, Jeong D, Lee K, Park HJ, Ko YG, and Lee JS (2019). mTOR kinase leads to PTEN-loss-induced cellular senescence by phosphorylating p53. Oncogene 38, 1639–1650. 10.1038/s41388-018-0521-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, and Dang CV (2006). Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 66, 8927–8930. 10.1158/0008-5472.CAN-06-1501. [DOI] [PubMed] [Google Scholar]
- King KS, Prodanov T, Kantorovich V, Fojo T, Hewitt JK, Zacharin M, Wesley R, Lodish M, Raygada M, Gimenez-Roqueplo AP, et al. (2011). Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: significant link to SDHB mutations. J. Clin. Oncol. 29, 4137–4142. 10.1200/JCO.2011.34.6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluckova K, Thakker A, Vettore L, Escribano-Gonzalez C, Hindshaw RL, Tearle JLE, Goncalves J, Kaul B, Lavery GG, Favier J, and Tennant DA (2020). Succinate dehydrogenase deficiency in a chromaffin cell model retains metabolic fitness through the maintenance of mitochondrial NADH oxidoreductase function. FASEB J 34, 303–315. 10.1096/fj.201901456R. [DOI] [PubMed] [Google Scholar]
- Koh J, Hogue JA, Wang Y, DiSalvo M, Allbritton NL, Shi Y, Olson JA Jr., and Sosa JA (2016). Single-cell functional analysis of parathyroid adenomas reveals distinct classes of calcium sensing behaviour in primary hyperparathyroidism. J. Cell Mol. Med. 20, 351–359. 10.1111/jcmm.12732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpershoek E, Favier J, Gaal J, Burnichon N, van Gessel B, Oudijk L, Badoual C, Gadessaud N, Venisse A, Bayley JP, et al. (2011). SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J. Clin. Endocrinol. Metab. 96, E1472–E1476. 10.1210/jc.2011-1043. [DOI] [PubMed] [Google Scholar]
- Korpershoek E, Pacak K, and Martiniova L (2012). Murine models and cell lines for the investigation of pheochromocytoma: applications for future therapies? Endocr. Pathol. 23, 43–54. 10.1007/s12022-012-9194-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs HA, and Johnson WA (1980). The role of citric acid in intermediate metabolism in animal tissues. FEBS Lett. 117, K1–K10. [DOI] [PubMed] [Google Scholar]
- Lee JH, Jang H, Lee SM, Lee JE, Choi J, Kim TW, Cho EJ, and Youn HD (2015). ATP-citrate lyase regulates cellular senescence via an AMPK- and p53-dependent pathway. FEBS J. 282, 361–371. 10.1111/febs.13139. [DOI] [PubMed] [Google Scholar]
- Li H, Huang K, Liu X, Liu J, Lu X, Tao K, Wang G, and Wang J (2014). Lithium chloride suppresses colorectal cancer cell survival and proliferation through ROS/GSK-3beta/NF-kappaB signaling pathway. Oxid. Med. Cell. Longev. 2014, 241864. 10.1155/2014/241864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maignan A, Guerin C, Julliard V, Paladino NC, Kim E, Roche P, Castinetti F, Essamet W, Mancini J, Imperiale A, et al. (2017). Implications of SDHB genetic testing in patients with sporadic pheochromocytoma. Langenbeck’s Arch. Surg. 402, 787–798. 10.1007/s00423-017-1564-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao D, and Hinds PW (2010). p35 is required for CDK5 activation in cellular senescence. J. Biol. Chem. 285, 14671–14680. 10.1074/jbc.M109.066118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martiniova L, Lai EW, Elkahloun AG, Abu-Asab M, Wickremasinghe A, Solis DC, Perera SM, Huynh TT, Lubensky IA, Tischler AS, et al. (2009). Characterization of an animal model of aggressive metastatic pheochromocytoma linked to a specific gene signature. Clin. Exp. Metastasis 26, 239–250. 10.1007/s10585-009-9236-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlac DM, Hadrava Vanova K, Bechmann N, Richter S, Folberth J, Ghayee HK, Ge GB, Abunimer L, Wesley R, Aherrahrou R, et al. (2021). Succinate mediates tumorigenic effects via succinate receptor 1: potential for new targeted treatment strategies in succinate dehydrogenase deficient paragangliomas. Front. Endocrinol. 12, 589451. 10.3389/fendo.2021.589451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClue SJ, and Stuart I (2008). Metabolism of the trisubstituted purine cyclin-dependent kinase inhibitor seliciclib (R-roscovitine) in vitro and in vivo. Drug Metab. Dispos. 36, 561–570. 10.1124/dmd.107.019232. [DOI] [PubMed] [Google Scholar]
- McDermott U, Sharma SV, Dowell L, Greninger P, Montagut C, Lamb J, Archibald H, Raudales R, Tam A, Lee D, et al. (2007). Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc. Natl. Acad. Sci. USA 104, 19936–19941. 10.1073/pnas.0707498104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer DA, Richer E, Benkovic SA, Hayashi K, Kansy JW, Hale CF, Moy LY, Kim Y, O’Callaghan JP, Tsai LH, et al. (2008). Striatal dysregulation of Cdk5 alters locomotor responses to cocaine, motor learning, and dendritic morphology. Proc. Natl. Acad. Sci. USA 105, 18561–18566. 10.1073/pnas.0806078105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijit M, Caracciolo V, Melillo A, Amicarelli F, and Giordano A (2020). Role of p53 in the regulation of cellular senescence. Biomolecules 10, E420. 10.3390/biom10030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Brown H, Pant HC, Pigino G, DeBoer S, Beffert U, and Brady ST (2004). A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J. 23, 2235–2245. 10.1038/sj.emboj.7600237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasr P, Gursahani HI, Pang Z, Bondada V, Lee J, Hadley RW, and Geddes JW (2003). Influence of cytosolic and mitochondrial Ca2+, ATP, mitochondrial membrane potential, and calpain activity on the mechanism of neuron death induced by 3-nitropropionic acid. Neurochem. Int. 43, 89–99. 10.1016/s0197-0186(02)00229-2. [DOI] [PubMed] [Google Scholar]
- Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, Buchta M, Franke G, Klisch J, Bley TA, et al. (2004). Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 292, 943–951. 10.1001/jama.292.8.943. [DOI] [PubMed] [Google Scholar]
- Oakhill JS, Steel R, Chen ZP, Scott JW, Ling N, Tam S, and Kemp BE (2011). AMPK is a direct adenylate charge-regulated protein kinase. Science 332, 1433–1435. 10.1126/science.1200094. [DOI] [PubMed] [Google Scholar]
- Pang Z, Bondada V, Sengoku T, Siman R, and Geddes JW (2003). Calpain facilitates the neuron death induced by 3-nitropropionic acid and contributes to the necrotic morphology. J. Neuropathol. Exp. Neurol. 62, 633–643. 10.1093/jnen/62.6.633. [DOI] [PubMed] [Google Scholar]
- Pap M, and Cooper GM (1998). Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J. Biol. Chem. 273, 19929–19932. 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, and Tsai LH (1999). Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402, 615–622. 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- Pinter K, Grignani RT, Watkins H, and Redwood C (2013). Localisation of AMPK gamma subunits in cardiac and skeletal muscles. J. Muscle Res. Cell Motil. 34, 369–378. 10.1007/s10974-013-9359-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner F, Angelo M, and Giese KP (2006). The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J. Biol. Chem. 281, 25457–25465. 10.1074/jbc.M603469200. [DOI] [PubMed] [Google Scholar]
- Pollard PJ, El-Bahrawy M, Poulsom R, Elia G, Killick P, Kelly G, Hunt T, Jeffery R, Seedhar P, Barwell J, et al. (2006). Expression of HIF-1alpha, HIF-2alpha (EPAS1), and their target genes in paraganglioma and pheochromocytoma with VHL and SDH mutations. J. Clin. Endocrinol. Metab. 91, 4593–4598. 10.1210/jc.2006-0920. [DOI] [PubMed] [Google Scholar]
- Pozo K, and Bibb JA (2016). The emerging role of Cdk5 in cancer. Trends Cancer 2, 606–618. 10.1016/j.trecan.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozo K, Castro-Rivera E, Tan C, Plattner F, Schwach G, Siegl V, Meyer D, Guo A, Gundara J, Mettlach G, et al. (2013). The role of Cdk5 in neuroendocrine thyroid cancer. Cancer Cell 24, 499–511. 10.1016/j.ccr.2013.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai SK, Bril F, Hatch HM, Xu Y, Shelton L, Kalavalapalli S, Click A, Lee D, Beecher C, Kirby A, et al. (2020). Targeting pheochromocytoma/paraganglioma with polyamine inhibitors. Metabolism 110, 154297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajgopal Y, and Vemuri MC (2002). Calpain activation and alpha-spectrin cleavage in rat brain by ethanol. Neurosci. Lett. 321, 187–191. 10.1016/s0304-3940(02)00063-0. [DOI] [PubMed] [Google Scholar]
- Ranganayaki S, Jamshidi N, Aiyaz M, Rashmi SK, Gayathri N, Harsha PK, Padmanabhan B, and Srinivas Bharath MM (2021). Inhibition of mitochondrial complex II in neuronal cells triggers unique pathways culminating in autophagy with implications for neurodegeneration. Sci. Rep. 11, 1483. 10.1038/s41598-020-79339-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross ME, Evinger MJ, Hyman SE, Carroll JM, Mucke L, Comb M, Reis DJ, Joh TH, and Goodman HM (1990). Identification of a functional glucocorticoid response element in the phenylethanolamine N-methyltransferase promoter using fusion genes introduced into chromaffin cells in primary culture. J. Neurosci. 10, 520–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossitti HM, Dutta RK, Larsson C, Ghayee HK, Söderkvist P, and Gimm O. (2020). Activation of RAS signalling is associated with altered cell adhesion in phaeochromocytoma. Int. J. Mol. Sci. 21, E8072. 10.3390/ijms21218072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, and Cantley LC (2004). The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 101, 3329–3335. 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis DC, Burnichon N, Timmers HJLM, Raygada MJ, Kozupa A, Merino MJ, Makey D, Adams KT, Venisse A, Gimenez-Roqueplo AP, and Pacak K (2009). Penetrance and clinical consequences of a gross SDHB deletion in a large family. Clin. Genet. 75, 354–363. 10.1111/j.1399-0004.2009.01157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockholm D, Bartoli M, Sillon G, Bourg N, Davoust J, and Richard I (2005). Imaging calpain protease activity by multiphoton FRET in living mice. J. Mol. Biol. 346, 215–222. 10.1016/j.jmb.2004.11.039. [DOI] [PubMed] [Google Scholar]
- Sun KH, de Pablo Y, Vincent F, and Shah K (2008). Deregulated Cdk5 promotes oxidative stress and mitochondrial dysfunction. J. Neurochem. 107, 265–278. 10.1111/j.1471-4159.2008.05616.x. [DOI] [PubMed] [Google Scholar]
- Sundstrom L, Greasley PJ, Engberg S, Wallander M, and Ryberg E (2013). Succinate receptor GPR91, a Galpha(i) coupled receptor that increases intracellular calcium concentrations through PLCbeta. FEBS Lett. 587, 2399–2404. 10.1016/j.febslet.2013.05.067. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Bridges D, Nakada D, Skiniotis G, Morrison SJ, Lin JD, Saltiel AR, and Inoki K (2013). Inhibition of AMPK catabolic action by GSK3. Mol. Cell 50, 407–419. 10.1016/j.molcel.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang QL, Xie XB, Wang J, Chen Q, Han AJ, Zou CY, Yin JQ, Liu DW, Liang Y, Zhao ZQ, et al. (2012). Glycogen synthase kinase-3beta, NF-kappaB signaling, and tumorigenesis of human osteosarcoma. J. Natl. Cancer Inst. 104, 749–763. 10.1093/jnci/djs210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Li C, Kang B, Gao G, Li C, and Zhang Z (2017). GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45, W98–W102. 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen CC, and Sabatini DM (2005). AMPK and p53 help cells through lean times. Cell Metab. 1, 287–288. 10.1016/j.cmet.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varghese F, Bukhari AB, Malhotra R, and De A (2014). IHC Profiler: an open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS One 9, e96801. 10.1371/journal.pone.0096801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vial G, Detaille D, and Guigas B (2019). Role of mitochondria in the mechanism(s) of action of metformin. Front. Endocrinol. 10, 294. 10.3389/fendo.2019.00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Liang B, Shirwany NA, and Zou MH (2011). 2-Deoxy-D-glucose treatment of endothelial cells induces autophagy by reactive oxygen species-mediated activation of the AMP-activated protein kinase. PLoS One 6, e17234. 10.1371/journal.pone.0017234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Thatcher SE, and Cassis LA (2017). Measuring blood pressure using a noninvasive tail cuff method in mice. Methods Mol. Biol. 1614, 69–73. 10.1007/978-1-4939-7030-8_6. [DOI] [PubMed] [Google Scholar]
- Wen Y, Planel E, Herman M, Figueroa HY, Wang L, Liu L, Lau LF, Yu WH, and Duff KE (2008). Interplay between cyclin-dependent kinase 5 and glycogen synthase kinase 3 beta mediated by neuregulin signaling leads to differential effects on tau phosphorylation and amyloid precursor protein processing. J. Neurosci. 28, 2624–2632. 10.1523/JNEUROSCI.5245-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JY, Huang TW, Hsieh YT, Wang YF, Yen CC, Lee GL, Yeh CC, Peng YJ, Kuo YY, Wen HT, et al. (2020). Cancer-derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol. Cell 77, 213–227.e5. 10.1016/j.molcel.2019.10.023. [DOI] [PubMed] [Google Scholar]
- Xie Z, Dong Y, Scholz R, Neumann D, and Zou MH (2008). Phosphorylation of LKB1 at serine 428 by protein kinase C-zeta is required for metforminenhanced activation of the AMP-activated protein kinase in endothelial cells. Circulation 117, 952–962. 10.1161/CIRCULATIONAHA.107.744490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Gray A, Hardie DG, Uzun A, Shaw S, Padbury J, Phornphutkul C, and Tseng YT (2017). A novel, de novo mutation in the PRKAG2 gene: infantile-onset phenotype and the signaling pathway involved. Am. J. Physiol. Heart Circ. Physiol. 313, H283–H292. 10.1152/ajpheart.00813.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, and Lowe SW (2007). Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660. 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Matro JC, Huntoon KM, Ye DY, Huynh TT, Fliedner SMJ, Breza J, Zhuang Z, and Pacak K (2012). Missense mutations in the human SDHB gene increase protein degradation without altering intrinsic enzymatic function. FASEB J 26, 4506–4516. 10.1096/fj.12-210146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadra G, Batista JL, and Loda M (2015). Dissecting the dual role of AMPK in cancer: from experimental to human studies. Mol. Cancer Res. 13, 1059–1072. 10.1158/1541-7786.MCR-15-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y, Sun X, Li B, Cai H, Xu C, Liang Q, Lu C, Qian R, Chen S, Yin L, et al. (2018). Establishment of a PRKAG2 cardiac syndrome disease model and mechanism study using human induced pluripotent stem cells. J. Mol. Cell. Cardiol. 117, 49–61. 10.1016/j.yjmcc.2018.02.007. [DOI] [PubMed] [Google Scholar]
- Zhang S, Lu Z, Mao W, Ahmed AA, Yang H, Zhou J, Jennings N, Rodriguez-Aguayo C, Lopez-Berestein G, Miranda R, et al. (2015). CDK5 regulates paclitaxel sensitivity in ovarian cancer cells by modulating AKT activation, p21Cip1- and p27Kip1-mediated G1 cell cycle arrest and apoptosis. PLoS One 10, e0131833. 10.1371/journal.pone.0131833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. (2001). Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108, 1167–1174. 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.