

Abstract
Objective:
We aimed to characterize cortical superficial siderosis, its determinants and sequel, in community-dwelling older adults.
Methods:
The sample consisted of Framingham (n = 1724; 2000–2009) and Rotterdam (n = 4325; 2005–2013) study participants who underwent brain MRI. In pooled individual-level analysis, we compared baseline characteristics in patients with cortical superficial siderosis to two reference groups: (i) persons without hemorrhagic MRI markers of cerebral amyloid angiopathy (no cortical superficial siderosis and no microbleeds) and (ii) those with presumed cerebral amyloid angiopathy based on the presence of strictly lobar microbleeds but without cortical superficial siderosis.
Results:
Among a total of 6049 participants, 4846 did not have any microbleeds or cortical superficial siderosis (80%), 401 had deep/mixed microbleeds (6.6%), 776 had strictly lobar microbleeds without cortical superficial siderosis (12.8%) and 26 had cortical superficial siderosis with/without microbleeds (0.43%). In comparison to participants without microbleeds or cortical superficial siderosis and to those with strictly lobar microbleeds but without cortical superficial siderosis, participants with cortical superficial siderosis were older (OR 1.09 per year, 95% CI 1.05, 1.14; p < 0.001 and 1.04, 95% CI 1.00, 1.09; p = 0.058, respectively), had overrepresentation of the APOE ε4 allele (5.19, 2.04, 13.25; p = 0.001 and 3.47, 1.35, 8.92; p = 0.01), and greater prevalence of intracerebral hemorrhage (72.57, 9.12, 577.49; p < 0.001 and 81.49, 3.40, >999.99; p = 0.006). During a mean follow-up of 5.6 years, 42.4% participants with cortical superficial siderosis had a stroke (five intracerebral hemorrhage, two ischemic strokes and four undetermined strokes), 19.2% had transient neurological deficits and 3.8% developed incident dementia.
Conclusion:
Our study adds supporting evidence to the association between cortical superficial siderosis and cerebral amyloid angiopathy within the general population. Community-dwelling persons with cortical superficial siderosis may be at high risk for intracerebral hemorrhage and future neurological events.
Keywords: Brain microbleeds, cerebral amyloid angiopathy, cerebral hemorrhage, community, cortical superficial siderosis, stroke facilities
Introduction
Cortical superficial siderosis (cSS) is increasingly recognized as an imaging marker of cerebral amyloid angiopathy (CAA) in clinical settings. In these hospital-based cohorts, cSS seems to be a robust indicator of increased risk of future intracerebral hemorrhage (ICH)1 and may mark underlying vasculopathic changes prone to vessel rupture.2,3
Lobar cerebral microbleeds (CMBs) are another MRI marker of CAA, and incorporated in the clinico-diagnostic Boston Criteria to diagnose patients 55 years of age or greater with ‘probable CAA’ without tissue biopsy.4 Using lobar CMBs to fulfill these criteria has demonstrated an 88% positive predictive value for histopathologically confirmed CAA in a hospital-based cohort.5 Conversely, the diagnostic value of lobar CMBs for CAA in community-dwelling populations seems rather limited (25% positive predictive value), highlighting the need to identify more specific MRI markers of CAA in the community.5
The modified Boston Criteria have demonstrated improved sensitivity for CAA diagnosis in hospital cohorts through the addition of cSS.6 Preliminary reports have suggested a cSS prevalence of 0.5–0.9% in community-dwelling individuals7–9; however, the underlying pathology and prognostic implications of cSS in the general population remain elusive. We aimed to characterize cSS, its determinants and sequela, in community-dwelling older adults and in particular in comparison to individuals with strictly lobar CMBs, by combining individual-level data from two large population cohorts.
Materials and methods
Sample
Framingham Original and Offspring Cohort participants and Rotterdam Study participants greater than 55 years of age who underwent brain MRI with T2*-weighted imaging allowing for cSS and CMB detection were eligible. Patients with a reported prior history of traumatic brain injury were excluded. The sample consisted of Framingham Study participants (n = 1724) who underwent brain MRI between 2000 and 2009, and Rotterdam Study participants (n = 4325) who underwent brain MRI between 2005 and 2013, for a total of 6049 participants. The institutional review boards of the Boston University Medical Center and Erasmus MC approved the study protocol and informed consent was obtained from all participants.
Vascular risk factors
In the Framingham Study and Rotterdam Study, vascular risk factors were assessed at the exam cycle closest to MRI. In the Framingham Study, hypertension was defined by the JNC-7 classification (SBP≥140 mm Hg and/or DBP≥90 mm Hg, or use of antihypertensive medications). Total cholesterol was measured on fasting specimens in the Offspring cohort, and random samples in the Original cohort. Medication use was assessed by self-report. In the Rotterdam Study, blood pressure measurements were averaged over two readings using a random zero sphygmomanometer. Hypertension was defined as a blood pressure of >140 mmHg systolic or >90 mmHg diastolic, or the use of blood pressure lowering medication. Serum total cholesterol was measured using an automated enzymatic procedure. Lipid-lowering medication use was assessed in home interviews. Pharmacy records were used to determine the use of antithrombotic medication (ATC code B01A).
MRI acquisition
Framingham Heart Study participants were imaged using a 1.5-tesla MRI scanner (Siemens Magnetom). T2*gradient echo sequences were obtained with the following parameters: repetition time 656 ms, echo time 26 ms, acquisition matrix 144×256, field of view 22 cm, 30° flip angle, and 19 slices of 5 mm thickness, and 2 mm gap. Rotterdam Study participants were imaged using a 1.5-Tesla MRI scanner (GE Healthcare, Milwaukee, WI). 3D T2*-gradient echo weighted images were obtained with the following parameters: repetition time 45 ms, echo time 31 ms, acquisition matrix 320×244, field of view 25×17.5 cm2, 13° flip angle, 96 slices of 1.6 mm thickness zero padded to 192 slices of 0.8 mm.
MRI analysis
We determined the volume of WMH according to previously published methods.10 We manually determined lacunes on the basis of their size (>3 mm, <15 mm) and imaging characteristics, as previously described.11 Total cerebral brain volumes as a percentage of cranial volume were calculated in semi-manual (Framingham Heart Study) or fully automated (Rotterdam Study) methods described elsewhere.10–13
Cerebral microbleeds were defined and cSS rated as per previously described methods.2,7,14,15 Sulci with cSS congruent with region of previous macrohemorrhage were excluded. Investigators in both cohorts have previously demonstrated excellent inter-rater reliability for the presence of CMBs and cSS.2,14,15 Participants were categorized into four groups according to their cSS and CMB profiles. Group A consisted of participants without cSS or CMBs on MRI; Group B consisted of participants with mixed or deep CMBs and without cSS, Group C consisted of participants with strictly lobar CMBs and without cSS and Group D consisted of participants with cSS with or without concurrent CMBs.
Operators were blinded to the subject’s demographic, clinical, and genetic characteristics. A cSS topography map was created through manual delineation of visualized cSS in each participant, followed by merger of all images into one probability map.
Apolipoprotein E status
Genotyping for apolipoprotein E (APOE) status was performed as previously described and available in a total of 5745 (95%) participants.14,16 APOE allele frequencies were calculated by determining the proportion of a given allele among all APOE alleles within the particular subgroup of interest. For multivariate regression analysis, APOE status was categorized as any ε2 (one or more ε2), any ε4 (1 or more ε4) and ε3/ε3 (reference).
Clinical outcomes
Stroke and transient ischemic attack (TIA) surveillance methods and protocol for determining the diagnosis and type of stroke (ischemic versus hemorrhagic) have previously been published for the Framingham and Rotterdam Studies.17–19 Strokes that occurred before the first research MRI were coded as prevalent strokes. New strokes that occurred after the first research MRI were identified as incident strokes.
Mild cognitive impairment (MCI) was classified as involving memory, executive function, or either one, using standardized scores. Memory was assessed using logical memory delayed recall and impairment was defined as performance below 1.5 standard deviations (SD). Executive function was assessed using Trails B-A (the difference in time between Trail-making B and Trail-making A tests) and impairment was defined as performance below 1.5 SD. MCI was defined by the presence of any combination of impairment.
Statistical analysis
Baseline characteristics of study participants were evaluated by cSS status, presented in Table 1.
Table 1.
Demographic variables according to CMB/cSS grouping (participants’ age: 55+ years)
| Group A: No CMBs/No cSS |
Group B: CMBs: Deep/infra and Mixed/No cSS |
Group C: CMBs Lobar ONLY/No cSS |
Group D: cSS/With or without CMBsa |
P b | |
|---|---|---|---|---|---|
| Number | 4846 | 401 | 776 | 26 | |
| Risk factors | |||||
| Age at MRI, years (mean, SD) | 67.9 ± 9.0 | 74.2 ± 8.7 | 71.8 ± 9.4 | 75.9 ± 8.3 | |
| Time between MRI and Exam, years (mean, SD) | 0.9 ± 1.9 | 1.3 ± 2.4 | 1.0 ± 2.2 | 0.1 ± 2.7 | |
| Male (n, %) | 2159 (45%) | 208 (52%)* | 357 (46%) | 15 (58%) | 0.005 |
| Hypertension (n, %) | 3060 (64%) | 306 (78%)* | 533 (70%) | 20 (87%) | 0.008 |
| Total cholesterol, mg/dL (mean, SD) | 207.5 ± 41.4 | 196.9 ± 42.8*,*** | 205.2 ± 44.2 | 206.0 ± 52.5 | 0.025 |
| Medications | |||||
| Antithrombotic use (antiplatelet and/or anticoagulation) (n, %) | 1507 (31%) | 228 (57%)*,*** | 330 (43%)* | 11 (42%) | <0.001 |
| Statin use (n, %) | 1398 (29%) | 160 (41%)* | 263 (34%) | 10 (45%) | <0.001 |
| Prevalent clinical outcomes | |||||
| Ischemic stroke (n, %) | 15 (3%) | 41 (10%) | 37 (5%) | 1 (4%) | 0.107 |
| ICH (n, %) | 13 (0.2%) | 0 | 3 (0.4%) | 5 (19%)*,*** | <0.001 |
| TIA (n, %) | 253/3431 (7%) | 54/354 (15%) | 60/694 (9%) | 4/22 (18%) | 0.079 |
| MCI Executive Dysfunction (n, %) | 299 (6%) | 36 (10%) | 46 (6%) | 2 (8%) | 0.211 |
| MCI Memory impairment (n, %) | 185 (4%) | 19 (5%) | 32 (5%) | 2 (8%) | 0.951 |
| APOE genotype (n, %) | |||||
| ε22 | 23 (0.5%) | 2 (0.5%) | 11 (1.5%)* | 0 | 0.001 |
| ε23 | 619 (13.4%) | 40 (10.5%) | 84 (11.4%) | 3 (13.1%) | |
| ε24 | 104 (2.3%) | 13 (3.4%) | 18 (2.5%) | 2 (8.7%) | |
| ε33 | 2785 (60.4%) | 235 (61.7%) | 407 (55.7%) | 7 (30.4%) | |
| ε34 | 980 (21.3%) | 85 (22.3%) | 190 (26.0%) | 7 (30.4%) | |
| ε44 | 99 (2.1%) | 6 (1.6%) | 21 (2.9%) | 4 (17.4%) | |
| ε2 minor allele frequency | 769 (8.3%) | 57 (7.5%) | 124 (8.5%)* | 5 (10.9%)* | |
| ε4 minor allele frequency | 1282 (13.9%) | 110 (14.4%) | 250 (17.1%) | 17 (37.0%) | |
| ε3 minor allele frequency | 7169 (77.8%) | 595 (78.1%) | 1088 (74.4%) | 24 (52.2%) | |
| MRI markers | |||||
| Extensive WMH (n, %) | 760 (16%) | 130 (32%)*,*** | 153 (20%)* | 5 (19%) | <0.001 |
| Lacunes (n, %) | 415 (9%) | 114 (28%)*,*** | 92 (12%) | 4 (15%) | <0.001 |
| TCBV(Mean, SD) | 80.6 ± 4.4 | 79.2 ± 4.5 | 80.5 ± 4.6a | 77.9 ± 4.2 | <0.001 |
APOE: apolipoprotein E; CMB: cerebral microbleed; cSS: cortical superficial siderosis; ICH: intracerebral hemorrhage; MCI: mild cognitive impairment; MRI: magnetic resonance imaging; SD: standard deviation; TCBV: total intracranial volume; TIA: transient ischemic attack; WMH: white matter hyperintensities.
No CMB 12/24; Any CMB = 12/24 (only deep = 0, strictly lobar = 10, mixed = 2).
Age-adjusted global p-values from global tests comparing the four groups using a series of age-adjusted linear (for continuous outcomes) or logistic (for categorical outcomes) models. Results of post-hoc pairwise comparisons made using the Tukey-Kramer method are indicated as per below where significant.
P < 0.05 relative to Group A.
P < 0.05 relative to Group B.
P < 0.05 relative to Group C.
We natural log transformed the ratio of WMH volume to total cranial volume and calculated SDUs by standardizing within 5-year age groups norms. Those with SDU>1 were classified as having extensive WMH.11 Age-adjusted analyses were performed to assess for differences in baseline characteristics across all four groups using linear or logistic models as appropriate. Intergroup multiple comparisons were made using Tukey-Kramer method. We then used separate logistic regression analyses to obtain odds ratios (OR) and 95% confidence intervals (95% CI) for determinants of cSS presence using two reference groups: (i) persons without hemorrhagic MRI markers of CAA (group A) and (ii) those with presumed CAA based on presence of strictly lobar microbleeds in the absence of cSS (group C). Analyses were adjusted for age, sex, and cohort (Table 2). We further used a series of logistic regression analyses to assess relationships between participants in group D versus group A and group C, respectively, and each of prevalent ischemic stroke (IS), intracerebral hemorrhage (ICH), TIA and MCI. Two multivariable models were evaluated: model 1, adjusted for age and sex; model 2, additionally adjusted for ischemic cerebral small vessel disease markers on MRI (lacunes and extensive WMH). All statistical analyses were performed using SAS version 9.4 (SAS Institute Inc., Cary, NC). Two-tailed p < 0.05 was considered statistically significant for the analysis.
Table 2.
Determinants of cSS presence
| Global P-valuesa | Group D vs. group A | Group D vs. group C | |||||
|---|---|---|---|---|---|---|---|
| ORa | 95% CI | P | ORa | 95% CI | P | ||
| Age (per year) | <0.001 | 1.09 | 1.05, 1.14 | <0.001 | 1.04 | 1.00, 1.09 | 0.058 |
| Male | <0.001 | 1.70 | 0.78, 3.71 | 0.183 | 1.43 | 0.64, 3.20 | 0.379 |
| Hypertension | <0.001 | 2.64 | 0.77, 9.06 | 0.123 | 2.47 | 0.71, 8.56 | 0.154 |
| Total cholesterol (per mg/dL) | <0.001 | 1.00 | 0.99, 1.01 | 0.735 | 1.01 | 1.00, 1.02 | 0.366 |
| Antithrombotic use | <0.001 | 0.93 | 0.41,2.10 | 0.855 | 0.76 | 0.34, 1.73 | 0.517 |
| Statin use | <0.001 | 1.88 | 0.80, 4.40 | 0.146 | 1.41 | 0.59, 3.34 | 0.436 |
| APOE ε2bAny ε2 vs. ε33 | <0.001 | 2.73 | 0.86,8.67 | 0.088 | 2.65 | 0.82,8.55 | 0.103 |
| APOE ε4cAny ε4 vs. ε33 | <0.001 | 5.19 | 2.04,13.15 | 0.001 | 3.47 | 1.35,8.92 | 0.010 |
| Extensive WMH | <0.001 | 1.41 | 0.53, 3.78 | 0.494 | 0.87 | 0.32, 2.39 | 0.794 |
| Lacunes | <0.001 | 1.21 | 0.41, 3.61 | 0.731 | 1.00 | 0.33, 3.05 | 0.996 |
| TCBV | <0.001 | 0.97 | 0.86, 1.10 | 0.645 | 0.96 | 0.85, 1.08 | 0.476 |
Note: For description of groups see Table 1. OR: odds ratio; CI: confidence interval; APOE: apolipoprotein E; cSS: cortical superficial siderosis; TCBV: total cranial brain volume; WMH: white matter hyperintensities.
Model is adjusted for age at MRI, sex and cohort.
Coded as 0 for ε33 and 1 for any ε2.
Coded as 0 for ε33 and 1 for any ε4.
Data availability
Anonymized data will be shared by request from any qualified investigator.
Results
Among a total of 6049 participants, 4846 did not have any CMBs or cSS (Group A; 80%), 401 had deep/mixed CMBs suggested of hypertensive arteriopathy (Group B; 6.6%), 776 had strictly lobar CMBs without cSS (Group C; 12.8%) and 26 had cSS with or without CMBs (Group D; 0.43%). CMBs were present in 12 participants with cSS (46%), and were strictly lobar in most cases (10 out of 12) and had mixed deep/lobar topography in two cases. None of the participants with cSS had a strictly deep CMB pattern. Maps of brain distribution of cSS amongst all affected participants demonstrated a parieto-occipital predominant distribution of cSS (Figure 1).
Figure 1.
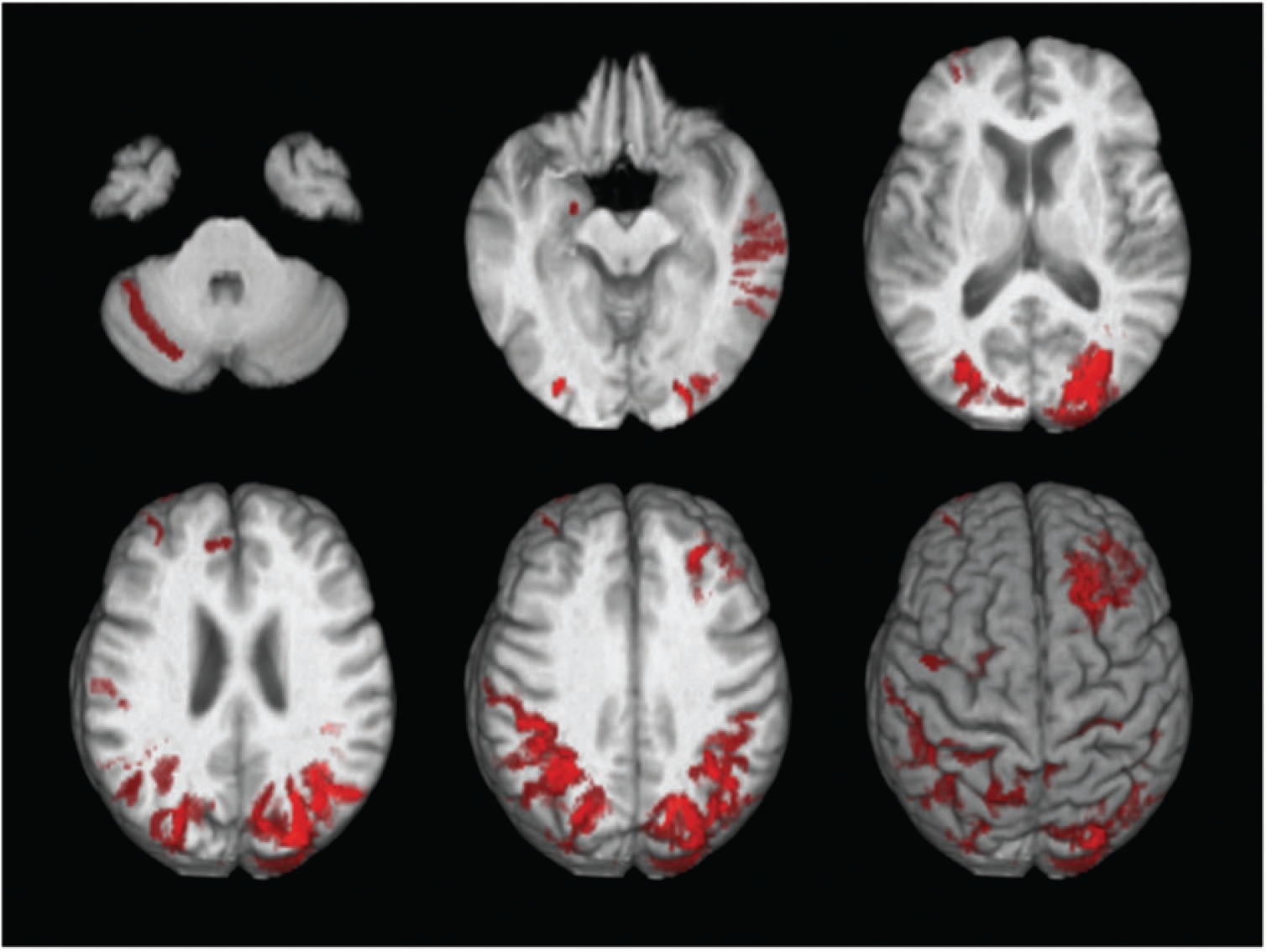
Map of brain distributions of superficial siderosis in 26 participants.
Baseline characteristics are listed in Table 1. Participants in Group B were more often male (p = 0.007), hypertensive (p = 0.015), and using statins (p = 0.002) relative to participants in Group A. They additionally had higher prevalence of antithrombotic use (p < 0.001 compared with A; p = 0.002 compared with C), extensive white matter disease (p < 0.001 compared with both A and C), lacunes on MRI (p < 0.001 compared with both A and C) and lower cholesterol levels (p = 0.014 compared with A; p = 0.048 compared with C). Participants in Group C had higher rates of antithrombotic use (0.007) and extensive white matter disease (p = 0.022), as well as reduced total cranial brain volume (p < 0.001) relative to Group A. Participants in group D had a higher prevalence of ICH relative to the other groups (p < 0.001). Both groups C (p = 0.016) and D (p = 0.043) had greater APOE ε2 and ε4 minor allele frequencies relative to Group A. None of the other intergroup comparisons was statistically significant. There was a numerical trend for greater prevalence of TIA in group D participants (18%), relative to the other three groups (7% group A; 15% group B, 9% group C; p = 0.078).
Multiple regression analyses demonstrated that in comparison to participants without CMBs or cSS (Group A) or those with strictly lobar CMBs without cSS (Group C), participants with cSS (Group D) were older (D vs. A: OR 1.09 per year, 95% CI 1.05, 1.14; p < 0.001 and D vs. C: OR 1.04, 95% CI 1.00, 1.09; p = 0.058), and had overrepresentation of the APOE ε4 allele (D vs. A: OR 5.19 for any ε4 allele relative to ε3/ε3, 95% CI 2.04, 13.15; p = 0.001 and D vs. C: OR 3.47, 95% CI 1.35, 8.92; p = 0.01). The APOE ε2 allele tended to also be overrepresented in Group D (Table 2).
Further adjusted analyses (Table 3) demonstrated persistent associations between higher prevalence of previous ICH (D vs. A: OR 76.11, 95% CI 9.58, 604.88; p < 0.001 and D vs. C: OR 66.42, 95% CI 3.36, > 999.99; p = 0.006) and lower prevalence of ischemic stroke (D vs. A: OR 0.06, 95% CI 0.01, 0.52; p = 0.011 and D vs. C: OR 0.04, 95% CI 0.003, 0.61; p = 0.020) in Group D participants, relative to participants in Groups A and C.
Table 3.
Multivariate analyses of association of cSS with prevalent clinical outcomes
| Modela | Ischemic stroke | TIA | ICHb | MCI any | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR | 95% CI | P | OR | 95% CI | P | OR | 95% CI | P | OR | 95% CI | P | ||
| D vs. A | 1 | 0.06 | 0.01, 0.52 | 0.011 | 1.03 | 0.30, 3.53 | 0.964 | 44.77 | 7.34, 272.96 | <0.001 | 1.48 | 0.50, 4.36 | 0.477 |
| 2 | 0.04 | 0.004, 0.39 | 0.006 | 0.99 | 0.29, 3.43 | 0.990 | 76.11 | 9.58, 604.88 | <0.001 | 1.44 | 0.49, 4.26 | 0.509 | |
| D vs. C | 1 | 0.05 | 0.004, 0.67 | 0.024 | 1.32 | 0.39, 4.47 | 0.661 | 26.89 | 3.42, 211.35 | 0.002 | 1.22 | 0.40, 3.72 | 0.730 |
| 2 | 0.04 | 0.003, 0.61 | 0.020 | 1.35 | 0.39, 4.58 | 0.636 | 66.42 | 3.36, >999.99 | 0.006 | 1.22 | 0.39, 3.78 | 0.731 | |
| D vs. A-B-C | 1 | 0.07 | 0.01, 0.61 | 0.016 | 0.98 | 0.29, 3.36 | 0.978 | 50.65 | 8.77, 292.58 | <0.001 | 1.33 | 0.45, 3.92 | 0.601 |
| 2 | 0.06 | 0.01, 0.57 | 0.014 | 0.98 | 0.28, 3.37 | 0.970 | 92.56 | 11.98, 714.95 | <0.001 | 1.31 | 0.44, 3.88 | 0.626 | |
Note: For description of groups see Table 1. OR: odds ratio; CI: confidence interval; APOE: apolipoprotein E; cSS: cortical superficial siderosis; TCBV: total cranial brain volume; WMH: white matter hyperintensities.
Model 1 is adjusted for age at MRI, sex and cohort; Model 2 is additionally adjusted for extensive WMH volume and lacunes.
With firth bias correction.
Post-hoc exploratory descriptive analysis demonstrated that during a mean follow-up of 5.6 years, 11 of the 26 (42.4%) participants with cSS had a stroke (five ICH, two ischemic strokes and four undetermined strokes), five (19.2%) had transient neurological deficits and one (3.8%) developed incident dementia. All five ICH occurred within the same hemisphere of cSS, as did at least two of the four undetermined strokes. Only eight of 26 (31%) participants with cSS remained free of future neurological events during follow-up (Table 4).
Table 4.
Prevalent and new neurological events in participants with cSS
| Case | Cohort | Location of cSS | Neurological preceding MRI | Subsequent neurological events | Follow-up (years) since MRI | Topographic correlation between cSS and new events |
|---|---|---|---|---|---|---|
| 1 | FHS | Left occipital | None | Left occipital ICH | 5.8 | Yes |
| 2 | FHS | Bifrontal | Remote SAH in 1980 TIA 1996-speech disturbance and L face weakness | AD/Dementia | 5.6 | uncertain |
| 3 | FHS | Left parietal | Episode non-focal weakness | Death from left parietal ICH | 2.3 | Yes |
| 4 | FHS | Right frontal | No | subsequent seizures (episodes of non-focal unresponsiveness and amnesia of events) | 6 | uncertain |
| 5 | FHS | Left parietal | No | No | 8.6 | – |
| 6 | FHS | Left frontal | Episode of paresthesias | No | 3.7 | – |
| 7 | FHS | Cerebellum, brainstem, and frontal | Episodes of diplopia | Episodes of vertical diplopia | 1 | Yes |
| 8 | FHS | Left frontal | No | Vascular death due to stroke of unspecified type (focal deficits noted preceding death, but no imaging or autopsy available) | 3.8 | uncertain |
| 9 | RS | Right parietal; right occipital | None | ICH right hemisphere. Dementia | 3 | Yes |
| 10 | RS | Right occipital | 1999 TIA | None | 2 | – |
| 11 | RS | Right frontal | Amaurosis fugax | Unspecified stroke left hemisphere. Dementia | 6 | No |
| 12 | RS | Right frontal | Collapse. Ischemic stroke right hemisphere | Stroke unspecified left hemisphere | 5.5 | Yes |
| 13 | RS | Right parietal and left occipital | None | ICH left hemisphere | 7.5 | Yes |
| 14 | RS | Left parietal and right parietal/occipital | None | Ischemic stroke right hemisphere | 7 | Yes |
| 15 | RS | Left parietal | None | None | 5 | – |
| 16 | RS | Left parietal | ICH Left parietal | None | 9 | – |
| 17 | RS | Left frontal/parietal/occipital and right frontal/occipital | None | None | 8 | |
| 18 | RS | Left frontal/temporal/parietal/occipital and right frontal/parietal | ICH left occipital | Dizziness | 9.5 | No |
| 19 | RS | Left frontal/occipitaloccipital and right frontal | Perceptive hearing loss Seizure | Unspecified stroke right hemisphere | 1 | Yes |
| 20 | RS | Bioccipital | Dizziness | ICH left hemisphere | 3.5 | Yes |
| 21 | RS | Bioccipital | None | Primary CNS lymphoma | 6 | uncertain |
| 22 | RS | Left occipital | None | MCI | 9 | No |
| 23 | RS | Left occipital | None | Hearing loss. Balance disturbance TIA left hemisphere. Ischemic stroke left hemisphere | 5 | Yes |
| 24 | RS | Left frontal and right occipital | None | None | 5 | – |
| 25 | RS | Left frontal | None | None | 10 | – |
| 26 | RS | Right frontal/parietal | None | TIA vertebrobasilar | 7 | No |
cSS: cortical superficial siderosis; FHS: Framingham Heart Study; ICH: intracerebral hemorrhage; MCI: mild cognitive impairment; RS: Rotterdam Study; SAH: subarachnoid hemorrhage; TIA: transient ischemic attack.
Discussion
Combining population-based imaging data from two large longitudinal cohorts, we studied determinants and sequela of cSS in the general elderly population. Our findings suggest that cSS is an infrequent imaging finding (prevalence 0.43%) in community-dwelling older populations. Our results, however, further the notion that cSS may be a potent marker for CAA in the general population, as evidenced by its (i) parieto-occipital predominant distribution, a known hallmark of clinical CAA20 and associations with (ii) higher age, (iii) overrepresentation of the APOE ε2 and ε4 alleles, and (iv) higher prevalence ICH compared to strictly lobar CMBs. In a clinical setting, it has been argued that cSS may reflect a more delayed manifestation of CAA or a marker of a distinct CAA phenotype at greater risk for ICH.1,2 Our data suggest that this may be true in the general population as well, as we found a higher prevalence of ICH in persons who had cSS than in persons with strictly lobar CMBs, another imaging hallmark of CAA, which remained present after taking into account other imaging markers of small vessel disease. Other compelling evidence for this argument from our data is the greater frequency of CAA-related APOE alleles observed in participants with cSS, in comparison to those with strictly lobar CMBs without cSS. In particular, a recent meta-analysis suggests that APOE ε2 might have a more important role in the pathophysiology and severity of cSS.21 Of note is that this does not preclude the possibility that cSS in the general population also reflects hemorrhage risk due to other underlying pathology than CAA.
Participants with cSS were reported to have numerically greater prevalence of “TIA” compared to all three other groups (Table 1; p ~0.08), but paradoxically were at less risk of ischemic strokes. An intriguing observation, which in combination with the existing literature, would suggest that CAA-related transient neurological episodes or ‘amyloid spells’, which have been consistently associated with cSS or—its precursor—convexity subarachnoid hemorrhage,22,23 may be being misdiagnosed as TIA in persons with cSS. Participants with cSS were additionally noted to have a high frequency of future neurological events, and ICH cases often demonstrated a topographic relationship with regions noted to have cSS at baseline. This supports the notion that cSS may mark regions of advanced CAA-related vasculopathic changes vulnerable to vessel rupture.2 This is particularly of clinical interest since many participants with cSS had been prescribed medications that could increase the risk of ICH (baseline 42% antithrombotic medication; 45% statin) for presumed concomitant thromboembolic/vaso-occlusive diseases.
CAA pathology preferentially involves the parieto-occipital lobes,20,24 and fittingly MRI markers of CAA, including cerebral microbleeds and white matter hyperintensities,25,26 have demonstrated posterior predominance. Our findings are the first to our knowledge demonstrating a similar posterior parieto-occipital predominant distribution of cSS, which further supports its association with CAA in these older community dwelling individuals.
The greatest limitation of our study is the small sample size of participants with cSS due to its infrequent occurrence within the general population. Additional limitations include possible heterogeneous CMB detection rates between the Rotterdam and Framingham Heart studies in view of their differing MRI parameters, and our inability to account for unreported traumatic brain injuries that could have contributed to cSS or CMBs. Moreover, we could not systematically exclude other factors that may have contributed to cSS, such as history of reversible cerebral vasoconstriction syndrome or distal aneurysmal rupture, for instance in the case of infective endocarditis—although both are rare occurrences in the general population. Our observational study cannot establish temporality in the associations or causation, and we cannot exclude the possibility of residual confounding. The predominant European descent of both cohorts limits the generalizability of our findings to other ethnic groups. In the absence of pathological specimens, we could not differentiate with definite certainty participants with CAA pathology versus those without. Most notably, as we did not adjust for multiple comparisons, our findings could have occurred merely by chance, are exploratory in nature, and require replication in an external sample to ensure validity. It is reassuring, however, that our observed associations with cSS in the community are consistent with prior reported associations in hospital cohorts. Lastly, we did not assess cSS progression over time, which has been reported to occur in ~30% of patients with CAA.27,28
Our study adds supporting evidence to the association between cSS and CAA, and suggests that cSS may be a potent MRI marker for CAA in the general population. Community-dwelling older persons with cSS may be at higher risk for ICH and future neurological events.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Framingham Heart Study’s National Heart, Lung, and Blood Institute contract (N01-HC-25195; HHSN268201500001I) and by grants from the National Institute of Neurological Disorders and Stroke (R01 NS17950), the National Institute on Aging (R01 AG008122; R01 AG16495; K23AG038444; R03 AG048180-01A1; AG033193), NIH grants (1RO1 HL64753; R01 HL076784; 1 R01 AG028321, P30 AG010129, NS017950), and NHLBI grants (HL67288 and 2K24HL04334). The Rotterdam Study is supported by Erasmus Medical Center and Erasmus University Rotterdam; the Netherlands Organisation for Scientific Research (NWO); the Netherlands Organisation for Health Research and Development (ZonMW); the Research Institute for Diseases in the Elderly (RIDE); the Netherlands Genomics Initiative (NGI); the Ministry of Education, Culture and Science; the Ministry of Health, Welfare and Sports; the European Commission (DG XII); and the Municipality of Rotterdam.
Footnotes
Declaration of conflicting interests
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Charidimou A, Boulouis G, Xiong L, Jessel MJ, Roongpiboonsopit D, Ayres A, et al. Cortical superficial siderosis and first-ever cerebral hemorrhage in cerebral amyloid angiopathy. Neurology 2017; 88(17): 1607–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shoamanesh A, Martinez-Ramirez S, Oliveira-Filho J, Reijmer Y, Falcone GJ, Ayres A, et al. Interrelationship of superficial siderosis and microbleeds in cerebral amyloid angiopathy. Neurology 2014; 83: 1838–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Charidimou A, Martinez-Ramirez S, Shoamanesh A, Oliveira-Filho J, Frosch M, Vashkevich A, et al. Cerebral amyloid angiopathy with and without hemorrhage: Evidence for different disease phenotypes. Neurology 2015; 84: 1206–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knudsen KA, Rosand J, Karluk D and Greenberg SM. Clinical diagnosis of cerebral amyloid angiopathy: validation of the boston criteria. Neurology 2001; 56: 537–539. [DOI] [PubMed] [Google Scholar]
- 5.Martinez-Ramirez S, Romero JR, Shoamanesh A, McKee AC, Van Etten E, Pontes-Neto O, et al. Diagnostic value of lobar microbleeds in individuals without intracerebral hemorrhage. Alzheimers Dement 2015; 11: 1480–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Linn J, Halpin A, Demaerel P, Ruhland J, Giese AD, Dichgans M, et al. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology 2010; 74: 1346–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vernooij MW, Ikram MA, Hofman A, Krestin GP, Breteler MM and van der Lugt A. Superficial siderosis in the general population. Neurology 2009; 73: 202–205. [DOI] [PubMed] [Google Scholar]
- 8.Wollenweber FA, Baykara E, Zedde M, Gesierich B, Achmuller M, Jouvent E, et al. Cortical superficial siderosis in different types of cerebral small vessel disease. Stroke 2017; 48(5): 1404–1407. [DOI] [PubMed] [Google Scholar]
- 9.Pichler M, Vemuri P, Rabinstein AA, Aakre J, Flemming KD, Brown Jr. RD, et al. Prevalence and natural history of superficial siderosis: A populationbased study. Stroke 2017; 48: 3210–3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeCarli C, Massaro J, Harvey D, Hald J, Tullberg M, Au R, et al. Measures of brain morphology and infarction in the framingham heart study: establishing what is normal. Neurobiol Aging 2005; 26: 491–510. [DOI] [PubMed] [Google Scholar]
- 11.Pikula A, Beiser AS, DeCarli C, Himali JJ, Debette S, Au R, et al. Multiple biomarkers and risk of clinical and subclinical vascular brain injury: the Framingham Offspring Study. Circulation 2012; 125: 2100–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ikram MA, Vrooman HA, Vernooij MW, van der Lijn F, Hofman A, van der Lugt A, et al. Brain tissue volumes in the general elderly population. The Rotterdam Scan Study. Neurobiol Aging 2008; 29: 882–890. [DOI] [PubMed] [Google Scholar]
- 13.Vrooman HA, Cocosco CA, van der Lijn F, Stokking R, Ikram MA, Vernooij MW, et al. Multi-spectral brain tissue segmentation using automatically trained k-nearest-neighbor classification. Neuroimage 2007; 37: 71–81. [DOI] [PubMed] [Google Scholar]
- 14.Romero JR, Preis SR, Beiser A, DeCarli C, Viswanathan A, Martinez-Ramirez S, et al. Risk factors, stroke prevention treatments, and prevalence of cerebral microbleeds in the Framingham Heart Study. Stroke 2014; 45: 1492–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akoudad S, Portegies ML, Koudstaal PJ, Hofman A, van der Lugt A, Ikram MA, et al. Cerebral microbleeds are associated with an increased risk of stroke: the Rotterdam Study. Circulation 2015; 132: 509–516. [DOI] [PubMed] [Google Scholar]
- 16.Sedaghat S, Cremers LG, de Groot M, Hofman A, van der Lugt A, Niessen WJ, et al. Lower microstructural integrity of brain white matter is related to higher mortality. Neurology 2016; 87: 927–934. [DOI] [PubMed] [Google Scholar]
- 17.Wolf PA, D’Agostino RB, O’Neal MA, Sytkowski P, Kase CS, Belanger AJ, et al. Secular trends in stroke incidence and mortality. The Framingham Study. Stroke 1992; 23: 1551–1555. [DOI] [PubMed] [Google Scholar]
- 18.Petrea RE, Beiser AS, Seshadri S, Kelly-Hayes M, Kase CS and Wolf PA. Gender differences in stroke incidence and poststroke disability in the Framingham Heart Study. Stroke 2009; 40: 1032–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wieberdink RG, Ikram MA, Hofman A, Koudstaal PJ and Breteler MM. Trends in stroke incidence rates and stroke risk factors in Rotterdam, the Netherlands from 1990 to 2008. Eur J Epidemiol 2012; 27: 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vinters HV and Gilbert JJ. Cerebral amyloid angiopathy: incidence and complications in the aging brain. II. The distribution of amyloid vascular changes. Stroke 1983; 14: 924–928. [DOI] [PubMed] [Google Scholar]
- 21.Charidimou A, Zonneveld HI, Shams S, Kantarci K, Shoamanesh A, Hilal S, et al. Apoe and cortical superficial siderosis in caa: meta-analysis and potential mechanisms. Neurology 2019; 93: e358–e371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Charidimou A, Peeters A, Fox Z, Gregoire SM, Vandermeeren Y, Laloux P, et al. Spectrum of transient focal neurological episodes in cerebral amyloid angiopathy: multicentre magnetic resonance imaging cohort study and meta-analysis. Stroke 2012; 43: 2324–2330. [DOI] [PubMed] [Google Scholar]
- 23.Kumar S, Goddeau RP Jr., Selim MH, Thomas A, Schlaug G, Alhazzani A, et al. Atraumatic convexal subarachnoid hemorrhage: clinical presentation, imaging patterns, and etiologies. Neurology 2010; 74: 893–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson KA, Gregas M, Becker JA, Kinnecom C, Salat DH, Moran EK, et al. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol 2007; 62: 229–234. [DOI] [PubMed] [Google Scholar]
- 25.Rosand J, Muzikansky A, Kumar A, Wisco JJ, Smith EE, Betensky RA, et al. Spatial clustering of hemorrhages in probable cerebral amyloid angiopathy. Ann Neurol 2005; 58: 459–462. [DOI] [PubMed] [Google Scholar]
- 26.Thanprasertsuk S, Martinez-Ramirez S, Pontes-Neto OM, Ni J, Ayres A, Reed A, et al. Posterior white matter disease distribution as a predictor of amyloid angiopathy. Neurology 2014; 83: 794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Charidimou A, Boulouis G, Xiong L, Pasi M, Roongpiboonsopit D, Ayres A, et al. Cortical superficial siderosis evolution. Stroke 2019; 50: 954–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pongpitakmetha T, Fotiadis P, Pasi M, Boulouis G, Xiong L, Warren AD, et al. Cortical superficial siderosis progression in cerebral amyloid angiopathy: Prospective MRI Study. Neurology 2020; 94: e1853–e1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data will be shared by request from any qualified investigator.