

Abstract
PURA-related neurodevelopmental disorders (PURA-NDDs) include 5q31.3 microdeletion syndrome and PURA syndrome. PURA has been proposed as a candidate gene responsible for 5q31.3 microdeletion syndrome. Phenotype comparisons between patients with PURA mutations and 5q31.3 microdeletions encompassing more than PURA gene are lacking. A total of 25 previously undescribed Mainland China patients were evaluated. Clinical data were obtained from medical record review and standardized medical history questionnaire. Clinical profile and genetic spectrum of the patients with PURA syndrome and genotype–phenotype correlations between PURA mutations group and 5q31.3 microdeletions group were analyzed. Our identified seventeen de nove PURA variants were novel, and two recurrent frameshift variants, c.697_699del (p.F233del) and c.159dup (p.L54Afs*147) were detected in the four independent pedigrees. One patient with 5q31.3 microdeletion further supported the shortest overlapping region only contains PURA and IGIP gene. Developmental delay/intellectual disability, neonatal hypotonia, neonatal feeding difficulties, hypersomnolence and dysmorphic features were prominent clinical features in PURA syndrome. There was no significant difference between two groups in incidence of neonatal problems, developmental delay and common medical comorbidities. We observed a higher frequency of abnormal brain MRI and specific facial dysmorphism in 5q31.3 microdeletion group. This is the first work describing a largest cohort of Mainland China patients broaden the clinical and molecular spectrum of PURA-NDDs. Our findings not only demonstrated that PURA haploinsufficiency was a major contributor to the important phenotypes of 5q31.3 microdeletion, but also implied that additional genes still played a role in the 5q31.3 microdeletion.
Subject terms: Genetic association study, Genetics of the nervous system, Genetic testing, Neurodevelopmental disorders
Introduction
PURA-related neurodevelopmental disorders (PURA-NDDs) include 5q31.3 deletion syndrome and PURA syndrome caused by a heterozygous pathogenic sequence variant in PURA [1]. 5q31.3 deletion syndrome is characterized by neonatal hypotonia, feeding difficulties, respiratory distress, severe global developmental delay/intellectual disability (GDD/ID) and neuroimaging abnormalities [2–6]. To date at least ten affected patients with overlapping 5q31.2q31.3 deletions, ranging from 352 kb to 5.0 Mb, have been reported. The shortest overlapping region between all published patients [2–6] harbors three genes: purine-rich element binding protein A (PURA), IgA-inducing protein (IGIP), and cysteine-rich transmembrane module containing 1 (CYSTM1). The PURA encodes a highly conserved transcriptional activator protein Pur-α that plays a critical role in brain development, neural progenitor cell proliferation, neuronal differentiation and maturation of dendrites [7–9]. Currently, the hypothesis is that PURA is a leading candidate gene responsible for neurological manifestations in 5q31.3 deletion syndrome [4, 10]. Lalani et al. reported PURA pathogenic variants as a cause of neonatal hypotonia, seizures, and encephalopathy in 5q31.3 deletion syndrome [10]. Hunt et al. and Reijnders et al. revealed mutations in PURA cause neurodevelopmental delay and neonatal problems [11, 12]. In addition, genotype–phenotype analysis suggested no strong correlation between PURA mutation classes and phenotypic variability. However, genotype–phenotype analysis has largely focused on patients with PURA mutations, none have compared clinical features in patients with PURA mutations to 5q31.3 microdeletion, which hinder exploring the role of PURA pathogenic variants in the important phenotypes and identifying additional genetic causes of phenotype in 5q31.3 microdeletion syndrome.
To date, only one patient with PURA syndrome have been described, whereas 5q31.3 microdeletion syndrome have never been reported in Mainland China [13]. Here, we reported 25 previously unreported Mainland Chinese patients with PURA-NDDs and presented clinical profile and genetic spectrum of these patients. Importantly, the shortest region gene of 5q31.3 deletion further supported by our study only including PURA and IGIP, thus, this finding significantly corroborated the importance of PURA in 5q31.3 deletion syndrome. Additionally, genotype–phenotype analysis showed pathogenic variants in PURA may contribute to the majority of important phenotypes of 5q31.3 microdeletion such as neonatal problems, developmental delay and common medical comorbidities. We also demonstrate additional genes or regions except for PURA with a high likelihood for causing other clinical phenotypes such as neonatal respiratory difficulty, abnormal brain MRI and specific dysmorphic features.
Methods
Cohort
The study recruited 25 previously unreported patients (P1–P25) in Mainland China from the China League of PURA-NDDs Rare Disease from 2020 to 2022. All patients [60% female, mean age = 2.12 years (range 0.1–9.3; SD = 2.4)] were diagnosed PURA-NDDs clinically and genetically. Perinatal events, developmental milestones, neurological features, gastrointestinal abnormalities, ophthalmological abnormality, skeletal abnormality, cardiovascular abnormality, endocrine abnormalities and urogenital system abnormalities were abstracted from medical records and a standardized medical history questionnaire completed by parents or guardians. Moreover, 18 families also provided pictures of their affected children. Additionally, in order to analyze patients’ morphological characteristics, two clinicians independently evaluated their pictures.
The study was approved by Ethics Committee of Xinhua Hospital, School of Medicine, Shanghai Jiao Tong University (XHEC-D-2022-064) and informed consent were signed by parents for participation and publication.
Evaluation of previously published cases
Eleven patients with deletions at 5q31.3 encompassing PURA have been described in six different reports [2–6, 13], and more than 100 patients with PURA mutations have been reported in 17 different publications [6, 10–12, 14–26]. We abstracted phenotypic and genetic data from patients reported in publications. The detailed statistical results of clinical phenotype with 5q31.3 microdeletion and facial dysmorphic features with PURA syndrome were provided in Supplementary Tables 1, 2.
Genetic testing
All patients have a mutation in PURA or deletion at 5q31.3 encompassing PURA. For Patient 25, Affymetrix CytoScan 750k array was utilized to detect genomic CNVs following the manufacturer’s guide (Thermo-Fisher Scientific, United States). Genomic coordinates were established according to the hg19 version of the human reference genome. PURA mutations were identified by exome sequencing (ES) followed by Sanger sequencing in two patients. Briefly, ES was performed using Agilent SureSelect V5 capture probe (Agilent, Santa Clara, CA, USA) following the manufacturer’s protocol. The captured DNA fragments were sequenced by Illumina NovaSeq (Illumina). De novo sequence variants were validated using targeted Sanger sequencing or by trio exome. Other genetic tests were conducted by outsourced laboratories. The PURA variants were named following Human Genome Variation Society nomenclature guideline. The PURA mRNA (NM_005859.4) and protein (NP_005850.1) were used as a reference. We reassessed pathogenicity of all PURA variants in our cohort according to the ACMG variant interpretation guidelines. PURA variants listed in Table 1 complied with following criteria: (1) loss-of function variants including frameshift, nonsense or splice site, (2) De novo (both maternity and paternity confirmed) in a subject with the disease and no family history, and (3) absent from normal individual population variant databases (EVS and gnomAD). The detailed process of ACMG variant interpretation were presented in the Supplementary Table 3.
Table 1.
Summarization of PURA gene variants in 24 patients with PURA syndrome.
| Patient | Variant locationa | Variant type | Protein changeb | Inheritance | Literature report | ACMG classification |
|---|---|---|---|---|---|---|
| P1 | c.159dup | Frameshift | p.L54Afs*147 | De novo | No | Pathogenic |
| P2 | c.42_43del | Frameshift | p.L15Gfs*185 | De novo | No | Likely pathogenic |
| P3 | c.159dup | Frameshift | p.L54Afs*147 | De novo | No | Pathogenic |
| P4 | c.697_699del | Deletion | p.F233del | De novo | # | Pathogenic |
| P5 | c.449delG | Frameshift | p.R150Pfs*75 | De novo | No | Pathogenic |
| P6 | c.159dup | Frameshift | p.L54Afs*147 | De novo | No | Pathogenic |
| P7 | c.697_699del | Deletion | p.F233del | De novo | # | Pathogenic |
| P8 | c.159dup | Frameshift | p.L54Afs*147 | De novo | No | Pathogenic |
| P9 | c.692T>G | Missense | p.F231C | De novo | No | Likely pathogenic |
| P10 | c.575C>T | Missense | p.A192V | De novo | No | Likely pathogenic |
| P11 | c.458G>C | Missense | p.R153P | De novo | No | Uncertain significance |
| P12 | c.531del | Frameshift | p.P178Lfs*47 | De novo | No | Pathogenic |
| P13 | c.10C>T | Nonsense | p.R4X | De novo | No | Likely pathogenic |
| P14 | c.583C>G | Missense | p.L195V | De novo | No | Uncertain significance |
| P15 | c.865delC | Frameshift | p.R289fs*39 | De novo | No | Likely pathogenic |
| P16 | c.812T>C | Missense | p.F271S | De novo | No | Uncertain significance |
| P17 | c.72delC | Frameshift | p.G25Afs*53 | De novo | No | Pathogenic |
| P18 | c.506G>C | Missense | p.R169P | De novo | No | Likely pathogenic |
| P19 | c.697_699del | Deletion | p.F233del | De novo | # | Pathogenic |
| P20 | c.697_699del | Deletion | p.F233del | De novo | # | Pathogenic |
| P21 | c.218T>G | Missense | p.F73C | De novo | No | Likely pathogenic |
| P22 | c.550C>T | Nonsense | p.Q184X | De novo | No | Likely pathogenic |
| P23 | c.149_156dup | Frameshift | p.G53Pfs*28 | De novo | No | Likely pathogenic |
| P24 | c.430A>T | Nonsense | p.K144X | De novo | No | Likely pathogenic |
*The stop codon; #: ref. 11.
ACMG American College of Medical Genetics and Genomic.
Three-dimensional structural model of PURA (GenBank: NP_005850.1) with de nove missense variants were predicted by alphafold.
Statistical analysis
Patients with PURA-NDDs were divided into two groups to investigate the role of PURA pathogenic variant in the important phenotypes and to identify likely additional genetic causes of important phenotype in 5q31.3 microdeletion syndrome. Group 1 (n = 11) from reports consisted of patients with 5q31.3 deletions encompassing PURA gene. Group 2 included patients with PURA mutations (24 patients from the present study). Fisher’s exact two-sided test was performed to assess statistical significance in important phenotypes between two groups. A P value <0.05 is judged to be statistically significant. The statistical analysis was done with SPSS 2.0.
Results
There were 24 patients with PURA syndrome and one patient with 5q31.3 microdeletion syndrome in our cohort. Detailed clinical features and genetic information of the enrolled patients were summarized in Supplementary Table 4.
Genetic characterization of PURA-related neurodevelopmental disorders in our cohort
We reported 24 patients with PURA syndrome identified by ES and one patient with 5q31.3 microdeletion syndrome by CMA. In all patients with PURA syndrome where segregation analysis was completed, the PURA variants arose de novo. No additional contributing mutations in other genes were identified in the 24 patients. PURA variants included seven missense, seven frameshift, three nonsense and one deletion (see Table 1, Fig. 1). It is worth pointing out that a recurrent frameshift variant, c.697_699del (p.F233del) was identified in the four independent pedigrees. Also, another recurrent frameshift variant, c.159dup (p.L54Afs*147) appeared across four unrelated patients, respectively. We discovered that three variants were located in N-terminal glycine-rich domain, three variants were in Pur repeat I domain, five variants were in Pur repeat II domain, three variants were in amphipathic helix, two variants were in Pur repeat III domain, and two variants were in C-terminal glutamine-glutamate-rich domain (Fig. 1).
Fig. 1. Genetic characterization of PURA syndrome in our cohort.
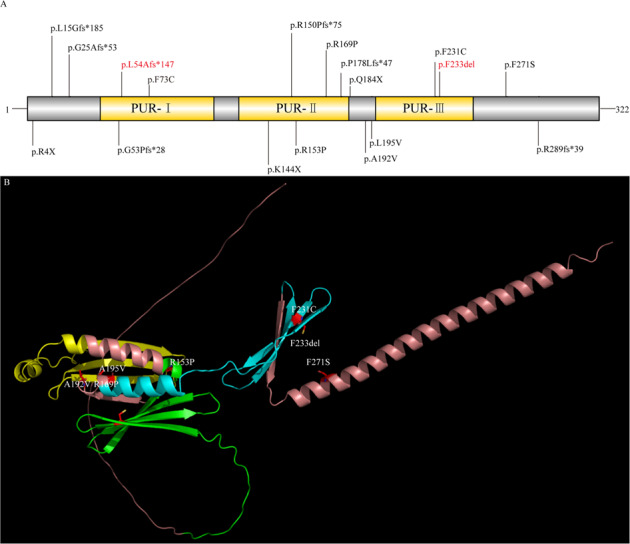
A Distribution of PURA mutations in our patients. Recurrent mutations are indicated in red color. Protein domains are from UniProt. B Three-dimensional Structural model of PURA with missense variants. The representation of the structure of human PURA (GenBank: NP_005850.1) was predicted by alphafold. PUR-I domain, PUR-II domain and PUR-III domain is respectively shown in green, yellow and cyan. β sheets are illustrated as flat directional arrows.
The representation of the structure of human PURA was constructed, and then we predicted novel variants effects on the protein structure by alphafold (Fig. 2, Fig. S1). Besides, we reassessed pathogenicity of PURA variants in our cohort according to the ACMG variant interpretation guidelines. A total of ten variants were likely pathogenic, three variants were uncertain significance, and the rest variants were pathogenic (Table 1). Over all, seventeen heterozygous variants, c.159dup, c.42_43del, c.449delG, c.692T>G, c.575C>T, c.458G>C, c.531del, c.10C>T, c.583C>G, c.865delC, c.812T>C, c.72delC, c.506G>C, c.218T>G, c.550C>T, c.149_156dup, c.430A>T were firstly reported. In addition, CMA detected a 1102 kb deletion in 5q31.2-q32 [arr 5q31.2-q32(138,449,368-139,551,713) x1] in P25 (Fig. 3). The deletion was not present in the parents of P25 indicating de novo origin, and the deletion was defined as pathogenic copy-number variants according to consensus recommendation of the ACMG [27]. Remarkably, the deletion partially overlapped or encompassed SIL1, MATR3, PAIP2, SLC23A1, MZB1, TMEM173, UBE2D2, CXXC5, NRG2, IGIP and PURA gene.
Fig. 2. The effects of PURA variants located in different domain on protein structure.

A The effects of loss-of-function variants on protein structure. R4X, Q184X, R150Pfs*35, and G53Pfs*48 variants, all give rise to varying degrees of truncated PURA proteins. B The characteristics of the representative structural changes of missense variants located in different domain. The phenylalanine 73 in Pur I domain forms stable three-dimension structures with leucine 190 which is destabilized by F73C variant. The Arg 169 in Pur II domain forms hydrogen bonds with Asp 158, R169P variant breaks the hydrogen bond. Besides, Proline appeared in β-sheet structures could eventually lead the β-sheet to disassemble. For Pur repeat III, we presented F233del and F231C variants. The phenylalanine (F233) residues and Val 246 located in β-sheet normally stabilize the domain through hydrophobic interactions, F233del could influence stable β-sheet structures. F231C disrupts a continuous hydrophobic interface and readily lead to protein accumulation. The red arrow indicates the H-bonds.
Fig. 3. Genetic characterization of 5q31.3 microdeletion syndrome in our cohort and previously reported cohort.

A Schematic representation of chromosome 5q31.3 indicating the deleted regions in Patient1. B Genome map captured from the UCSC genome browser (https://genome.ucsc.edu/). The custom tracks reveal the deletion regions from one currently reported patient and six previously reported patients depicted as red lines and a blue line, respectively. C Magnified view of the shortest region of overlap (SRO) depicted as the yellow box. SRO contains PURA and IGIP genes.
Overall prevalence of clinical phenotype with PURA syndrome
Considering the lack of detailed clinical data of P25 with 5q31.3 microdeletion, he was not considered in the following statistical analyses. Accordingly, the 24 patients with PURA syndrome were primarily analyzed. The most common clinical phenotype in our cohort were global developmental delay including DD/ID (23/23, 100%), speech delay (23/23, 100%) and motor delay (22/23, 95.7%), neonatal hypotonia (21/23, 91.3%), neonatal feeding difficulties (19/23, 82.6%), hypersomnolence (17/22, 77.2%), facial dysmorphism (12/17, 70.6%) and neonatal respiratory difficulty (15/23, 65.2%).
Neonatal problems
The patients with PURA syndrome in our cohort born at 39 weeks (average value) of gestation by spontaneous vaginal delivery (50%) and cesarean section (50%), one of which had a history of intrauterine growth retardation. The average weight of patients was approximately 3.3 ± 0.45 kg and average length was 49.7 ± 1.2 cm in born, and they were within the normal range according to the respective weight-for-age and length-for-age growth charts. These patients nearly universally had neonatal onset hypotonia, often with feeding difficulties or respiratory difficulties in the newborn period resulting in necessary hospital nursing. Moreover, twelve of the twenty-three patients developed hypoventilation requiring mechanical ventilation and eleven patients required nasogastric tube feeding. Seventeen (17/22, 77.2%) patients had hypersomnolence and five (5/20, 11.3%) had hypothermia. Another symptom which was less frequent was gastroesophageal reflux (P1 and P12). Notably, newly discovered neonatal signs and symptoms included anemia (P2, P4 and P12), septicemia (P3 and P10), thrombocytopenia (P1) and coagulation disorder (P18). With the exception of one patient (P6) died at the age of 1 months after continuous neonatal respiratory failure, the remaining 23(95.8%) patients were alive. Lack of longitudinal
follow-up in our patients, however, is a limitation in the assessment of the actual mortality.
Developmental delay
Gross motor was delayed in all patients with a vast majority (18/19, 94.7%) never attaining independent ambulation. The oldest patient walked independently by age 4 years. Regression of achieved skills has not been discovered. Among them, speech and language delay were notable, with 88.9% were nonverbal. Only two patients (P9 and P15) could speak, of which P9 speak single word and P15’s speech was meaningful and understandable. Moreover, all patients showed varying degrees of intellectual disability from mild to profound. Notably, P15 presented mild intellectual disability, and she received normal primary school education with her peers although academic performance was not satisfactory.
Neurological abnormalities
Among patients with PURA syndrome, 17/23 (73.9%) had varying degrees of neurological manifestations. The most common initial symptom was hypotonia, more prominent in newborn. One out of 21(4.8%) was mentioned to have a seizure at the time of evaluation. The patient had recurrent generalized tonic-clonic seizure which was first observed at postnatal day one. Following that seizure lasted for about 30 s on average. The EEGs confirmed seizure-like activity, which resolved after antiepileptic treatment. Accessible records of EEG showed that nine patients had an abnormal EEG (9/15, 60%). Among them, eight patients were described as non-specifically abnormal of EEG in the absence of clinical seizures. Approximately 80% of patients underwent brain MRI. In total, 15/18 (83.3%) patients demonstrated visible abnormalities on the MRI, whereas the remaining three patients (3/18,16.7%) had no abnormalities observed on the MRI. Widened subarachnoid space (8/18, 44.4%) was the most frequently identified brain abnormality. Moreover, delayed myelination (3/18,16.7%) and white matter abnormalities (1/18, 5.5%) were observed in several patients. In addition, we noted that these patients with PURA variants were prone to startle (10/20, 50.0%). Guardians generally described that patient was timid and easily frightened. Stereotypic hand movements have not been discovered up to now.
Gastrointestinal abnormalities
Gastrointestinal abnormalities were described frequently in patients with PURA syndrome especially in infant period. Excessive drooling (9/19, 47.4%) was the most commonly reported phenotype followed by swallowing problems (8/20, 40%). Besides, constipation was often observed (7/20, 35%) and may be severe, requiring the use of suitable laxatives to remedy.
Ophthalmological abnormalities
Strabismus and nystagmus as most commonly discovered ophthalmological findings were present in respectively 17.6% (3/17) of the patients and could possibly be related to strabismus-associated refractive errors including myopia or hyperopia. It is likely that these abnormalities are more common than described in the present study, since not all patients had undergone professional eye examination. Besides, cortical visual impairment has not been discerned in our cohort, despite previously reported patients were noted to have related eye abnormality [11, 14, 17].
Facial dysmorphisms
Facial dysmorphology examinations of our patients were performed by two clinicians. No apparent facial dysmorphic features were reported in five patients (5/17, 29.4%), however, the remainder had at least one dysmorphic feature. The most frequent features were myopathic face (13/17,76.5%), depressed nasal bridge (12/17, 70.6%), telecanthus (12/17, 70.6%) and prominent forehead (7/17, 41.2%), followed by high-arched palate (6/17, 35.3%) and microcephaly (4/17, 23.5%). For patients’ photographs and dysmorphic facial features, please refer to Fig. 4.
Fig. 4. Representative images of seven patients with PURA mutations.

A–G represent different patients.They shared facial dysmorphism including myopathic face, depressed nasal bridge and telecanthus.
Other clinical features
The common skeletal abnormality was hip dysplasia, which happened in 23% (3/13) of the patients. But more than that, less frequently observed abnormalities in the previously reported publications such as scoliosis and joint laxity [12, 17], were not found in any of our patients. As for endocrine abnormalities, only two patients (P3 and P8) were reported low vitamin D (13.3%), whereas aberrant thyroid hormone levels were not noted in any patients. Despite not frequently observed, a proportion of patients had congenital malformations of the heart, urogenital or respiratory system. Congenital cardiovascular system abnormalities included atrial septal defect in four patients and patent ductus arteriosus in two patients. Otherwise, myocarditis and myocardial damage were detected in one patient, respectively. Abnormalities of the urogenital system were reported in six patients: cryptorchidism in three, bilateral renal pyelectasis in two, unilateral renal pyelectasis in one.
As shown in Supplementary Fig. S2, we found, for the first time, P7 presented with skin symptoms at the proximal left arm and chest, mainly manifesting as large areas of lightened skin hypopigmentation. In addition, P4 exhibited hypomagnesemia (Mg2+ < 0.7 mmol/L) and hypocalcemia (Ca2+ < 2.0 mmol/L) in the newborn period. No additional pathogenic variants in other genes were identified in aforementioned two patients. None of the other patients shared above similar clinical symptoms.
Overview of clinical features of PURA syndrome
In order to delineate the clinical spectrum of PURA syndrome, we summarized the incidence of common clinical phenotypes in known patients with PURA syndrome according to previously published literature as well as patients in our cohort. An overview of clinical information available in Table 2, the result suggested that neonatal problems were prominent in all known patients with PURA syndrome including neonatal hypotonia (135/157, 86.0%), neonatal feeding difficulties (128/157, 81.5%) and neonatal respiratory difficulty (87/157, 55.4%). All patients with PURA variants have intellectual disability with language and motor delay. Facial dysmorphism (104/159, 65.4%) were detected in a significant number of patients. A variety of neurological problems were reported. Eighty-six out of 163 (52.8%) were diagnosed with epilepsy including different seizure type such as generalized tonic-colonic seizures, absence seizures or tonic seizures. In addition, abnormal brain MRI (88/160, 55.0%) and electroencephalography (39/82, 47.6%) were detected in patients with PURA syndrome. For other abnormalities, excessive drooling was most commonly reported (34/55, 47%), and less frequently observed abnormalities including low vitamin D (10/34, 29.4%), urogenital system abnormalities (15/61, 24.6%), cardiac abnormalities (14/62, 22.6%), constipation (34/154, 22.1%) and strabismus (31/151, 20.5%).
Table 2.
Percentage of clinical features reported in patients in our study (n = 24) and with previously published PURA patients (n = max 142).
| Literature report numbera, b | Percentage (%) | Our report number | Percentage (%) | Total number | Percentage (%) | |
|---|---|---|---|---|---|---|
| General information | ||||||
| Sex, female/male (%) | 93/64 | 59.2%/40.8% | 11/13 | 45.8%/54.2% | ||
| Age at inclusion, median (range) | 6 y (5mo–48 y) | 2y3m (1m–9y) | ||||
| Neonatal problems | ||||||
| Neonatal hypotonia | 114/134 | 85.2 | 21/23 | 91.3 | 135/157 | 86.0 |
| Neonatal respiratory difficulty | 72/134 | 53.5 | 15/23 | 65.2 | 87/157 | 55.4 |
| Neonatal feeding difficulties | 109/134 | 81.3 | 19/23 | 82.6 | 128/157 | 81.5 |
| Hypothermia | 15/134 | 11.3 | 5/20 | 25.0 | 20/154 | 13.0 |
| Hypersomnolence | 35/134 | 26.1 | 17/22 | 77.2 | 52/156 | 33.3 |
| Developmental delay | ||||||
| Speech delay | NA | 93.5a | 23/23 | 100 | ||
| Motor delay | NA | 100a | 22/23 | 95.7 | ||
| ID/DD | 32/32 | 100b | 23/23 | 100 | 55/55 | 100 |
| Facial dysmorphisms | 92/142 | 64.8 | 12/17 | 70.6 | 104/159 | 65.4 |
| No dysmorphisms | 5/17 | 29.4 | ||||
| Myopathic face | 13/17 | 76.5 | ||||
| Depressed nasal bridge | 12/17 | 70.6 | ||||
| Telecanthus | 12/17 | 70.6 | ||||
| Prominent forehead | 7/17 | 41.2 | ||||
| High-arched palate | 6/17 | 35.3 | ||||
| Microcephaly | 4/17 | 23.5 | ||||
| Neurological abnormalities | ||||||
| Epilepsy | 84/142 | 59.2 | 1/21 | 4.8 | 85/163 | 52.1 |
| Stereotypic hand movements | 32/134 | 23.9 | 0/20 | 0.0 | 32/154 | 20.8 |
| Pathological startle response | 24/134 | 17.9 | 10/20 | 50.0 | 34/154 | 22.1 |
| Abnormal Brain MRI | 73/142 | 51.4 | 15/18 | 83.3 | 88/160 | 55.0 |
| Abnormal Electroencephalography | 30/67 | 44.7 | 9/15 | 60.0 | 39/82 | 47.6 |
| Gastrointestinal abnormalities | ||||||
| Swallowing problems | 15/134 | 11.2 | 8/20 | 40.0 | 23/154 | 14.9 |
| Excessive drooling | 25/36 | 69.4 | 9/19 | 47.4 | 34/55 | 61.8 |
| Constipation | 27/134 | 20.1 | 7/20 | 35.0 | 34/154 | 22.1 |
| Ophthalmological abnormalities | ||||||
| Strabismus | 28/134 | 20.9 | 3/17 | 17.6 | 31/151 | 20.5 |
| Cortical visual impairment | 15/134 | 11.2 | 0/17 | 0.0 | 15/151 | 9.9 |
| Nystagmus | 22/134 | 16.4 | 3/17 | 17.6 | 25/151 | 16.6 |
| Skeletal abnormalities | ||||||
| Hip dysplasia | 21/134 | 15.7 | 3/13 | 23.1 | 24/147 | 16.3 |
| Scoliosis | 26/134 | 19.4 | 0/11 | 0.0 | 26/145 | 17.9 |
| Endocrine abnormalities | ||||||
| Low vitamin D | 8/19 | 42.1 | 2/15 | 13.3 | 10/34 | 29.4 |
| Cardiac abnormalities | 6/40 | 15.0 | 8/22 | 36.4 | 14/62 | 22.6 |
| Urogenital system abnormalities | 9/42 | 21.4 | 6/19 | 31.6 | 15/61 | 24.6 |
Comparison of the phenotypes between 5q31.3 microdeletion syndrome and PURA syndrome
The deletion sizes of 5q31.3 microdeletion syndrome were highly variable, ranging from 352 kb to 5.0 Mb. The shortest overlapping region among all published patients harbors three genes: PURA, IGIP, and CYSTM1. It is worth noting that P25 with 5q31.3 microdeletion partially overlapped or encompassed SIL1, MATR3, PAIP2, SLC23A1, MZB1, TMEM173, UBE2D2, CXXC5, NRG2, IGIP and PURA further supported the shortest overlapping region only contains PURA and IGIP gene (Fig. 2). PURA has been described as the primary candidate for the neurodevelopmental features observed in 5q31.3 microdeletion syndrome [4, 10]. To further clarify the role of PURA in 5q31.3 microdeletion syndrome, we analyzed the genetic and clinical phenotype of 5q31.3 microdeletion syndrome (group 1) and PURA syndrome patients (group 2) (see Table 3). Overall, we found that there was no significant difference between two groups in incidence of neonatal hypotonia, neonatal feeding difficulties and developmental delay (p > 0.05). Besides that, no significant difference in skeletal abnormalities, ophthalmological abnormalities as well as cardiovascular abnormalities, were observed between the two groups (p > 0.05). These results suggested the pathogenic variants in PURA may contribute to the neonatal hypotonia, neonatal feeding difficulties, GDD/ID and common complication. However, the frequency of the neonatal respiratory difficulty and abnormal brain MRI in group 1 were higher compared to that in the group 2 (100 % vs. 55.4% and 90.9% vs. 55% respectively), with statistically significant (P = 0.011 and P = 0.025 respectively), suggesting the role of additional genes or regulatory regions proximal to PURA.
Table 3.
Comparison of the prevalence of phenotypes between previously published 5q31.3 microdeletion syndrome and PURA syndrome (including our patients).
| 5q31.3 microdeletion syndrome number | Percentage (%) | PURA syndrome number | Percentage (%) | P valuea | |
|---|---|---|---|---|---|
| Neonatal problems | |||||
| Neonatal hypotonia | 11/11 | 100 | 135/157 | 86.0 | 0.363 |
| Neonatal respiratory difficulty | 9/9 | 100 | 87/157 | 55.4 | 0.011 |
| Neonatal feeding difficulties | 9/9 | 100 | 128/157 | 81.5 | 0.362 |
| Developmental delay | 11/11 | 100 | 90/90 | 100 | 1.00 |
| Neurological abnormalities | |||||
| Epilepsy | 8/11 | 72.7 | 85/163 | 52.1 | 0.226 |
| Abnormal Brain MRI | 10/11 | 90.9 | 88/160 | 55.0 | 0.025 |
| Abnormal EEG | 9/11 | 81.8 | 39/82 | 47.6 | 0.052 |
| Ophthalmological abnormalities | |||||
| Strabismus | 3/5 | 60.0 | 31/151 | 20.5 | 0.069 |
| Skeletal abnormalities | |||||
| Scoliosis | 1/4 | 25.0 | 26/145 | 17.9 | 0.555 |
| Cardiovascular abnormalities | |||||
| Congenital malformations of the heart | 1/3- | 33.3 | 14/62 | 22.6 | 0.551 |
| Craniofacial features | |||||
| No dysmorphic | 0/10 | 0.0 | 6/89 | 6.7 | 1.000 |
| Myopathic face | 8/10 | 80.0 | 36/80 | 48.2 | 0.047 |
| Narrow forehead | 5/8 | 62.5 | 0/5 | 0.0 | 0.075 |
| High forehead | 1/8 | 12.5 | 21/45 | 46.6 | 0.120 |
| Hypertelorism | 6/9 | 66.7 | 3/30 | 10.0 | 0.002 |
| Microcephaly | 2/8 | 25.0 | 3/14 | 21.4 | 1.000 |
| Micrognathia | 5/8 | 62.5 | 2/24 | 8.3 | 0.005 |
| High-arched palate | 6/8 | 75.0 | 15/73 | 20.5 | 0.003 |
| Open-tended mouth | 8/10 | 80.0 | 12/34 | 35.3 | 0.027 |
| Depressed nasal bridge | 7/11 | 63.6 | 11/68 | 16.2 | 0.002 |
| Anteverted nares | 4/10 | 40.0 | 9/72 | 12.5 | 0.048 |
| Upslanting palpebral fissures | 4/8 | 50.0 | 7/61 | 11.5 | 0.019 |
| Low-set or abnormal ears | 2/8 | 25.0 | 7/67 | 10.4 | 0.244 |
| Long face | 1/5 | 20.0 | 5/63 | 7.9 | 0.379 |
| Dolichocephaly | 1/5 | 20.0 | 4/30 | 13.3 | 0.561 |
| Epicanthic fold | 1/5 | 20.0 | 13/67 | 19.4 | 1.000 |
| Telecanthus | 6/8 | 75.0 | 3/36 | 8.3 | 0.000 |
| Ptosis | 6/10 | 60.0 | 4/56 | 7.1 | 0.000 |
| Prominent cheeks | 1/5 | 20.0 | NA | NA | NA |
| Abnormal dentition | 1/3 | 33.3 | 4/28 | 14.3 | 0.422 |
| Long philtrum | 6/10 | 60.0 | 7/30 | 23.3 | 0.052 |
| Tented upper vermilion border | 5/7 | 71.4 | 1/11 | 9.1 | 0.013 |
| Sparse eyebrows | 2/8 | 25.0 | 2/24 | 8.3 | 0.254 |
| Almond-shaped palpebral fissure | NA | NA | 2/24 | 8.3 | NA |
| Full cheeks | NA | NA | 7/24 | 29.2 | NA |
aAll P values are Fisher’s Exact.
Furthermore, a comparative analysis of craniofacial features was firstly performed between two groups. As shown in Table 3, a marked higher frequency of the telecanthus, ptosis, high-arched palate, depressed nasal bridge, micrognathia and hypertelorism were observed in group 1 (p < 0.01). In addition, the incidence of other common features such as myopathic face, open-tended mouth, anteverted nares, upslanting palpebral fissures and tented upper vermilion border were higher significantly as well compared with group 2 (0.01 < p < 0.05), suggesting additional genes or regions of chromosome 5q31.2q31.3 besides PURA may contribute to these craniofacial features. As for no dysmorphic, however, did not differ significantly between two groups (p > 0.05).
Discussion
To date, only few patients with PURA variants have been reported, whereas 5q31.3 microdeletion syndrome have never been reported in Mainland China [18, 20]. To fill in the gap, we report 24 patients with PURA variants and one patient with 5q31.3 microdeletion, as a largest cohort in Chinese ethic group, to elucidate the clinical and genetic spectrum of PURA-related neurodevelopmental disorders.
Mutations in the PURA gene located on 5q31.2, were reported as a cause of dominant form of PURA syndrome. The ubiquitously product of PURA gene, Pur-a, regulates a variety of cellular processes including DNA replication, gene transcription, RNA transport and mRNA translation [28]. The function of Pur-a relies on the three conserved PUR domain [29]. Pur repeat I and Pur repeat II participate in binding of singles stranded DNA or RNA, whereas the Pur repeat III is involved in the dimerization of Pur-a [30]. Two PURA knockout mice demonstrate that mice appear normal at birth but develop neurological features, whereas heterozygous mice appeared normal but exhibited neurologic and myeloid defects [7, 8]. We found that our identified variants are interspersed throughout PURA and the majority are novel. Of the 18 variants, however, the previously reported c.697_699del (p.Phe233del) variant was recurrent. Interestingly, by means of summarizing all reported PURA variants, both Liu et al. and Cinquina et al. pointed out that the most frequently reported mutation site was c.697_699del affecting seven cases as well [18, 24]. The above-mentioned evidence supported that c.697_699del variant was a mutant hot spot in PURA syndrome. The variant occurs within highly conserved regions of Pur repeat III which is necessary for dimerization and binding to linearized DNA thus favoring DNA replication and gene transcription. Strikingly, the previously unreported variant, c.159dup (p.L54Afs*147), was repeatedly identified in four independent pedigrees. The duplication occurs at the 54th amino acid (the first quarter of the protein). It altered reading frame and generated a premature stop codon. Additionally, it falls with Pur repeat I, a highly conserved residue, and presumably has potential to interfere with the formation of the ssDNA/ssRNA binding domain. The functional effect at a molecular level is not yet clear, but it seems to be associated with a severe clinical phenotype. In silico prediction tools, PolyPhen-2 and SIFT, predict that all missense mutations in our study have damaging effects. Given that PURA is a single-exon gene and thus not likely subject to nonsense-mediated decay, all truncating mutations found in our cohort probably result in loss or partial loss of PUR domain leading to PURA haploinsufficiency. Notably, similar to previous studies, we could not find correlations between the types or locations of PURA variants and clinical severity [6, 12].
PURA syndrome is a developmental encephalopathy characterized by early hypotonia, global developmental delay and neurological symptom [17]. To clarify the phenotypic spectrum in our cohort, twenty-four previously unreported patients with PURA syndrome were comprehensively evaluated. The most common clinical phenotype in our patients were in line with previously reported [19]. Other common phenotypes suggested there are higher rates of digestive and cardiac morbidity. Consequently, with the diagnosis of PURA syndrome, it is crucial to keep a strict watch over all the patient’s organ systems in a multidisciplinary team to monitor the possible multisystem complications. The incidence of epilepsy was low in our cohort, we hypothesized it might be associated with younger age of enrolled patients. We cannot exclude that a proportion of nonepileptic patients will develop epilepsy later in the course of their disease, thus, a longer-term follow-up is necessary to obtain actual incidences of epilepsy. In addition to the well-established phenotypes associated with variation in PURA, our patients also exhibit skin symptoms, hypomagnesemia or hypocalcemia. As these phenotypes are from a single patient only, it is not possible to be confirm these characters are a clinical trait of PURA syndrome. Nevertheless, it would be valuable to look for these phenotypes in more patients with PURA variants to confirm whether these phenotypes are related, or due to an unidentified cause. Otherwise, a small proportion of patients were able to achieve independent walking with significant therapy implying physical therapy can help improve symptoms. Since there is a broad spectrum of clinical features and variability in clinical severity within PURA syndrome challenging to diagnose clinically, an overview of common clinical phenotypes in known patients with PURA syndrome are highly valuable and essential. Our result demonstrated that neonatal problems and GDD/ID were prominent in all known PURA patients. Meanwhile, epilepsy, and facial deformities could not be ignored. Thus, we suggest that PURA syndrome need to be considered as a differential diagnosis in patients who have neonatal hypotonia with feeding and respiratory difficulty, followed by profound GDD/ID and facial deformities. Widened subarachnoid space, delayed myelination or white matter abnormalities, moreover, were noted on brain-imaging studies in 15/18 patients which consistent with those previously described [10, 11, 17]. In combination with the abnormal brain MRI and one or more of the above syndromes, thus, early targeted genetic testing for PURA syndrome should be performed to achieve early diagnosis.
PURA-related neurodevelopmental disorders (PURA-NDDs) include 5q31.3 deletion syndrome and PURA syndrome [10]. Within the 5q31.3 region, PURA has been proposed as a critical causative gene, since Hunt et al. firstly provided definitive evidence that mutations limited to PURA are indeed sufficient to cause obvious neurodevelopmental delay [11]. Additionally, Brown et al. reported two patients have a shortest region (101 kb) of 5q31.3 microdeletion only encompassing three genes: PURA, IGIP and CYSTM1 [4]. It is important to point out that the shortest region gene of 5q31.3 deletion further supported by our study only including PURA and IGIP, nevertheless, IGIP has not been reported to be involved in neural development to date. The important finding thus significantly corroborated the importance of PURA in 5q31.3 deletion. The concrete role of the other genes in 5q31.3 deletion, however, is yet to be characterized. We therefore compared phenotypes between known patients with PURA variants alone and that with 5q31.3 deletions encompassing more than PURA gene. Fisher’s exact two-sided test were performed to assess statistical significance in important phenotypes between two groups to explore genotype–phenotype correlations. The results showed that there was no significant difference between two groups in the common clinical features including neonatal problems, GDD/ID and epilepsy which are in line with previous research [6]. Meanwhile, there was no difference in the frequency of medical comorbidities including skeletal abnormalities, ophthalmological abnormalities and cardiovascular abnormalities between the two group. All the aforementioned results suggested that PURA haploinsufficiency affects these common phenotypes and complication. We observed a slightly higher frequency of abnormal MRI in 5q31.3 deletion group. This finding suggested to us that several other overlapping candidate genes probably participate in abnormal brain morphology. Brown et al. proposed that PURA and NRG2 may account for a more severe phenotype in 5q31.3 deletion syndrome [4], and NRG2, one of the members of the neuregulin gene family, related to growth and differentiation of neurons and glial cells [31]. Therefore, we speculated whether NRG2 may play a role in pathogenetic mechanisms of 5q31.3 deletion. Of course, further studies are needed to obtain insight into virtual underlying genetic and biological mechanisms. Given many syndromes have recognizable facial dysmorphism features that are highly informative to clinical geneticists. Thus, a statistical analysis of facial dysmorphism was firstly performed between two groups. As for normal facial appearance, although, did not differ significantly between two groups. We noted, however, that an obvious higher frequency of certain facial dysmorphism in patients with 5q31.3 microdeletion. The above results raised an intriguing question of whether there might be other overlapping gene haploinsufficiency causing more pronounced facial abnormalities. Of course, given the relatively small number of patients with 5q31.3 microdeletion, this finding should be interpreted cautiously. On the whole, our findings support the hypothesis that the deletion of PURA contributes to, but is not the sole cause of, the 5q31.3 microdeletion phenotype.
There were several limitations to this analysis. We obtained medical history by medical record review or questionnaires completed by parents, and the collected data may be subject to recall or information bias. Besides, the phenotype comparisons identified in our analysis are heavily influenced by sample size, future studies could be done in larger samples to provide a clear role of PURA haploinsufficiency in the important phenotypes in 5q31.3 microdeletion syndrome.
In summary, we further expand the clinical and genetic characteristics in the largest series of patient in Mainland China with PURA-related neurodevelopmental disorders published to date. We presented 18 novel variants expanding the genetic landscape of PURA as well as some new or rare phenotypic characteristics of patients with PURA-syndrome. The results suggested that in cases of neonatal hypotonia with feeding and respiratory difficulty, followed by profound GDD/ID, further gene testing is warranted to rule out PURA-related neurodevelopmental disorders. Through firstly detailed comparison of the phenotypes between 5q31.3 microdeletion syndrome and PURA syndrome, our findings further supported the hypothesis that the deletion of PURA gene contributes to most of important phenotype with 5q31.3 microdeletion syndrome, but additional genes or regions of chromosome 5q31.2q31.3 besides PURA contribute to a few of phenotype of 5q31.3 microdeletion syndrome as well.
Supplementary information
Acknowledgements
We would like to acknowledge the affected patients and their families for the participation in the study. We also acknowledged Yue Tao for structural protein model.
Author contributions
KS, YY and NX: conception and design of the study; WD and NX: drafting the paper or figures; YS: review and editing the paper; BX and WD: evaluation of the pictures of individuals for the morphological analysis; YF, YG, LW and YZ: acquisition and analysis of data. BX, WQ and XG: providing partial clinical data. All authors read and approved the final paper.
Funding
This work was supported by the National Key R&D Program of China (No. 2019YFC1005100, to YGY); the National Natural Science Foundation of China (No. 82101950, to NX); Shanghai Municipal Commission of Health and Family Planning (No. 20204Y0453, to NX); the National Natural Science Foundation of China (No.81873671 and No. 82070914, to YGY); the Shanghai Science and Technology Commission (No. 19140904500, to YGY); Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant Support (No. 20191908, to YGY).
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files).
Competing interests
The authors declare no competing interests.
Ethics approval
The study was conducted with the approval of Ethics Committee of Xinhua Hospital, School of Medicine, Shanghai Jiao Tong University (XHEC-D-2022-064).
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Kun Sun, Yongguo Yu, Na Xu.
Contributor Information
Kun Sun, Email: sunkun@xinhuamed.com.cn.
Yongguo Yu, Email: yuyongguo@shsmu.edu.cn.
Na Xu, Email: xuna@xinhuamed.com.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41431-022-01217-4.
References
- 1.Reijnders MRF, Leventer RJ, Lee BH, Baralle D, Selber P, Paciorkowski AR, et al. PURA-Related Neurodevelopmental Disorders. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, et al., editors. GeneReviews(®). Seattle (WA): University of Washington, Seattle Copyright © 1993-2022, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.; 1993.
- 2.Shimojima K, Isidor B, Le Caignec C, Kondo A, Sakata S, Ohno K, et al. A new microdeletion syndrome of 5q31.3 characterized by severe developmental delays, distinctive facial features, and delayed myelination. Am J Med Genet Part A. 2011;155a:732–6. doi: 10.1002/ajmg.a.33891. [DOI] [PubMed] [Google Scholar]
- 3.Hosoki K, Ohta T, Natsume J, Imai S, Okumura A, Matsui T, et al. Clinical phenotype and candidate genes for the 5q31.3 microdeletion syndrome. Am J Med Genet Part A. 2012;158a:1891–6. doi: 10.1002/ajmg.a.35439. [DOI] [PubMed] [Google Scholar]
- 4.Brown N, Burgess T, Forbes R, McGillivray G, Kornberg A, Mandelstam S, et al. 5q31.3 Microdeletion syndrome: clinical and molecular characterization of two further cases. Am J Med Genet Part A. 2013;161a:2604–8. doi: 10.1002/ajmg.a.36108. [DOI] [PubMed] [Google Scholar]
- 5.Bonaglia MC, Zanotta N, Giorda R, D’Angelo G, Zucca C. Long-term follow-up of a patient with 5q31.3 microdeletion syndrome and the smallest de novo 5q31.2q31.3 deletion involving PURA. Mol Cytogenet. 2015;8:89. doi: 10.1186/s13039-015-0193-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi SA, Lee HS, Park TJ, Park S, Ko YJ, Kim SY, et al. Expanding the clinical phenotype and genetic spectrum of PURA-related neurodevelopmental disorders. Brain Dev. 2021;43:912–8. doi: 10.1016/j.braindev.2021.05.009. [DOI] [PubMed] [Google Scholar]
- 7.Khalili K, Del Valle L, Muralidharan V, Gault WJ, Darbinian N, Otte J, et al. Puralpha is essential for postnatal brain development and developmentally coupled cellular proliferation as revealed by genetic inactivation in the mouse. Mol Cell Biol. 2003;23:6857–75. doi: 10.1128/MCB.23.19.6857-6875.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hokkanen S, Feldmann HM, Ding H, Jung CK, Bojarski L, Renner-Müller I, et al. Lack of Pur-alpha alters postnatal brain development and causes megalencephaly. Hum Mol Genet. 2012;21:473–84. doi: 10.1093/hmg/ddr476. [DOI] [PubMed] [Google Scholar]
- 9.Kuo CJ, Lee KH, Huang CC, Wang IF, Hsieh CC, Lin HC, et al. Purα regulates the induction of Znf179 transcription during neuronal differentiation. Biochem Biophys Res Commun. 2020;533:1477–83. doi: 10.1016/j.bbrc.2020.10.047. [DOI] [PubMed] [Google Scholar]
- 10.Lalani SR, Zhang J, Schaaf CP, Brown CW, Magoulas P, Tsai AC, et al. Mutations in PURA cause profound neonatal hypotonia, seizures, and encephalopathy in 5q31.3 microdeletion syndrome. Am J Hum Genet. 2014;95:579–83. doi: 10.1016/j.ajhg.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hunt D, Leventer RJ, Simons C, Taft R, Swoboda KJ, Gawne-Cain M, et al. Whole exome sequencing in family trios reveals de novo mutations in PURA as a cause of severe neurodevelopmental delay and learning disability. J Med Genet. 2014;51:806–13. doi: 10.1136/jmedgenet-2014-102798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reijnders MRF, Janowski R, Alvi M, Self JE, van Essen TJ, Vreeburg M, et al. PURA syndrome: clinical delineation and genotype-phenotype study in 32 individuals with review of published literature. J Med Genet. 2018;55:104–13. doi: 10.1136/jmedgenet-2017-104946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimojima K, Okamoto N, Ohmura K, Nagase H, Yamamoto T. Infantile spasms related to a 5q31.2-q31.3 microdeletion including PURA. Hum Genome Var. 2018;5:18007. doi: 10.1038/hgv.2018.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka AJ, Bai R, Cho MT, Anyane-Yeboa K, Ahimaz P, Wilson AL, et al. De novo mutations in PURA are associated with hypotonia and developmental delay. Cold Spring Harb Mol case Stud. 2015;1:a000356. doi: 10.1101/mcs.a000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okamoto N, Nakao H, Niihori T, Aoki Y. Patient with a novel purine-rich element binding protein A mutation. Congenit Anom. 2017;57:201–4. doi: 10.1111/cga.12214. [DOI] [PubMed] [Google Scholar]
- 16.Rezkalla J, Von Wald T, Hansen KA. Premature Thelarche and the PURA Syndrome. Obstet Gynecol. 2017;129:1037–9. doi: 10.1097/AOG.0000000000002047. [DOI] [PubMed] [Google Scholar]
- 17.Lee BH, Reijnders MRF, Abubakare O, Tuttle E, Lape B, Minks KQ, et al. Expanding the neurodevelopmental phenotype of PURA syndrome. Am J Med Genet Part A. 2018;176:56–67. doi: 10.1002/ajmg.a.38521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Liu R, Xu T, Zhou YX, Zhang SC. Neonatal PURA syndrome: a case report and literature review. Transl Pediatr. 2021;10:194–203. doi: 10.21037/tp-20-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johannesen KM, Gardella E, Gjerulfsen CE, Bayat A, Rouhl RPW, Reijnders M, et al. PURA-related developmental and epileptic encephalopathy: phenotypic and genotypic spectrum. Neurol Genet. 2021;7:e613. doi: 10.1212/NXG.0000000000000613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin SJ, Lin YF, Tsai CH, Huang CH, Ho F, Tsai SF, et al. Complex movement disorders in a boy with PURA syndrome. Mov Disord Clin Pract. 2021;8:1137–9. doi: 10.1002/mdc3.13272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spangenberg L, Guecaimburú R, Tapié A, Vivas S, Rodríguez S, Graña M, et al. Novel frameshift mutation in PURA gene causes severe encephalopathy of unclear cause. Mol Genet Genom Med. 2021;9:e1622. doi: 10.1002/mgg3.1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwong AK, Tsang MH, Fung JL, Mak CC, Chan KL, Rodenburg RJT, et al. Exome sequencing in paediatric patients with movement disorders. Orphanet J Rare Dis. 2021;16:32. doi: 10.1186/s13023-021-01688-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mroczek M, Zafeiriou D, Gurgel-Gianetti J, Vilela Morais de Azevedo B, Roos A, Bartels E, et al. Three individuals with PURA syndrome in a Cohort of patients with neuromuscular disease. Neuropediatrics. 2021;52:390–3. doi: 10.1055/s-0040-1715625. [DOI] [PubMed] [Google Scholar]
- 24.Cinquina V, Ciaccio C, Venturini M, Masson R, Ritelli M, Colombi M. Expanding the PURA syndrome phenotype: A child with the recurrent PURA p.(Phe233del) pathogenic variant showing similarities with cutis laxa. Mol Genet Genom Med. 2021;9:e1562. doi: 10.1002/mgg3.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boczek NJ, Macke EL, Kemppainen J, Klee EW, Renaud DL, Gavrilova RH. Expansion of PURA-related phenotypes and discovery of a novel PURA variant: a case report. Child Neurol Open. 2020;7:2329048x20955003. doi: 10.1177/2329048X20955003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodríguez-García ME, Cotrina-Vinagre FJ, Arranz-Canales E, Aragón AM, Hernández-Sánchez L, Rodríguez-Fornés F, et al. A novel de novo mutation in the PURA gene associated with a new clinical finding: large brainstem. J Genet. 2020;99:7. [PubMed]
- 27.Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen) Genet Med. 2020;22:245–57. doi: 10.1038/s41436-019-0686-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weber J, Bao H, Hartlmüller C, Wang Z, Windhager A, Janowski R, et al. Structural basis of nucleic-acid recognition and double-strand unwinding by the essential neuronal protein Pur-alpha. eLife. 2016;5:e11297. [DOI] [PMC free article] [PubMed]
- 29.Aumiller V, Graebsch A, Kremmer E, Niessing D, Förstemann K. Drosophila Pur-α binds to trinucleotide-repeat containing cellular RNAs and translocates to the early oocyte. RNA Biol. 2012;9:633–43. doi: 10.4161/rna.19760. [DOI] [PubMed] [Google Scholar]
- 30.Graebsch A, Roche S, Niessing D. X-ray structure of Pur-alpha reveals a Whirly-like fold and an unusual nucleic-acid binding surface. Proc Natl Acad Sci USA. 2009;106:18521–6. doi: 10.1073/pnas.0907990106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Falls DL. Neuregulins: functions, forms, and signaling strategies. Exp Cell Res. 2003;284:14–30. doi: 10.1016/S0014-4827(02)00102-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files).