

Abstract
BACKGROUND:
Excess cholesterol accumulation in lesional macrophages elicits complex responses in atherosclerosis. Epsins, a family of endocytic adaptors, fuel the progression of atherosclerosis; however, the underlying mechanism and therapeutic potential of targeting Epsins remains unknown. In this study, we determined the role of Epsins in macrophage-mediated metabolic regulation. We then developed an innovative method to therapeutically target macrophage Epsins with specially-designed S2P-conjugated lipid nanoparticles (NPs), which encapsulate small interfering RNAs to suppress Epsins.
METHODS:
We used single cell RNA sequencing (scRNA-seq) with our newly developed algorithm MEBOCOST to study cell-cell communications mediated by metabolites from sender cells and sensor proteins on receiver cells. Biomedical, cellular and molecular approaches were utilized to investigate the role of macrophage Epsins in regulating lipid metabolism and transport. We performed this study using myeloid-specific Epsin double knockout (LysM-DKO) mice and mice with a genetic reduction of ABCG1 (LysM-DKO-ABCG1fl/+). The NPs targeting lesional macrophages were developed to encapsulate interfering RNAs to treat atherosclerosis.
RESULTS:
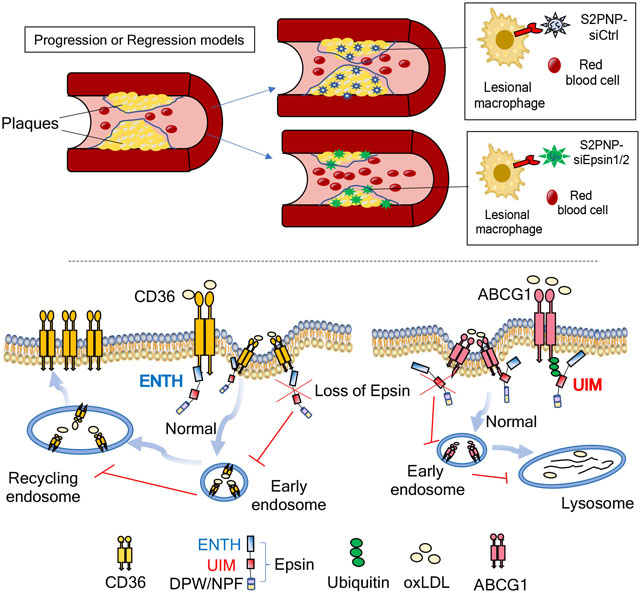
We revealed that Epsins regulate lipid metabolism and transport in atherosclerotic macrophages. Inhibiting Epsins by nanotherapy halts inflammation and accelerates atheroma resolution. Harnessing lesional macrophage-specific NP delivery of Epsin siRNAs, we showed that silencing of macrophage Epsins diminished atherosclerotic plaque size and promoted plaque regression. Mechanistically, we demonstrated that Epsins bound to CD36 to facilitate lipid uptake by enhancing CD36 endocytosis and recycling. Conversely, Epsins promoted ABCG1 degradation via lysosomes and hampered ABCG1-mediated cholesterol efflux and reverse cholesterol transport. In a LysM-DKO-ABCG1fl/+ mouse model, enhanced cholesterol efflux and reverse transport due to Epsin deficiency was suppressed by the reduction of ABCG1.
CONCLUSIONS:
Our findings suggest that targeting Epsins in lesional macrophages may offer therapeutic benefits for advanced atherosclerosis by reducing CD36-mediated lipid uptake and increasing ABCG1-mediated cholesterol efflux.
Keywords: Epsin, nanoparticle, lipid metabolism, atherosclerosis, CD36, ABCG1, Basic Science Research, Metabolism, Computational Biology, Lipids and Cholesterol, Vascular biology
Graphical Abstract
INTRODUCTION
Atherosclerosis is a complex and chronic condition that results in a buildup of arterial fatty deposits, called plaques that are the root cause of coronary heart disease, ischemic stroke, and peripheral artery disease 1,2. Despite effective lipid-lowering therapies including new antibody and siRNA drugs targeting PCSK9, atherosclerosis is still the leading cause of mortality worldwide. Atherosclerotic lesion formation involves interactions between oxidized low-density lipoprotein (oxLDL) and constituents of the arterial wall including monocyte-derived macrophages, immune cells and vascular smooth muscle cells 3,4. Mounting evidence indicates that factors with the potential to promote macrophage egress from plaques (e.g., regulatory T cells) play a critical role in regulating macrophage pro-resolving functions during atherosclerosis regression 5. Furthermore, the pro-inflammatory effects of oxLDL leads to endothelial activation and macrophage recruitment 6. Invading macrophages in the sub-endothelium ingest modified LDL and are transformed into lipid-laden foam cells 1,3. This escalates arterial inflammation—a prime contributor to the transition from a stable to vulnerable atheroma 7–10. Accordingly, patients that receive a therapeutic cocktail aimed at alleviating dyslipidemia and hypertension still develop deleterious cardiovascular diseases 2,11. This accentuates the urgent need for next-generation therapies geared toward eradicating inflammation in the arterial wall.
Epsins are a family of highly-conserved endocytic adaptor proteins that associate with the plasma membrane through interactions between their N-terminal homology (ENTH) domain and membrane phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) 12–14, where it recognizes and recruits ubiquitinated cell surface receptors to clathrin-coated pits for internalization via its ubiquitin-interacting motif (UIM) and clathrin/AP-2 binding sites, respectively 15–18. Epsins 1 and 2 are ubiquitously expressed, redundant in function, and essential for embryonic survival 19. The embryonic lethality of Epsin 1 and 2 double knockout mice (DKO) prompted us to generate conditional Epsin1fl/fl; Epsin2−/− mice. Combining DKO strains with specific Cre deleter mouse strains creates tissue and cell type-specific DKO mice 19,20. Despite their multifaceted functions, our previous work demonstrated that Epsins possess a vast degree of specificity and selectivity in choosing their binding partners. Therefore, the actions of Epsins are cell context-dependent 19–26. Notably, we demonstrate that myeloid-specific Epsin loss downregulates inflammation and impedes atheroma formation in an atherogenic mouse model by stabilizing macrophage LRP-1 26. However, this model failed to address the role of epsins in hindering atheroma regression and promoting plaque rapture and the resulting debilitating cardiovascular events.
Lesional macrophages internalize lipids primarily through scavenger receptor-mediated endocytosis 27, which is also responsible for foam cell formation 28–32. Mice lacking both Scavenger Receptor-A (SR-A) and CD36 revealed that CD36 is the major oxLDL receptor required for foam cell formation 29–32. Our previous studies revealed that Epsins 1 and 2 are upregulated in lesional macrophages 26. Epsin-deficient (DKO) macrophages exhibited a striking reduction in foam cell formation 26. As robust lipid uptake by macrophages is one of major responsible factors that drive foam cell formation, we posit a central role for Epsins in regulating lipid uptake in macrophages; however, how Epsins facilitate lipid uptake and whether this occurs through scavenger receptor-mediated uptake of modified LDL during foam cell formation remains unknown.
Reverse cholesterol transport (RCT) is responsible for removing excess cholesterol from peripheral tissues to the liver, where it is reused or removed from the body 33. A critical part of RCT is cholesterol efflux, where accumulated cholesterol removed from macrophages in the subintima is mediated by ATP-binding membrane cassette transporters (ABCA1 and ABCG1) and other molecules 34–36. Among the regulators of RCT, ABCG1 plays a prominent role in macrophage cholesterol and phospholipid transport to maintain cellular lipid homeostasis by using high density lipoprotein (HDL) as a cholesterol acceptor 37. HDL is thought to facilitate macrophage re-programming to be less inflammatory through an ATF-3-dependent pathway 38. Several clinical trials and more recent studies suggest that increased HDL-cholesterol concentrations fail to improve cardiovascular disease outcomes 39–41; yet, new data indicate that rather than the steady-state HDL-cholesterol (HDL-C) levels 42, the ability of HDL particles to transport cholesterol from atherosclerotic plaques to the liver is perhaps more important for combating atherosclerosis 43,44.
Consequently, a shift in focus from increasing HDL-cholesterol concentrations to raising RCT function by increasing cholesterol efflux capacity (CEC), the HDL-ABCG1-mediated cholesterol efflux from macrophages, has become a prospective therapy for coronary artery disease (CAD), especially in severe acute myocardial infarction (AMI) patients 45–48. As lack of effective cholesterol efflux in macrophages hinders the reversal of foam cells to healthy macrophages, we envisioned a major role for macrophage Epsins in regulating cholesterol efflux. Given the importance of cholesterol efflux and RCT in atheroma resolution and the aforementioned risk reduction in AMI patients 34–36, it is crucial to determine whether Epsins inhibit cholesterol efflux and RCT to promote atherosclerotic plaque progression and impede lesion regression.
In this study, we harnessed innovative single-cell bioinformatics technology and therapeutic nanotechnology to better understand atherosclerosis. This allowed us to determine the detrimental role of epsins in prohibiting atheroma regression and promoting plaque rupture using a preclinical atherosclerosis regression model. Mechanistically, we utilized scRNA-seq analysis of macrophages and newly-developed bioinformatic techniques to identify novel targets involved in lipid metabolism during atherosclerosis. Based on these targets, we generated a myeloid-specific-deficient ABCG1 murine model to uncover how epsins regulate lipid metabolism to propel atherosclerosis. Specifically, we showed that Epsins facilitate CD36-mediated lipid uptake and inhibit ABCG1-mediated cholesterol efflux. We also employed a novel macrophage-specific nanoparticle delivery system to investigate the therapeutic treatment of atherosclerosis. Lastly, using an early-stage progression model, an advanced-stage progression model, and a regression model of atherosclerosis, we established the therapeutic benefit of treating this disease with a nanoparticle delivery system. This comprehensive mechanistic study on lipid metabolism and clinically-relevant nanoparticle-mediated RNAi therapies represents a potential breakthrough for treating this devastating disease.
METHODS
The detailed methods and materials are available in the Supplemental Materials. Antibodies, primers, and reagents are listed in Major Resources Table.
Data availability:
The authors declare that all supporting data are available within the article and its supplementary files. Additional methods or data related to this study are available from the corresponding authors upon reasonable request. The RNA-seq and scRNA-Seq data generated in this study were deposited into the GEO database with the accession number GSE214414.
RESULTS
Single-cell RNA-seq revealed that macrophage Epsins regulate cholesterol metabolism and efflux pathways
For an unbiased analysis of all pathways regulated by Epsins in macrophages, we performed single-cell RNA sequencing (scRNA-seq) analysis of aortic tissue isolated from wild type (WT) and macrophage-specific LysM-DKO (DKO) mice on a normal diet (Figure S1A–S1B). We identified a diverse range of cell types, including vascular smooth muscle cells (VSMCs), fibroblasts, endothelial cells, macrophages, and other cells (Figure 1A). Known cell markers facilitated annotation of cell types, such as Tagln and Acta2 for VSMCs, Pecam1 and Cdh5 for endothelial cells, Cd14 and Cd68 for macrophages49–51 (Figure S2A). Macrophage populations were further subclustered to investigate their heterogeneity among subpopulations (Figure 1B). Multiple well-known macrophage subpopulations were recaptured, including M1 and M2 macrophages. Markers reported in the literature were used to annotate the subtypes (e.g., Mrc1 [Cd206] and Cd163 for M2 macrophages)52 (Figure S2B–S2D). The composition of these aortic cell populations showed differences between WT and DKO mice (Figure S3).
Figure 1. scRNA-seq data revealed down-regulation of cholesterol-related cell-cell communications mediated by metabolites and their sensor proteins in macrophage subpopulations in LysM-DKO aorta compared to WT.
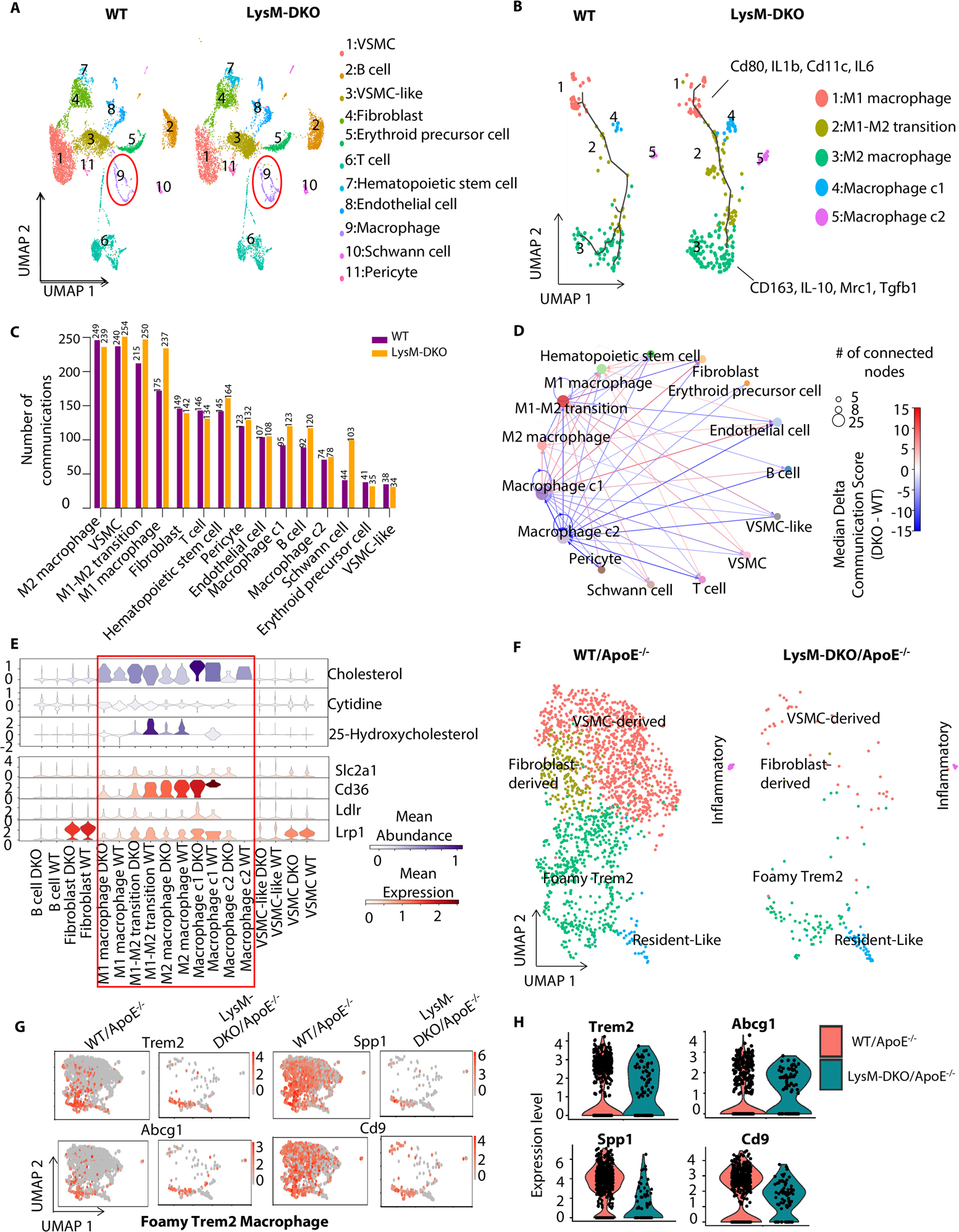
Aortas from WT and LysM-DKO mice (n=3 mice/each group) were isolated, digested and mixed for scRNA sequencing. (A) UMAP plot of cell clusters in aortas from WT and LysM-DKO mice on normal diet. Macrophage populations are indicated in the red circle. (B) UMAP of subclusters of major macrophage populations in (A). Trajectory inferred by Monocle3 was displayed. (C) A bar plot showing the number of communication events in cell groups in WT and LysM-DKO. (D) A circle plot showing the differential communications between cell groups in LysM-DKO compared to WT. The arrows indicate directions of communication from sender cells to receiver cells. The size of nodes positively correlates with the number of connected nodes. Line colors indicate the difference in communication scores of LysM-DKO compared to WT. (E) Violin plots of the abundance of representative metabolites and sensors across cell types in WT and LysM-DKO. Metabolite abundance (upper), sensor abundance (below). (F) ScRNA-seq identified the different macrophage subpopulations in aortas from WT/ApoE−/− and LysM-DKO/ApoE−/− mice on Western diet for 16 weeks. UMAP plot of each macrophage subpopulation in WT/ApoE−/− and LysM-DKO/ApoE−/−. (G-H) Feature plots (G) and Violin plots (H) showing the global expression level of marker genes in the macrophage sub types (e.g., Foamy Trem2 macrophage).
The molecular mechanisms underlying Epsin-mediated regulation of foam cell formation and the promotion of lipid uptake by macrophages are poorly understood. We therefore employed the algorithm MEBOCOST, which is the first algorithm developed to detect cell communications mediated by metabolites and their sensor proteins such as receptors and transporters (metabolite-sensor cell communications) 53. MEBOCOST predicted each metabolite-mediated cell-cell communication for which the metabolite is secreted by one cell and travels to interact with the sensor protein of another cell. The cell type that expresses the enzyme that generates the metabolite is defined as the “sender”, while the cell type that expressed the sensor protein is defined as the “receiver”. The results showed that macrophage subpopulations and VSMCs were the top cell types ranked by the number of metabolite-sensor cell communications from the largest to the smallest (Figure 1C). Furthermore, the numbers of metabolite-sensor cell communications in these cell populations increased in DKO compared to WT samples (Figure 1C). Macrophage subpopulations were found to not only send metabolites to other cell types, but also receive metabolites from other cell types. They could send and receive metabolites in an autocrine manner as well (Figure 1D). The communications between a pair of cell populations can be mediated by multiple metabolite-sensor pairs. The overall changes in communication were estimated by the median differences of communication scores between any two cell groups. We observed widespread changes of cell-cell communications in DKO when compared to WT, with both strengthened and weakened communications (Figure 1D). To gain a deeper insight into the changed metabolite-sensor cell communications related to macrophages, we inspected individual sender cell types of metabolites and receiver cell types of sensor proteins. First, we observed a decrease of cholesterol-Cd36 cell communications from macrophages, especially in macrophage cluster 2 (c2), compared with other cell types in DKO and WT samples (Figure S4A); second, cholesterol-Cd36 cell communications from macrophage c2 to macrophage c1 were reduced by Epsin DKO (Figure S4B and S5A–E). As for the specificity of metabolite and sensor abundance in different cell types, we observed that cholesterol and its derivative, 25-hydroxycholesterol, were highly-enriched in macrophage populations. Cd36, the receptor of cholesterol, was also highly expressed in macrophage populations (Figure 1E and S6A–B). The cell communications through cholesterol and Cd36 in macrophages was supported by their abundance in these cells. Finally, the most variable communications in macrophages mediated by cholesterol appeared to be mostly decreased rather than increased (Figure S4C).
We sequenced and analyzed the macrophage subpopulations in aortas from WT/ApoE−/− and LysM-DKO/ApoE−/− mice on a Western Diet for 16 weeks. We identified multiple macrophage subpopulations such as Foamy Trem2 macrophages, Resident-Like macrophages, Inflammatory macrophages, VSMS-derived macrophages, and Fibroblast-derived macrophages (Figure 1F). The composition of these subpopulations showed differences between WT/ApoE−/− and LysM-DKO/ApoE−/− mice on Western diet for 16 weeks (Figure 1F). Known markers54,55 were used to annotate the macrophage subtypes, such as Foamy Trem2 macrophages (Trem2, Abcg1, Spp1 and Cd9) (Figure 1G and 1H).
We subsequently performed bulk RNA-seq analysis of peritoneal macrophages isolated from WT and LysM-DKO mice on normal diet and revealed that loss of Epsins 1 and 2 significantly altered the expression of 2208 genes (Figure S7A). These include 1138 downregulated and 1070 upregulated genes (Figure S7A). Notably, the differentially regulated genes were associated with cholesterol efflux and metabolism, suggesting that Epsins play a role in regulating lipid cholesterol metabolism and efflux pathways (Figure S7B). Consistently, Gene Set Enrichment Analysis (GSEA) showed that genes related to fatty acid metabolism tended to be downregulated in DKO macrophages (Figure S7C). In contrast, genes associated with cholesterol transport and efflux (Figure S7D), and negative regulation of macrophage foam cell formation (Figure S7E), tended to be upregulated by Epsin deficiency.
We next compared DKO peritoneal macrophage RNA-seq data from WT and LysM-DKO mice on a normal diet with RNA-seq data from wild type and CD36 knockout (CD36KO) macrophages from an open-source database 56. We observed a significant overlap of differentially expressed genes induced by Epsin and CD36 deletion from mouse macrophages. There were 169 genes significantly upregulated by both Epsin-DKO and CD36KO (4-fold enrichment compared to the 41 genes expected by random chance, Fisher’s exact test P value 9.73 × 10−25) (Figure S7F left panels); 29 genes (6-fold enrichment, Fisher’s exact test P value 6.15 × 10−9) were downregulated by both Epsin-DKO and CD36KO (Figure S7F right panels). GO analysis revealed multiple pathways regulated by both Epsin and CD36 deficiency. These include the downregulation of inflammation pathways, lipid biosynthetic processes, triglyceride metabolic processes, and upregulation of the small GTPase-mediated signal transduction pathway (Figure S7G). These results indicated that Epsins and CD36 may have superimposing roles in regulating molecular pathways involved in lipid metabolism and inflammation.
Epsin promotes CD36-mediated lipid uptake by promoting CD36 endocytosis and recycling
Based on the bioinformatic results, we hypothesized that Epsins regulate CD36 internalization and, thus, lipid uptake. We performed qRT-PCR, western blot (WB) and flow cytometry to check the RNA, total protein, and surface protein levels of CD36. Interestingly, RNA (Figure 2A) and total protein (Figure 2B) expression of CD36 showed no significant differences between WT and DKO macrophages; however, the surface protein level of CD36 significantly decreased with the treatment of oxLDL in clathrin-dependent manner in WT, but not in DKO macrophages (Figure 2C and S8A–C). Immunofluorescence staining (IF) revealed oxLDL-induced CD36 trafficking to early endosomes. We observed colocalization of CD36 with EEA1 (early endosome antigen 1, Figure 2D) and Rab11 (recycling endosome marker, Figure 2E) in WT macrophages treated with oxLDL, which was not observed in DKO macrophages.
Figure 2. Epsin facilitates CD36-mediated lipid uptake by promoting CD36 endocytosis and recycling.
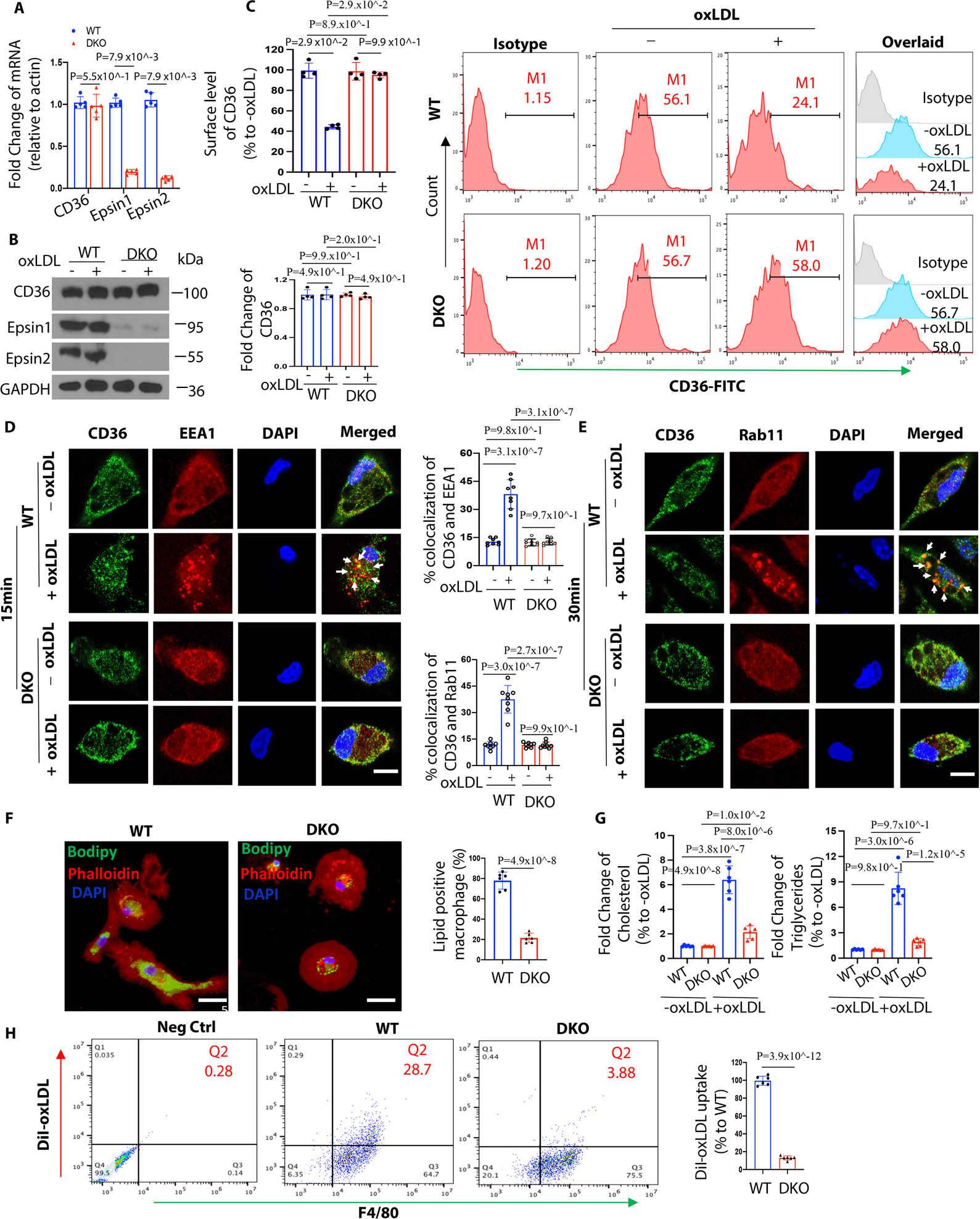
(A-C) Thioglycolate (TG) induced peritoneal macrophages from WT/ApoE−/− (n=5) and LysM-DKO/ApoE−/− (n=5) mice on normal diet (ND) were incubated in lipid-deficient medium for 24h and treated with or without 100μg/mL oxLDL for 1h at 370C. qRT-PCR analysis of CD36 expression (A), western blot (WB) analysis for total protein level of CD36 (B) and flow cytometry for surface level of CD36 (C). (D-E) Elicited TG-induced peritoneal macrophages from WT/ApoE−/− and LysM-DKO/ApoE−/− mice on ND were incubated in lipid-deficient medium for 24h and treated with or without 100μg/mL oxLDL for 15min (D) or 30min (E) at 370C. Macrophages were co-stained with CD36 (green), the early endosome marker EEA1 (red) or the recycling endosome marker Rab11 (Red) and DAPI (bule), and imaged using confocal microscope. White arrows indicate the endocytic vesicles, scale bar=5μm, n=8/group. (F) BODIPY staining of peritoneal macrophages from WT and LysM-DKO mice on normal diet were pre-incubated with 25μg/mL oxLDL for 24h in lipid-deficient medium, n=6/group, scale bar=10μm. (G) Cholesterol and triglycerides levels in WT and LysM-DKO macrophages treated with 25μg/mL oxLDL for 24h in lipid-deficient medium (n=6). (H) Peritoneal macrophages isolated from WT and LysM-DKO mice on normal diet were incubated in lipid-deficient medium for 24h followed by the treatment with DiI-oxLDL for 2h at 370C to assess the lipoprotein uptake by flow cytometry, n=6/group. Data from A-H are presented as mean ± SD. Mann-Whitney U test was utilized in A-C. Two-way ANOVA followed by Sidak post hoc multiple comparisons test was conducted in D-E and G. Unpaired t test was conducted in F and H.
To further study the role of Epsins in regulating macrophage lipid uptake, we stained oxLDL-treated WT and DKO macrophages with BODIPY and Oil Red O (ORO). Deficiency of Epsins impaired oxLDL uptake and foam cell formation (Figure 2F and S9A–B) and reduced cholesterol and triglyceride levels (Figure 2G) in DKO macrophages. Flow cytometry confirmed that macrophage lipid uptake was reduced in DKO macrophages using DiI-oxLDL treatment (Figure 2H and S8B). Together, these results indicate that Epsins are crucial for CD36-mediated lipid uptake and cholesterol metabolism.
Epsin ENTH domain is required for CD36-mediated lipid uptake and foam cell formation
Next, we investigated how Epsins and CD36 interact to cause the disparity in CD36 surface and total protein levels using immunoprecipitation (IP) and WB. We determined that CD36 binds Epsin1 in WT, but not in DKO macrophages (Figure 3A). This indicated that Epsin1 and CD36 interact endogenously. To determine the Epsin domains responsible for this interaction, we created Flag-tagged mammalian Epsin1 wild type full length and ENTH or UIM deletion constructs (FLAG-Epsin1WT, FLAG-Epsin1ΔENTH or FLAG-Epsin1ΔUIM) (Figure 3B). We transfected these constructs to HEK 293T cells and performed co-IP with CD36. The interaction between Epsin1 and CD36 was dependent on the ENTH domain as the binding between Epsin1ΔENTH and CD36 was reduced in FLAG-Epsin1ΔENTH-expressing cells (Figure 3B). As expected, we did not detect ubiquitinated CD36 in response to oxLDL treatment in the presence of the proteasome inhibitor MG132 (Figure 3C). In addition, we transfected the Epsin full length and deletion Epsin constructs into WT and DKO macrophages treated with oxLDL. The transfection of full-length Epsin restored lipid uptake in DKO macrophages, which was not observed in cells expressing FLAG-Epsin1ΔENTH (Figure 3D, 3E and S10).
Figure 3. Epsin interacts with CD36 via Epsin-ENTH domain which is critical for CD36-mediated lipid uptake and foam cell formation.
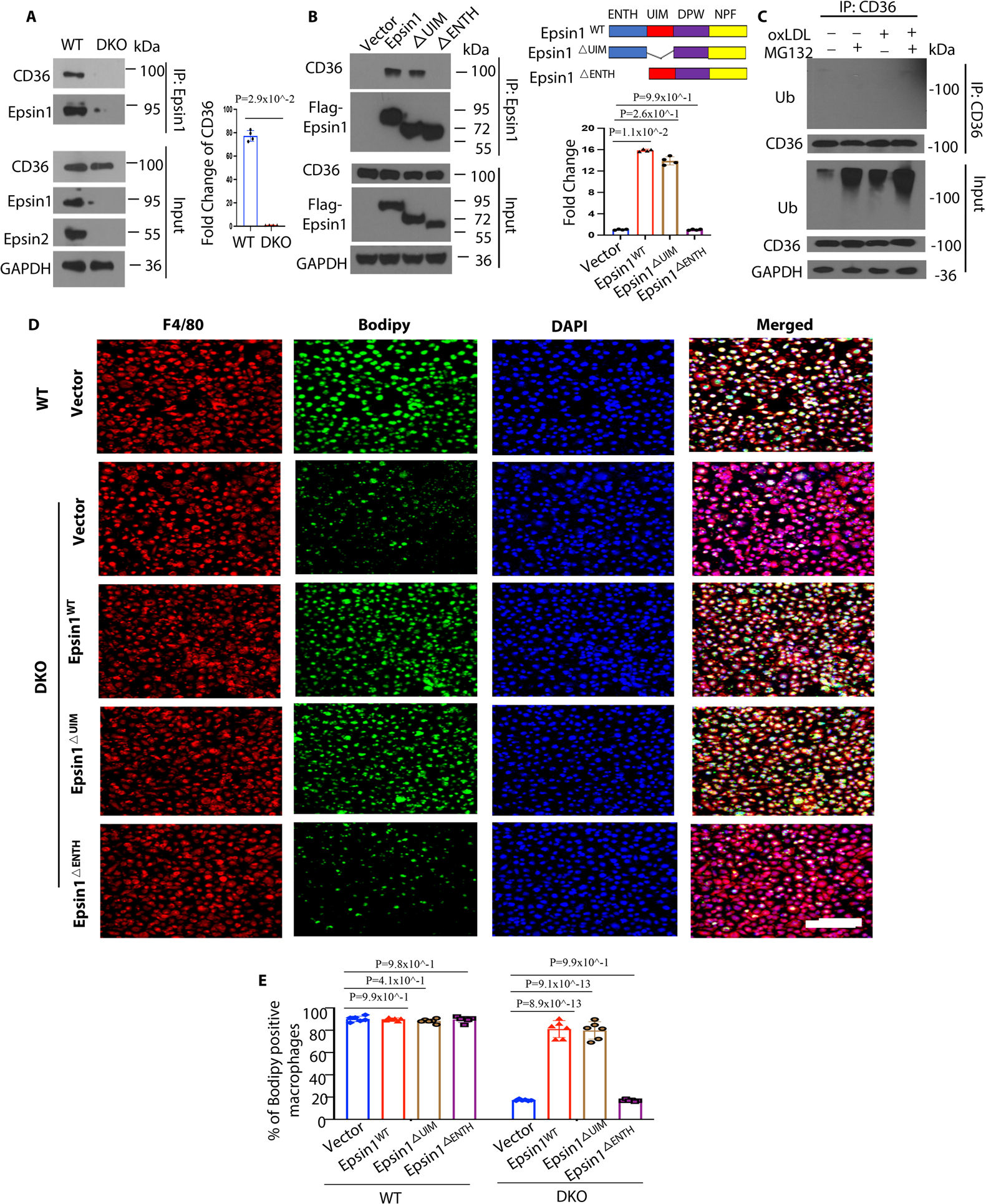
(A) Bone marrow derived macrophages (BMDM) isolated from WT and LysM-DKO mice on normal diet were incubated in lipid-deficient medium for 24h followed by the treatment with 100μg/mL oxLDL for 1h, IP and WB analysis of Epsin1 and CD36 (n=4). (B) CD36 plasmids and full length (FLAG-Epsin1WT) or domain-deletion constructs (FLAG-Epsin1ΔENTH or FLAG-Epsin1ΔUIM) in the pcDNA3 vector were transfected into HEK 293T cells for 48 h and then treated with 100μg/mL oxLDL for 1h, followed by immunoprecipitation (IP) and WB analysis using antibodies against FLAG tags and CD36 (n=4). (C) Peritoneal macrophages isolated from WT/ApoE−/− mice on normal diet were incubated in lipid-deficient medium for 24h followed by treatment with 5 μM MG132 for 3h. Cells were subsequently treated with 100 μg/mL oxLDL for 1h followed by IP and WB for ubiquitin and CD36. (D) FLAG-Epsin1WT, FLAG-Epsin1ΔENTH, and FLAG-Epsin1ΔUIM constructs were transfected into LysM-DKO/ApoE−/− macrophages for 48 h and treated with 100μg/mL oxLDL for 1 h, followed by staining with F4/80 (red), BODIPY (green) and DAPI (blue). Scale bar= 200μm. (E) Statistics for (D and Figure S10), n=6. Data from A-D are presented as mean ± SD. Mann-Whitney U test was utilized in A. Kruskal-Wallis test followed by Dunn’s post hoc multiple comparisons test was conducted in B. One-way ANOVA followed by Tukey post hoc multiple comparisons test was conducted in E.
RNA-seq analyses of WT, Epsin-DKO and ABCG1-KO macrophages reveal that Epsins and ABCG1 reciprocally regulate inflammation and lipid metabolism pathways
Emerging evidence supports raising RCT function by increasing HDL-cholesterol efflux capacity (CEC) to treat recurrent heart attacks. To study this, we generated myeloid-specific ABCG1-knock out (ABCG1-KO) mice. Given the intimate relationship of HDL with ABCG1 in regulating cholesterol efflux, and the involvement of Epsins in cholesterol efflux regulation, we performed RNA-seq analysis of WT, ABCG1-KO and Epsin-DKO macrophages. The loss of ABCG1 in macrophages significantly altered the expression of 1733 genes, including 986 downregulated and 747 upregulated genes (Figure S11A–S11C). GSEA indicated that genes associated with the inflammatory response (Figure 4A) and TNFα signaling via NF-κB (Figure 4B) are upregulated by ABCG1-KO in macrophages. In contrast, genes associated with cholesterol homeostasis are downregulated by ABCG1-KO (Figure 4C). We then performed GO enrichment analysis for genes up- and down-regulated in ABCG1-KO relative to WT macrophages. Positive regulation of cytokine production, tumor necrosis factor production and acute inflammatory responses were upregulated by ABCG1-KO in macrophages (Figure 4D). Negative regulation of lipid storage and cholesterol biosynthetic processes were downregulated in ABCG1-KO compared to WT macrophages (Figure 4D). Interestingly, GSEA analyses showed that genes associated with cell activation involved in immune response (Figure S11D), regulation of leukocyte migration (Figure S11E) and leukocyte proliferation (Figure S11F) are markedly upregulated by ABCG1 deficiency in macrophages. In contrast, these genes are downregulated in DKO macrophages (Figure S11D–S11F).
Figure 4. RNA-seq analyses of WT, DKO and ABCG1 knockout (ABCG1KO) peritoneal macrophages indicate inverse regulation of lipid metabolism and inflammatory response.
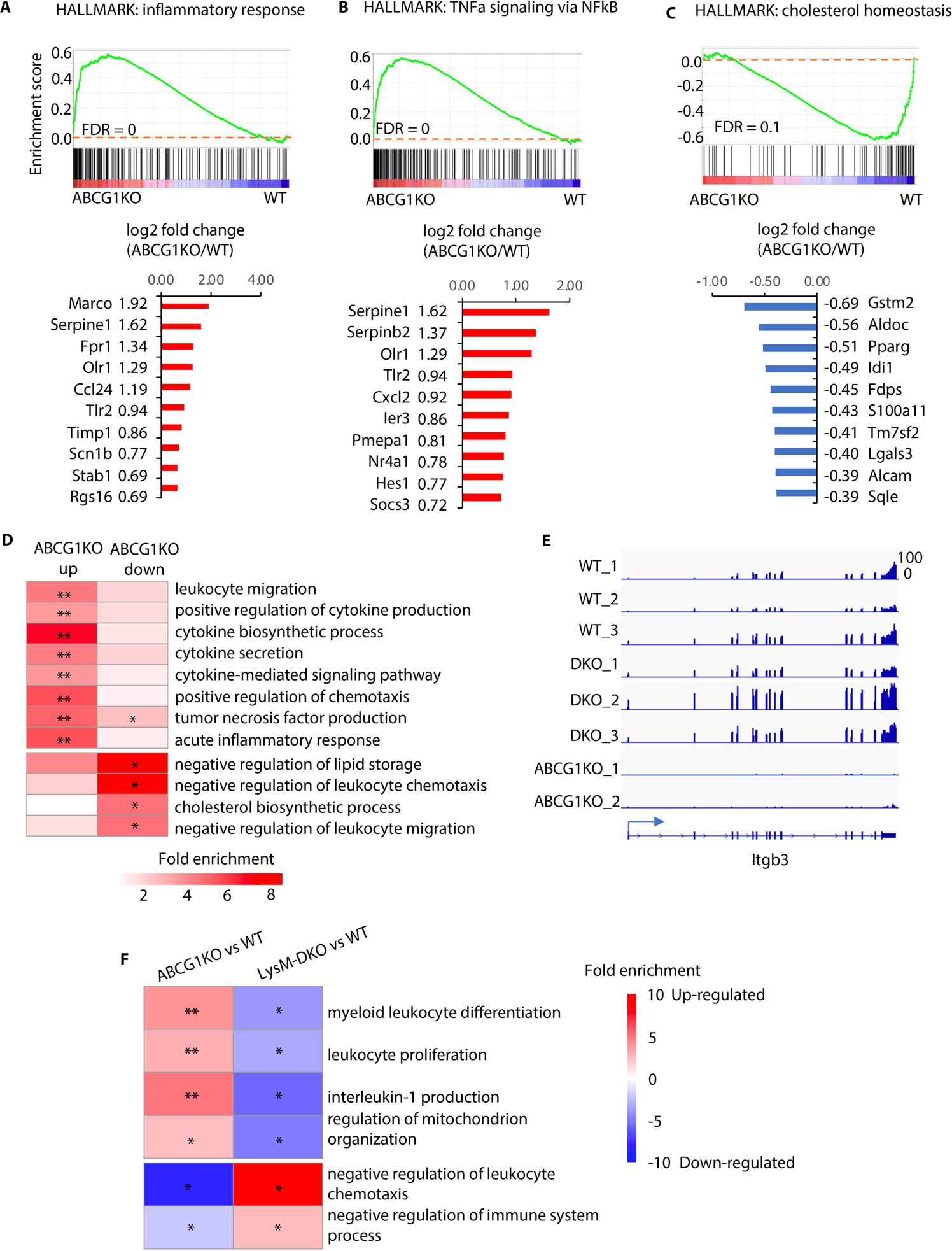
(A-C) The Gene Set Enrichment Analysis (GSEA) (top panels) indicates the tendency of individual pathways to be up or down regulated in ABCG1KO macrophages compared to wild type. Genes associated with inflammatory response (HALLMAKR) (A), TNFα signaling via NFκB (HALLMARK) (B), and cholesterol homeostasis (HALLMARK) (C) are analyzed. The bar plots (bottom panels) showing log2 fold changes of expression of altered genes. (D) GO enrichment analysis for up- and down- regulated genes in ABCG1KO relative to WT. (E) Genome browser tracks to show expression of Itgb3 in individual samples. (F) The GO enrichment analysis revealed pathways reversely regulated in ABCG1 knock out and Epsin deficient peritoneal macrophages, compared to wild type. n=3/each group, * Adjusted P<0.05, ** adjusted P<1×10−5.
These results suggest that Epsins and ABCG1 reciprocally regulate inflammation pathways. Examination of individual expressed genes confirmed common target genes were oppositely regulated by Epsins and ABCG1. For instance, Itgb3, which prevents differentiation of macrophages and monocytes into foam cells 57, was inversely regulated by DKO and ABCG1-KO, respectively (Figure 4E). In addition, GO enrichment analysis revealed pathways were opposingly regulated by ABCG1-KO and DKO in macrophages (Figure 4F). Therefore, our RNA-seq analyses from ABCG1-KO and Epsin-DKO mouse lines further supports our findings showing that Epsins and ABCG1 play opposing roles in regulating cholesterol efflux, inflammatory responses, and foam cell formation.
Epsin loss augments macrophage cholesterol efflux in vitro and RCT in vivo
RNA-seq analysis of WT, DKO and ABCG1-KO macrophages indicated that the loss of Epsins affects cholesterol efflux. At the same time, we wanted to determine if Epsins inhibit cholesterol efflux and RCT to promote atherosclerosis. WT and DKO macrophages were pretreated with a liver X receptor (LXR) agonist (T0901317, 3μmol/L) which can increase ABCG1-mediated cholesterol efflux to HDL58, then incubated with [3H]-cholesterol and ac-LDL. We found that in vitro cholesterol efflux to HDL, but not ApoA-1, was markedly enhanced in DKO versus WT macrophages (Figure 5A and 5B). Consistently, in vivo RCT (Figure 5C) was elevated in mice injected with [3H]-cholesterol-loaded DKO macrophages when compared to WT macrophages. Our results showed that [3H]-radioactivity in liver, plasma, feces, and intestinal contents all showed significant increases in mice injected with DKO macrophages, but not those injected with WT macrophages (Figure 5D–5I and S12). These findings suggest that Epsin-deficient macrophages have higher cholesterol efflux and RCT.
Figure 5. Epsins deficiency in macrophages shows increased cholesterol efflux in vitro and reverse cholesterol transport (RCT) in vivo.
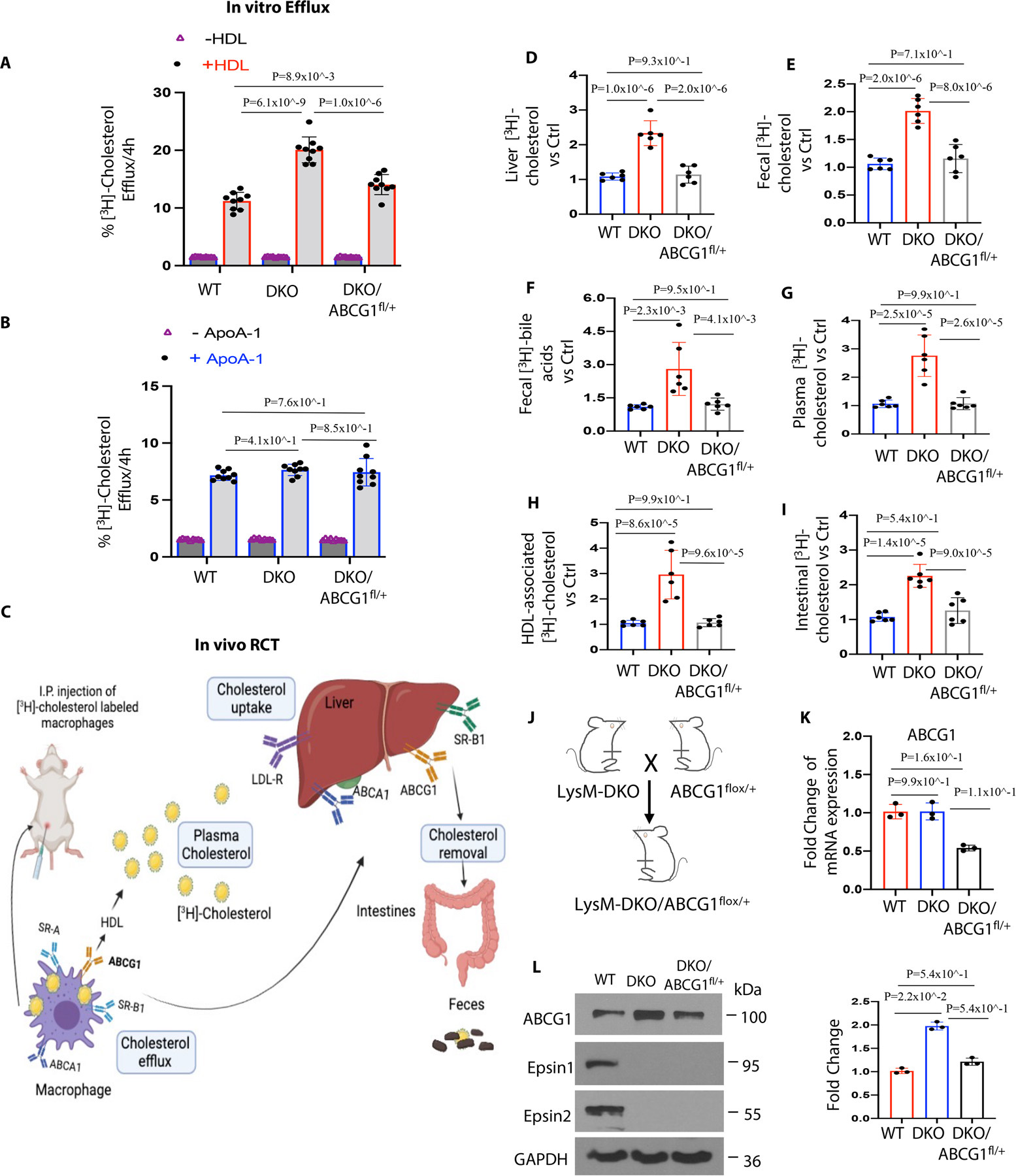
(A-B) Peritoneal macrophages were isolated from WT, LysM-DKO and LysM-DKO/ABCG1fl/+ mice on normal diet. In vitro [3H]-cholesterol labeled WT, LysM-DKO or LysM-DKO/ABCG1fl/+ macrophages were incubated in the presence or absence of HDL (25μg/mL) and ApoA-1 (10μg/mL) in the presence of 3μmol/L LXR agonist (T0901317) (n=9). (C) Schematic of [3H]-cholesterol loaded macrophage-RCT pathway. WT, LysM-DKO or LysM-DKO/ABCG1fl/+ peritoneal macrophages were treated with 4μCi/mL [3H]-cholesterol and 50μg/mL ac-LDL for 48h followed by the injection of radiolabeled foam cells to C57BL/6/WT mice. After 48h, blood, liver and feces were collected and measured using a scintillation counter. (D-I) Distribution of [3H]-radioactivity counts in serum, HDL, liver feces and intestinal contents were determined by scintillation counter (n=6). (J) LysM-DKO mice were crossed with ABCG1fl/+ mice to generate LysM-DKO-ABCG1fl/+ mice. (K-L) Bone marrow derived macrophages isolated from WT, LysM-DKO and LysM-DKO/ABCG1fl/+ mice on normal diet (n=6) were analyzed by qRT-PCR (K) and WB (L) (n≥3). Data from A-L are presented as mean ± SD. One-way ANOVA followed by Tukey post hoc multiple comparisons test was conducted in A, B, and D-I; Kruskal-Wallis test followed by Dunn’s post hoc multiple comparisons test was conducted in K and L.
Epsin-UIM domain binds ABCG1 to promote degradation via the lysosome
To investigate the molecular mechanisms underlying Epsin-mediated ABCG1 regulation, we tested whether Epsins interact with ABCG1 and governed its turnover in macrophages. We first assessed protein levels of surface receptors such as ABCG1, ABCA1, SR-A1, and LDLR in macrophages involved in the progression of atherosclerosis. WB results showed that ABCG1, but not ABCA1, SR-A1 and LDLR, were upregulated in primary macrophages from DKO mice when compared to WT mice (Figure 6A). The mRNA levels of ABCG1 in both WT and DKO macrophages were not significantly different (Figure S13). Because ABCG1 is known to mediate cholesterol efflux to HDL 48, we hypothesized that Epsins bind ABCG1 under atherosclerotic conditions and promote ABCG1 ubiquitin-dependent degradation to inhibit cholesterol efflux in macrophages and facilitate the development of atherosclerosis. When the isolated peritoneal macrophages from DKO and WT mice were pretreated with a LXR activator followed by a cell surface biotinylation assay, we observed significantly elevated levels of biotinylated ABCG1 in DKO macrophages (Figure 6B). We also performed flow cytometry and observed that only WT macrophages showed a decreased surface level of ABCG1 with oxLDL treatment in a clathrin-dependent way (Figure 6C and S14A–C). Therefore, decreased total and surface levels of ABCG1 in WT macrophages implied that Epsins promote ABCG1 degradation.
Figure 6. Epsins bind to ABCG1 and facilitate the internalization and degradation of ABCG1 via lysosomes.
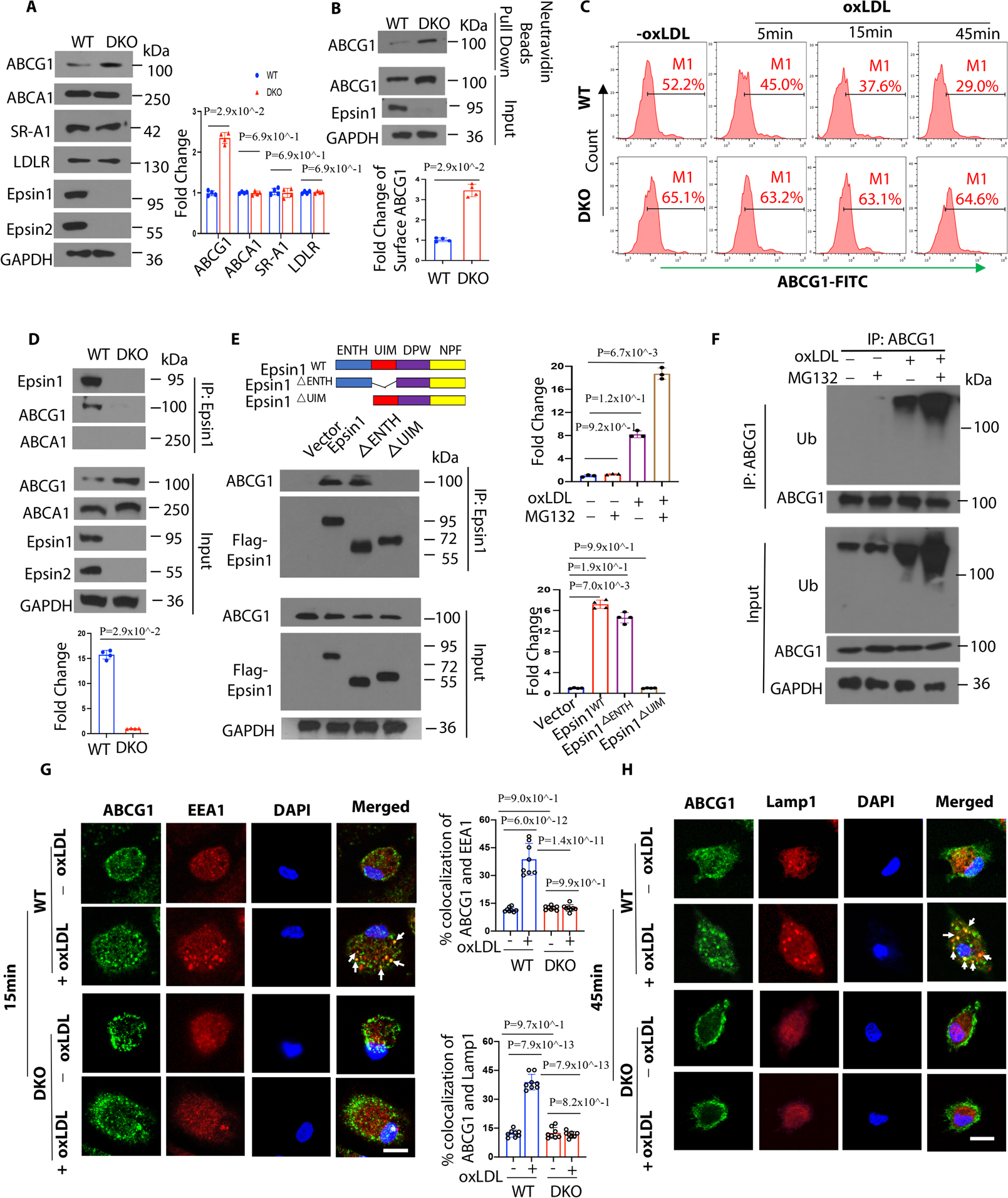
(A) Peritoneal macrophages from WT/ApoE−/− and LysM-DKO/ApoE−/− mice on normal diet (ND) were lysed for WB analysis (n=4). (B) Peritoneal macrophages from WT/ApoE−/− and LysM-DKO/ApoE−/− mice on ND were pretreated with a liver X receptor (LXR) activator and followed by a cell surface biotinylation assay to evaluate the cell surface ABCG1 levels (n=4). (C) LXR agonist activated WT and LysM-DKO peritoneal macrophages were incubated in lipid-deficient medium for 24h and treated with or without 100μg/mL oxLDL for 5, 15 and 45 min followed by staining with anti-ABCG1 and analyzed by flow cytometry. (D) LXR agonist pretreated peritoneal macrophages isolated from WT and LysM-DKO mice on ND were treated with 100μg/mL oxLDL for 1h followed by IP and WB for Epsin1 and ABCG1 or ABCA1 (n=4). (E) ABCG1 plasmids and full length (FLAG-Epsin1WT) or domain-deletion constructs (FLAG-Epsin1ΔENTH or FLAG-Epsin1ΔUIM) in the pcDNA3 vector were transfected into HEK 293T cells for 24 h in the presence of LXR agonist. Cells were then treated with 100μg/mL oxLDL for 1h, followed by IP and WB analysis using antibodies against FLAG tags and ABCG1 (n=4). (F) Peritoneal macrophages isolated from WT mice on ND were cultured in serum-free medium for 24h followed by treatment with 5 μM MG132 for 3h. Cells were then treated with 100μg/mL oxLDL for 1h followed by IP and WB for ubiquitin and ABCG1. (G-H) WT and LysM-DKO peritoneal macrophages were incubated in lipid-deficient medium for 24h followed by incubation with or without 100μg/mL oxLDL for 15min (G) or 45min (H) at 370C. Macrophages were stained with ABCG1 (green), early endosome marker EEA1 (red) or lysosome marker Lamp1 (red) and DAPI (blue), then assessed by confocal microscopy. White arrows indicate the endocytic vesicles, scale bar=5μm, n=8/group. Data from A-H are presented as mean ± SD. Unpaired non-parametric Mann-Whitney U test was conducted in A, B and D; Kruskal-Wallis test followed by Dunn’s post hoc multiple comparisons test was conducted in C, E and F. Two-way ANOVA followed by Sidak post hoc multiple comparisons test was conducted in G and H.
We next determined how Epsins cause increases in total and surface protein levels of ABCG1 in macrophages from DKO mice. When peritoneal- or bone marrow-derived macrophages from WT and DKO mice were lysed and processed for IP with anti-Epsin1 antibodies, we observed ABCG1 in WT macrophages, but not in DKO macrophages (Figure 6D). This suggested that Epsins bind to ABCG1. Accordingly, we showed that Epsin interacted with ABCG1 through the Epsin UIM domain, which was confirmed by transfection of Flag-tagged mammalian Epsin wild type full length, ENTH or UIM deletion constructs as well as ABCG1 plasmid to HEK 293T cells (Figure 6E). We showed that ABCG1 is ubiquitinated in response to oxLDL, allowing Epsins to interact with ABCG1 through their UIM, which promotes ABCG1 lysosomal degradation (Figure 6F). In addition, we observed oxLDL-induced ABCG1 trafficking to early endosomes (Figure 6G) and lysosomes (Figure 6H) in WT macrophages, but this was not evident in DKO cells.
To confirm that elevated cholesterol efflux and RCT were a result of increased total and surface ABCG1 levels in ApoE−/−/LysM-DKO macrophages, we created ApoE−/−/LysM-DKO-ABCG1fl/+ mice by crossing the ApoE−/−/LysM-DKO mouse strain with ABCG1 (flox/+) heterozygous mice to generate a novel mouse model (ApoE−/−/LysM-DKO/ABCG1myeloid-het) with genetically-reduced ABCG1 levels in Epsin-deficient macrophages (Figure 5J). We observed reduced gene (Figure 5K) and protein (Figure 5L) expression of ABCG1 in ApoE−/−/LysM-DKO-ABCG1fl/+ macrophages. The LysM-DKO-ABCG1fl/+ macrophages showed less cholesterol efflux to HDL and RCT compared to ApoE−/−/LysM-DKO macrophages (Figure 5A, 5B and 5D–5I). These studies demonstrated that Epsin deficiency in macrophages specifically enhanced ABCG1-mediated cholesterol efflux to HDL and RCT, which is inversely related to the risk for atherosclerotic cardiovascular disease34. Immunostaining of human patient aortic arch (Figure S15A) and mouse aortic root (Figure S15B) sections revealed that ABCG1 expression is inversely associated with atherosclerotic plaque severity (i.e., there is less ABCG1 expression in patients with more severe atheromas).
S2PNP-siEpsin1/2 hinders progression of early and advanced stages of atherosclerosis
Because we found that Epsins facilitate CD36-mediated lipid uptake and promote ABCG1 degradation to inhibit ABCG1-mediated cholesterol efflux, we determined whether targeting Epsins could prevent the progression of atherosclerosis. We used a targeted NP platform 59 modified with a lesional macrophage-specific targeting peptide (S2P) for systemic delivery of Epsin 1 and 2 siRNAs (S2PNP-siEpsin1/2) (Figure S16A).
Isolated macrophages from WT mice were treated with S2PNP-siControl (S2PNP-siCtrl) or S2PNP-siEpsin1/2. RNA and protein were then isolated to check the in vitro silencing efficacy of the NPs (Figure S16B and S16C). To assess the effect of NPs on in vitro peritoneal macrophage lipid loading, cells isolated from ApoE−/− mice were treated with S2PNP-siCtrl or S2PNP-siEpsin1/2 for 24h followed by treatment with oxLDL or plasma isolated from ApoE−/− mice fed a WD for 8 weeks. The treatment of oxLDL or plasma demonstrated decreased lipid uptake by S2PNP-siEpsin1/2-treated macrophages compared to S2PNP-siCtrl treated cells (Figure S17A and S17B).
ApoE−/− mice fed a WD for 8 weeks (early stage, Figure S18) or 17 weeks (advanced stage, Figure 7) were divided into three groups including baseline (before NP injection), S2PNP-siCtrl, and S2PNP-siEpsin1/2. The baseline group was sacrificed before the injection of NPs. S2PNP-siCtrl or S2PNP-siEpsin1/2 NPs were intravenously injected twice a week for 3 weeks (Figure S18A and 7A). Digested lesions from S2PNP-siCtrl or S2PNP-siEpsin1/2 treated mice were used for WB to check in vivo silencing of Epsins 1 and 2 (Figure S18B). Additionally, the administration of S2PNP-siEpsin1/2 NPs resulted in a decrease in lesional macrophage accumulation and Epsins 1 and 2 expressions within the atherosclerotic plaques as shown by diminished CD68 and Epsin staining in aortic root sections (Figure S18C and S18D). En face ORO staining of aortas, aortic roots, and brachiocephalic artery (BCA) sections (Figure S18E, 7B and 7C) showed reduced lesion size, lesion number and retarded progression of both early (11 weeks on WD) and advanced (20 weeks on WD) stages of atherosclerosis in ApoE−/− mice treated with S2PNP-siEpsin1/2 NPs opposed to the S2PNP-siCtrl treated group.
Figure 7. S2PNP-siEpsin1/2 delivery inhibits atheroma progression, decreases necrotic core content, and increases smooth muscle cell content in advanced stage of atherosclerosis.
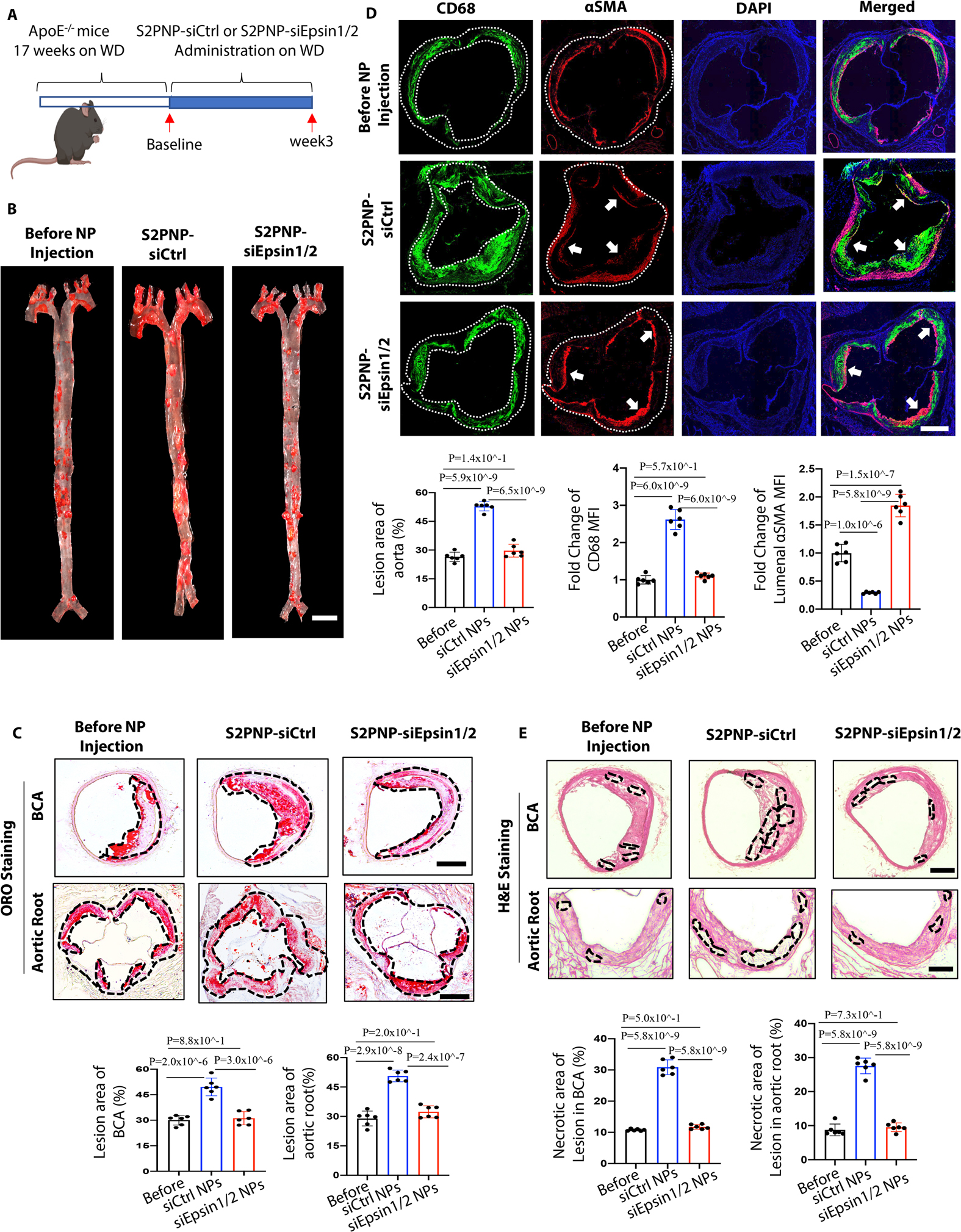
(A) Male ApoE−/− mice fed a Western Diet (WD) for 17 weeks followed by treatment of S2PNP-siCtrl or S2PNP-siEpsin1/2 for 3 weeks (2 doses per week). (B) En face ORO staining of aortas from baseline, control siRNA NP-treated ApoE−/− or Epsin1/2 siRNA NP treated male ApoE−/− mice fed a WD, lesion areas were analyzed with NIH ImageJ, scale bar=5mm. (C) ORO staining of brachiocephalic artery (BCA) and aortic root sections of above three groups. Lesional area (black dash line outlined) were analyzed by NIH ImageJ. (D) Aortic roots from S2PNP-siCtrl-treated ApoE−/− or S2PNP-siEpsin1/2 treated male ApoE−/− mice were stained with the macrophage marker CD68 (dashed white line outlined) and αSMA (white arrow). (E) H&E staining of BCA and aortic root sections of the above three groups. Necrotic areas (black dash line outlined) were analyzed by NIH ImageJ. Data from B-E (n=6) are presented as mean ± SD. Scale bar: B=5mm; C, D, E=500μm. One-way ANOVA followed by Tukey’s post hoc multiple comparisons test was conducted in B-E.
Inhibiting the transition of atheromas from stable lesions to vulnerable plaques is paramount for preventing heart attacks and strokes. We observed elevated smooth muscle cell area and reduced macrophage accumulation (Figure 7D), increased elastic fibers and collagen content (Figure S19), and reduced necrotic core area (Figure 7E) within the atherosclerotic lesions in S2PNP-siEpsin1/2 treated mice compared to the S2PNP-siCtrl NP group. In addition, we observed no changes in plasma total cholesterol, triglycerides, or HDL and non-HDL cholesterol levels between S2PNP-siCtrl and S2PNP-siEpsin1/2 treated mice (Figure S20). These features suggest that the administration of lesion macrophage-specific targeting NPs containing Epsin1/2 siRNA can stabilize atherosclerotic plaques without affecting plasma cholesterol levels.
S2PNP-siEpsin1/2 promotes atheroma resolution by diminishing inflammation and enhancing lesion stability
Given the role of macrophage cholesterol efflux and RCT in atheroma resolution, we also wanted to assess the therapeutic effects of S2PNP-siEpsin1/2 NPs on atheroma resolution. We established another mouse model by injecting an adeno-associated virus-8 carrying a mutant PCSK9 gene (AAV8-PCSK9D377Y) into C57BL/6 mice. This generated an atherosclerotic mouse model harboring a PCSK9 gain-of-function 60, which mimics LDLR null mice 26 and requires no additional genetic modification 60. Another advantage of this mouse line is that a single AAV injection is sufficient to generate atherosclerotic mice by WD feeding compared to long-term crossbreeding with ApoE−/− or LDLR−/− mice.
To test if S2PNP-siEpsin1/2 treatment can promote atheroma resolution, PCSK9-AAV8 mice fed a WD for 16 weeks followed by 4 weeks of normal diet feeding combined with S2PNP-siCtrl or S2PNP-siEpsin1/2 treatment (Figure 8A). The latter group showed a reduction of lesion size and lesional regression as evidenced in the IF and ORO staining of the whole aorta, aortic root and BCA sections (Figure 8B, 8C, 8D and S21A). Van Gieson’s trichrome staining of BCA sections showed increased collagen content and elastic fibers in the S2PNP-siEpsin1/2 treated group compared to the S2PNP-siCtrl treated or baseline groups (Figure S21B). In addition, the IF co-staining of CD68 and α-SMA showed reduced macrophage accumulation in the fibrous cap, reduced size of atheromatous cores, and increased smooth muscle cell area within the fibrous caps of atheromas in the S2PNP-siEpsin1/2 group (Figure 8C).
Figure 8. S2PNP-siEpsin1/2 promotes the resolution of atheroma, reduces inflammation, and stabilizes atherosclerotic plaques.
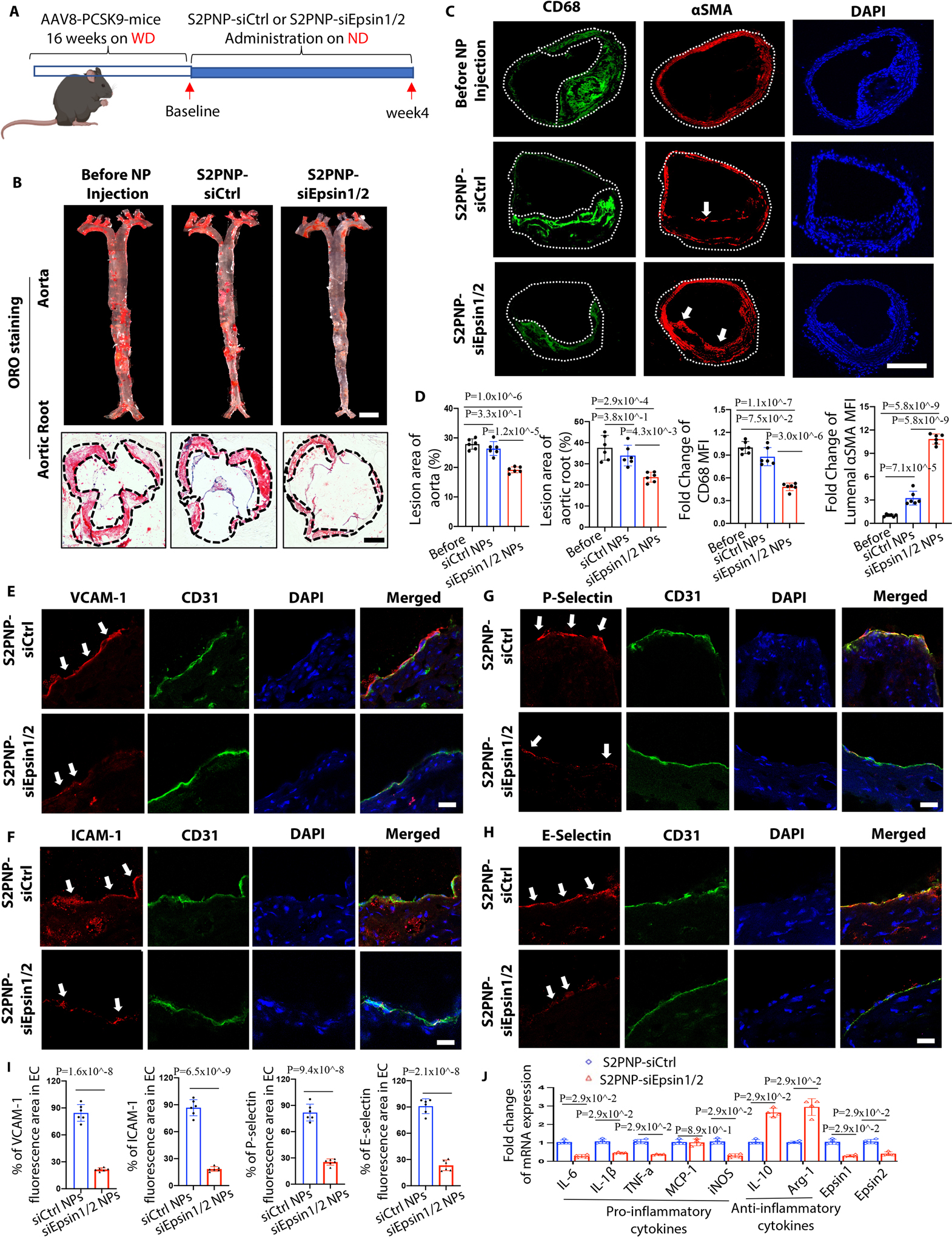
(A) Male C57BL/6 WT mice were injected twice with PCSK9-AAV8 (D377Y) virus and fed a WD for 16 weeks and followed by normal diet feeding with the treatment of S2PNP-siCtrl or S2PNP-siEpsin1/2 for 4 weeks (2 doses per week). (B) En face ORO staining of aortas from above mice. Lesion area was analyzed using NIH ImageJ, scale bar=5mm. (C) Immunostaining of CD68 (green) and αSMA (red) in the above groups. White arrows indicate the luminal αSMA. For (B) and (C), n=6/group, scale bar: B (aorta)=5mm, B (aortic root) =500μm, C=500μm. (D) Statistics for ORO staining (B) and IF staining (C). (E-H) Co-staining of CD31 (endothelial cell marker, green) with vascular cell adhesion molecule-1 (VCAM-1, red, E), intercellular adhesion molecule-1 (ICAM-1, red, F), P-selectin (red, G) or E-selectin (red, H) and DAPI on the luminal surface of aortic root sections. White arrows indicate CD31/ICAM-1, CD31/VCAM-1, CD31/E-selectin or CD31/P-selectin colocalization. Scale bars=20μm. (I) Statistical analysis of E-H, n=6/group. (J) Isolated macrophages from PCSK9-mice were treated with S2PNP-siCtrl or S2PNP-siEpsin1/2 for 24h, RNA was isolated, and qRT-PCR was performed (n=4). B-J are presented as mean ± SD. and were analyzed using one-way ANOVA (B and C) and the unpaired Student’s t-test (E-J). One-way ANOVA followed by Tukey’s post hoc multiple comparisons test was conducted in D; Unpaired t test was conducted in I; Mann-Whitney U test was conducted in J.
Adhesion molecules play a crucial role in attaching monocytes to activated endothelial cells (EC) and recruiting monocytes to the subendothelial layer. Co-staining of CD31 with vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), P-selectin or E-selectin (Figure 8E–8I) was performed using aortic root sections. Administration of S2PNP-siEpsin1/2 NPs significantly suppressed these adhesion molecules when compared to the S2PNP-siCtrl treated group. In addition, the expression of the macrophage egressing gene CCR7 was significant increased, while the retention genes Sema3a and Netrin1 were decreased, in DKO macrophages (Figure S21C). This was consistent with our RNA-seq analysis that showed the deficiency of macrophage Epsins facilitates immune cell migration. Furthermore, macrophages treated with S2PNP-siEpsin1/2 NPs showed decreased expression of pro-inflammatory genes (iNOS, IL-6, IL-1β, and TNF-α) and increased expression of anti-inflammatory markers (IL-10 and Arg1) when compared to the S2P-siCtrl treated group (Figure 8J). These decreased pro-inflammatory cytokines lead to reduced ICAM-1, VCAM-1, P- and E-selectin expression in ECs (Figure 8E–8H), hindering recruitment of additional macrophages to the lesion.
To confirm our in vivo findings in both early/advanced progression and regression atherosclerosis models, we separately analyzed female and male mice. The results from these studies did not show differences between females and males (Figure 7B, 8B, S18E and S22A–C). Taken together, these results indicate that silencing of Epsins in lesional macrophages may offer therapeutic benefits in preventing or treating advanced atherosclerosis.
In addition, to confirm our in vitro studies in mouse macrophages, we performed Epsin1/2 siRNA-mediated knockdown in human monocyte-derived macrophages (THP1 macrophages). We observed similar molecular mechanisms and phenotypes in Epsin1/2-deficient THP1 macrophages and LysM-DKO macrophages with respect to lipid uptake, neutral lipid accumulation, and cholesterol efflux. In addition, Epsin-deficiency in these macrophages similarly regulated of the expression of CD36, ABCG1, and LRP126 (Figure S23A–D and S24A–G).
DISCUSSION
We discovered that Epsins 1 and 2 govern cholesterol transport (Figure S25) in atherosclerotic macrophages by regulating CD36-mediated lipid uptake (Figure 1–3 and S2–S10) and ABCG1-dependent cholesterol efflux (Figure 4–6 and S11–S14). Importantly, we demonstrated that targeting lesional macrophage Epsins using a nanotechnology-based siRNA-mediated therapy halted inflammation, impeded early and late atherogenesis (Figure 7 and S16–S20), and accelerated atheroma resolution (Figure 8 and S21). By silencing lesional macrophage Epsins, we observed a reduction in necrotic core area in atherosclerotic lesions and attenuation of inflammation (Figure 7 and Figure 8). In addition, our scRNA-seq data provided a high-resolution view of the macrophage landscape within the lesion and highlighted that CD36-mediated lipid uptake was diminished at the same time ABCG1 expression was upregulated in Trem2+ macrophages, a sub-cluster of macrophages that play a role in atheroma resolution. Our scRNA-seq data further showed that lesional macrophage clusters are reduced by macrophage epsin-deficiency. Together, our studies point toward a promising new strategy for the treatment of atherosclerosis.
The advent of therapies targeting PCSK9 to treat atherosclerosis, including monoclonal anti-PCSK9 antibodies and PCSK9 siRNAs, has been considered to be a game-changing development 61–63; however, these and other therapies largely ignore the rampant inflammation that occurs in atherosclerotic arteries. Arterial inflammation is a prime contributor to the transition from a stable to vulnerable atheroma 7–10. While anti-inflammatory medicines such as anti-Interleukin-1β antibodies diminish the risk of recurrent myocardial infarction, these treatments have side effects 64,65. In addition, the inability to reduce lipid ingestion while concomitantly reinforcing cholesterol efflux in lesional macrophages has impeded the battle against atherosclerosis.
Epsins may interact with different receptors in macrophages at different stages of atheroma progression or regression. We previously reported that without Epsins, the surface levels of LRP-1 are abundant in macrophages, causing enhanced efferocytosis and an anti-inflammatory macrophage phenotype that resulted in reduced foam cell formation. Given that macrophage LRP1 has opposing effects on plaque biogenesis, depending on whether the plaque is growing or shrinking, and as LRP1 plays an accessory role in lipid uptake and metabolism, the phenotype of blunted oxLDL uptake in Epsin-deficient macrophages prompted us to investigate the mechanisms beyond LRP1-mediated regulation.
In this study, we showed that Epsins bind to CD36 to facilitate lipid uptake by enhancing CD36 endocytosis and recycling. Conversely, Epsins promote ABCG1 degradation via lysosomes and hamper ABCG1-mediated cholesterol efflux and reverse cholesterol transport. Thus, these two pathways greatly reduced foam cell formation and, ultimately, reduce atherosclerosis. By harnessing the latest developments in nanomedicine and designing S2P-NPs carrying Epsin1/2 siRNAs (S2PNP-siEpsin1/2), we showed a reduction in plaque formation in both early and advanced atherosclerosis (Figure S18 and 7). Furthermore, in a pre-clinical atheroma resolution model, this approach reduced lesional macrophage accumulation, decreased pro-inflammatory cytokine and adhesion molecule expression, and accelerated plaque regression (Figure 8). Taken together, our study uncovered a novel and critical role for Epsin in regulating lipid metabolism and cholesterol efflux as well as offering a potential new nanotherapy for treating atherosclerosis by targeting macrophage Epsins.
As we showed that silencing of lesional macrophage Epsins reduces necrotic core size, we reasoned that inhibiting Epsins may reinforce efferocytosis in macrophages, analogous to our observations in DKO macrophages 26 because efferocytosis can regulate necrotic core formation 66–68. Despite the fact that S2P-NPs have been shown to only negligibly target the endothelium 59, whether S2PNP-siEpsin1/2 inhibits endothelial Epsins to synergistically curb atherosclerosis will require further investigation as deletion of endothelial Epsins also reduces inflammation and atherogenesis 24,25. Interestingly, we did not observe significant changes in the lipid profiles of plasma obtained from S2PNP-siCtrl and S2PNP-siEpsin1/2 treated ApoE−/− mice, suggesting either minor liver targeting by S2PNP-siEpsin1/2 or that S2PNP-siEpsin1/2-mediated silencing of Epsins in liver was inconsequential. Either way, determining if S2PNP-siEpsin1/2 targets liver Epsins warrants further investigation.
While autophagy can eliminate excess lipid droplet-associated cholesterol esters taken up by macrophages 69–72, this process is attenuated during atheroma progression, leading to defective lipolysis, impaired efferocytosis, and escalated inflammation, which increases the likelihood of vulnerable lesion rupture 69–75. A recent report suggests that macrophage SR-B1 modulates autophagy through the VPS34 complex and PPARα transcription of Tfeb in atherosclerosis 76. SR-B1 is a multifunctional membrane receptor, which is not only responsible for selective uptake of HDL- derived cholesteryl esters into cells, but also for transferring HDL-cholesterol efflux from peripheral tissues to back to liver 77. Given that Epsins are implicated in autophagy function in Drosophila 78, the question of whether these proteins suppress autophagic activity by binding to SR-B1 and inhibiting SR-B1-regulated function in macrophages remains unanswered.
Our studies also revealed that LXR-activated macrophage surface expression of ABCG1 was significantly reduced with oxLDL treatment of WT, but not DKO macrophages, owing to their internalization and degradation mediated by the Epsins (Figure 6). It is thought that basal macrophage ABCG1 is largely located at an intracellular location 79 and then redistributed to the cell surface following LXR activation 58. Our immunostaining and biotinylation studies (Figure 6) are consistent with this supposition. More importantly, staining of ABCG1 in both human patient aortic arch and mouse aortic root sections, showed a reduction in ABCG1 expression in severe atherosclerotic lesions (Figure S15), suggesting the loss of Epsins would maintain the abundance of ABCG1 to limit plaque accumulation. Recent clinical trials suggest that enhanced cholesterol efflux capacity (CEC) considerably reduces the risk of repeated acute myocardial infarctions (MIs) 39–41. In addition, cholesterol efflux capacity has few demonstrable links to health risks, in contrast to that observed upon alteration of HDL levels 45. Our results show that targeting Epsins augments cholesterol efflux (Figure 5 and S23) and, consequently, may potentially enhance cholesterol efflux capacity, suggesting that inhibition of Epsins represents a promising new strategy for treating patients prone to recurrent MIs and severe CAD.
In conclusion, our results provide a powerful proof-of-concept for the therapeutic potential of Epsin siRNA-containing NPs to precisely target lesional macrophages to abolish inflammation and resolve atheroma. By illustrating that targeting Epsin expression in macrophages restricts lipid ingestion in foam cells and concurrently enhances cholesterol efflux, we believe this study provides a foundation for the development of therapies that precisely resolve lesional inflammation in advanced atherosclerosis.
Supplementary Material
Novelty and Significance.
What Is Known?
Epsin endocytic adaptor proteins are upregulated in human and mouse atherosclerotic lesions
Lesional macrophages internalize lipids primarily through scavenger receptor-mediated endocytosis such as CD36 and SR-A
Macrophage cholesterol efflux and reverse cholesterol transport is crucial to atheroma resolution
What New Information Does This Article Contribute?
ScRNA-seq combined with the newly-developed algorithm MEBOCOST reveals that Epsins are involved in macrophage-mediated lipid metabolic regulation
Macrophage epsins promote lipid uptake by targeting CD36 endocytosis and membrane recycling via the Epsin ENTH domain
Epsins bind ubiquitinated ABCG1—resulting in the endocytosis and lysosomal degradation of this cholesterol transporter, which reduces cholesterol efflux
Macrophage-specific, nanoparticle-mediated RNAi delivery exhibits a therapeutic benefit for the treatment of atherosclerosis
Atherosclerotic plaque regression using this nanoparticle delivery platform represents a clinically-relevant approach for the treatment of advanced atherosclerosis
Atherosclerotic plaque regression is a crucial process in the treatment of many cardiovascular diseases. Despite the development of new cholesterol-lowering therapies, including the recently approved PCSK9 small interfering RNA (siRNA) antagonists, patients still face a risk of developing major acute cardiovascular events resulting from chronic inflammation in the plaque. We employed a novel nanomedicine platform containing a stabilin-2 targeting peptide (S2P) to deliver Epsin-specific siRNAs to lesional macrophages. We discovered that inhibition of these adaptor proteins in lesional macrophages significantly diminished plaque size and necrotic core area, increased fibrous cap thickness, and promoted plaque regression.
Acknowledgments:
We appreciate GEO database access and we thank the Flow Cytometry Core at Boston Children’s Hospital for the use of the LSRII, the Viral Core at Boston Children’s Hospital for providing viral stocks, and the Biopolymers Facility at Harvard Medical School for quality control analysis of RNA samples.
Sources of Funding:
This work was supported by R01HL146134 (H.C., S.S., M.F.L.) and American Heart Association Established Investigator Award and Transformational Project Award (H.C.), and in part by NIH grants R01HL137229, R01HL141853, R01HL156362, R01HL158097, R01HL162367, (H.C.), AHA 17SDG33410868 (H.W.), R01GM125632 (K.F.C.), and P01 HL116263 (M.F.L.).
Nonstandard Abbreviations and Acronyms
- AAV8
adeno-associated virus-8
- ABCG1
ATP binding cassette subfamily G member 1
- ACAT
acyl-CoA cholesterol acyltransferase
- AMI
acute myocardial infarction
- BCA
brachiocephalic artery
- CAD
coronary artery disease
- CEC
cholesterol efflux capacity
- DKO
double knockout
- ENTH
Epsin N-terminal homology
- GEM
gel beads-in-emulsion
- GO
gene ontology
- GSEA
gene set enrichment analysis
- HDL
high density lipoprotein
- ICAM-1
intercellular adhesion molecule-1
- IF
immunofluorescence staining
- IP
immunoprecipitation
- LXR
liver X receptor
- M-CSF
macrophage colony stimulating factor
- NP
nanoparticle
- ORO
oil red O
- oxLDL
oxidized low-density lipoprotein
- PCSK9
proprotein convertase subtilisin/kexin type 9 serine protease
- RCT
reverse cholesterol transport
- scRNA-seq
single-cell RNA sequencing
- siRNA
small interfering RNA
- SR-A
scavenger receptor-A
- S2PNP-siEpsin1/2
S2P-conjugated Epsins 1 and 2 siRNA nanoparticle
- S2PNP-siCtrl
S2P-conjugated Control nanoparticle
- TG
thioglycolate
- UIM
ubiquitin-interacting motif
- VCAM-1
vascular cell adhesion molecule-1
- VSMCs
vascular smooth muscle cells
- WB
western blot
- WD
western diet
Footnotes
Disclosures: All authors declare that they have no competing interests.
Supplemental Material:
REFERRENCES
- 1.Libby P Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2045–2051. doi: 10.1161/ATVBAHA.108.179705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17:1410–1422. doi: 10.1038/nm.2538 [DOI] [PubMed] [Google Scholar]
- 3.Glass CK, Witztum JL. Atherosclerosis. the road ahead. Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0 [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Nanda V, Direnzo D, Ye J, Xiao S, Kojima Y, Howe KL, Jarr KU, Flores AM, Tsantilas P, et al. Clonally expanding smooth muscle cells promote atherosclerosis by escaping efferocytosis and activating the complement cascade. Proc Natl Acad Sci U S A. 2020;117:15818–15826. doi: 10.1073/pnas.2006348117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma M, Schlegel MP, Afonso MS, Brown EJ, Rahman K, Weinstock A, Sansbury BE, Corr EM, van Solingen C, Koelwyn GJ, et al. Regulatory T Cells License Macrophage Pro-Resolving Functions During Atherosclerosis Regression. Circ Res. 2020;127:335–353. doi: 10.1161/CIRCRESAHA.119.316461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steinberg D The LDL modification hypothesis of atherogenesis: an update. J Lipid Res. 2009;50 Suppl:S376–381. doi: 10.1194/jlr.R800087-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hansson GK, Libby P, Tabas I. Inflammation and plaque vulnerability. J Intern Med. 2015;278:483–493. doi: 10.1111/joim.12406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doring Y Not growth but death: GM-CSF/IL-23 axis drives atherosclerotic plaque vulnerability by enhancing macrophage and DC apoptosis. Circ Res. 2015;116:222–224. doi: 10.1161/CIRCRESAHA.114.305674 [DOI] [PubMed] [Google Scholar]
- 9.Alsheikh-Ali AA, Kitsios GD, Balk EM, Lau J, Ip S. The vulnerable atherosclerotic plaque: scope of the literature. Ann Intern Med. 2010;153:387–395. doi: 10.7326/0003-4819-153-6-201009210-00272 [DOI] [PubMed] [Google Scholar]
- 10.Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47:C13–18. doi: 10.1016/j.jacc.2005.10.065 [DOI] [PubMed] [Google Scholar]
- 11.Charo IF, Taub R. Anti-inflammatory therapeutics for the treatment of atherosclerosis. Nat Rev Drug Discov. 2011;10:365–376. doi: 10.1038/nrd3444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Camilli P, Chen H, Hyman J, Panepucci E, Bateman A, Brunger AT. The ENTH domain. FEBS Lett. 2002;513:11–18. doi: 10.1016/s0014-5793(01)03306-3 [DOI] [PubMed] [Google Scholar]
- 13.Itoh T, Koshiba S, Kigawa T, Kikuchi A, Yokoyama S, Takenawa T. Role of the ENTH domain in phosphatidylinositol-4,5-bisphosphate binding and endocytosis. Science. 2001;291:1047–1051. doi: 10.1126/science.291.5506.1047 [DOI] [PubMed] [Google Scholar]
- 14.Rosenthal JA, Chen H, Slepnev VI, Pellegrini L, Salcini AE, Di Fiore PP, De Camilli P. The epsins define a family of proteins that interact with components of the clathrin coat and contain a new protein module. J Biol Chem. 1999;274:33959–33965. doi: 10.1074/jbc.274.48.33959 [DOI] [PubMed] [Google Scholar]
- 15.Cui K, Dong Y, Wang B, Cowan DB, Chan SL, Shyy J, Chen H. Endocytic Adaptors in Cardiovascular Disease. Front Cell Dev Biol. 2020;8:624159. doi: 10.3389/fcell.2020.624159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen H, De Camilli P. The association of epsin with ubiquitinated cargo along the endocytic pathway is negatively regulated by its interaction with clathrin. Proc Natl Acad Sci U S A. 2005;102:2766–2771. doi: 10.1073/pnas.0409719102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shih SC, Katzmann DJ, Schnell JD, Sutanto M, Emr SD, Hicke L. Epsins and Vps27p/Hrs contain ubiquitin-binding domains that function in receptor endocytosis. Nat Cell Biol. 2002;4:389–393. doi: 10.1038/ncb790 [DOI] [PubMed] [Google Scholar]
- 18.Polo S, Sigismund S, Faretta M, Guidi M, Capua MR, Bossi G, Chen H, De Camilli P, Di Fiore PP. A single motif responsible for ubiquitin recognition and monoubiquitination in endocytic proteins. Nature. 2002;416:451–455. doi: 10.1038/416451a [DOI] [PubMed] [Google Scholar]
- 19.Chen H, Ko G, Zatti A, Di Giacomo G, Liu L, Raiteri E, Perucco E, Collesi C, Min W, Zeiss C, et al. Embryonic arrest at midgestation and disruption of Notch signaling produced by the absence of both epsin 1 and epsin 2 in mice. Proc Natl Acad Sci U S A. 2009;106:13838–13843. doi: 10.1073/pnas.0907008106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tessneer KL, Pasula S, Cai X, Dong Y, McManus J, Liu X, Yu L, Hahn S, Chang B, Chen Y, et al. Genetic reduction of vascular endothelial growth factor receptor 2 rescues aberrant angiogenesis caused by epsin deficiency. Arterioscler Thromb Vasc Biol. 2014;34:331–337. doi: 10.1161/ATVBAHA.113.302586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang B, Tessneer KL, McManus J, Liu X, Hahn S, Pasula S, Wu H, Song H, Chen Y, Cai X, et al. Epsin is required for Dishevelled stability and Wnt signalling activation in colon cancer development. Nat Commun. 2015;6:6380. doi: 10.1038/ncomms7380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu X, Pasula S, Song H, Tessneer KL, Dong Y, Hahn S, Yago T, Brophy ML, Chang B, Cai X, et al. Temporal and spatial regulation of epsin abundance and VEGFR3 signaling are required for lymphatic valve formation and function. Sci Signal. 2014;7:ra97. doi: 10.1126/scisignal.2005413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pasula S, Cai X, Dong Y, Messa M, McManus J, Chang B, Liu X, Zhu H, Mansat RS, Yoon SJ, et al. Endothelial epsin deficiency decreases tumor growth by enhancing VEGF signaling. J Clin Invest. 2012;122:4424–4438. doi: 10.1172/JCI64537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong Y, Wang B, Cui K, Cai X, Bhattacharjee S, Wong S, Cowan DB, Chen H. Epsins Negatively Regulate Aortic Endothelial Cell Function by Augmenting Inflammatory Signaling. Cells. 2021;10. doi: 10.3390/cells10081918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong Y, Lee Y, Cui K, He M, Wang B, Bhattacharjee S, Zhu B, Yago T, Zhang K, Deng L, et al. Epsin-mediated degradation of IP3R1 fuels atherosclerosis. Nat Commun. 2020;11:3984. doi: 10.1038/s41467-020-17848-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brophy ML, Dong Y, Tao H, Yancey PG, Song K, Zhang K, Wen A, Wu H, Lee Y, Malovichko MV, et al. Myeloid-Specific Deletion of Epsins 1 and 2 Reduces Atherosclerosis by Preventing LRP-1 Downregulation. Circ Res. 2019;124:e6–e19. doi: 10.1161/CIRCRESAHA.118.313028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moore KJ, Freeman MW. Scavenger receptors in atherosclerosis: beyond lipid uptake. Arterioscler Thromb Vasc Biol. 2006;26:1702–1711. doi: 10.1161/01.ATV.0000229218.97976.43 [DOI] [PubMed] [Google Scholar]
- 28.Ouimet M, Ediriweera H, Afonso MS, Ramkhelawon B, Singaravelu R, Liao X, Bandler RC, Rahman K, Fisher EA, Rayner KJ, et al. microRNA-33 Regulates Macrophage Autophagy in Atherosclerosis. Arterioscler Thromb Vasc Biol. 2017;37:1058–1067. doi: 10.1161/ATVBAHA.116.308916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kunjathoor VV, Febbraio M, Podrez EA, Moore KJ, Andersson L, Koehn S, Rhee JS, Silverstein R, Hoff HF, Freeman MW. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem. 2002;277:49982–49988. doi: 10.1074/jbc.M209649200 [DOI] [PubMed] [Google Scholar]
- 30.Podrez EA, Febbraio M, Sheibani N, Schmitt D, Silverstein RL, Hajjar DP, Cohen PA, Frazier WA, Hoff HF, Hazen SL. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. J Clin Invest. 2000;105:1095–1108. doi: 10.1172/JCI8574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, Hoff HF, Sharma K, Silverstein RL. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest. 2000;105:1049–1056. doi: 10.1172/JCI9259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nozaki S, Kashiwagi H, Yamashita S, Nakagawa T, Kostner B, Tomiyama Y, Nakata A, Ishigami M, Miyagawa J, Kameda-Takemura K, et al. Reduced uptake of oxidized low density lipoproteins in monocyte-derived macrophages from CD36-deficient subjects. J Clin Invest. 1995;96:1859–1865. doi: 10.1172/JCI118231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohashi R, Mu H, Wang X, Yao Q, Chen C. Reverse cholesterol transport and cholesterol efflux in atherosclerosis. QJM. 2005;98:845–856. doi: 10.1093/qjmed/hci136 [DOI] [PubMed] [Google Scholar]
- 34.Kennedy MA, Barrera GC, Nakamura K, Baldan A, Tarr P, Fishbein MC, Frank J, Francone OL, Edwards PA. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005;1:121–131. doi: 10.1016/j.cmet.2005.01.002 [DOI] [PubMed] [Google Scholar]
- 35.Westerterp M, Tsuchiya K, Tattersall IW, Fotakis P, Bochem AE, Molusky MM, Ntonga V, Abramowicz S, Parks JS, Welch CL, et al. Deficiency of ATP-Binding Cassette Transporters A1 and G1 in Endothelial Cells Accelerates Atherosclerosis in Mice. Arterioscler Thromb Vasc Biol. 2016;36:1328–1337. doi: 10.1161/ATVBAHA.115.306670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yvan-Charvet L, Ranalletta M, Wang N, Han S, Terasaka N, Li R, Welch C, Tall AR. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J Clin Invest. 2007;117:3900–3908. doi: 10.1172/JCI33372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sag D, Cekic C, Wu R, Linden J, Hedrick CC. The cholesterol transporter ABCG1 links cholesterol homeostasis and tumour immunity. Nat Commun. 2015;6:6354. doi: 10.1038/ncomms7354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moore KJ, Fisher EA. High-density lipoproteins put out the fire. Cell Metab. 2014;19:175–176. doi: 10.1016/j.cmet.2014.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nicholls SJ, Puri R, Ballantyne CM, Jukema JW, Kastelein JJP, Koenig W, Wright RS, Kallend D, Wijngaard P, Borgman M, et al. Effect of Infusion of High-Density Lipoprotein Mimetic Containing Recombinant Apolipoprotein A-I Milano on Coronary Disease in Patients With an Acute Coronary Syndrome in the MILANO-PILOT Trial: A Randomized Clinical Trial. JAMA Cardiol. 2018;3:806–814. doi: 10.1001/jamacardio.2018.2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tardif JC, Gregoire J, L’Allier PL, Ibrahim R, Lesperance J, Heinonen TM, Kouz S, Berry C, Basser R, Lavoie MA, et al. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA. 2007;297:1675–1682. doi: 10.1001/jama.297.15.jpc70004 [DOI] [PubMed] [Google Scholar]
- 41.Keene D, Price C, Shun-Shin MJ, Francis DP. Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: meta-analysis of randomised controlled trials including 117,411 patients. BMJ. 2014;349:g4379. doi: 10.1136/bmj.g4379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hewing B, Moore KJ, Fisher EA. HDL and cardiovascular risk: time to call the plumber? Circ Res. 2012;111:1117–1120. doi: 10.1161/CIRCRESAHA.112.280958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Josefs T, Basu D, Vaisar T, Arets B, Kanter JE, Huggins LA, Hu Y, Liu J, Clouet-Foraison N, Heinecke JW, et al. Atherosclerosis Regression and Cholesterol Efflux in Hypertriglyceridemic Mice. Circ Res. 2021;128:690–705. doi: 10.1161/CIRCRESAHA.120.317458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, Neeland IJ, Yuhanna IS, Rader DR, de Lemos JA, et al. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;371:2383–2393. doi: 10.1056/NEJMoa1409065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Silvain J, Kerneis M, Guerin M, Montalescot G. Modulation of cholesterol efflux capacity in patients with myocardial infarction. Curr Opin Cardiol. 2019;34:714–720. doi: 10.1097/HCO.0000000000000677 [DOI] [PubMed] [Google Scholar]
- 47.Capodanno D, Mehran R, Gibson CM, Angiolillo DJ. CSL112, a reconstituted, infusible, plasma-derived apolipoprotein A-I: safety and tolerability profiles and implications for management in patients with myocardial infarction. Expert Opin Investig Drugs. 2018;27:997–1005. doi: 10.1080/13543784.2018.1543399 [DOI] [PubMed] [Google Scholar]
- 48.Ouimet M, Barrett TJ, Fisher EA. HDL and Reverse Cholesterol Transport. Circ Res. 2019;124:1505–1518. doi: 10.1161/CIRCRESAHA.119.312617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tombor LS, John D, Glaser SF, Luxan G, Forte E, Furtado M, Rosenthal N, Baumgarten N, Schulz MH, Wittig J, et al. Single cell sequencing reveals endothelial plasticity with transient mesenchymal activation after myocardial infarction. Nat Commun. 2021;12:681. doi: 10.1038/s41467-021-20905-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Franzen O, Gan LM, Bjorkegren JLM. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database (Oxford). 2019;2019. doi: 10.1093/database/baz046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dobnikar L, Taylor AL, Chappell J, Oldach P, Harman JL, Oerton E, Dzierzak E, Bennett MR, Spivakov M, Jorgensen HF. Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels. Nat Commun. 2018;9:4567. doi: 10.1038/s41467-018-06891-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Atri C, Guerfali FZ, Laouini D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int J Mol Sci. 2018;19. doi: 10.3390/ijms19061801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zheng R, Zhang Y, Tsuji T, Zhang L, Tseng Y-H, Chen K. MEBOCOST: Metabolic Cell-Cell Communication Modeling by Single Cell Transcriptome. bioRxiv. 2022. [Google Scholar]
- 54.Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, Wolf D, Saliba AE, Zernecke A. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ Res. 2018;122:1661–1674. doi: 10.1161/CIRCRESAHA.117.312509 [DOI] [PubMed] [Google Scholar]
- 55.Zernecke A, Winkels H, Cochain C, Williams JW, Wolf D, Soehnlein O, Robbins CS, Monaco C, Park I, McNamara CA, et al. Meta-Analysis of Leukocyte Diversity in Atherosclerotic Mouse Aortas. Circ Res. 2020;127:402–426. doi: 10.1161/CIRCRESAHA.120.316903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen Y, Yang M, Huang W, Chen W, Zhao Y, Schulte ML, Volberding P, Gerbec Z, Zimmermann MT, Zeighami A, et al. Mitochondrial Metabolic Reprogramming by CD36 Signaling Drives Macrophage Inflammatory Responses. Circ Res. 2019;125:1087–1102. doi: 10.1161/CIRCRESAHA.119.315833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Antonov AS, Kolodgie FD, Munn DH, Gerrity RG. Regulation of macrophage foam cell formation by alphaVbeta3 integrin: potential role in human atherosclerosis. Am J Pathol. 2004;165:247–258. doi: 10.1016/s0002-9440(10)63293-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang N, Ranalletta M, Matsuura F, Peng F, Tall AR. LXR-induced redistribution of ABCG1 to plasma membrane in macrophages enhances cholesterol mass efflux to HDL. Arterioscler Thromb Vasc Biol. 2006;26:1310–1316. doi: 10.1161/01.ATV.0000218998.75963.02 [DOI] [PubMed] [Google Scholar]
- 59.Tao W, Yurdagul A Jr., Kong N, Li W, Wang X, Doran AC, Feng C, Wang J, Islam MA, Farokhzad OC, et al. siRNA nanoparticles targeting CaMKIIgamma in lesional macrophages improve atherosclerotic plaque stability in mice. Sci Transl Med. 2020;12. doi: 10.1126/scitranslmed.aay1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oppi S, Luscher TF, Stein S. Mouse Models for Atherosclerosis Research-Which Is My Line? Front Cardiovasc Med. 2019;6:46. doi: 10.3389/fcvm.2019.00046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fitzgerald K, White S, Borodovsky A, Bettencourt BR, Strahs A, Clausen V, Wijngaard P, Horton JD, Taubel J, Brooks A, et al. A Highly Durable RNAi Therapeutic Inhibitor of PCSK9. N Engl J Med. 2017;376:41–51. doi: 10.1056/NEJMoa1609243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.German CA, Shapiro MD. Small Interfering RNA Therapeutic Inclisiran: A New Approach to Targeting PCSK9. BioDrugs. 2020;34:1–9. doi: 10.1007/s40259-019-00399-6 [DOI] [PubMed] [Google Scholar]
- 63.Feingold KR. Cholesterol Lowering Drugs. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, Dungan K, Hershman JM, Hofland J, Kalra S, et al. , eds. Endotext. South Dartmouth (MA); 2000. [Google Scholar]
- 64.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914 [DOI] [PubMed] [Google Scholar]
- 65.De Benedetti F, Gattorno M, Anton J, Ben-Chetrit E, Frenkel J, Hoffman HM, Kone-Paut I, Lachmann HJ, Ozen S, Simon A, et al. Canakinumab for the Treatment of Autoinflammatory Recurrent Fever Syndromes. N Engl J Med. 2018;378:1908–1919. doi: 10.1056/NEJMoa1706314 [DOI] [PubMed] [Google Scholar]
- 66.Kojima Y, Weissman IL, Leeper NJ. The Role of Efferocytosis in Atherosclerosis. Circulation. 2017;135:476–489. doi: 10.1161/CIRCULATIONAHA.116.025684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe−/− mice. Arterioscler Thromb Vasc Biol. 2008;28:1421–1428. doi: 10.1161/ATVBAHA.108.167197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gerlach BD, Ampomah PB, Yurdagul A Jr., Liu C, Lauring MC, Wang X, Kasikara C, Kong N, Shi J, Tao W, et al. Efferocytosis induces macrophage proliferation to help resolve tissue injury. Cell Metab. 2021;33:2445–2463 e2448. doi: 10.1016/j.cmet.2021.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sergin I, Evans TD, Zhang X, Bhattacharya S, Stokes CJ, Song E, Ali S, Dehestani B, Holloway KB, Micevych PS, et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat Commun. 2017;8:15750. doi: 10.1038/ncomms15750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Evans TD, Sergin I, Zhang X, Razani B. Target acquired: Selective autophagy in cardiometabolic disease. Sci Signal. 2017;10. doi: 10.1126/scisignal.aag2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Le Guezennec X, Brichkina A, Huang YF, Kostromina E, Han W, Bulavin DV. Wip1-dependent regulation of autophagy, obesity, and atherosclerosis. Cell Metab. 2012;16:68–80. doi: 10.1016/j.cmet.2012.06.003 [DOI] [PubMed] [Google Scholar]
- 72.Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011;13:655–667. doi: 10.1016/j.cmet.2011.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012;15:534–544. doi: 10.1016/j.cmet.2012.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Butcher MJ, Herre M, Ley K, Galkina E. Flow cytometry analysis of immune cells within murine aortas. J Vis Exp. 2011. doi: 10.3791/2848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Purcell-Huynh DA, Farese RV Jr., Johnson DF, Flynn LM, Pierotti V, Newland DL, Linton MF, Sanan DA, Young SG. Transgenic mice expressing high levels of human apolipoprotein B develop severe atherosclerotic lesions in response to a high-fat diet. J Clin Invest. 1995;95:2246–2257. doi: 10.1172/JCI117915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tao H, Yancey PG, Blakemore JL, Zhang Y, Ding L, Jerome WG, Brown JD, Vickers KC, Linton MF. Macrophage SR-BI modulates autophagy via VPS34 complex and PPARalpha transcription of Tfeb in atherosclerosis. J Clin Invest. 2021;131. doi: 10.1172/JCI94229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Van Eck M, Bos IS, Hildebrand RB, Van Rij BT, Van Berkel TJ. Dual role for scavenger receptor class B, type I on bone marrow-derived cells in atherosclerotic lesion development. Am J Pathol. 2004;165:785–794. doi: 10.1016/S0002-9440(10)63341-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Csikos G, Lippai M, Lukacsovich T, Juhasz G, Henn L, Erdelyi M, Maroy P, Sass M. A novel role for the Drosophila epsin (lqf): involvement in autophagy. Autophagy. 2009;5:636–648. doi: 10.4161/auto.5.5.8168 [DOI] [PubMed] [Google Scholar]
- 79.Tarling EJ, Edwards PA. ATP binding cassette transporter G1 (ABCG1) is an intracellular sterol transporter. Proc Natl Acad Sci U S A. 2011;108:19719–19724. doi: 10.1073/pnas.1113021108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587 e3529. doi: 10.1016/j.cell.2021.04.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566:496–502. doi: 10.1038/s41586-019-0969-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhu X, Xu Y, Solis LM, Tao W, Wang L, Behrens C, Xu X, Zhao L, Liu D, Wu J, et al. Long-circulating siRNA nanoparticles for validating Prohibitin1-targeted non-small cell lung cancer treatment. Proc Natl Acad Sci U S A. 2015;112:7779–7784. doi: 10.1073/pnas.1505629112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cui K, Podolnikova NP, Bailey W, Szmuc E, Podrez EA, Byzova TV, Yakubenko VP. Inhibition of integrin alphaDbeta2-mediated macrophage adhesion to end product of docosahexaenoic acid (DHA) oxidation prevents macrophage accumulation during inflammation. J Biol Chem. 2019;294:14370–14382. doi: 10.1074/jbc.RA119.009590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Maess MB, Wittig B, Lorkowski S. Highly efficient transfection of human THP-1 macrophages by nucleofection. J Vis Exp. 2014:e51960. doi: 10.3791/51960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037 [DOI] [PubMed] [Google Scholar]
- 91.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen H, Boutros PC. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics. 2011;12:35. doi: 10.1186/1471-2105-12-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Spangenburg EE, Pratt SJP, Wohlers LM, Lovering RM. Use of BODIPY (493/503) to visualize intramuscular lipid droplets in skeletal muscle. J Biomed Biotechnol. 2011;2011:598358. doi: 10.1155/2011/598358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Robinet P, Wang Z, Hazen SL, Smith JD. A simple and sensitive enzymatic method for cholesterol quantification in macrophages and foam cells. J Lipid Res. 2010;51:3364–3369. doi: 10.1194/jlr.D007336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sankaranarayanan S, Kellner-Weibel G, de la Llera-Moya M, Phillips MC, Asztalos BF, Bittman R, Rothblat GH. A sensitive assay for ABCA1-mediated cholesterol efflux using BODIPY-cholesterol. J Lipid Res. 2011;52:2332–2340. doi: 10.1194/jlr.D018051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Escola-Gil JC, Lee-Rueckert M, Santos D, Cedo L, Blanco-Vaca F, Julve J. Quantification of In Vitro Macrophage Cholesterol Efflux and In Vivo Macrophage-Specific Reverse Cholesterol Transport. Methods Mol Biol. 2015;1339:211–233. doi: 10.1007/978-1-4939-2929-0_15 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all supporting data are available within the article and its supplementary files. Additional methods or data related to this study are available from the corresponding authors upon reasonable request. The RNA-seq and scRNA-Seq data generated in this study were deposited into the GEO database with the accession number GSE214414.