

Abstract
RNA therapeutics have had a tremendous impact on medicine, recently exemplified by the rapid development and deployment of mRNA vaccines to combat the COVID-19 pandemic. In addition, RNA-targeting drugs have been developed for diseases with significant unmet medical needs through selective mRNA knockdown or modulation of pre-mRNA splicing. Recently, RNA editing, particularly antisense RNA-guided adenosine deaminase acting on RNA (ADAR)-based programmable A-to-I editing, has emerged as a powerful tool to manipulate RNA to enable correction of disease-causing mutations and modulate gene expression and protein function. Beyond correcting pathogenic mutations, the technology is particularly well suited for therapeutic applications that require a transient pharmacodynamic effect, such as the treatment of acute pain, obesity, viral infection, and inflammation, where it would be undesirable to introduce permanent alterations to the genome. Furthermore, transient modulation of protein function, such as altering the active sites of enzymes or the interface of protein-protein interactions, opens the door to therapeutic avenues ranging from regenerative medicine to oncology. These emerging RNA-editing-based toolsets are poised to broadly impact biotechnology and therapeutic applications. Here, we review the emerging field of therapeutic RNA editing, highlight recent laboratory advancements, and discuss the key challenges on the path to clinical development.
Keywords: RNA editing, RNA therapeutics, ADAR, ADAR1, ADAR2, gene therapy, ASO, precision medicine
Graphical abstract
ADAR-based RNA editing has emerged as a powerful tool to engineer RNAs, enable correction of disease-causing mutations, and modulate protein functions. We review the emerging field of therapeutic RNA editing, highlight recent laboratory advancements, and discuss the key challenges on the path to clinical development.
Introduction
Large-scale genome sequencing has progressively revealed the causal genetic variation underlying many human diseases.1,2 This information has driven significant innovation in biotechnology and ushered in the modern era of DNA and RNA therapeutics. While DNA targeting can result in durable and potentially permanent cures, RNA-targeting modalities enable tunability and reversibility. The lack of permanent off-targets offers unique advantages in specific therapeutic settings. Here we focus on recently emerging precision RNA-editing approaches, especially those based on adenosine deaminases acting on RNA (ADARs), that are enabling programmable endogenous RNA modulation beyond RNA knockdown or overexpression.
ADARs represent a family of enzymes that deaminates RNA adenosines (A) into inosines (I) within double-stranded RNA (dsRNA). Inosine is functionally recognized by the cellular machineries as guanosine (G), thereby allowing the enzyme to modulate translation, splicing, or any regulatory mechanism reliant upon an adenosine-containing motif. A-to-I RNA editing was discovered in the late 1980s,3,4 and leveraging ADARs for therapeutic purposes was first proposed in 1995.5 Over the past decade there has been a renewed interest in the development of this RNA-targeting modality, with numerous groups demonstrating the redirection of endogenous ADAR activity for site-specific A>G editing using guide RNA (gRNA) antisense to a target messenger RNA of interest in human cells and in vivo animal models,6,7,8,9,10,11,12,13,14 as well delivery of exogenous ADARs to enable targeted RNA editing.15,16
Notably, G-to-A missense and nonsense mutations account for 28% of pathogenic single-nucleotide variants (SNVs) reported on ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/, accessed on April 13, 2022) and can be targeted for ADAR-mediated restoration of the wild-type sequence. More broadly, adenosines are critical for many functional sites within RNA, such as translation initiation sites (TISs), splice acceptor and donor sites, microRNA-binding sites, and polyadenylation signals (PAS). This further expands the therapeutic potential for RNA editing to regulate protein expression levels and splicing and may be additive with current approaches that utilize antisense oligonucleotides (ASOs) for masking TISs, splice sites, or polyadenylation signals. Furthermore, A-to-G changes can result in 17 different amino acid substitutions, enabling the modulation of protein function and protein-protein interactions. Indeed, natural ADAR function has been shown to modulate proteins with 55 editing sites identified in coding regions,17 many of which are conserved across species.18 Nonsense mutations (UAG, UGA, UAA) can be recoded to a tryptophan (UGG), which may be tolerable to a protein, depending on the exact position of the nonsense mutation.19,20 Altogether, RNA editing opens a wide range of opportunities for therapeutic and protective benefits to patients. Correspondingly there is a growing interest in clinical translation, with many academic labs and biotechnology companies now focused on refining and tuning this technology with a goal of enabling human therapeutic applications.
Several challenges, however, must be overcome to bring the therapeutic potential of RNA editing to patients. ADAR is inherently promiscuous and has the potential to deaminate any adenosine within a dsRNA structure. Thus, gRNA-directed RNA editing has the potential for bystander and off-target editing as well as possible unintended impact on splicing and translation. Furthermore, ADAR has natural sequence preferences that may not align with a chosen therapeutically relevant adenosine. These challenges highlight the need for exquisite gRNA engineering that enables highly efficient and specific RNA editing. Additionally, non-clinical and clinical assays to quantify editing efficiency and transcriptome integrity are necessary to establish safety metrics to support clinical development.
Beyond RNA-editing-specific challenges, issues of delivery and manufacturing that broadly impact the fields of gene therapy and ASO therapy must also be addressed. For example, while delivering payloads with an adeno-associated virus (AAV) vector has significant clinical precedent, issues persist related to manufacturing, quality control, and safety, while the possibility for immunogenicity and transgene silencing may hinder efficacy. Furthermore, the narrow tropism of wild-type AAVs and biodistribution of ASOs limits delivery to the liver and muscle, and direct injection into the central nervous system (CNS), while ASOs are also readily absorbed in the kidney.21 Solutions to each of these stated challenges are in development as the field of RNA editing advances toward the clinic. We will first review the underlying biology of ADAR-mediated RNA editing and how it can inform its therapeutic application.
Factors that affect endogenous RNA editing
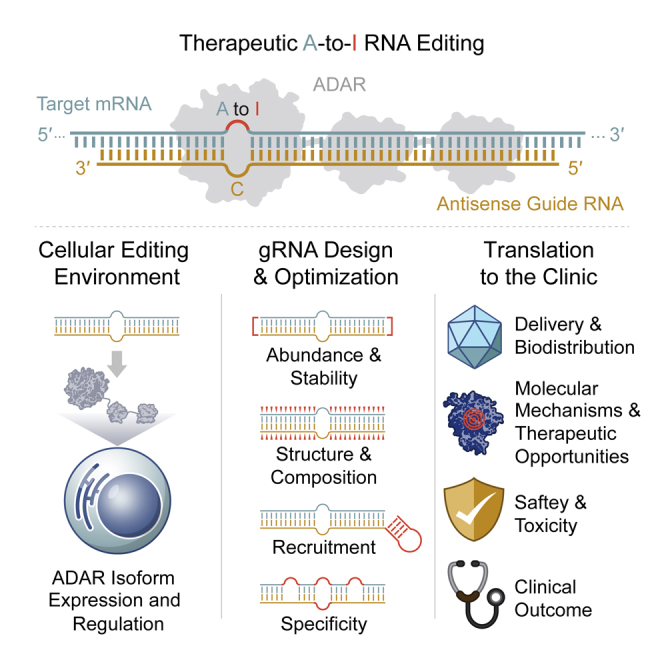
Since the discovery of ADAR in 1987,3,4 much progress has been made in understanding the natural biological functions of this enzyme group. Understanding fundamental ADAR biology, including various isoforms and structures, expression and regulation, and cellular and subcellular localization, is critical to unlocking the therapeutic potential of RNA editing. Thus, we begin by reviewing key aspects of ADAR biology that can inform drug design, development, and translation to the clinic. Key variables that impact A-to-I RNA editing are also depicted in Figure 1.
Figure 1.

Factors affecting ADAR-mediated RNA editing
ADAR isoforms and structure
The ADAR family is composed of three genes that encode five different protein isoforms: ADAR1p110, ADAR1p150, ADAR2a, ADAR2b, and ADAR3. Each isoform contains N-terminal dsRNA-binding domains (dsRBDs) followed by a C-terminal deaminase domain. All isoforms possess a nuclear localization signal (NLS), while ADARp150 also has a nuclear export signal (NES) that promotes cytosolic localization. ADAR2 is spliced in several isoforms, with only ADAR2a and ADAR2b being translated into proteins. ADAR2b contains an Alu insertion in the deaminase domain, which may explain the 50% reduction in activity compared with ADAR2a.22 ADAR3 lacks deaminase activity but may play a role in regulating RNA editing through competitive antagonism with ADAR1 and ADAR2.23,24,25 The structural differences between ADAR isoforms are responsible for subtle differences in their substrate preferences that must be taken into consideration during the gRNA design process to ensure efficient and selective editing depending on the isoform present in the tissue and cell type of interest.
The substrate preferences of ADAR can be mechanistically traced back to its structure. The dsRBDs engage a 12- to 14-bp stretch of dsRNA with specificity to the A-form helix and ribose 2′ hydroxyl groups that distinguish it from double-stranded DNA.26 The shallow minor groove of the A-form helix provides access to the bases and allows for sequence-specific contacts, which can explain how dsRBDs from various proteins have unique binding preferences. Indeed, ADAR dsRBD-binding selectivity has been shown to influence editing selectivity,27 and replacing the dsRBDs of ADAR1 with PKR significantly alters the editing activity.28 One of the most well-studied ADAR substrates is the GRIA2 R/G site, which forms an evolutionarily conserved hairpin structure driven by hybridization of exon 13 to the downstream intron and contains three mismatches within the RNA duplex that are key to efficient and selective editing.29 The solution structure of the dsRBDs of ADAR2 bound to the GRIA2 R/G substrate reveals sequence-specific contacts at one of the mismatches and within the hairpin loop.30 This leaves open the possibility of designing gRNAs that form dsRNA structures that are preferentially bound to ADAR1 and/or ADAR2 dsRBDs.
Crystal structures of the deaminase domain of ADAR2 have also revealed many characteristics that explain the nuances of ADAR editing. Before the availability of structural data, many deep sequencing studies of A-to-I editing demonstrated that ADARs have certain motif preferences, with the “UAG” sequence motif being favored and a 5′ G being disfavored. Like the dsRBDs, the deaminase domain crystal structure detailed dsRNA-specific engagement via 5′ and 3′ binding loops with contacts stretching from 10 bp upstream to 8 bp downstream of the target adenosine.31,32 The structure also revealed a disordered 5′ binding loop that becomes ordered upon binding to a dsRNA substrate.31 Interestingly, the ADAR2 5′ binding loop is highly conserved across species yet differs significantly when compared with the ADAR1 5′ binding loop, which may explain differences in their substrate specificities.32 Additionally, the crystal structure revealed that ADAR2 acts through a common base-flipping mechanism,33 in which the edited adenosine is flipped out of the duplex and the vacant position is occupied by residue E488, which directly contacts the orphan base. Base flipping allows exposure of the adenosine to the active site to drive deamination.
Initial observations indicated that a hyperactive ADAR2 E488Q mutant gained activity through improved base flipping, not improved binding affinity.34 The crystal structure revealed hydrogen bonding between E488 and the orphan cytidine, and the pH independence of E488Q may explain the improved base flipping. The crystal structure also provided an explanation for ADAR2 disfavoring of a 5′ G neighbor, as a 5′ G or C could result in a steric clash with ADAR2 G489.31 Interestingly, a recent crystal structure revealed that a 5′ G-G mismatch adopts a non-canonical Gsyn:Ganti hydrogen bonding that alleviates the steric clash and enhances editing of a 5′ G adenosine,35 as had been previously reported.36 These insights into base flipping and deamination in turn impact gRNA design (which we discuss in more depth in “considerations for gRNA design and optimization”). More recently, the first crystal structure of the deaminase domain and dsRBD2 engaged to a substrate revealed an asymmetric dimerization via the deaminase domain, and the authors showed that many substrates are dimerization dependent.37 This highlighted a surprising and novel mode of engagement, as previous data provided evidence of dimerization through the dsRBDs.38 Further work is required to better understand the more complex quaternary structures formed through the deaminase domain and/or the dsRBDs, and how they could inform gRNA design.
ADAR expression and regulation
ADAR1p110 is ubiquitously expressed. A-to-I editing has been detected at millions of sites within the transcriptome and is present in all tissues and cell types.39,40 A-to-I editing of self-dsRNA mediated by ADAR1 can prevent activation of the cytoplasmic immune sensor, MDA-5.41 As such, ADAR1 expression is essential for maintaining homeostasis and regulating innate immunity, as evidenced by the severe phenotype of patients with partial-loss-of-function ADAR1 mutations leading to Aicardi-Goutières syndrome.42,43 Full loss-of-function mutations to the deaminase domain have not yet been identified in humans, suggesting that such mutations would be lethal. Indeed, ADAR1 knockout in mice is embryonic lethal.41,44 Conversely, ADAR1 overexpression is associated with certain cancers,45,46 highlighting a potential risk of introducing exogenous ADAR to promote therapeutic RNA editing.
ADAR1p150 expression is transcriptionally controlled by an interferon-responsive element in the promoter region47 and possesses an N-terminal Z-DNA-binding domain as well as an NES. As such, interferon stimulation induces ADAR1p150 expression and localization to the cytoplasm, where it can edit cytosolic dsRNA substrates and alter the RNA editome,48 playing a key role in viral immunity.49 Interferon stimulation in vitro has been used to improve RNA-editing efficiency,6 and the possibility of transient ADAR1p150 induction in vivo, for instance, due to innate immune responses to viral infection or drug-delivery systems, should be considered when evaluating the specificity of therapeutic RNA editing.
ADAR2 protein and enzymatic activity are limited to select tissues, such as the brain and heart.50,51 ADAR2 plays a key role in site-specific editing for the recoding of amino acids. Murine ADAR2 knockout leads to death several weeks after birth,52 while the lethal phenotype is rescued by encoding a key RNA-editing site within the GRIA2 gene at the genomic level, highlighting the importance of ADAR2 for site-specific editing.
In contrast to ADAR1 and ADAR2, ADAR3 is exclusively expressed in the brain and lacks deaminase activity.23,53 ADAR3 expression negatively correlates with editing and is believed to repress A-to-I editing by competitive antagonism of ADAR1 and ADAR2. This mechanism has been further characterized in glioblastomas, where ADAR3 competes against ADAR2 for the binding of GRIA2 transcripts and negatively modulates its editing.24 ADAR3 knockout mice displayed impaired learning and memory; however, RNA editing at most sites within the transcriptome was unaffected by ADAR3 knockout, with only ten sites showing a statistical difference from wild type, suggesting that the regulation of editing may be substrate specific.54
ADAR subcellular localization and transport influence enzyme accessibility to dsRNA substrates and subsequent A-to-I editing. ADARp110- and ADAR2-mediated RNA editing happen co-transcriptionally, and enzyme localization is reported in the nucleus and nucleolus.55,56 Alternatively, ADARp150 localizes to the cytosol upon interferon stimulation, where it can access and edit cytosolic dsRNA substrates.57 It should be noted that this distinction of ADARp110 as a nuclear protein and ADARp150 as a cytosolic protein is an oversimplification, as both isoforms are known to shuttle between nucleus and cytoplasm. Nuclear import is mediated by transportin-1 (Trn1), which interacts with an atypical NLS sequence found in the third dsRBD of ADAR1 isoforms.58 The third dsRBD cannot bind dsRNA and Trn1 simultaneously, which makes ADAR1 nuclear import dependent on dsRNA cytoplasmic content. On the other hand, nuclear export of ADAR1p110 and ADAR1p150 are regulated differently, with ADAR1p110 exported by exportin-5 (XPO5) while the p150 isoform is bound by exportin-1 (XPO1) on its NES. Overall, dsRNA content and accessibility in the cytoplasm or nuclear compartment play an important role in subcellular localization and subsequent A-to-I editing. Unlike cytoplasmic antisense approaches using RNAi- or RNase-H-mediated knockdown, gRNAs that rely on ADARp110- or ADAR2-mediated RNA editing must localize to the nucleus.
Beyond the ADAR1/2/3 dsRBD proteins, the human genome encodes for more than 1,000 RNA-binding proteins (RBPs), 16 of which contain dsRBDs that may directly compete with ADAR binding: ADAD1, ADAD2, CDKN2AIP, DGCR8, DHX9, DICER, DROSHA, ILF3, MRLP44, PKR, PRKRA, SON, STAU1, STAU 2, STRBP, and TARBP2. Not surprisingly, these double-stranded RBPs (dsRBPs) are found to be in the same interactome59,60 and share roles in various RNA-related biological processes, such as innate immune response, microRNA processing, apoptosis, and cell cycle. They can act with ADAR either synergistically or antagonistically depending on the cellular context.61 Beyond the biological functions, the crosstalk between different dsRBPs and ADAR highlight the importance of the RBP landscape in A-to-I editing. The interaction landscape can be modulated by the cellular context, such as viral infections, UV light, cell cycle, and tissue expression. Thus, the expression levels of dsRBPs are a contributing factor to RNA editing,40 emphasizing the importance of assaying RNA editing within model systems that reflect the dsRBP expression profile of the therapeutically relevant target cell.
A comprehensive picture of the A-to-I editing landscape in human tissues was captured by profiling A-to-I editing in over 50 organs from 8,551 samples of the Genotype-Tissue Expression (GTEx) consortium.40 ADAR1 and ADAR2 are the only A-to-I mRNA editors known in humans, yet their mRNA expression shows only a moderate correlation with A-to-I editing, depending on the tissue (R2 = 0.2–0.25 across all tissues with a higher correlation of 0.55 in the brain). This suggests that additional factors regulate editing. A-to-I regulation can arise from various factors such as RNA splicing, RNA expression levels, and the RBP landscape, which can restrict accessibility to the targeted adenosines. Despite similar editing activity in most tissues, outliers were detected including the cerebellum and arteries with the highest editing levels (potentially explained by high co-expression of ADAR1 and ADAR2) and skeletal muscles demonstrating the lowest editing levels and low expression of ADAR1. Additionally, the authors identified a transregulatory mechanism in skeletal muscle via aminoacyl tRNA synthetase complex interacting multifunctional protein 2 (AIMP2), which negatively impacts the stability of both ADAR1 and ADAR2 and may further explain the low editing levels detected in skeletal muscle.40 Additionally, 3,710 tissue-specific edited sites were identified, and it is widely documented that ADAR1 and ADAR2 have overlapping but unique editing profiles,34,62 highlighting the need to engineer and screen gRNAs within disease-relevant models to best reflect the in vivo editing environment.
The ubiquitous expression and activity of ADAR in all human tissues opens the door to many therapeutic applications; however, more work is needed to assess the feasibility of endogenous ADAR recruitment in various tissues. Furthermore, the editing environment within human cell lines is often less active than in the corresponding tissues in vivo,63 and certain models may have limited utility for assessing in vivo activity of therapeutic gRNAs.
Considerations for gRNA design and optimization
As detailed in the preceding section, the underlying biology and structure of ADAR are crucial to developing an RNA-editing therapeutic. At the cellular level, a detailed understanding of the expressed ADAR isoforms and transregulators is needed to select model systems that reflect the in vivo editing environment, while structural knowledge can be leveraged to inform the optimization of gRNA efficiency and specificity. The application of this knowledge and how it can be leveraged to inform gRNA design and engineering is described in this section and outlined in Figure 2.
Figure 2.

Factors influencing gRNA efficiency and selectivity
Broadly speaking, two main delivery approaches for ADAR-recruiting gRNAs have been described: ASOs, which include in vitro transcribed or chemically synthesized gRNAs that are delivered directly to the cell; or DNA-encoded gRNAs that are delivered with viral or non-viral technologies, where the gRNA is transcribed upon entry of the exogenous DNA template into the nucleus. Each approach has a set of considerations and ultimately, the delivery modality is influenced by the disease, tissue, and cell type of interest. gRNA design parameters are dependent upon the chosen delivery method and will be discussed independently in the following sections. Regardless of the delivery method, engineering gRNA efficiency and specificity is perhaps the most important element of developing an RNA-editing therapeutic and is complicated by the promiscuous activity and innate sequence preferences of the ADAR enzyme.
Delivery of a DNA-encoded gRNA relies on endogenous cellular transcription to produce the functional gRNA molecule. This drives persistent, durable expression of the gRNA in a natural RNA state. Importantly, a gRNA transcribed from a DNA payload is not limited by the same size constraints as a chemically synthesized ASO, enabling a larger design space to create the ideal target-specific ADAR substrate. Additionally, DNA payloads are amenable to the use of promoters, regulatory elements, and RNA structural modifiers that can be used to tune gRNA expression, persistence, and subcellular localization. RBP sequence motifs can also be used to recruit and promote protein interactions that enhance RNA editing (see the foregoing discussion of RBPs). Since many human diseases affect terminally differentiated cell populations (e.g., neurons, muscle cells), delivery of a DNA-encoded gRNA carries the promise of long-term, durable treatment with a single administration of drug. However, like traditional gene therapy, DNA payloads cannot be simply “turned off” if an adverse event is experienced, highlighting the need for exquisite specificity and robust non-clinical development data. Depending on the exact delivery method (e.g., AAV, non-viral particles), immunogenicity and triggering of DNA-sensing pathways may limit the overall delivery efficiency and safety. These aspects are not unique to RNA editing and must be considered by the entire gene therapy field.
As an alternative to DNA-encoded gRNAs, ASOs can be used to recruit ADAR for RNA editing. ASOs can be chemically synthesized with chemical modifications or in vitro transcribed from a DNA template. In the case of chemical synthesis, ASOs may be limited by size because of synthesis capabilities, and there exists a delicate trade-off between chemical toxicity and drug efficacy. However, numerous advancements in ASO chemistry can improve stability, specificity, and efficiency. With direct administration of these molecules, redosing is necessary because of their relatively short half-life, but with certain chemical modifications molecules may persist for weeks to months.64 In some contexts, this transient aspect may be an added feature, for example in the transient modification of a pain receptor. Additionally, ASOs follow more traditional drug pharmacokinetic and pharmacodynamic (PK/PD) profiles, and dosing can be stopped if an adverse event is observed.
For both DNA-encoded gRNAs and ASOs, delivery of the drug to the target tissue and cell type remains a key challenge. Thus, regardless of gRNA design, continued innovation of delivery technologies is required to maximize the therapeutic potential of RNA editing.
DNA-encoded approaches
Recruitment of endogenous ADAR
Programmable RNA-editing systems typically consist of two components: the ADAR enzyme and a gRNA that hybridizes to a target mRNA of interest, thereby creating the dsRNA ADAR substrate. Initial efforts in the field of RNA editing relied on overexpression of exogenous ADAR or chimeric enzymes composed of the deaminase domain fused to RBPs with engineered gRNAs to recruit the enzyme to the target.9,15,65,66,67,68,69 Initially, the gRNA designs typically consisted of two domains: an antisense domain, typically 20–40 nucleotides (nt) in length bearing a C mismatch opposite the target adenosine, and a recruitment domain that brought the ADAR enzyme to the mRNA of interest via a protein-RNA interaction. DNA-encoded gRNAs consisted of a variety of recruitment domains, ranging from a portion of the naturally occurring GRIA2 pre-mRNA hairpin or crRNA:tracrRNA to BoxB and MS2 stem loops, and were utilized to recruit either the wild-type ADAR2 or fusions of the catalytic domains of ADAR to Cas13, λN-peptide and MS2 coat proteins, respectively.9,65,67,68 Proof-of-concept studies demonstrated the use of AAV-delivered adenosine deaminases in mouse models of Duchenne muscular dystrophy, ornithine transcarbamylase deficiency, and Rett syndrome.9,70 While ADAR-overexpression-based approaches demonstrated the therapeutic potential of RNA editing, the promiscuous nature of ADAR led to transcriptome-wide off-target A-to-I editing9,15,71 with potentially toxic effects seen in mice.9 To overcome this problem, it is important to restrict the catalytic activity of the overexpressed enzyme only to the target mRNA. By splitting the ADAR2 deaminase domain into two catalytically inactive fragments that are brought together by a chimeric gRNA at the given target mRNA to transiently form a functional enzyme, we achieved >100-fold more specific RNA editing as compared with full-length deaminase overexpression.72 This novel strategy resulted in greatly improved transcriptomic specificity, and the split-ADAR2 system was functional with RBPs of human origin to limit immunogenicity concerns. Further improvements to the enzymatic activity of the split-ADAR2 system or additional protein engineering strategies that enhance specificity may improve its therapeutic potential.
Even with enhanced specificity of engineered exogenous proteins, this approach will still be challenged by packaging limits of the delivery modalities (e.g., AAVs) and immunogenicity concerns. Therefore, recruitment of endogenous ADAR to perform targeted RNA editing is the preferred approach. We recently demonstrated the use of DNA-encoded gRNAs for the recruitment of endogenous ADAR to mediate RNA editing.9 While gRNAs with antisense domains as short as 20 nt sufficed to recruit overexpressed ADAR, increasing the length, for example, to 60 nt or more enabled recruitment of endogenous ADARs.9 This was an important advancement of the technology, as it opened the door to potential therapeutic applications.
gRNA expression, stability, and localization
DNA-encoded gRNAs can be further optimized by focusing on expression, stability, and localization. gRNAs are typically transcribed from pol III promoters (e.g., U6) and lack a 5′ cap and a 3′ poly(A) tail, leaving them vulnerable to 5′ and 3′ exonucleases, thereby reducing their half-lives. Given that RNA editing is a transient event that dilutes out with mRNA turnover, it is important to improve expression and stability of the U6 transcribed gRNA. Circularization of RNA is one strategy to prevent exonuclease digestion and increase RNA half-life. To this end, we created DNA-encoded circular gRNAs by flanking long antisense domains with twister ribozymes.8,73 Upon transcription, the twister ribozymes self-cleave, leaving specific overhangs that are recognized and ligated by the ubiquitously expressed RtcB RNA ligase to form a circular gRNA.74 The use of circular gRNA greatly improved the persistence of RNA editing over linear gRNAs both in vitro and in vivo. While no editing of the PCSK9 3′ untranslated region (UTR) in mouse livers was detectable via AAV-delivered linear gRNA, 11% editing was detectable via AAV-delivered circular gRNA. By packaging two copies of the U6 promoter and circular gRNA within an AAV, RNA-editing levels increased to 53% at 8 weeks post injection.74 Additionally, AAV-delivered circular gRNAs were utilized to repair a premature stop codon (W392X) in the α-L-iduronidase mRNA in the liver of a mouse model of Hurler syndrome via recruitment of endogenous ADAR enzymes, resulting in 12% RNA editing and partial restoration of enzyme activity.8,11 Although short-term studies showed no toxicity in mice with RNA-editing levels being maintained up to 8 weeks post injection, longer studies are needed to assess the safety and durability of AAV-delivered circular gRNAs.
An alternative strategy to improve gRNA stability is the use of natural exonuclease-resistant structures at the 5′ and/or 3′ ends of the gRNA.75,76,77 Advances in the field of short interfering RNA (siRNA) and CRISPR gRNAs have demonstrated the utility of this approach in enhancing the stability of U6 transcribed RNA.78,79 This knowledge from the fields of CRISPR gRNAs and antisense RNAs can be applied to enhance the performance of the ADAR-recruiting gRNAs. Furthermore, focused efforts need to be made to engineer spatiotemporal regulation of RNA editing. The use of tissue-specific enhancer elements will allow for modulation of RNA-editing activity in space, while engineering small-molecule-based regulation of gRNA activity could enable temporal control.80,81
gRNA structure and interaction with mRNA
While gRNA abundance is an important factor contributing to the efficiency of RNA editing, intrinsic characteristics of the gRNA, such as intramolecular secondary structure and nucleotide composition, also play a major role in influencing the activity of a gRNA. Most transcribed gRNAs are relatively long (greater than 40 bp) and can have complex secondary structures. The secondary structure of a gRNA affects its ability to bind its target, and the use of computational tools to predict intramolecular secondary structure can improve gRNA designs. Additionally, the editing of adenosines on the gRNA itself could impact editing of the target adenosine. RNA editing via ADARs can occur on both strands of an RNA duplex, thereby altering the sequence of the gRNA itself. This could, in turn, impact the ability to effect ADAR-mediated editing of additional target transcripts.82 Conversely, the secondary structure of the target pre-mRNA and position of the editing site within the transcript, such as the UTR versus coding region (CDS), may also impact editing. As observed in the ASO and RNAi fields, many regions within an mRNA are amenable or refractory to knockdown due to accessibility. However, these knockdown strategies have the luxury of tiling across the mRNA to identify the optimal location for knockdown. RNA editing may prove challenging if a target adenosine lies within a highly structured or inaccessible region of an mRNA, making it difficult to edit. It remains to be determined whether longer gRNAs or gRNAs that employ two or more discontinuous hybridization regions could modulate the target RNA structure to help access adenosines located in such regions.7 A more systematic approach comparing the accessibility of ASOs and gRNA-mediated RNA editing would help to better understand the limitations imposed by the target mRNA structure. Furthermore, the entire dsRNA stretch formed between the gRNA and target mRNA becomes a substrate for the ADAR enzyme. Thus, further engineering of the gRNA is essential in achieving specific editing of the target adenosine.
Engineering specificity
The ability to recruit endogenous ADAR limits the issue of transcriptome-wide off-target editing; however, bystander editing of non-target adenosines within the gRNA-target complex is commonly observed. As discussed, ADAR enzymes have promiscuous editing activity as evidenced by their role in regulating innate immune responses to dsRNAs and the millions of identified editing sites within the transcriptome.51,83,84 Despite the promiscuous nature of ADAR, many natural substrates have been identified that are edited with high selectivity and efficiency for the purpose of modulating protein function by recoding at the amino acid level or altering pre-mRNA splicing.85,86,87 It is hypothesized that secondary structural features within the dsRNA can drive efficient and selective editing of these substrates. Secondary structural features downstream of the edited adenosine within the GRIA2 R/G substrate have been shown to increase editing efficiency,29 while the addition of secondary structures was shown to limit the promiscuous nature of ADAR activity within a dsRNA substrate.88 Mutagenesis and high-throughput screening of natural substrates within NEIL1, TTYH2, and AJUBA pre-mRNA have demonstrated the impact that secondary structure can have on editing efficiency.89 In addition, high-throughput screening of secondary structures within long dsRNA substrates mapped ADAR activity 30 nt upstream of secondary structure disruptions and displayed a periodicity to editing.82 Furthermore, co-immunoprecipitation and RNA sequencing showed a periodicity to ADAR engagement to natural substrates occurring in 50-nt increments.90 These observations may be leveraged to engineer gRNAs with improved specificity required for therapeutic applications. However, these features are observed within a cis RNA interaction, and it remains to be seen how easily they will port into the trans interaction of a gRNA and target RNA.
We recently used secondary structural features to address the issue of bystander editing. We first demonstrated that a perfect complementary gRNA containing a C mismatch across the target adenosine mediates numerous bystander editing events driven by endogenous ADAR. Others have shown that incorporation of a G mismatch positioned at bystander adenosines can reduce off-target ADAR activity, but the RNA-editing efficiency of the target adenosine may be negatively impacted.10 As an alternative approach, we incorporated internal loops in specific positions along the entire length of the gRNA. This eliminated promiscuous RNA editing without affecting the efficiency of the target adenosine,8 and a similar approach using discontinuous stretches of hybridization also improved specificity.7 Another approach demonstrated that precise nucleotide deletions across bystander adenosines can lead to improved specificity of circular and linear gRNAs.11 We anticipate that additional refinements to gRNA design will further reduce bystander editing and boost target editing efficiency.
Delivery of DNA-encoded gRNAs
Currently there is a limited clinically validated toolset for the delivery of DNA payloads; thus, despite challenges, the gene therapy field relies heavily on adenoviruses and AAVs. Preclinical in vivo proof-of-concept studies for ADAR-based RNA editing have used AAVs to deliver DNA-encoded gRNAs to mouse livers. The natural tropism of many AAV serotypes lends itself to targeting disorders of the liver, muscle, CNS, and eye. However, ADARs are ubiquitously expressed, and ongoing efforts to expand the tropism and specificity of AAV serotypes is an active area of research that might enable the delivery of gRNA to additional tissue types.91,92,93,94,95,96 For example, efficient delivery of AAV to the CNS requires invasive techniques such as direct injection into the brain parenchyma. Delivery vectors with the ability to efficiently cross the blood-brain barrier and transduce the CNS would increase safety and simplify the design and execution of preclinical and clinical studies. However, systemic injection of AAV results in high transduction of the liver. Thus, reducing liver uptake while increasing transduction of the target organ may improve safety and efficacy. In addition to viral delivery, non-viral approaches, such as lipid nanoparticles (LNPs), can be used to deliver DNA payloads but, as with AAV, primary uptake is in the liver. As delivery technologies improve, new therapeutic opportunities will emerge for RNA editing.
Antisense oligonucleotides for RNA editing
ASOs are another widely used approach for therapeutic RNA editing that builds on decades of work in the oligonucleotide chemistry field. ASOs have progressively undergone three major improvements: the introduction of phosphorothioate backbone chemistry, the use of sugar modifications such as 2′-O-methyl, and the use of nucleic acid analogs, such as locked nucleic acids (LNAs).97 In combination, these improvements have enhanced stability, efficiency, biodistribution, cell penetrance, and safety, resulting in enormous growth in oligonucleotide-based therapeutics in the last two decades. Currently, there are more than 15 ASO-based therapies that have reached late-stage clinical testing or received Food and Drug Administration approval.98,99 Importantly, lessons learned from the ASO field can be leveraged for the design and clinical application of chemically synthesized gRNAs for therapeutic RNA editing.
Many of the challenges shared broadly by the ASO field, including delivery, biodistribution, cell penetrance, and safety, are similarly applicable to RNA editing. Additionally, a few challenges unique to RNA editing exist and include gRNA length, potentially distinct interactions of the ADAR enzyme with ASO chemistry, and nuclear delivery and localization. In the contexts of RNase-H-mediated degradation or exon skipping and siRNAs for RNAi-mediated knockdown, short oligos of ∼20 nt are effective. However, ASOs to mediate RNA editing will likely require at least 30 nt,12,13 and the use of recruitment domains could further increase the length to 60–90 nt.6 In addition to length, ideal gRNA structures that balance stability while promoting ADAR binding and enzymatic activity will be key to maximizing RNA-editing efficiency and specificity. Similarly, ASO stability was optimized with “gapmers” that modified structural features while still retaining RNase-H-directed activity.100 Lastly, while RNase-H activity can occur in the nucleus or cytoplasm101 and RNAi-mediated knockdown occurs in the cytoplasm,102 most RNA editing occurs co-transcriptionally in the nucleus.103 Thus, nuclear delivery and localization of the chemically modified gRNA are important parameters in achieving efficient RNA editing.
Key advancements for ASOs and RNA editing
Many key ASO advancements have been adopted by the field of RNA editing, and numerous labs have used ASOs to recruit endogenous ADAR to edit target adenosines in vitro and in vivo.104,105,106 An early application was demonstrated using an exogenous ADAR deaminase domain covalently linked to an ASO that directed the deaminase domain to a target mRNA.66 Building on this early work, recruitment of endogenous ADAR was achieved using chemically modified ASOs with an antisense domain attached to a portion of the GRIA2 R/G hairpin.6 This method demonstrated RNA editing across multiple mRNA targets in cell lines and primary cells. Chemical modifications included 2′OMe groups throughout much of the ASO, aside from the 3-nt motif across from the target and select locations within the GRIA2 R/G hairpin; phosphorothioates at the 5′ and 3′ ends, and LNAs at the 3′ end. The use of chemical modifications was crucial for the recruitment of endogenous ADAR, as an unmodified ASO resulted in no detectable editing unless the cells were treated with interferon-α to induce ADAR p150 expression. Editing of two therapeutically relevant targets was also demonstrated: introduction of T701C in STAT1 to prevent phosphorylation and downstream signaling of the JAK-STAT pathway,107 and correction of the PiZZ mutation (E342K) in SERPINA1, the most common cause of α1-antitrypsin deficiency (AATD).108 Twenty-one percent editing of STAT1 and 10%–18% editing of the E342K codon within a SERPINA1 cDNA minigene was achieved using chemically modified ASOs. Interestingly, the optimal ASO design was 91 nt in length, which included a 38-nt antisense domain and 53-nt hairpin structure. This is far longer than the ∼20-nt ASOs used for RNase-H-mediated knockdown and exon skipping, and longer designs may complicate delivery, manufacturing, and safety. It is also important to note that bystander editing was observed at the neighboring 3′ adenosine of the SERPINA1 target. The introduction of a 2′OMe group on the paired uracil within the ASO was able to reduce this bystander editing, albeit with a concurrent reduction of editing at the target adenosine. A similar trade-off was observed with the use of A-G mismatches to reduce bystander editing for DNA-encoded gRNAs.10 Further work is needed to understand the basic design principles to optimize both efficiency and selectivity of editing. An alternative strategy used a much longer ASO of 100 nt and utilized 2′OMe modifications on the 5′ and 3′ ends. Using this design to target the PPIB transcript yielded 20% editing in human T cells, while the unmodified ASO failed to produce detectable editing.10 These studies clearly demonstrated the potential of ASOs in eliciting efficient and specific RNA editing using the endogenous human ADAR enzyme.
Given the known structural details of the ADAR footprint and the size of many natural substrates that result in recoding of amino acids, which often contain less than 30 bp of dsRNA, it is not surprising that recent publications have significantly shortened the length of ASOs. One recent publication demonstrated the use of a 30-nt stereopure ASO with phosphorothioate backbone.12 The footprint of the design matches the canonical asymmetric footprint of ADAR, with approximately 5 bp on the 5′ side of the target to accommodate the 5′ binding loop of the deaminase domain, and approximately 25 bp on the 3′ side of the target to accommodate the 3′ binding loop of the deaminase domain, along with the dsRBDs. Additionally, their ASO design contained extensive use of 2′-fluoro modifications on the 5′ end, 2′-O-methyl on the 3′ end, and deoxyribonucleotides across from the edit site, indicating that ADAR is tolerant of these modifications in their respective locations. The stereopure ASOs achieved robust editing in tissue culture and in vivo. A liver-targeting GalNac-ASO conjugate was intravenously administered to non-human primates (NHPs) and achieved up to 50% editing for a non-clinical target in the 3′ UTR of the endogenous ACTB transcript. While the target adenosine lies in an ADAR-favored UAG motif, these data in NHPs supports the translatability of RNA editing. A single dose showed persistent RNA editing 50 days post injection, further highlighting the therapeutic potential of GalNac-ASO conjugates. In the context of a disease-relevant target, stereopure ASOs achieved ∼75% editing of the SERPINA1 E342K mutation in vitro. Shortening the gRNA to 30 nt simplified the manufacturing, and the lack of a hairpin recruitment structure means the ASO engagement with ADAR is dependent on target hybridization and is less likely to perturb natural ADAR function. Furthermore, shorter length reduces the risk of chemical toxicity that appears to be a class effect with high-dose, chemically modified ASOs.109
Knowledge of ADAR structure and function can also be leveraged to better inform ASO design. A clever “bump-hole” design paired an engineered ADAR2 E488Y mutant with an ASO containing an abasic site across from the target adenosine.69 Owing to a steric clash, the ADAR2 E488Y mutant had low enzymatic activity; however, the abasic site resolved this clash and restricted its activity to the ASO-target complex formed upon hybridization to the target mRNA. This strategy could enable the use of exogenous ADAR while minimizing off-target editing but comes with the complication of delivering a non-human protein with the risk of an antidrug response to the ADAR2 E488Y. More recently, the same group detailed the rational design of ASOs for the recruitment of endogenous ADAR.13 The ADAR2 E488Q mutation has been well documented to improve editing through hydrogen bonding of Q488 to the orphan base in a pH-independent manner.31,110 Inspired by this observation, the researchers sought to improve hydrogen bonding from the orphan base on the ASO with the wild-type ADAR2 E488. Indeed, incorporation of the cytidine analog 2′-deoxy Benner’s base Z (dZ), which was hypothesized to have a favorable hydrogen bond pattern with E488, improved the biochemical reaction rate kinetics of both wild-type ADAR1 and ADAR2 3-fold. When tested in human ARPE-19 cells, incorporation of dZ at the orphan position of the ASO improved editing of a γ-secretase cleavage site within the APP transcript from 6% to 19%. There is still much to be learned about the principles behind chemical modifications and how they impact ADAR substrate engagement and deamination, but these results highlight the potential of rational ASO design to augment the interaction and enzyme kinetics of endogenous ADAR.
These data indicate that ASOs are a viable and promising path for therapeutic RNA editing. In the short term, ASO delivery to the liver, muscle, or kidney, or direct injection to the CNS, are viable options. Unlike the long-term persistence of DNA-encoded gRNAs resulting from AAV delivery, ASO half-life allows for transient editing and redosing as needed, and the dose can be optimized to fine-tune the desired amount of editing required for the therapeutic effect. Ongoing work within the ASO field to address the challenges associated with delivery, biodistribution, and cell penetrance will quickly be adopted and applied to RNA editing. Meanwhile, additional work to optimize and standardize ASO designs for the recruitment of endogenous ADAR is needed.
Therapeutic opportunities
RNA editing provides many attractive therapeutic applications, the most logical being correction of G-to-A missense and nonsense mutations, of which ∼7,000 pathogenic G-to-A mutations are reported in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/, accessed on April 13, 2022). In support of therapeutic RNA editing, several proof-of-concept in vivo studies using ADAR-mediated RNA editing to correct missense and nonsense mutations have been described. In a mouse model of Hurler syndrome, endogenous ADAR was recruited to correct a nonsense mutation in the IDUA transcript and restore protein function.8,11 In two mouse models of Rett syndrome, RNA editing using exogenous ADAR was able to correct both nonsense (MECP2W104X) and missense (MECP2R106Q) mutations.20,70 Correction of a nonsense mutation in the mdx mouse model of Duchenne muscular dystrophy was achieved with exogenous ADAR recruitment.9 Additionally, RNA editing of a 5′ splice site missense mutation in the spfash mouse model of ornithine transcarbamylase deficiency restored correct splicing in vivo,9 highlighting an ability of the technology to function at the pre-mRNA level. Lastly, significant attention has been directed to the SERPINA1 E342K mutation that causes AATD, and two independent groups have demonstrated >40% RNA editing of mutant SERPINA1 within human cells using ASOs.6,12
Beyond correction of point mutations, targeting adenosine-containing motifs such as splice acceptor sites, TISs, polyadenylation signals, and microRNA-binding sites can modulate mRNA and/or protein levels for therapeutic purposes. ADAR plays a natural role in the regulation of splicing,111 and genomic editing of splice sites is able to modulate splicing,112,113,114 strengthening the rationale for therapeutic splice site targeting. Furthermore, ASOs and DNA-encoded antisense RNAs have been used to mask and block the function of splice sites,115 polyadenylation sites,116,117,118 TISs,119 upstream open reading frames,120 and microRNA-binding sites.121 Therefore, gRNAs designed to both mask and edit these regions may provide an additive effect. Indeed, many of these motifs have been hardwired at the genomic level by DNA editing122,123,124 and have conferred the desired molecular effect.
Further applications for RNA editing can also be envisioned. The advent of monoclonal antibodies125 created a new instrument to block protein function and signaling by binding to soluble proteins and membrane proteins, such as tumor necrosis factor α126 and HER2,127 respectively. However, intracellular proteins are inaccessible to antibodies, and complex membrane proteins pose a challenge to antibody discovery, such as ion channels and G-protein-coupled receptors. ADAR-mediated RNA editing can introduce 17 different amino acid substitutions that can be used to modulate protein function and abolish or enhance protein-protein interactions. This may be of particular interest for proteins that are not amenable to antibody therapy. For example, RNA editing of the BACE cleavage site on APP was demonstrated in ARPE-19 cells,13 a potential target for the treatment of Alzheimer’s disease.128 Additionally, endogenous ADAR2 plays a central role in modulating ion channel permeability,129 and extending this function to therapeutic regulation of ion channels, such as Nav1.7, is of great interest.130,131
The therapeutic targets mentioned above could also be corrected at the genomic level using DNA-editing technologies; therefore, one must consider the risk/benefit profile of DNA versus RNA editing when selecting a therapeutic approach for any given disease indication. First and foremost, DNA-modifying enzymes create permanent changes that impact 100% of transcribed RNAs at both on- and off-target sites. In contrast, RNA editing is transient in nature for the life of the edited RNA molecule and can be tuned to the desired fraction of RNA molecules to be edited within a cell. For therapeutic applications that require a transient pharmacodynamic effect, such as the treatment of acute pain, obesity, viral infection, and inflammation, it would be undesirable to introduce permanent alterations to the genome. Thus, the transient modulation of protein expression or function by RNA editing is advantageous. Additionally, the tunability of RNA editing can be exploited where partial knockdown or partial protein modulation is desired. In fact, many endogenous ADAR dsRNA substrates that are edited for the purpose of recoding show a significant range in editing efficiency, from single-digit to 100%.18 Some organisms have even evolved techniques to fine-tune RNA editing based on their environment.132,133,134 Mutagenesis studies have demonstrated that altering the secondary structure of natural substrates can increase or decrease editing,89 highlighting once again the importance of gRNA design for therapeutic application of RNA editing.
RNA editing offers unique safety and delivery advantages over DNA editing. Despite the potential and early clinical success of CRISPR-Cas DNA-editing technologies,135 safety concerns persist.136 RNA editing does not cause permanent alterations at the genomic level, avoiding the oncogenic risk associated with DNA editing and, as discussed above, allowing for transient treatment of acute conditions. Additionally, a single gRNA payload is sufficient to recruit endogenous ADAR. This is in contrast to DNA-editing systems that rely on the use bacterial proteins or hyperactive enzymes that carry the risk of immunogenicity137,138 and present delivery challenges due to their size. Because ADAR is ubiquitously expressed, its potential within any organ or cell type is only limited by the delivery of the gRNA to the target cell. This includes non-dividing cells, such as neurons in the CNS, where the lack of homology-directed repair (HDR) pathways limits the use of certain DNA-editing technologies. ADAR-mediated RNA editing is limited to A-to-G changes, and efforts to utilize APOBEC1 for C-to-U RNA editing are less advanced.139 In contrast, improved DNA-editing technologies, such as base editing and prime editing, can introduce mutations not feasible with current RNA-editing technologies and circumvent the need for HDR required by traditional CRISPR-Cas9 methods. The ability for permanent genomic alterations also makes DNA editing particularly attractive in rapidly dividing cells or progenitor cells and has been extensively used in ex vivo cell therapy applications. Overall, given the many differentiators highlighted above, RNA editing has great potential as a therapeutic modality across a wide range of challenging diseases and has become an important part of the biotechnology molecular toolkit.
The field of RNA editing will continue to gain traction from advances in delivery technology as new AAV capsids and ASO modifications expand the tropism and penetrance of different tissues. Meanwhile, advances to gRNA discovery and design may open new opportunities. Increased knowledge of ADAR and gRNA structure will allow for more sophisticated design, such as the recoding of multiple codons within a transcript, as seen for natural substrates.87 The limited cargo size of a gRNA expression cassette could easily allow for multiplexing and editing of multiple transcripts. One could envision targeting multiple pathways or engineering both interfaces of a protein-protein interaction. The potential for new modalities has also emerged. RNA editing is being leveraged for RNA sensing, allowing expression of a payload to be gated on the transcriptional stage of the target cell.140,141,142 However, to make any of these possibilities a reality, early proof-of-concept studies may need to be improved to translate the results into the clinic.
Translation to the clinic
Increased understanding of the fundamental biology and control of RNA editing has advanced this technology to the cusp of clinical application. Successful translation to the clinic requires addressing remaining challenges. Several regulatory guidance documents are available that broadly address many of the challenges facing sponsors during the development of gene therapies and regenerative medicines.143,144,145,146,147,148,149,150,151,152,153 These guidance documents encompass novel platform technologies such as RNA editing and represent current regulatory thinking on research pharmacology, non-clinical safety, product manufacture/characterization, and clinical assessment. While these guidance documents can generally be applied to RNA editing, there remain technology-specific issues requiring careful consideration during development. A broad overview of the clinical considerations is outlined in Figure 3 and will be discussed in more detail here.
Figure 3.

Considerations to support the clinical development of RNA editing
RNA editing must be exquisitely selective for the intended RNA target, with biologically negligible off-target editing. This is required to achieve the intended pharmacological activity and minimize safety risks. Validated methods for screening on- and off-target editing are required to determine the specificity profile of any given gRNA. Deep RNA sequencing can characterize global alterations in the cellular transcriptome and establish the editing signature of a gRNA.51,154 Establishing this signature across the relevant cell and tissue types, influenced by the expected biodistribution of a given delivery modality, will be critical to predicting clinical safety risk. Previous murine studies that introduced exogenous ADAR or hyperactive forms of ADAR showed a significant increase in off-target editing,9,15,20,70 while recent publications that redirect endogenous ADAR to the target of interest have minimal off-target editing.8,11,12 Should specific transcripts demonstrate elevated levels of off-target editing, further characterization toward understanding any physiological or toxicological consequences may be required. In some instances, off-target editing may not affect protein-level expression or function, such as in the case of an edit leading to a synonymous codon change that does not alter the structure of the translated protein. In other cases, off-target edits could be significantly disruptive, for example in the introduction of a non-synonymous mutation leading to a gain or loss of function to the protein. Given the spectrum of outcomes from potential off-target editing, it is important to consider the impact of those edits on a case-by-case basis, particularly since different pathways will have varying tolerance for perturbation. At a minimum, the relationship between off-target editing and protein expression should be established and followed up by functional studies to investigate the impact to known downstream pathways. The broader consequences of off-target editing at the tissue and organism levels will be evaluated in the toxicology studies required by regulatory agencies, but analyses in relevant human cells may aid in interpretation of findings.
Similar to strategies used to assess DNA-editing off-target events,155 deep sequencing methods can also be used to determine whether there have been changes to the endogenous editome of the cell as a result of preferential ADAR recruitment to gRNA-targeted sequences.81 Long-term disruption of natural ADAR function could have immunological consequences and impact a number of cellular pathways. It should be noted that the transcriptome has millions of A-to-I editing sites39 spread across thousands of dsRNA substrates.156 It is unlikely that the addition of a single substrate would perturb ADAR activity; however, gRNA designs that include a recruitment domain6,7 are capable of binding ADAR independent of hybridization to the target. This poses a greater risk of perturbing ADAR activity, especially if expressed at high levels.
Additionally, when assessing the potential for off-target effects it is important to consider the relative contributions of ADAR1 and ADAR2 toward therapeutic editing. Each enzyme is capable of efficient and selective editing of natural substrates for recoding at the amino acid level, yet subtle differences in their preferential editing based on sequence context and secondary structure exist.26,62 For example, therapeutic editing in the liver would primarily rely on ADAR1, while biodistribution to tissues with high ADAR2 expression (e.g., brain)40 may result in altered editing efficiency or specificity of the target mRNA. Ensuring the gRNA is selective for the target adenosine in both an ADAR1 and ADAR2 environment is an important consideration, especially when the delivery modalities may lack specificity for the target tissue(s). Engineered cell lines that express ADAR1 and ADAR2 in isolation can be a valuable tool to assess the relative specificity of each enzyme for gRNA-mediated editing of the target mRNA.7
Much attention is focused on quantifying RNA editing at the transcript level, but equally crucial is ensuring this leads to a corresponding change in protein that imparts the desired phenotypic outcome. It is often assumed that correction of a missense mutation will lead to a corresponding level of corrected protein; however, this may not always be the case. Although inosine is interpreted as guanosine, there is a small loss in fidelity that can vary based on sequence context, and the presence of more than one inosine can stall translation.157 Therefore, it is desirable to quantify both RNA editing and protein restoration in preclinical safety and efficacy assessments.
gRNA delivery, whether through an ASO or DNA-encoded approach, is an important factor in maximizing exposure and activity in the cells of interest and minimizing off-target exposure and expression that could contribute to unwanted side effects. An optimal delivery approach should enable efficient tropism, cellular uptake, and cell-type-specific expression and function. The method of delivery will impact non-clinical, manufacturing, clinical, and regulatory considerations for RNA-editing drug development. The gRNA itself is sufficiently compact to be developed as a chemically modified oligonucleotide, analogous in many ways to several commercially approved ASO examples.158 This approach would likely involve repeated dosing to achieve a persistent effect and may be restricted in its therapeutic application based on the natural pharmacokinetic and biodistribution properties of the ASO. Alternatively, viral vectors such as AAV can be used to deliver DNA-encoded gRNAs; this offers the potential for persistent gRNA expression with just a single dose. AAV vectors have shown promise for durable gene expression across a range of indications in the clinic, with approved products in the United States for inherited retinal dystrophy in 2017 and spinal muscular atrophy in 2019.159 AAV capsids can be engineered to drive tissue-specific tropism that would enable vectorized delivery of gRNAs with targeted biodistribution.160 Translation of AAV-based drug products comes with well-known challenges in manufacturing and safety that must be taken into consideration during development.161,162 In particular, the immune response to AAV vectors generally precludes repeated administration in the same patient, and some high-dose clinical trials have led to severe adverse events. However, there are several strategies currently being explored to circumvent or lessen the impact of this immune response, including immunosuppression regimens, use of immune orthogonal AAVs, and capsid engineering to enable lower doses.163,164 Achieving maximum payload delivery to the target cells while minimizing exposure of non-target cells can reduce drug manufacturing costs and patient dosing requirements, which could translate into reduced toxicity risks.
Regardless of the delivery method selected, a comprehensive characterization of vector and gRNA tissue biodistribution and expression profile in relevant non-clinical models is expected to enable first-in-human dosing. Because the transcriptome differs across tissues, the biodistribution data can highlight cells and tissues of particular interest when assessing efficacy and tolerability. Biodistribution/expression data can be used in conjunction with on- and off-target editing data in relevant model systems to project dosing requirements needed to achieve a therapeutic benefit and a safety margin to derive an initial clinical dose and dose escalation strategy. In some cases, these data may need to be extrapolated across multiple model systems. For example, healthy large animal models typically used for non-clinical toxicology studies may not have the desired target mutations that enable a readout of on-target RNA-editing efficiency, but they can inform on dose response and gRNA biodistribution. A dose response for editing could then be extrapolated from a disease model that has a relevant on-target mutation, using biodistribution/expression data in the target tissue to connect the model system readouts. Therapeutic dosing strategies will thus be highly dependent on indication and model systems available and will be a critical topic for discussion with regulatory agencies during preclinical development.
Clinical development
From a clinical perspective, each disease indication, target organ, and delivery modality will influence the clinical development plan and ultimately the information that can be learned from early-phase clinical trials. There is a desire to quantify both RNA editing and protein influence from tissue biopsies to inform dose selection; however, the complexity of clinical biopsies differs across tissues. For instance, biopsies of the liver come with risks and are less frequently done. Similarly, biopsies from the CNS are generally not feasible. In the case of muscle, biopsies are more routinely performed and may enable a comparative analysis of RNA editing, protein correction or restoration, and phenotypic change in the clinical study. This information will be key in dose escalation studies to identify the minimal dose required for a therapeutic impact. In tissues where biopsies are not feasible, understanding the relationship between RNA editing, protein modulation, and phenotypic outcome must be clearly established in preclinical studies, and careful consideration is required when selecting the appropriate dose and readouts for human studies.
Correction of missense and nonsense mutations is the most logical application for RNA editing. Numerous groups have demonstrated preclinical data targeting a missense mutation in SERPINA1 leading to AATD6,12 (as reviewed above). The G-to-A SNV encoding the E342K mutation affects more than 100,000 people worldwide,108 creating a large unmet medical need. The mutation causes a toxic gain of function, and aggregated protein accumulates in hepatocytes. Additionally, reduced α1-antitrypsin (AAT) secretion from hepatocytes into the serum causes neutrophil elastase, the AAT natural substrate, to accumulate in the lungs.108 Because of this combined gain and loss of function in the liver and lungs, attempts at knockdown, gene replacement, or protein therapy have fallen short, since they do not address both aspects of the disease. AATD is well suited for therapeutic RNA editing. The liver is an ideal target organ for delivery of DNA or RNA payloads, and correction of the SNV at the RNA level can retain endogenous expression levels while reducing toxicity in the liver and increasing secretion to the serum. Lastly, a clear benchmark of >11 μM AAT in the serum has been established to restore its function in the lungs. This provides a great opportunity to establish RNA editing as a new therapeutic modality and address a large unmet medical need.
As RNA editing becomes established in the clinic, we anticipate refined use to treat indications with smaller patient cohorts, eventually enabling truly personalized medicine, similar to recent examples with ASOs. In one case study, deep sequencing of a pediatric patient suffering from Batten’s disease revealed a pathogenic splice variant in the MFSD8 gene leading to a premature termination codon. An ASO was quickly designed to mask the cryptic splice acceptor site and restore the use of the canonical splice acceptor site. Within 1 year of diagnosis, the drug was designed, manufactured, and administered to a single patient, who displayed reduced symptoms after treatment.165 Current delivery technologies for DNA payloads are not yet amenable to individualized treatment, but the ease of ASO synthesis may facilitate the small-scale manufacturing needed for wider adoption of personalized medicine. As knowledge of gRNA design principles improves, we anticipate similar scenarios unfolding for patients with rare, pathogenic G-to-A SNVs.
Conclusion
RNA therapeutics based on ASOs and RNAi that enable programmable RNA knockdown are already having considerable impact on human medicine. The recent advent of ADAR-based technologies that add programmable RNA editing to the molecular toolkit has created new possibilities in transcriptome engineering. By enabling direct nucleotide-level modulation of endogenous RNA transcripts and, correspondingly, an ability to modulate RNA substrates or translated proteins thereof at levels that match native stoichiometric levels, temporal dynamics, and in situ spatial distributions, this modality is opening new avenues in precision therapeutics. Additionally, the approach leverages the cells’ existing RNA-editing machinery, thereby alleviating the need for exogenous and immunogenic proteins to drive editing. In addition to enabling direct repair of G-to-A disease-causing mutations and nonsense mutations, targeted RNA edits can also enable modulation of RNA stability and splicing. Furthermore, transiently modulating protein function, such as the active sites of proteins or modulation of protein-protein interaction interfaces, opens the door to therapeutic avenues ranging from regenerative medicine to oncology. Combined with the intrinsic advantages that RNA-based therapeutics possess tunability and reversibility and that off-targets are not permanent, these emerging ADAR-based toolsets, coupled with rapidly improving viral and non-viral delivery modalities, are poised to broadly impact biotechnology and therapeutic applications.
Acknowledgments
The authors would like to thank Rafael Ponce and Drew Dietz for their thoughtful input regarding non-clinical and clinical development, Ron Hause for curating G>A mutations from ClinVar, Eric Smith for graphics support, and Patrice Ferriola for providing editorial assistance. P.M. and S.N. were generously supported by UCSD Institutional Funds, a Department of Defense grant (W81XWH-22-1-0401), NIH grants (OT2OD032742, R01GM123313), and a CIRM training grant EDUC4-12804 (to S.N.).
Author contributions
All authors were involved in conceptualization, writing, review, and approval of the manuscript.
Declaration of interests
B.J.B., D.K., Y.S., D.B., T.J.L., and D.J.H. are employees of Shape Therapeutics. P.M. is a scientific co-founder of Shape Therapeutics, Boundless Biosciences, Navega Therapeutics, and Engine Biosciences. The terms of these arrangements have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.
Contributor Information
David J. Huss, Email: david@shapetx.com.
Prashant Mali, Email: pmali@ucsd.edu.
References
- 1.Consortium G.T. The genotype-tissue expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bass B.L., Weintraub H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell. 1988;55:1089–1098. doi: 10.1016/0092-8674(88)90253-x. [DOI] [PubMed] [Google Scholar]
- 4.Bass B.L., Weintraub H. A developmentally regulated activity that unwinds RNA duplexes. Cell. 1987;48:607–613. doi: 10.1016/0092-8674(87)90239-x. [DOI] [PubMed] [Google Scholar]
- 5.Woolf T.M., Chase J.M., Stinchcomb D.T. Toward the therapeutic editing of mutated RNA sequences. Proc. Natl. Acad. Sci. USA. 1995;92:8298–8302. doi: 10.1073/pnas.92.18.8298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Merkle T., Merz S., Reautschnig P., Blaha A., Li Q., Vogel P., Wettengel J., Li J.B., Stafforst T. Precise RNA editing by recruiting endogenous ADARs with antisense oligonucleotides. Nat. Biotechnol. 2019;37:133–138. doi: 10.1038/s41587-019-0013-6. [DOI] [PubMed] [Google Scholar]
- 7.Reautschnig P., Wahn N., Wettengel J., Schulz A.E., Latifi N., Vogel P., Kang T.W., Pfeiffer L.S., Zarges C., Naumann U., et al. CLUSTER guide RNAs enable precise and efficient RNA editing with endogenous ADAR enzymes in vivo. Nat. Biotechnol. 2022;40:759–768. doi: 10.1038/s41587-021-01105-0. [DOI] [PubMed] [Google Scholar]
- 8.Katrekar D., Yen J., Xiang Y., Saha A., Meluzzi D., Savva Y., Mali P. Efficient in vitro and in vivo RNA editing via recruitment of endogenous ADARs using circular guide RNAs. Nat. Biotechnol. 2022;40:938–945. doi: 10.1038/s41587-021-01171-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katrekar D., Chen G., Meluzzi D., Ganesh A., Worlikar A., Shih Y.R., Varghese S., Mali P. In vivo RNA editing of point mutations via RNA-guided adenosine deaminases. Nat. Methods. 2019;16:239–242. doi: 10.1038/s41592-019-0323-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qu L., Yi Z., Zhu S., Wang C., Cao Z., Zhou Z., Yuan P., Yu Y., Tian F., Liu Z., et al. Programmable RNA editing by recruiting endogenous ADAR using engineered RNAs. Nat. Biotechnol. 2019;37:1059–1069. doi: 10.1038/s41587-019-0178-z. [DOI] [PubMed] [Google Scholar]
- 11.Yi Z., Qu L., Tang H., Liu Z., Liu Y., Tian F., Wang C., Zhang X., Feng Z., Yu Y., et al. Engineered circular ADAR-recruiting RNAs increase the efficiency and fidelity of RNA editing in vitro and in vivo. Nat. Biotechnol. 2022;40:946–955. doi: 10.1038/s41587-021-01180-3. [DOI] [PubMed] [Google Scholar]
- 12.Monian P., Shivalila C., Lu G., Shimizu M., Boulay D., Bussow K., Byrne M., Bezigian A., Chatterjee A., Chew D., et al. Endogenous ADAR-mediated RNA editing in non-human primates using stereopure chemically modified oligonucleotides. Nat. Biotechnol. 2022;40:1093–1102. doi: 10.1038/s41587-022-01225-1. [DOI] [PubMed] [Google Scholar]
- 13.Doherty E.E., Wilcox X.E., van Sint Fiet L., Kemmel C., Turunen J.J., Klein B., Tantillo D.J., Fisher A.J., Beal P.A. Rational design of RNA editing guide strands: cytidine analogs at the orphan position. J. Am. Chem. Soc. 2021;143:6865–6876. doi: 10.1021/jacs.0c13319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nose K., Hidaka K., Yano T., Tomita Y., Fukuda M. Short-chain guide RNA for site-directed A-to-I RNA editing. Nucleic Acid Ther. 2021;31:58–67. doi: 10.1089/nat.2020.0866. [DOI] [PubMed] [Google Scholar]
- 15.Cox D.B.T., Gootenberg J.S., Abudayyeh O.O., Franklin B., Kellner M.J., Joung J., Zhang F. RNA editing with CRISPR-Cas13. Science. 2017;358:1019–1027. doi: 10.1126/science.aaq0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rauch S., He E., Srienc M., Zhou H., Zhang Z., Dickinson B.C. Programmable RNA-guided RNA effector proteins built from human parts. Cell. 2019;178:122–134.e12. doi: 10.1016/j.cell.2019.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J.B., Levanon E.Y., Yoon J.K., Aach J., Xie B., Leproust E., Zhang K., Gao Y., Church G.M. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science. 2009;324:1210–1213. doi: 10.1126/science.1170995. [DOI] [PubMed] [Google Scholar]
- 18.Pinto Y., Cohen H.Y., Levanon E.Y. Mammalian conserved ADAR targets comprise only a small fragment of the human editosome. Genome Biol. 2014;15:R5. doi: 10.1186/gb-2014-15-1-r5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sinnamon J.R., Kim S.Y., Corson G.M., Song Z., Nakai H., Adelman J.P., Mandel G. Site-directed RNA repair of endogenous Mecp2 RNA in neurons. Proc. Natl. Acad. Sci. USA. 2017;114:E9395–E9402. doi: 10.1073/pnas.1715320114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sinnamon J.R., Jacobson M.E., Yung J.F., Fisk J.R., Jeng S., McWeeney S.K., Parmelee L.K., Chan C.N., Yee S.P., Mandel G. Targeted RNA editing in brainstem alleviates respiratory dysfunction in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2206053119. e2206053119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geary R.S., Norris D., Yu R., Bennett C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015;87:46–51. doi: 10.1016/j.addr.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 22.Gerber A., O'Connell M.A., Keller W. Two forms of human double-stranded RNA-specific editase 1 (hRED1) generated by the insertion of an Alu cassette. RNA. 1997;3:453–463. [PMC free article] [PubMed] [Google Scholar]
- 23.Chen C.X., Cho D.S., Wang Q., Lai F., Carter K.C., Nishikura K. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA. 2000;6:755–767. doi: 10.1017/s1355838200000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oakes E., Anderson A., Cohen-Gadol A., Hundley H.A. Adenosine deaminase that acts on RNA 3 (ADAR3) binding to glutamate receptor subunit B pre-mRNA inhibits RNA editing in glioblastoma. J. Biol. Chem. 2017;292:4326–4335. doi: 10.1074/jbc.M117.779868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raghava Kurup R., Oakes E.K., Manning A.C., Mukherjee P., Vadlamani P., Hundley H.A. RNA binding by ADAR3 inhibits adenosine-to-inosine editing and promotes expression of immune response protein MAVS. J. Biol. Chem. 2022;298:102267. doi: 10.1016/j.jbc.2022.102267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Masliah G., Barraud P., Allain F.H.T. RNA recognition by double-stranded RNA binding domains: a matter of shape and sequence. Cell. Mol. Life Sci. 2013;70:1875–1895. doi: 10.1007/s00018-012-1119-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stephens O.M., Haudenschild B.L., Beal P.A. The binding selectivity of ADAR2's dsRBMs contributes to RNA-editing selectivity. Chem. Biol. 2004;11:1239–1250. doi: 10.1016/j.chembiol.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y., Lei M., Samuel C.E. Chimeric double-stranded RNA-specific adenosine deaminase ADAR1 proteins reveal functional selectivity of double-stranded RNA-binding domains from ADAR1 and protein kinase PKR. Proc. Natl. Acad. Sci. USA. 2000;97:12541–12546. doi: 10.1073/pnas.97.23.12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Källman A.M., Sahlin M., Öhman M. ADAR2 A→I editing: site selectivity and editing efficiency are separate events. Nucleic Acids Res. 2003;31:4874–4881. doi: 10.1093/nar/gkg681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stefl R., Oberstrass F.C., Hood J.L., Jourdan M., Zimmermann M., Skrisovska L., Maris C., Peng L., Hofr C., Emeson R.B., Allain F.H.T. The solution structure of the ADAR2 dsRBM-RNA complex reveals a sequence-specific readout of the minor groove. Cell. 2010;143:225–237. doi: 10.1016/j.cell.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matthews M.M., Thomas J.M., Zheng Y., Tran K., Phelps K.J., Scott A.I., Havel J., Fisher A.J., Beal P.A. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat. Struct. Mol. Biol. 2016;23:426–433. doi: 10.1038/nsmb.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park S., Doherty E.E., Xie Y., Padyana A.K., Fang F., Zhang Y., Karki A., Lebrilla C.B., Siegel J.B., Beal P.A. High-throughput mutagenesis reveals unique structural features of human ADAR1. Nat. Commun. 2020;11:5130. doi: 10.1038/s41467-020-18862-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brooks S.C., Adhikary S., Rubinson E.H., Eichman B.F. Recent advances in the structural mechanisms of DNA glycosylases. Biochim. Biophys. Acta. 2013;1834:247–271. doi: 10.1016/j.bbapap.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuttan A., Bass B.L. Mechanistic insights into editing-site specificity of ADARs. Proc. Natl. Acad. Sci. USA. 2012;109:E3295–E3304. doi: 10.1073/pnas.1212548109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doherty E.E., Karki A., Wilcox X.E., Mendoza H.G., Manjunath A., Matos V.J., Fisher A.J., Beal P.A. ADAR activation by inducing a syn conformation at guanosine adjacent to an editing site. Nucleic Acids Res. 2022;50:10857–10868. doi: 10.1093/nar/gkac897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schneider M.F., Wettengel J., Hoffmann P.C., Stafforst T. Optimal guideRNAs for re-directing deaminase activity of hADAR1 and hADAR2 in trans. Nucleic Acids Res. 2014;42:e87. doi: 10.1093/nar/gku272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thuy-Boun A.S., Thomas J.M., Grajo H.L., Palumbo C.M., Park S., Nguyen L.T., Fisher A.J., Beal P.A. Asymmetric dimerization of adenosine deaminase acting on RNA facilitates substrate recognition. Nucleic Acids Res. 2020;48:7958–7972. doi: 10.1093/nar/gkaa532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poulsen H., Jorgensen R., Heding A., Nielsen F.C., Bonven B., Egebjerg J. Dimerization of ADAR2 is mediated by the double-stranded RNA binding domain. RNA. 2006;12:1350–1360. doi: 10.1261/rna.2314406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bazak L., Haviv A., Barak M., Jacob-Hirsch J., Deng P., Zhang R., Isaacs F.J., Rechavi G., Li J.B., Eisenberg E., Levanon E.Y. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014;24:365–376. doi: 10.1101/gr.164749.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tan M.H., Li Q., Shanmugam R., Piskol R., Kohler J., Young A.N., Liu K.I., Zhang R., Ramaswami G., Ariyoshi K., et al. Dynamic landscape and regulation of RNA editing in mammals. Nature. 2017;550:249–254. doi: 10.1038/nature24041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liddicoat B.J., Piskol R., Chalk A.M., Ramaswami G., Higuchi M., Hartner J.C., Li J.B., Seeburg P.H., Walkley C.R. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science. 2015;349:1115–1120. doi: 10.1126/science.aac7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rice G.I., Kasher P.R., Forte G.M.A., Mannion N.M., Greenwood S.M., Szynkiewicz M., Dickerson J.E., Bhaskar S.S., Zampini M., Briggs T.A., et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat. Genet. 2012;44:1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lamers M.M., van den Hoogen B.G., Haagmans B.L. ADAR1: “Editor-in-Chief” of cytoplasmic innate immunity. Front. Immunol. 2019;10:1763. doi: 10.3389/fimmu.2019.01763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Q., Miyakoda M., Yang W., Khillan J., Stachura D.L., Weiss M.J., Nishikura K. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J. Biol. Chem. 2004;279:4952–4961. doi: 10.1074/jbc.M310162200. [DOI] [PubMed] [Google Scholar]
- 45.Baker A.R., Slack F.J. ADAR1 and its implications in cancer development and treatment. Trends Genet. 2022;38:821–830. doi: 10.1016/j.tig.2022.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gannon H.S., Zou T., Kiessling M.K., Gao G.F., Cai D., Choi P.S., Ivan A.P., Buchumenski I., Berger A.C., Goldstein J.T., et al. Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nat. Commun. 2018;9:5450. doi: 10.1038/s41467-018-07824-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.George C.X., Samuel C.E. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc. Natl. Acad. Sci. USA. 1999;96:4621–4626. doi: 10.1073/pnas.96.8.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kleinova R., Leuchtenberger A.F., Giudice C.L., Tanzer A., Derdak S., Picardi E., Jantsch M.F. The ADAR1 editome reveals drivers of editing-specificity for ADAR1-isoforms. bioRxiv. 2021 doi: 10.1101/2021.11.24.469911. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pfaller C.K., George C.X., Samuel C.E. Adenosine deaminases acting on RNA (ADARs) and viral infections. Annu. Rev. Virol. 2021;8:239–264. doi: 10.1146/annurev-virology-091919-065320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jain M., Mann T.D., Stulić M., Rao S.P., Kirsch A., Pullirsch D., Strobl X., Rath C., Reissig L., Moreth K., et al. RNA editing of Filamin A pre-mRNA regulates vascular contraction and diastolic blood pressure. EMBO J. 2018;37:e94813. doi: 10.15252/embj.201694813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roth S.H., Levanon E.Y., Eisenberg E. Genome-wide quantification of ADAR adenosine-to-inosine RNA editing activity. Nat. Methods. 2019;16:1131–1138. doi: 10.1038/s41592-019-0610-9. [DOI] [PubMed] [Google Scholar]
- 52.Higuchi M., Maas S., Single F.N., Hartner J., Rozov A., Burnashev N., Feldmeyer D., Sprengel R., Seeburg P.H. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406:78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- 53.Melcher T., Maas S., Herb A., Sprengel R., Higuchi M., Seeburg P.H. RED2, a brain-specific member of the RNA-specific adenosine deaminase family. J. Biol. Chem. 1996;271:31795–31798. doi: 10.1074/jbc.271.50.31795. [DOI] [PubMed] [Google Scholar]
- 54.Mladenova D., Barry G., Konen L.M., Pineda S.S., Guennewig B., Avesson L., Zinn R., Schonrock N., Bitar M., Jonkhout N., et al. Adar3 is involved in learning and memory in mice. Front. Neurosci. 2018;12:243. doi: 10.3389/fnins.2018.00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maas S., Gommans W.M. Identification of a selective nuclear import signal in adenosine deaminases acting on RNA. Nucleic Acids Res. 2009;37:5822–5829. doi: 10.1093/nar/gkp599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Licht K., Kapoor U., Amman F., Picardi E., Martin D., Bajad P., Jantsch M.F. A high resolution A-to-I editing map in the mouse identifies editing events controlled by pre-mRNA splicing. Genome Res. 2019;29:1453–1463. doi: 10.1101/gr.242636.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Desterro J.M.P., Keegan L.P., Lafarga M., Berciano M.T., O'Connell M., Carmo-Fonseca M. Dynamic association of RNA-editing enzymes with the nucleolus. J. Cell Sci. 2003;116:1805–1818. doi: 10.1242/jcs.00371. [DOI] [PubMed] [Google Scholar]
- 58.Barraud P., Banerjee S., Mohamed W.I., Jantsch M.F., Allain F.H.T. A bimodular nuclear localization signal assembled via an extended double-stranded RNA-binding domain acts as an RNA-sensing signal for transportin 1. Proc. Natl. Acad. Sci. USA. 2014;111:E1852–E1861. doi: 10.1073/pnas.1323698111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Freund E.C., Sapiro A.L., Li Q., Linder S., Moresco J.J., Yates J.R., 3rd, Li J.B. Unbiased identification of trans regulators of ADAR and A-to-I RNA editing. Cell Rep. 2020;31:107656. doi: 10.1016/j.celrep.2020.107656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nourreddine S., Lavoie G., Paradis J., Ben El Kadhi K., Méant A., Aubert L., Grondin B., Gendron P., Chabot B., Bouvier M., et al. NF45 and NF90 regulate mitotic gene expression by competing with staufen-mediated mRNA decay. Cell Rep. 2020;31:107660. doi: 10.1016/j.celrep.2020.107660. [DOI] [PubMed] [Google Scholar]
- 61.Pfaller C.K., Li Z., George C.X., Samuel C.E. Protein kinase PKR and RNA adenosine deaminase ADAR1: new roles for old players as modulators of the interferon response. Curr. Opin. Immunol. 2011;23:573–582. doi: 10.1016/j.coi.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Y., Park S., Beal P.A. Selective recognition of RNA substrates by ADAR deaminase domains. Biochemistry. 2018;57:1640–1651. doi: 10.1021/acs.biochem.7b01100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schaffer A.A., Kopel E., Hendel A., Picardi E., Levanon E.Y., Eisenberg E. The cell line A-to-I RNA editing catalogue. Nucleic Acids Res. 2020;48:5849–5858. doi: 10.1093/nar/gkaa305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brown C.R., Gupta S., Qin J., Racie T., He G., Lentini S., Malone R., Yu M., Matsuda S., Shulga-Morskaya S., et al. Investigating the pharmacodynamic durability of GalNAc-siRNA conjugates. Nucleic Acids Res. 2020;48:11827–11844. doi: 10.1093/nar/gkaa670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fukuda M., Umeno H., Nose K., Nishitarumizu A., Noguchi R., Nakagawa H. Construction of a guide-RNA for site-directed RNA mutagenesis utilising intracellular A-to-I RNA editing. Sci. Rep. 2017;7:41478. doi: 10.1038/srep41478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stafforst T., Schneider M.F. An RNA-deaminase conjugate selectively repairs point mutations. Angew. Chem. Int. Ed. Engl. 2012;51:11166–11169. doi: 10.1002/anie.201206489. [DOI] [PubMed] [Google Scholar]
- 67.Montiel-Gonzalez M.F., Vallecillo-Viejo I., Yudowski G.A., Rosenthal J.J.C. Correction of mutations within the cystic fibrosis transmembrane conductance regulator by site-directed RNA editing. Proc. Natl. Acad. Sci. USA. 2013;110:18285–18290. doi: 10.1073/pnas.1306243110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wettengel J., Reautschnig P., Geisler S., Kahle P.J., Stafforst T. Harnessing human ADAR2 for RNA repair - recoding a PINK1 mutation rescues mitophagy. Nucleic Acids Res. 2017;45:2797–2808. doi: 10.1093/nar/gkw911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Monteleone L.R., Matthews M.M., Palumbo C.M., Thomas J.M., Zheng Y., Chiang Y., Fisher A.J., Beal P.A. A bump-hole approach for directed RNA editing. Cell Chem. Biol. 2019;26:269–277.e5. doi: 10.1016/j.chembiol.2018.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sinnamon J.R., Kim S.Y., Fisk J.R., Song Z., Nakai H., Jeng S., McWeeney S.K., Mandel G. In vivo repair of a protein underlying a neurological disorder by programmable RNA editing. Cell Rep. 2020;32:107878. doi: 10.1016/j.celrep.2020.107878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vallecillo-Viejo I.C., Liscovitch-Brauer N., Montiel-Gonzalez M.F., Eisenberg E., Rosenthal J.J.C. Abundant off-target edits from site-directed RNA editing can be reduced by nuclear localization of the editing enzyme. RNA Biol. 2018;15:104–114. doi: 10.1080/15476286.2017.1387711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Katrekar D., Xiang Y., Palmer N., Saha A., Meluzzi D., Mali P. Comprehensive interrogation of the ADAR2 deaminase domain for engineering enhanced RNA editing activity and specificity. Elife. 2022;11:e75555. doi: 10.7554/eLife.75555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Katrekar D., Yen J., Xiang Y., Saha A., Meluzzi D., Savva Y., Mali P. Robust RNA Editing via Recruitment of Endogenous ADARs Using Circular Guide RNAs. bioRxiv. 2021 doi: 10.1101/2021.01.12.426286. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Litke J.L., Jaffrey S.R. Highly efficient expression of circular RNA aptamers in cells using autocatalytic transcripts. Nat. Biotechnol. 2019;37:667–675. doi: 10.1038/s41587-019-0090-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhao M., Woodside M.T. Mechanical strength of RNA knot in Zika virus protects against cellular defenses. Nat. Chem. Biol. 2021;17:975–981. doi: 10.1038/s41589-021-00829-z. [DOI] [PubMed] [Google Scholar]
- 76.MacFadden A., O'Donoghue Z., Silva P.A.G.C., Chapman E.G., Olsthoorn R.C., Sterken M.G., Pijlman G.P., Bredenbeek P.J., Kieft J.S. Mechanism and structural diversity of exoribonuclease-resistant RNA structures in flaviviral RNAs. Nat. Commun. 2018;9:119. doi: 10.1038/s41467-017-02604-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang G., Liu Y., Huang S., Qu S., Cheng D., Yao Y., Ji Q., Wang X., Huang X., Liu J. Enhancement of prime editing via xrRNA motif-joined pegRNA. Nat. Commun. 2022;13:1856. doi: 10.1038/s41467-022-29507-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nelson J.W., Randolph P.B., Shen S.P., Everette K.A., Chen P.J., Anzalone A.V., An M., Newby G.A., Chen J.C., Hsu A., Liu D.R. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 2022;40:402–410. doi: 10.1038/s41587-021-01039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Paul C.P., Good P.D., Winer I., Engelke D.R. Effective expression of small interfering RNA in human cells. Nat. Biotechnol. 2002;20:505–508. doi: 10.1038/nbt0502-505. [DOI] [PubMed] [Google Scholar]
- 80.Disney M.D. Targeting RNA with small molecules to capture opportunities at the intersection of chemistry, biology, and medicine. J. Am. Chem. Soc. 2019;141:6776–6790. doi: 10.1021/jacs.8b13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pennacchio L.A., Loots G.G., Nobrega M.A., Ovcharenko I. Predicting tissue-specific enhancers in the human genome. Genome Res. 2007;17:201–211. doi: 10.1101/gr.5972507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Uzonyi A., Nir R., Shliefer O., Stern-Ginossar N., Antebi Y., Stelzer Y., Levanon E.Y., Schwartz S. Deciphering the principles of the RNA editing code via large-scale systematic probing. Mol. Cell. 2021;81:2374–2387.e3. doi: 10.1016/j.molcel.2021.03.024. [DOI] [PubMed] [Google Scholar]
- 83.Kim D.D.Y., Kim T.T.Y., Walsh T., Kobayashi Y., Matise T.C., Buyske S., Gabriel A. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 2004;14:1719–1725. doi: 10.1101/gr.2855504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Athanasiadis A., Rich A., Maas S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. Plos Biol. 2004;2:e391. doi: 10.1371/journal.pbio.0020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Egebjerg J., Kukekov V., Heinemann S.F. Intron sequence directs RNA editing of the glutamate receptor subunit GluR2 coding sequence. Proc. Natl. Acad. Sci. USA. 1994;91:10270–10274. doi: 10.1073/pnas.91.22.10270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yeo J., Goodman R.A., Schirle N.T., David S.S., Beal P.A. RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proc. Natl. Acad. Sci. USA. 2010;107:20715–20719. doi: 10.1073/pnas.1009231107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Niswender C.M., Sanders-Bush E., Emeson R.B. Identification and characterization of RNA editing events within the 5-HT2C receptora. Ann. N. Y. Acad. Sci. 1998;861:38–48. doi: 10.1111/j.1749-6632.1998.tb10171.x. [DOI] [PubMed] [Google Scholar]
- 88.Lehmann K.A., Bass B.L. The importance of internal loops within RNA substrates of ADAR111Edited by D. E. Draper. J. Mol. Biol. 1999;291:1–13. doi: 10.1006/jmbi.1999.2914. [DOI] [PubMed] [Google Scholar]
- 89.Liu X., Sun T., Shcherbina A., Li Q., Jarmoskaite I., Kappel K., Ramaswami G., Das R., Kundaje A., Li J.B. Learning cis-regulatory principles of ADAR-based RNA editing from CRISPR-mediated mutagenesis. Nat. Commun. 2021;12:2165. doi: 10.1038/s41467-021-22489-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Song Y., Yang W., Fu Q., Wu L., Zhao X., Zhang Y., Zhang R. irCLASH reveals RNA substrates recognized by human ADARs. Nat. Struct. Mol. Biol. 2020;27:351–362. doi: 10.1038/s41594-020-0398-4. [DOI] [PubMed] [Google Scholar]
- 91.Brown D., Altermatt M., Dobreva T., Chen S., Wang A., Thomson M., Gradinaru V. Deep parallel characterization of AAV tropism and AAV-mediated transcriptional changes via single-cell RNA sequencing. Front. Immunol. 2021;12:730825. doi: 10.3389/fimmu.2021.730825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Maheshri N., Koerber J.T., Kaspar B.K., Schaffer D.V. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat. Biotechnol. 2006;24:198–204. doi: 10.1038/nbt1182. [DOI] [PubMed] [Google Scholar]
- 93.Li W., Zhang L., Johnson J.S., Zhijian W., Grieger J.C., Ping-Jie X., Drouin L.M., Agbandje-McKenna M., Pickles R.J., Samulski R.J. Generation of novel AAV variants by directed evolution for improved CFTR delivery to human ciliated airway epithelium. Mol. Ther. 2009;17:2067–2077. doi: 10.1038/mt.2009.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chan K.Y., Jang M.J., Yoo B.B., Greenbaum A., Ravi N., Wu W.L., Sánchez-Guardado L., Lois C., Mazmanian S.K., Deverman B.E., Gradinaru V. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017;20:1172–1179. doi: 10.1038/nn.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tabebordbar M., Lagerborg K.A., Stanton A., King E.M., Ye S., Tellez L., Krunnfusz A., Tavakoli S., Widrick J.J., Messemer K.A., et al. Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell. 2021;184:4919–4938.e22. doi: 10.1016/j.cell.2021.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Becker J., Fakhiri J., Grimm D. Fantastic AAV gene therapy vectors and how to find them-random diversification, rational design and machine learning. Pathogens. 2022;11 doi: 10.3390/pathogens11070756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Agrawal S. The evolution of antisense oligonucleotide chemistry-A personal journey. Biomedicines. 2021;9:503. doi: 10.3390/biomedicines9050503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Alhamadani F., Zhang K., Parikh R., Wu H., Rasmussen T.P., Bahal R., Zhong X.B., Manautou J.E. Adverse drug reactions and toxicity of the food and drug administration-approved antisense oligonucleotide drugs. Drug Metab. Dispos. 2022;50:879–887. doi: 10.1124/dmd.121.000418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhu Y., Zhu L., Wang X., Jin H. RNA-based therapeutics: an overview and prospectus. Cell Death Dis. 2022;13:644. doi: 10.1038/s41419-022-05075-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Metelev V., Lisziewicz J., Agrawal S. Study of antisense oligonucleotide phosphorothioates containing segments of oligodeoxynucleotides and 2′-o- methyloligoribonucleotides. Bioorg. Med. Chem. Lett. 1994;4:2929–2934. [Google Scholar]
- 101.Karaki S., Paris C., Rocchi P. In: Antisense Therapy. Sharad S., Kapur S., editors. 2019. Antisense oligonucleotides, A novel developing targeting therapy. [DOI] [Google Scholar]
- 102.Zeng Y., Cullen B.R. RNA interference in human cells is restricted to the cytoplasm. RNA. 2002;8:855–860. doi: 10.1017/s1355838202020071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rodriguez J., Menet J.S., Rosbash M. Nascent-seq indicates widespread cotranscriptional RNA editing in Drosophila. Mol. Cell. 2012;47:27–37. doi: 10.1016/j.molcel.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yin H., Song C.Q., Suresh S., Wu Q., Walsh S., Rhym L.H., Mintzer E., Bolukbasi M.F., Zhu L.J., Kauffman K., et al. Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat. Biotechnol. 2017;35:1179–1187. doi: 10.1038/nbt.4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ryan D.E., Taussig D., Steinfeld I., Phadnis S.M., Lunstad B.D., Singh M., Vuong X., Okochi K.D., McCaffrey R., Olesiak M., et al. Improving CRISPR-Cas specificity with chemical modifications in single-guide RNAs. Nucleic Acids Res. 2018;46:792–803. doi: 10.1093/nar/gkx1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schubert M.S., Cedrone E., Neun B., Behlke M.A., Dobrovolskaia M.A. Chemical modification of CRISPR gRNAs eliminate type I interferon responses in human peripheral blood mononuclear cells. J. Cytokine Biol. 2018;3:121. doi: 10.4172/2576-3881.1000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.O'Shea J.J., Schwartz D.M., Villarino A.V., Gadina M., McInnes I.B., Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015;66:311–328. doi: 10.1146/annurev-med-051113-024537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Greene C.M., Marciniak S.J., Teckman J., Ferrarotti I., Brantly M.L., Lomas D.A., Stoller J.K., McElvaney N.G. alpha1-Antitrypsin deficiency. Nat. Rev. Dis. Primers. 2016;2:16051. doi: 10.1038/nrdp.2016.51. [DOI] [PubMed] [Google Scholar]
- 109.Frazier K.S. Antisense oligonucleotide therapies: the promise and the challenges from a toxicologic pathologist's perspective. Toxicol. Pathol. 2015;43:78–89. doi: 10.1177/0192623314551840. [DOI] [PubMed] [Google Scholar]
- 110.Malik T.N., Doherty E.E., Gaded V.M., Hill T.M., Beal P.A., Emeson R.B. Regulation of RNA editing by intracellular acidification. Nucleic Acids Res. 2021;49:4020–4036. doi: 10.1093/nar/gkab157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rueter S.M., Dawson T.R., Emeson R.B. Regulation of alternative splicing by RNA editing. Nature. 1999;399:75–80. doi: 10.1038/19992. [DOI] [PubMed] [Google Scholar]
- 112.Kluesner M.G., Lahr W.S., Lonetree C.L., Smeester B.A., Qiu X., Slipek N.J., Claudio Vázquez P.N., Pitzen S.P., Pomeroy E.J., Vignes M.J., et al. CRISPR-Cas9 cytidine and adenosine base editing of splice-sites mediates highly-efficient disruption of proteins in primary and immortalized cells. Nat. Commun. 2021;12:2437. doi: 10.1038/s41467-021-22009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Long C., Li H., Tiburcy M., Rodriguez-Caycedo C., Kyrychenko V., Zhou H., Zhang Y., Min Y.L., Shelton J.M., Mammen P.P.A., et al. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci. Adv. 2018;4:eaap9004. doi: 10.1126/sciadv.aap9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.García-Tuñón I., Alonso-Pérez V., Vuelta E., Pérez-Ramos S., Herrero M., Méndez L., Hernández-Sánchez J.M., Martín-Izquierdo M., Saldaña R., Sevilla J., et al. Splice donor site sgRNAs enhance CRISPR/Cas9-mediated knockout efficiency. PLoS One. 2019;14:e0216674. doi: 10.1371/journal.pone.0216674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Havens M.A., Hastings M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016;44:6549–6563. doi: 10.1093/nar/gkw533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Marsollier A.C., Joubert R., Mariot V., Dumonceaux J. Targeting the polyadenylation signal of pre-mRNA: a new gene silencing approach for facioscapulohumeral dystrophy. Int. J. Mol. Sci. 2018;19:1347. doi: 10.3390/ijms19051347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vickers T.A., Wyatt J.R., Burckin T., Bennett C.F., Freier S.M. Fully modified 2' MOE oligonucleotides redirect polyadenylation. Nucleic Acids Res. 2001;29:1293–1299. doi: 10.1093/nar/29.6.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Marsollier A.C., Ciszewski L., Mariot V., Popplewell L., Voit T., Dickson G., Dumonceaux J. Antisense targeting of 3' end elements involved in DUX4 mRNA processing is an efficient therapeutic strategy for facioscapulohumeral dystrophy: a new gene-silencing approach. Hum. Mol. Genet. 2016;25:1468–1478. doi: 10.1093/hmg/ddw015. [DOI] [PubMed] [Google Scholar]
- 119.Boiziau C., Kurfurst R., Cazenave C., Roig V., Thuong N.T., Toulmé J.J. Inhibition of translation initiation by antisense oligonucleotides via an RNase-H independent mechanism. Nucleic Acids Res. 1991;19:1113–1119. doi: 10.1093/nar/19.5.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liang X.H., Sun H., Shen W., Wang S., Yao J., Migawa M.T., Bui H.H., Damle S.S., Riney S., Graham M.J., et al. Antisense oligonucleotides targeting translation inhibitory elements in 5' UTRs can selectively increase protein levels. Nucleic Acids Res. 2017;45:9528–9546. doi: 10.1093/nar/gkx632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang Z. The principles of MiRNA-masking antisense oligonucleotides technology. Methods Mol. Biol. 2011;676:43–49. doi: 10.1007/978-1-60761-863-8_3. [DOI] [PubMed] [Google Scholar]
- 122.Chen S., Xie W., Liu Z., Shan H., Chen M., Song Y., Yu H., Lai L., Li Z. CRISPR start-loss: a novel and practical alternative for gene silencing through base-editing-induced start codon mutations. Mol. Ther. Nucleic Acids. 2020;21:1062–1073. doi: 10.1016/j.omtn.2020.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Winter J., Luu A., Gapinske M., Manandhar S., Shirguppe S., Woods W.S., Song J.S., Perez-Pinera P. Targeted exon skipping with AAV-mediated split adenine base editors. Cell Discov. 2019;5:41. doi: 10.1038/s41421-019-0109-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Šikrová D., Cadar V.A., Ariyurek Y., Laros J.F.J., Balog J., van der Maarel S.M. Adenine base editing of the DUX4 polyadenylation signal for targeted genetic therapy in facioscapulohumeral muscular dystrophy. Mol. Ther. Nucleic Acids. 2021;25:342–354. doi: 10.1016/j.omtn.2021.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Köhler G., Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 126.Mitoma H., Horiuchi T., Tsukamoto H., Ueda N. Molecular mechanisms of action of anti-TNF-alpha agents - comparison among therapeutic TNF-alpha antagonists. Cytokine. 2018;101:56–63. doi: 10.1016/j.cyto.2016.08.014. [DOI] [PubMed] [Google Scholar]
- 127.Costa R.L.B., Czerniecki B.J. Clinical development of immunotherapies for HER2(+) breast cancer: a review of HER2-directed monoclonal antibodies and beyond. NPJ Breast Cancer. 2020;6:10. doi: 10.1038/s41523-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang X., Song W. The role of APP and BACE1 trafficking in APP processing and amyloid-beta generation. Alzheimers Res. Ther. 2013;5:46. doi: 10.1186/alzrt211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tan B.Z., Huang H., Lam R., Soong T.W. Dynamic regulation of RNA editing of ion channels and receptors in the mammalian nervous system. Mol. Brain. 2009;2:13. doi: 10.1186/1756-6606-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.King G.F., Vetter I. No gain, no pain: NaV1.7 as an analgesic target. ACS Chem. Neurosci. 2014;5:749–751. doi: 10.1021/cn500171p. [DOI] [PubMed] [Google Scholar]
- 131.Moreno A.M., Alemán F., Catroli G.F., Hunt M., Hu M., Dailamy A., Pla A., Woller S.A., Palmer N., Parekh U., et al. Long-lasting analgesia via targeted in situ repression of NaV1.7 in mice. Sci. Transl. Med. 2021;13:eaay9056. doi: 10.1126/scitranslmed.aay9056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Riemondy K.A., Gillen A.E., White E.A., Bogren L.K., Hesselberth J.R., Martin S.L. Dynamic temperature-sensitive A-to-I RNA editing in the brain of a heterothermic mammal during hibernation. RNA. 2018;24:1481–1495. doi: 10.1261/rna.066522.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Garrett S., Rosenthal J.J.C. RNA editing underlies temperature adaptation in K+ channels from polar octopuses. Science. 2012;335:848–851. doi: 10.1126/science.1212795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Palavicini J.P., Correa-Rojas R.A., Rosenthal J.J.C. Extra double-stranded RNA binding domain (dsRBD) in a squid RNA editing enzyme confers resistance to high salt environment. J. Biol. Chem. 2012;287:17754–17764. doi: 10.1074/jbc.M112.366005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gillmore J.D., Gane E., Taubel J., Kao J., Fontana M., Maitland M.L., Seitzer J., O'Connell D., Walsh K.R., Wood K., et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 2021;385:493–502. doi: 10.1056/NEJMoa2107454. [DOI] [PubMed] [Google Scholar]
- 136.Leibowitz M.L., Papathanasiou S., Doerfler P.A., Blaine L.J., Sun L., Yao Y., Zhang C.Z., Weiss M.J., Pellman D. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat. Genet. 2021;53:895–905. doi: 10.1038/s41588-021-00838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Charlesworth C.T., Deshpande P.S., Dever D.P., Camarena J., Lemgart V.T., Cromer M.K., Vakulskas C.A., Collingwood M.A., Zhang L., Bode N.M., et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019;25:249–254. doi: 10.1038/s41591-018-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wagner D.L., Amini L., Wendering D.J., Burkhardt L.M., Akyüz L., Reinke P., Volk H.D., Schmueck-Henneresse M. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 2019;25:242–248. doi: 10.1038/s41591-018-0204-6. [DOI] [PubMed] [Google Scholar]
- 139.Bhakta S., Sakari M., Tsukahara T. RNA editing of BFP, a point mutant of GFP, using artificial APOBEC1 deaminase to restore the genetic code. Sci. Rep. 2020;10:17304. doi: 10.1038/s41598-020-74374-5. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 140.Jiang K., Koob J., Chen X.D., Krajeski R.N., Zhang Y., Volf V., Zhou W., Sgrizzi S.R., Villiger L., Gootenberg J.S., et al. Programmable eukaryotic protein synthesis with RNA sensors by harnessing ADAR. Nat. Biotechnol. 2022 doi: 10.1038/s41587-022-01534-5. online ahead of print. [DOI] [PubMed] [Google Scholar]
- 141.Kaseniit K.E., Katz N., Kolber N.S., Call C.C., Wengier D.L., Cody W.B., Sattely E.S., Gao X.J. Modular, programmable RNA sensing using ADAR editing in living cells. Nat. Biotechnol. 2022 doi: 10.1038/s41587-022-01493-x. online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Qian Y., Li J., Zhao S., Matthews E.A., Adoff M., Zhong W., An X., Yeo M., Park C., Yang X., et al. Programmable RNA sensing for cell monitoring and manipulation. Nature. 2022;610:713–721. doi: 10.1038/s41586-022-05280-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use . Draft Guidelines from the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use: Nonclinical Biodistribution Considerations for Gene Therapy Products S12. 2021. [Google Scholar]
- 144.U.S. Department of Health and Human Services, FDA, Center for Biologics Evaluation and Research, editors. Draft guidance for industry: human gene therapy for neurodegenerative diseases. 2021. [Google Scholar]
- 145.U.S. Department of Health and Human Services, FDA, Center for Biologics Evaluation and Research, editors. Guidance for industry: chemistry, manufacturing, and control (CMC) information for human gene therapy investigational new drug applications (INDs) 2020. [Google Scholar]
- 146.U.S. Department of Health and Human Services, FDA, Center for Biologics Evaluation and Research, editors. Guidance for industry: long term follow-up after administration of human gene therapy products. 2020. [Google Scholar]
- 147.U.S. Department of Health and Human Services, FDA, Center for Biologics Evaluation and Research, editors. Guidance for industry: human gene therapy for hemophilia. 2020. [Google Scholar]
- 148.U.S. Department of Health and Human Services, FDA, Center for Biologics Evaluation and Research, editors. Guidance for industry: human gene therapy for rare diseases. 2020. [Google Scholar]
- 149.U.S. Department of Health and Human Services, FDA, Center for Biologics Evaluation and Research, editors. Guidance for industry: human gene therapy for retinal disorders. 2020. [Google Scholar]
- 150.EuropeansMedicinesAgency, editor. Guideline on the quality, non-clinical and clinical aspects of gene therapy medicinal products. 2018. [Google Scholar]
- 151.U.S. Department of Health and Human Services, FDA, Center for Biologics Evaluation and Research, editors. Guidance for industry: considerations for the design of early-phase clinical trials of cellular and gene therapy products. 2015. [Google Scholar]
- 152.U.S. Department of Health and Human Services, FDA, Center for Biologics Evaluation and Research, editors. Guidance for industry: preclinical assessment of investigational cellular and gene therapy products. 2013. [Google Scholar]
- 153.U.S. Department of Health and Human Services, FDA, Center for Biologics Evaluation and Research, editors. Guidance for industry: potency tests for cellular and gene therapy products. 2011. [Google Scholar]
- 154.Lo Giudice C., Silvestris D.A., Roth S.H., Eisenberg E., Pesole G., Gallo A., Picardi E. Quantifying RNA editing in deep transcriptome datasets. Front. Genet. 2020;11:194. doi: 10.3389/fgene.2020.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Maeder M.L., Stefanidakis M., Wilson C.J., Baral R., Barrera L.A., Bounoutas G.S., Bumcrot D., Chao H., Ciulla D.M., DaSilva J.A., et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019;25:229–233. doi: 10.1038/s41591-018-0327-9. [DOI] [PubMed] [Google Scholar]
- 156.Reich D.P., Bass B.L. Mapping the dsRNA world. Cold Spring Harb. Perspect. Biol. 2019;11:a035352. doi: 10.1101/cshperspect.a035352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Licht K., Hartl M., Amman F., Anrather D., Janisiw M.P., Jantsch M.F. Inosine induces context-dependent recoding and translational stalling. Nucleic Acids Res. 2019;47:3–14. doi: 10.1093/nar/gky1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Crooke S.T., Baker B.F., Crooke R.M., Liang X.H. Antisense technology: an overview and prospectus. Nat. Rev. Drug Discov. 2021;20:427–453. doi: 10.1038/s41573-021-00162-z. [DOI] [PubMed] [Google Scholar]
- 159.OTAT, editor. Approved Cellular and Gene Therapy Products. 2022. [Google Scholar]
- 160.Korneyenkov M.A., Zamyatnin A.A., Jr. Next step in gene delivery: modern approaches and further perspectives of AAV tropism modification. Pharmaceutics. 2021;13:750. doi: 10.3390/pharmaceutics13050750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Srivastava A., Mallela K.M.G., Deorkar N., Brophy G. Manufacturing challenges and rational formulation development for AAV viral vectors. J. Pharm. Sci. 2021;110:2609–2624. doi: 10.1016/j.xphs.2021.03.024. [DOI] [PubMed] [Google Scholar]
- 162.Colella P., Ronzitti G., Mingozzi F. Emerging issues in AAV-mediated in vivo gene therapy. Mol. Ther. Methods Clin. Dev. 2018;8:87–104. doi: 10.1016/j.omtm.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mingozzi F., High K.A. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood. 2013;122:23–36. doi: 10.1182/blood-2013-01-306647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Moreno A.M., Palmer N., Alemán F., Chen G., Pla A., Jiang N., Leong Chew W., Law M., Mali P. Immune-orthogonal orthologues of AAV capsids and of Cas9 circumvent the immune response to the administration of gene therapy. Nat. Biomed. Eng. 2019;3:806–816. doi: 10.1038/s41551-019-0431-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kim J., Hu C., Moufawad El Achkar C., Black L.E., Douville J., Larson A., Pendergast M.K., Goldkind S.F., Lee E.A., Kuniholm A., et al. Patient-customized oligonucleotide therapy for a rare genetic disease. N. Engl. J. Med. 2019;381:1644–1652. doi: 10.1056/NEJMoa1813279. [DOI] [PMC free article] [PubMed] [Google Scholar]