

Abstract
The development of advanced genetic tools is boosting microbial engineering which can potentially tackle wide-ranging challenges currently faced by our society. Here we present SURE editing, a multi-recombinase engineering rationale combining oligonucleotide recombineering with the selective capacity of antibiotic resistance via transient insertion of selector plasmids. We test this method in Mycoplasma pneumoniae, a bacterium with a very inefficient native recombination machinery. Using SURE editing, we can seamlessly generate, in a single step, a wide variety of genome modifications at high efficiencies, including the largest possible deletion of this genome (30 Kb) and the targeted complementation of essential genes in the deletion of a region of interest. Additional steps can be taken to remove the selector plasmid from the edited area, to obtain markerless or even scarless edits. Of note, SURE editing is compatible with different site-specific recombinases for mediating transient plasmid integration. This battery of selector plasmids can be used to select different edits, regardless of the target sequence, which significantly reduces the cloning load associated to genome engineering projects. Given the proven functionality in several microorganisms of the machinery behind the SURE editing logic, this method is likely to represent a valuable advance for the synthetic biology field.
Graphical Abstract
Graphical Abstract.
Outline of SURE editing rationale that couples oligo recombineering with transient insertion of selector plasmids to efficiently edit bacterial genomes.
INTRODUCTION
The 21st century has been coined as ‘the century of biology’ based on the technological advances in synthetic biology, and its near-term prospective developments (1). Indeed, radically engineered microorganisms can be envisioned that at least partially address different challenges of humankind, including CO2 pollution (2), plastic degradation and valorization (3), and diagnose and treatment of different diseases (4). To boost the development of this field, genome engineering techniques should be as simple and standardized as possible. This would make microbial engineering accessible to a broader scientific community, and eventually expand the catalogue of editable microorganisms—a key prerequisite to transitioning this discipline from the laboratory to the field (5).
Early genetic engineering in bacteria mainly was based on transformation of customized DNA constructs (generally plasmids (6), but also linear double-stranded [ds] DNA fragments (7)) containing large regions of homology (200–2000 bp) into recombination-proficient strains. However, native recombination machinery is often inefficient, and only the introduction of an excisable selective marker (often an antibiotic resistance gene flanked by motifs that are recognized by site-specific recombinases, such as Cre (8)) into the customized DNA constructs enables the isolation of edited cells carrying the intended modification.
In 1998, a new logic for engineering bacterial genomes, called recombineering, was developed (9,10). This technology relies on phage-derived proteins, such as those encoded by the Red operon from λ phage (9) or RecET from the Rac prophage (10). Both systems code for at least two different protein activities: (i) a 5′ to 3′ dsDNA exonuclease (Exo (11) and RecE (12)) and (ii) a single-stranded (ss)DNA annealing protein (SSAP) (Beta (13) and RecT (14)). Thanks to the coordinated action of these two activities, recombineering protocols can use linear dsDNA fragments as a substrate for recombination, even with regions of homology as short as 40 nucleotides (nt) (15). However, recombineering with dsDNA substrates was mostly circumscribed to Escherichia coli genome engineering, and this technology only really flourished when it was found that synthetic ssDNA molecules (i.e. commercially available oligonucleotides) could be used as recombinogenic substrates (16). This process, known as oligonucleotide recombineering (hereafter, oligo-recombineering) only requires the activity of the phage-derived SSAP that mediates the homology-driven hybridization of the editing oligonucleotide with the lagging strand of the replication fork. Oligo-recombineering was initially set up in E. coli with the Beta protein of λ phage as SSAP (16), but attempts to directly transfer this technology to other bacteria gave limited results (17,18). In fact, the performance of phage-derived recombinases is not maintained across different bacterial genera and depends on the phylogenetic distance between the native host of the phage and the bacteria being engineered (18,19). These findings prompted a survey of phage genomes to identify efficient SSAPs for a wide variety of microbes. This has allowed oligo-recombineering protocols to be used in at least 29 different bacterial species (20), and the recent development of a method termed ‘Serial Enrichment for Efficient Recombineering’ (SEER) (21) promises to further expand the range of editable microorganisms.
However, oligo-recombineering has a major drawback associated with the small size of the recombinogenic substrate that it can use, which precludes the introduction of long pieces of DNA in a precise location. This limits the technology to gene deletions, nucleotide substitutions and the introduction of very small sequences. Importantly, this same limitation precludes the inclusion of a selective marker, although this would facilitate selection of edited cells. In some species, this is not a major issue, as small edits can be obtained at ultra-high frequencies (for instance, 5.1 × 10–1 in Escherichia coli to change a single nucleotide (21)), yet oligo-recombineering protocols adopted for most strains show only modest editing rates (10–2 to10–5) (20), even for small changes. Moreover, in all species, the oligo incorporation rate tends to drop in a power trend that correlates with the size of the attempted editing (22,23). This makes it cumbersome to select cells carrying large genetic changes, even for species for which highly efficient oligo-recombineering protocols are available. To solve this, oligo-recombineering protocols have been merged with a counterselection method based on CRISPR/Cas9, in which sgRNAs are designed to create a double-strand break (DSB) in only unmodified cells (23–28). Nonetheless, the genetic parts encoding the CRISPR/Cas9 system can accumulate mutations with a frequency (10–3 to 10–4) (23,25,26) that might exceed that of oligo-recombineering for large genetic changes, hampering selection of edited cells. For instance, in Mycoplasma pneumoniae, this approach is suitable to efficiently select clones carrying short edits (i.e. 50 bp), but larger modifications (starting around 1 kb) already require extensive screening to identify edited clones (23).
Recently, it has been shown that oligo-recombineering can be used to edit mycobacterial genomes while simultaneously introducing recombination sites (i.e. attB/attP sites) for site-specific recombinases (i.e. Bxb1) (29) at the modified area. This technology, termed ORBIT, facilitates the selection of edited clones through the targeted insertion of plasmids carrying an antibiotic resistance gene at the edited area. We have now expanded this logic to develop the ‘Selection of Ultra-rare Recombineering Events’ (SURE) editing method. Using this method in M. pneumoniae, a bacterium with a very inefficient native recombination machinery, resulted in a clear improvement over the previously available method based on oligo-recombineering coupled to CRISPR/Cas9 counterselection (23). Using SURE editing, we seamlessly generated several mutants carrying gene deletions (of up to 30 kb) in different loci, even in loci containing essential genes, thanks to targeted complementation. Further, we show that SURE editing is compatible with several pairs of recombination sites and site-specific recombinases, and is suitable for generating markerless and even scarless edits if required. Moreover, we set up a new inducible system for Mycoplasmas based on the regulatory elements of cmt operon from Pseudomonas putida, allowing us to create plasmids that mediate self-integration and excision in a controlled manner. In this way, markerless edits can be created on demand in a single-transformation step. We expect SURE editing to offer a rapid, versatile, and almost cloning-free strategy for engineering genomes of different bacterial species at will, given the proven functionality of the machinery behind this logic in several microorganisms.
MATERIALS AND METHODS
Bacterial strains and culture conditions
All strains used herein are summarized in Supplementary Table S1. The Mycoplasma pneumoniae wild-type (WT) strain M129-B7 (ATTC 29342, subtype 1, broth passage no. 35) and all its derivatives were grown at 37°C under 5% CO2 in tissue culture flasks or multiwell plates with Hayflick modified medium, as described elsewhere (30). Hayflick broth was supplemented with puromycin (3 μg/ml), gentamicin (100 μg/ml) or chloramphenicol (20 μg/ml) for cell selection, as needed. To induce the Ptet or Pcum systems, anhydrotetracycline (5 ng/ml) or p-isopropyl benzoate (cumate) (100 μM) were used unless otherwise indicated. When growth on a plate was required, Hayflick broth was supplemented with 0.8% bacto agar.
For cloning purposes, E. coli NEB® 5-alpha High Efficiency strain was grown at 37°C in LB broth or on LB agar plates supplemented with ampicillin (100 μg /ml).
Plasmids and oligonucleotides
All plasmids generated in this work were assembled following the Gibson method (31) and are listed in Supplementary Table S2. Gene amplifications were carried out with Phusion DNA polymerase. When required, IDT performed gene synthesis. Oligonucleotides used as substrate for recombineering (editing oligos) as well as those used to screen edited clones were synthesized by IDT (Supplementary Table S3). Editing oligos were designed as shown in Supplementary Table S4; briefly, this determines the number of times that every sub-sequence of 20 nucleotides present in the M129 strain genome is found in a different location of the chromosome when the search allows for 0, 1, 2 or 3 mismatches. This information was taken into consideration to select the sequences that were included in the 40 nucleotide homology arms (HAs) of the editing oligos, to ensure specificity and to minimize the number of off-target recombineering events. The correct identity of assembled plasmids and edited genomes was verified by Sanger sequencing (Eurofins genomics).
Editing transformation
Electrocompetent cells from the M129-GP35-PtetCre or M129-GP35 strain (depending on the presence or not of SSR-A coding gene in the used selector plasmid) were prepared as previously described (23). The resulting cell suspensions (70 μl) were mixed with 0.5 nmol of the editing oligo selected (i.e. 5 μl of a 100 μM oligo solution) and 2 μg of the desired selector vector, except for those used to compare performance of the three different SURE editing systems, in which the amount of plasmid was adjusted to transform equimolar quantities (i.e. 1.23, 2 or 2.7 μg for pLoxPuro, pLoxPuroCre or pLoxPuroCreVcre, respectively). In all cases, a control transformation only with the selector vector was carried out to estimate the frequency of spontaneous plasmid insertion in the absence of editing oligos. After the electroporation pulse, cells were harvested from the cuvette in Hayflick medium already supplemented with aTc and allowed to recover at 37ºC for 2 h. The entire transformation volume was then inoculated in a 75-cm2 flask containing 25-ml Hayflick supplemented with puromycin and aTc, to induce expression of SSR-A, which mediates plasmid integration. After 24 h, cells were scraped from the flasks and seeded onto puromycin-selective Hayflick-agar plates. For all edits, one-third of the transformation volume was seeded, except for Δ 90 bp edit, for which only 1% of the transformation volume was seeded. The total number of puromycin-resistant colonies in each transformation was calculated according to the seeded volumes for each edit.
Screening of edited clones
Colonies were picked from puromycin-selective Hayflick-agar plates and inoculated in 96-well plates filled with 200 μl of puromycin-supplemented Hayflick medium per well. Once the cells had grown and reached confluency, genomic DNA was extracted using MasterPure DNA purification kit (Lucigen) following manufacturer's instructions. For screening, around 30 ng the gDNA prep was used as template for the PCR reaction. PCR products were analysed by electrophoresis to estimate the size of the amplified products. To further confirm that the correct modifications were present, products were cleaned-up using QIAquick PCR purification kit and sequenced by Sanger method.
Vector backbone excision mediated by suicide plasmid coding for Vcre
Clones carrying any selector vector inserted at the edited area were grown in tissue culture flasks to prepare electrocompetent cells as previously described (23). The resulting cell suspensions (70 μl of each) were mixed with 2 μg of a suicide vector termed pGentaVcre. A control transformation with no plasmid was always performed in parallel. After the electroporation pulse, cells were allowed to recover at 37ºC for 2 h before inoculating one-fifth of the transformation in a 75-cm2 flask containing 25-ml Hayflick supplemented with gentamicin. Flasks were incubated at 37ºC for 5 days, a timeframe long enough to kill non-transformed cells and to excise the selector plasmid from the edited area in cells that received the pGentaVcre suicide vector. After this incubation, survivor cells were scraped from the flask and seeded onto non-selective Hayflick-agar plates. Colonies were grown on 96-well plates containing 200 μl of non-selective Hayflick medium; once expanded, genomic DNA was extracted as described above, with PCR confirming the excision of the selector plasmid from the edited area. Finally, to rule out possible integration events of the suicide plasmid into the genome, the resulting strains were grown in 96-well plates with Hayflick medium, or with Hayflick supplemented with gentamicin, using from each strain stock 1 μl as starting inoculum into a final volume of 200 ul of the corresponding medium. Growth was monitored by the 430nm/560nm absorbance ratio (30) that reflects pH changes in the medium thanks to the presence of phenol red in the Hayflick medium. To this end absorbance measures were taken every 20 min in a Tecan I-control 1.9.17.0 Infinite 200 device for around 5 days of growth at 37ºC.
Scarless editing
Electrocompetent cells from edited and resolved clones (i.e. strains carrying the intended modification with the vector backbone excised from the edited area) were prepared as previously described (23). The resulting cell suspensions (70 μl) were mixed with 0.5 nmol of the editing oligo intended to delete the scar at the modified locus (i.e. 5 μl of a 100 μM oligo solution) and 2 μg of a suicide vector termed pPuroPtet-I-SceI. Control transformations without oligo or without plasmid were carried out in parallel. After the electroporation pulse, cells recovered at 37ºC for 2 h before inoculating one-fifth of the transformation into a 75-cm2 flask containing 25 ml of Hayflick supplemented with puromycin. Flasks were incubated at 37ºC for 5 days; at 1 day post-inoculation, aTc was added into the medium to induce the expression of I-SceI. In this way, cells have a window of time of 24 h to incorporate the oligonucleotide and delete the scar before expressing the endonuclease to eliminate clones carrying the restriction site incorporated at the scar. After this incubation, survivor cells were scraped from the flask and seeded onto non-selective Hayflick-agar plates. Grown colonies were picked to 96-well plates filled with 200 μl of non-selective Hayflick; once expanded, genomic DNA was extracted as described above, and the excision of the selector plasmid from the edited area was confirmed by PCR. Growth curves of the resulting strain in Hayflick medium, or Hayflick supplemented with puromycin or gentamicin were carried out as described above, to rule out integration events of the suicide plasmids coding for I-SceI endonuclease or Vcre recombinase, respectively.
Rational design and screening of Pcum inducible systems with Venus reporter
Recently, a novel inducible system based on the regulatory components (i.e. CymR transcription factor and its operator sequence CuO) of the cmt operon from Pseudomonas putida has been successfully adapted to different organisms (32–36) including mammalian cells (37). Given its apparent portability and the fact that the system responds to cumate, a non-toxic, inexpensive and carbon-source independent compound, we decided to adapt this system to M. pneumoniae. Based on this system, we rationally designed different cumate responsive constructs for Mycoplasmas and tested their performance driving the expression of a reporter gene (i.e. the coding sequence of venus fluorescent protein).
In all designs, the CymR coding gene (i.e. the repressor of the system) is placed under control of SynMyco regulatory region (RR), a synthetic sequence that promotes efficient transcription and translation of coding sequences in different Mycoplasma species (38). To drive the expression of the reporter gene, three different sequences were used that were derived from PVeg, a strong constitutive promoter of B. subtilis already used in the cumate inducible system available for this strain (36) (Supplementary Figure S1A). Pcum1 design was based on the WT sequence of PVeg, which has been described to carry two different binding sites for RNA polymerase (RNApol) (39). Based on the almost constitutive behaviour of the Pcum1 design (Supplementary Figure S1B), PCum2 was generated, in which the RNA polymerase binding site more distant to the CuO was removed (Supplementary Figure S1A). Finally, given the limited strength of Pcum2 design at the induced condition (Supplementary Figure S1B), Pcum2.1 was generated; this is a derivative of PCum2 in which few nucleotides were changed to increase the affinity of RNApol complex towards this sequence (40) (Supplementary Figure S1A).
To screen the different inducible systems, transposons carrying the venus coding gene coupled to the Pcum designs or to the previously available Ptet system were transformed into the WT strain. The resulting strains were inoculated together in 96-well plates filled with Hayflick medium supplemented with the corresponding doses of cumate (4, 20 and 100 μM) or aTc (2, 10 and 50 ng/ml) as an inducer, as indicated. In addition, the WT strain (which did not carry the venus coding gene) was included as a control strain to determine autofluorescence. Each strain and inducer dose were assessed in three different wells (i.e. biological replicates). After 48 h of growth, medium was removed from the wells and washed twice with PBS to minimize interference of the medium with fluorescence measurements. The absorbance and fluorescence values were measured using Tecan I-control 1.9.17.0 Infinite 200. The settings were determined for optimal gain, 25 flashes and 20 μs of integration time. The fluorescence settings were ƛex = 514 nm and ƛem = 574 nm, whereas absorbance was determined at ƛ = 600 nm. All fluorescence levels were normalized by absorbance levels. To summarize all the results, the leakiness (Supplementary Figure S1C) and the inducibility (Supplementary Figure S1D) of all the inducible systems were calculated. Leakiness was determined by dividing the fluorescent signal in the absence of inducer for each strain by that observed in the WT strain. Inducibility was calculated by dividing the fluorescence signal observed in the optimal inducer concentration by that obtained in the absence of inducer for each strain. The values obtained for these two parameters in each inducible system were assessed for statistical significance using a one-way ANOVA Tukey's test.
Vector backbone excision mediated by cumate induction
Clones carrying pLoxPuroCreVcre selector vector inserted at the edited area were grown in 24-well plates in Hayflick medium supplemented with cumate at 100 μM final concentration. Additional medium compositions based on plain Hayflick, Hayflick supplemented with puromycin, or Hayflick supplemented with puromycin and cumate were included as controls (Figure 7C). After cells had reached confluency, genomic DNA was extracted using MasterPure DNA purification kit (Lucigen) following manufacturer's instructions. For screening, around 30 ng the gDNA prep was used as template for the PCR reaction. PCR products were analyzed by electrophoresis to estimate the size of the amplified products. To further confirm the intended modifications, these products were cleaned using QIAquick PCR purification kit and sequenced by Sanger method.
Figure 7.

Selection of small- and large-scale genome edits with an all-in-one selector plasmid for SURE editing. (A) Illustration depicting the main features included in the all-in-one selector plasmid capable to mediate its own integration and excision on demand. (B) Left side, scheme indicating the expected chromosomal conformations upon the indicated genome edits selected with pLoxPuroCreVcre plasmid. Black arrows represent the oligos used for the PCR screening and the expected size for each situation is indicated on top of the dashed line that connects them. Right side: Electrophoretic analysis of the PCRs conducted for the indicated edits in five different puromycin-resistant colonies (analysed colonies), a negative control and a WT sample are included as a reference. The ratio of edited clones at each locus is indicated at the bottom of each picture. (C) Picture of the plate in which three clones carrying the Δ30 kb edit selected with pLoxPuroCreVcre were inoculated and grown in the presence (+) or absence (–) of puromycin and cumate as indicated. Growth is monitored by the ability of M. pneumoniae to acidify and thus mediate a colour change on the medium containing phenol red. (D) Left, scheme indicating the expected chromosomal conformations of the area modified in the Δ30 kb edit, after excision of the vector. Black arrows represent the oligos used for the PCR screening, and the expected size is indicated above the dashed line. Right, electrophoretic analysis of the PCRs conducted in the three clones carrying the Δ30 kb edit selected with pLoxPuroCreVcre, after being grown in the presence of cumate (+) to induce Vcre expression. A non-induced clone (–) is shown as a reference.
Library construction and whole genome sequencing
Genomic DNA was quantified using Qubit® dsDNA BR (Invitrogen, Life Technologies) and its integrity was assayed with an agarose gel. gDNA fragmentation was done using the Covaris S2 instrument (Covaris Inc.) adjusting the settings as follows: 10% duty cycle, intensity 5, and 200 cycles per burst for 40 s (for 600 bp size distribution). Libraries were prepared using the NEBNext® Ultra DNA Library Prep for Illumina® kit (ref. E7370) according to the manufacturer's protocol. Briefly, 100 nanograms of DNA were subjected to end repair, addition of ‘A’ bases to 3’ ends, ligation of adapters and USER excision. All purification steps were performed using AgenCourt AMPure XP beads (ref. A63882, Beckman Coulter). Library amplification was performed by PCR using NEBNext® Multiplex Oligos for Illumina (Index Primers Set 1, ref. E7335), (Index Primers Set 2, ref. E7500), (Index Primers Set 3, ref. E7710) or/and (Index Primers Set 4, ref. E7730). Final libraries were analyzed using Agilent DNA 1000 chip to estimate the quantity and check size distribution, and were then quantified by qPCR using the KAPA Library Quantification Kit (ref. KK4835, KapaBiosystems) prior to amplification with Illumina's cBot. Libraries were sequenced 2 × 151 + 8 bp on Illumina's NextSeq500. Whole genome sequencing data are available at Array Express under accession E-MTAB-11600.
Bioinformatic analyses of whole-genome sequencing data
Raw DNA sequencing reads were mapped to the M. pneumoniae M129 reference genome (NC_000912.1) by using the Bowtie 2 alignment software in paired-end mode (41) after trimming the Illumina adapter sequences by running trimmomatic (42). Paired reads mapped unambiguously with a minimum quality of 30 were selected by SAMtools (43) to define the coverage depth profile associated with each sample. These coverage profiles were processed to find base pair positions with read counts equal to 0 (i.e. not present in the reference genome) and merged when contiguous regions were found, returning deletions matching the expected deleted regions on each sample (Supplementary Figure S2). Further analyses with freebayes (44) were performed over the previously filtered mapped reads to explore SNPs and indels when compared to the reference genome. All these variants are listed Supplementary Table S5. To further confirm the specificity of the edits performed, reads containing the loxP and lox72 sequences, that constitute the boundaries of the editing scar with the chromosomal region, were selected. These reads were processed to remove the lox sequences, and the remaining genomic DNA was mapped against the M. pneumoniae M129 reference genome by BlastN (45). Moreover, to rule out non-specific insertions of either editing oligos or selective plasmids, this protocol was repeated for the presence of lox66 and lox71 sequences, respectively. Whereas lox71 and lox66 sequences could not be found in any of the sequenced samples, loxP and lox72 were present in a single location that was sample-specific and matched with the expected edit for each sample (Supplementary Table S6). Remarkably, our initial attempt to find lox72 reads in the sample corresponding to M129-GP35-PtetCre Δ1kbmpn440::lox scar strain failed, so we refined the search for a mutated version of lox72 that was already revealed by Sanger sequencing (Supplementary Figure S3). In addition, genome positions where the GP35 transposon is inserted were defined for each strain using FASTQINS pipeline, which reports positions contiguous to an inverted repeat corresponding to the point of insertion (46). These points can be internally validated by the mapping of two positions for a same insertion point due to the duplication of 7 bp produced by the staggered cut of the transposase (Supplementary Table S6). Finally, for the samples corresponding to edits in mpn088, mpn256, mpn440 and mpn583 we checked if any of the raw sequenced reads (read length = 112 bp) mapped to the Vcre or GentaR sequences (1143 and 1488 bp, respectively) by using Bowtie 2 alignment software (41) with the single-end configuration and a minimal alignment length set to 20 bp allowing up to two mismatches. This procedure did not return any sequence mapped indicating that the suicide plasmid employed to excise the selector vector from the edited loci did not get inserted into the genome of the resulting strains (Supplementary Table S6).
RESULTS
SURE editing rationale and validation of the system at four different loci
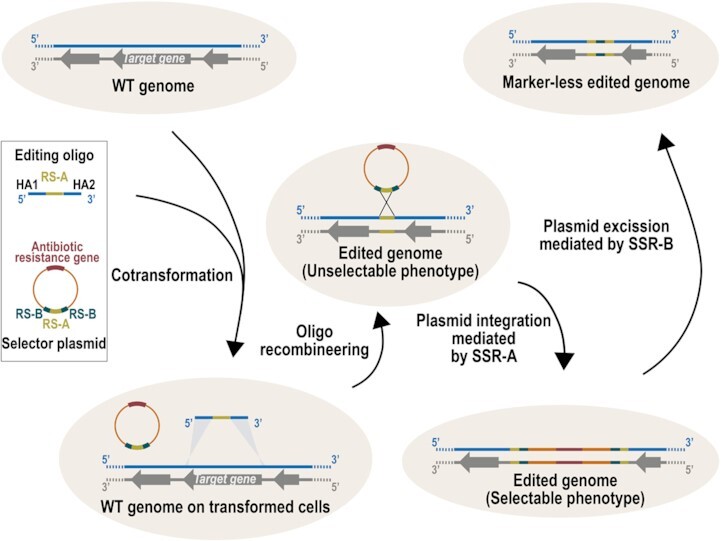
To overcome the main drawbacks associated with oligo-recombineering protocols, we developed a system based on two components: an oligo and a non-replicative plasmid (herein, selector plasmid). First, the oligo performs the genetic modification through homology arms (HA) and introduces a recognition site (RS) for site-specific recombinases (SSR). The editing oligos would create the desired modification with a non-selectable phenotype, while leaving in the edited locus a RS of SSR that can act as a landing pad. Second, the selector plasmid carries an antibiotic resistance gene and a RS compatible with that placed in the landing pad. Thus, co-transformation of the oligo with the selector plasmid results in plasmid integration, generating a selectable phenotype. Once the clones carrying the desired modification have been selected the plasmid might be removed from the edited area thanks to the inclusion of two RS incompatible with those used for the integration of the vector. We termed this engineering rationale SURE editing (Figure 1).
Figure 1.

SURE editing rationale. Cells carrying a wild-type (WT) genome are co-transformed with an editing oligo and a selector plasmid. The editing oligo is composed of two homology arms (HA1 and HA2) that hybridize at the regions flanking the target gene and a site-specific recombinase A recognition site (RS-A). The selector plasmid carries one copy of RS-A and two copies of site-specific recombinase B recognition site (RS-B), as well as an antibiotic resistance gene. After an oligo-recombineering event catalysed by a phage-derived SSAP, edited genomes with unselectable phenotype are generated. Site-specific recombinase A can then mediate the recombination between the RS-A introduced in the edited locus and the RS-A present in the selector plasmid, thereby integrating the vector and generating a selectable phenotype for edited cells. Finally, site-specific recombinase B can drive the excision of the vector through recombination between the two RS-Bs.
We tested this hypothesis using M. pneumoniae, as this bacterium has been traditionally difficult to engineer (similar to many other Mycoplasmas) (47).
The basic enzymes required as components in SURE editing are: (i) GP35, as a SSAP; (ii) SSR-A, to catalyse plasmid integration and (iii) SSR-B, to perform vector deletion from edited clones. For SSR-A, we selected Cre, a tyrosine recombinase derived from P1 bacteriophage that is well-characterized and extensively used (48,49). For SSR-B, we focused on Vcre, a lesser-known tyrosine recombinase encoded by a plasmid present in several Vibrio species (50). In line with these choices, we used lox motifs as RS-A and vlox sequences as RS-B. Of note, the Cre/lox and Vcre/vlox systems do not cross-react with each other (50). We selected these recombinase enzymes and SSR sites as they work in a broad variety of microorganisms.
First, using transposon delivery, we obtained a strain expressing both GP35 (SSAP) and Cre (SSR-A) with a constitutive and inducible Tet promoter, respectively (M129-GP35-PtetCre strain). Then, we transformed this strain with oligos designed to delete 1 kb in four distinct, non-essential locations of the chromosome (i.e. the mpn088, mpn256, mpn440 and mpn583 loci) and to insert a lox site at the edited locus (Figure 2A). Each oligo was co-transformed with a selector plasmid (termed pLoxPuro) carrying a puromycin resistance cassette, one lox site (as RS-A) and two vlox sites (as RS-Bs) (Figure 2A). A high number of puromycin-resistant colonies were obtained in all four transformations, ranging from 890 to 1820 colony forming units (CFU) depending on the edited locus (Supplementary Figure S3A). We found only 40 CFUs in the negative control (transformation with only selector plasmid but not oligos), indicating that the insertion of the plasmid is mainly dependent on the placement of a landing pad by the editing oligo. To confirm the intended deletions, five colonies from each transformation were analysed by PCR. For instance, the scheme in Figure 2B shows an example of the expected chromosomal conformations of mpn088 locus before and after editing. The PCR screen showed that 60% of cells were edited at both the mpn088 and mpn583 loci (three of five tested clones), 80% (four of five clones) at the mpn256 locus and 100% (five of five clones) at the mpn440 locus (Figure 2C).
Figure 2.

Validation of SURE editing at four different loci. (A) Scheme depicting the chromosomal locations of the four loci selected to test the functionality of the system. Insets: a more detailed scheme of the target area indicating the coding sequences present in either the plus strand (blue) or the minus strand (grey), as well as the molecules transformed into the cells. Homology arms (HA) of the editing oligo follow the colour code of the chromosome to reflect if its sequence is that of the plus strand or the minus strand. The shadowed triangles indicate the places in which the sequences used as HA are present in the chromosome, which are at a 1-kb distance to each other. (B) Scheme showing as an example the chromosomal conformation of mpn088 locus before editing (WT genome) and after editing with the plasmid inserted (edited genome) or excised (edited + resolved genome). Black arrows represent the oligos used for the PCR screening; the expected size for each situation is indicated above the dashed line connecting them. (C) Top: electrophoretic analysis of PCRs conducted at the indicated loci in five different puromycin-resistant colonies (analysed colonies); a negative control and a WT sample are included as references. The ratio of edited clones at each locus is based on the observed size of the PCR products. Bottom: electrophoretic analysis of PCR products from the indicated loci in five different colonies (analysed colonies) obtained after transforming one clone carrying the plasmid inserted at the edited locus with a suicide plasmid coding for Vcre. A negative control and a WT sample are included as references.
To conclude the protocol, we carried out a second transformation step with a suicide plasmid encoding SSR-B (i.e. Vcre). Transient expression of SSR from suicide plasmids has been already reported in different Mycoplasma species (51,52) and in a Coxiella burnetii strain (53). In this framework, transformed cells are incubated with antibiotic for a timeframe long enough to kill cells that did not receive the suicide plasmid, and to allow the SSR to catalyse its reaction in most of the cells carrying the suicide plasmid although they cannot proliferate. Specifically, transformation of the pGentaVcre suicide plasmid led to the excision of the selector vector in the 90% of the colonies analysed (i.e. 18 of 20; Figure 2C). All edited loci were confirmed by Sanger sequencing (Supplementary Figure S3B). Moreover, we did whole-genome sequencing of one clone for each of the 1-kb genome edits performed. This allowed us to confirm the desired edits, as inferred from the absence of reads mapping to the specific region deleted on each strain (Supplementary Figure S2). In addition, when compared to the reference genome, the variants detected were for the most part (>90%) present in more than one sample, suggesting that they were fixed in the population before any round of editing. Indeed, variants that were detected in just one sample (and hence might have occurred during the editing process) were always found in the same strain (M129-GP35-PtetCre Δ1kbmpn088::lox scar), which might indicate some sort of sample-specific sequencing phenomenon (Supplementary Table S5). In addition, the off-target modifications that were more likely to occur with this editing method (e.g. due to unspecific integration of the selector vector and/or the editing oligo) were not detected in any of the samples (Supplementary Table S6). Finally, we also ruled out possible integration events of the suicide plasmid carrying Vcre recombinase, as inferred from the absence of reads mapping to the main elements of the plasmid (Supplementary Table S6), and the lack of growth of the resulting strains in medium supplemented with gentamicin (Supplementary Figure S3C). Altogether, these results demonstrate that SURE editing allows clones carrying 1 kb, markerless edits to be generated and selected with high efficiency in a rapid, simple and specific manner, using a single selector plasmid, irrespective of the position of the genome being edited. Hence, once this plasmid is assembled, SURE editing can be considered as a cloning-free protocol and applied to different chromosome locations.
Assessing the limits of SURE editing
We next evaluated whether the selective capacity of SURE editing enables clones that carry large genome modifications to be isolated. Based on the availability of a high coverage essentiality study for M. pneumoniae (54), we identified the largest chromosomal region that can be deleted without affecting any essential function for this bacterium. This region encompasses more than 30 kb and accounts for up to 5.48% of the non-essential (NE) genome (54), containing 25 NE coding genes (from mpn490 to mpn514) (Figure 3A). We designed a battery of editing oligos in which the 5′ HA is constantly located at the 3′ end of mpn490 gene; in each oligo, the 3′ HA is displaced by different distances from the 5′-HA (ranging from 90 bp to 30 kb) (Figure 3A). In separate reactions, each of these oligos was transformed together with pLoxPuro (as the selector plasmid) into the M129-GP35-PtetCre strain. We observed a trend in which the number of puromycin-resistant colonies decreased with the increasing size of the attempted deletion; however, all transformations showed a higher number of colonies than the control without oligo (Supplementary Figure S4A). An example of a scheme depicting the expected chromosomal conformations before and after the smallest and largest edits is given in Figure 3B. For all the attempted deletions, more than 50% of all cells (i.e. 29 of the 35 colonies analysed by PCR) carried the expected modification (Figure 3C). Furthermore, for some edits, this percentage increased up to 100% of the screened colonies (Δ90 bp, Δ10 kb and Δ30 kb edits). Sequencing of the PCR products confirmed the accuracy of the designed deletions and the consequent integration of the selector plasmid between the regions selected as HA in the editing oligos (Supplementary Figure S4B). In addition, we performed whole-genome sequencing of one the clones carrying the largest deletion (i.e. 30 kb), to verify that the only modification that it carried was the planned edit (Supplementary Figure S2 and Supplementary Tables S5 and S6). These results demonstrate that SURE editing is able to execute small as well as large changes, and hence that this method offers a simple way to carry out target editing, not only at a gene level but also at a genome scale.
Figure 3.

SURE editing performance in the largest deletable region of M. pneumoniae genome. (A) Chromosome section representing the largest deletable region in M. pneumoniae genome and showing the battery of designed editing oligos. The coding sequences present in either the plus strand (blue) or the minus strand (grey) are represented by arrows and the gene identifiers associated to them are written in red for essential genes, and green for non-essential genes. The light blue shadow triangles between the chromosome and the oligos indicate the places in which the sequences used as HA are present in the genome. All editing oligos share a common 5′ HA, whereas the 3′ HA binds to chromosomal regions placed at different distances from the 5′ HA, as indicated above each editing oligo. (B) Scheme showing as an example the expected chromosomal rearrangements upon the Δ90 bp (left) and Δ30 kb (right) genome edits. Non-edited genomes for both cases are also shown. Black arrows represent the oligos used for the PCR screening; the expected size for each situation is indicated above the dashed line that connects them. For all edits, modified genomes should produce a PCR product slightly bigger than that of the selector plasmid (3.4 kb), whereas WT genomes should generate a PCR product slightly bigger than that of the attempted deletion. Note that for deletions of 10 kb and larger, the processivity of the polymerase is not efficient enough to amplify the product. (C) Electrophoretic analysis of the PCRs conducted for the indicated edits in five different puromycin-resistant colonies (i.e. the analysed colonies); a negative control and a WT sample are included as references. The ratio of edited clones at each locus is indicated at the bottom of each picture.
Turning SURE editing into a scarless genome modification method
The inclusion of a lox66 site between the HAs of the editing oligo allows us to rescue ultra-rare events of oligo-recombineering, due to integration of a selector plasmid at the edited locus that carries a lox71 site compatible with that introduced by the oligo (55). When these two lox sites recombine to mediate the integration of the selector plasmid, a scar in the edited area containing a double mutant inactive lox72 site and a wild-type active loxP site is generated, even after vector removal (Figure 2B). In this scenario, no further rounds of editing are possible; here, the likelihood is high that a lox-based selector plasmid would be preferentially inserted into this scar rather than into a novel lox landing site created by a new event of oligo-recombineering.
To solve this limitation, we evaluated whether SURE editing can be turned into a scarless genome modification method. To this end, we repeated the 90-bp deletion performed in Figure 3 using a new oligo: besides introducing a lox site, it also mediates the insertion at the edited area of 18 bp that constitute the restriction site of I-SceI homing endonuclease (56) (Figure 4A). The extended length of its restriction site makes this endonuclease a non-cutting enzyme in most bacterial genomes, which has favoured its use as a counter selective marker in some genome editing methods (57). As expected, the number of colonies obtained was significantly higher in the co-transformation of the editing oligo and the selector vector than in the control transformation with only pLoxPuro plasmid (Supplementary Figure S5A). Of the five puromycin-resistant colonies analysed, four showed PCR amplification products compatible with the insertion of the selector plasmid at the target site (Figure 4B). One of these clones was expanded and transformed with a non-replicative plasmid coding for Vcre, leading to the excision of the vector in most of the analysed colonies (Figure 4C). Of note, the editing scar retained after Vcre excision contained one vlox site, two lox sites and the I-SceI restriction site (Figure 4C). We took advantage of this to co-transform a suicide plasmid that expresses I-SceI endonuclease in an inducible manner along with a new editing oligo designed to delete the scar from the edited cells. Specifically, this oligo has the exact same sequence of the one used in the first editing step, but without the lox or the I-SceI restriction site. In this framework, upon the co-transformation and induction of I-SceI expression, only those cells that have incorporated the editing oligo and consequently removed the restriction site from the chromosome are expected to survive (Figure 4D and E). Accordingly, five survivor colonies analysed by PCR showed an amplification product compatible with deletion of the editing scar (Figure 4D) and this deletion of the scar was confirmed by sequencing the PCR products (Supplementary Figure S5B). In addition, the resulting strain was unable to grow in gentamicin or puromycin, which discards possible integration events of the suicide plasmid employed in the protocol coding for Vcre and I-SceI, respectively (Supplementary Figure S5C). Altogether, these results demonstrated that SURE editing generated scarless modifications in a simple and cloning-free manner, which will eventually allow iterative rounds of editing with a single selector vector (i.e. pLoxPuro). While inclusion of the I-SceI restriction site in this example is mediated by an editing oligo, it could be also included through the selector plasmid, which would reduce the length of the editing oligo and thereby decrease its cost and improve its editing capacity.
Figure 4.

Scarless SURE editing. (A) Cells carrying a WT genome were co-transformed with the SSR-A-dependent plasmid selector and an editing oligo that contained a restriction site for the I-SceI enzyme and an RS-A site between the two homology arms (HA1 and HA2). (B) Upon integration of the vector, edited cells had the resistance marker included in the genome. After picking colonies and isolating genomic DNA, cells were analysed by PCR. Four out of the five analysed clones showed a positive band of 3681 bp, consistent with the edited genome. (C) Positive clones were transformed with the pSSR-B vector, which mediated the excision of the integrated vector, thereby eliminating the resistance marker and leaving a scar that contained the I-SceI site and the lox sequences from the recombination event. Four out five analysed clones were positive by PCR. (D, E) The cells were transformed with an oligo that eliminated the scar from the genome and a selector vector that allowed the expression of I-SceI enzyme. (D) If the cells were edited, and the scar had been eliminated, the I-SceI enzyme did not cut. NT, cells that were non-transformed and carried the editing scar (cells that were not cotransformed with oligo and I-SceI coding plasmid). (E) If the cells were not edited by the oligo, the genome was cut, and the cells died. All five clones analysed had the I-SceI site excised out, confirming that the scar has been eliminated
Expanding SURE editing logic to different site-specific recombinases
We demonstrated that we could overcome the main limitation of SURE editing technology (i.e. the unavoidable presence a scar containing an active RS at the end of the editing protocol) by developing a ‘scarless’ editing protocol. This approach will allow iterative rounds of genome editing to be carried out but involves an additional transformation step.
To accelerate eventual iterative rounds of genome modifications and increase the modularity of SURE editing logic, we created a collection of selector plasmids based on different SSRs. As a first step, using the pLoxPuro vector as a reference, we created four additional selector vectors (termed pSloxPuroScre, pRoxPuroDre, pVoxPuroVika and pAttBPuroBxb1) based on the Scre (50), Dre (58), Vika (59) and Bxb1 (60,61) recombinases, respectively; each of these carries the corresponding RS (i.e. slox, rox, vox and attB/attP, respectively). In contrast to our previous assays, these selector plasmids now contain the SSR-A coding gene under the control of the Tet inducible promoter, thus avoiding the requirement of obtaining in advance strains expressing the specific SSR-A. We then compared the ability of these selector plasmids (i.e. this set and pLoxPuroCre) (Figure 5A) to select an oligo-recombineering event that mediates a 0.84-kb deletion at the mpn507 locus (Figure 5B). After co-transforming the editing oligo with each of these selector plasmids into a strain that solely expresses GP35 as SSAP (M129-GP35 strain), we observed variable numbers of puromycin-resistant colonies, with pLoxPuroCre being the most productive vector, and pSloxPuroScre, the least productive, for colonies obtained (Supplementary Figure S6A). Control transformations with most of the selector plasmids showed only a low number of colonies. In contrast, the control transformation with pVoxPuroVika gave approximatively one-third as many colonies as obtained from the co-transformation of this vector with the editing oligo (Supplementary Figure S6A). This suggests the existence of a vox-like sequence within the genome of M. pneumoniae that led to unspecific integration of the vector (i.e. independent of the editing oligo). A scheme showing the expected chromosomal conformations before and after the intended edit is shown in Figure 5C, using pLoxPuroCre selector vector as an example. PCR screening revealed that all plasmids mediated the selection of edited cells at high efficiencies (4 or 5 out of 5 screened cells; Figure 5D), except from pVoxPuroVika, in which only 3 out of 5 of the screened colonies carried the intended modification (Figure 5D), probably as result of the relaxed specificity of the Vika/vox system in M. pneumoniae (Supplementary Figure S6A). Sequencing the PCR products showed integration of the different selector plasmids and deletion of the target area in all cases (Supplementary Figure S6B). Altogether, these results demonstrated that SURE editing logic is compatible with several SSRs regardless of the family to which they belong. This expanded modularity offers a faster approach to perform iterative rounds of genome editing as compared to the scarless approach.
Figure 5.

SURE editing compatibility with different SSRs. (A) The structure of the different constructed selector plasmids, together with the editing oligos used to test their activity. All oligos shared the same homology arms (HA1 and HA2) that flank the indicated RS. (B) Scheme depicting the chromosomal location of the locus selected to compare the efficiency of the different selector plasmids. A more detailed view of the target area is shown within the square, indicating the coding sequences present in either the plus strand (blue) or the minus strand (grey), as well as the molecules transformed into the cells. HAs of the editing oligo (blue) have a sequence of the chromosome plus strand, whereas the RS present in the editing oligo and the selector plasmid are depicted in multicolour, to reflect that they change their sequence depending on the particular RS/SSR pair used. The light-blue shadowed triangles indicate the places in which the sequences used as HA are present in the chromosome, which are at a 0.84-kb distance to each other. (C) Scheme showing the chromosomal conformation of the locus before editing (WT genome) and after editing with the plasmid inserted there (edited genome). Black arrows represent the oligos used for the PCR screening and the expected size for each situation is indicated on top of the dashed line that connects them. Note that while the size shown corresponds to that expected using pLoxPuroCre as selector plasmid, all selector plasmids will generate a similar-sized PCR product. (D) Electrophoretic analysis of the PCRs conducted for each selector plasmid in five different puromycin-resistant colonies (analysed colonies); a negative control and a WT sample are included as a reference. The ratio of edited clones at each locus is indicated at the bottom of each image.
SURE editing enables gene platforms to be introduced at the target locus
A second major drawback of oligo-recombineering protocols (besides the inability to directly select edited clones in the absence of clear phenotype) is the lack of a single-step process for introducing gene platforms. Given that SURE-editing technology involves the transient integration of a plasmid to facilitate the selection of edited clones, we reasoned that cloning the desired platform into a specific location of the vector might overcome this limitation of classical recombineering protocols.
We tested the ability of SURE-editing to introduce gene platforms within a framework of genome streamlining. As a proof of concept, we focused our attention on a chromosome region close to the ori that contains non-essential genes (mpn633, mpn634, mpn635 and mpn638) and flanks an E operon composed of two essential genes (mpn636 and mpn637) (54) (Figure 6A). For this, we designed i) an editing oligo to delete the entire area (5.5 kb; from mpn633 to mpn638) and ii) a selector plasmid (termed pLoxPuroCreCOMP636-637) in which we cloned the two E genes to complement the planned deletion (Figure 6B). Co-transformation of both molecules into a M129-GP35 strain produced a substantially higher number of colonies than that observed in the control transformation with only selector plasmid (Supplementary Figure S7A). We analysed five puromycin-resistant colonies from the co-transformation by PCR; for three of these, we obtained PCR products of a size compatible with the deletion of the whole area and the insertion of pLoxPuroCreCOMP636-637 vector (Figure 6C and D). One of these clones was subsequently transformed with a suicide vector encoding Vcre, thereby excising the vector backbone but leaving at the edited area the E operon (Figure 6C). PCR analyses (Figure 6D) and sequencing of the amplification products (Supplementary Figure S7B) confirmed that the vector had been successfully removed from the target area. Moreover, whole-genome sequencing of the resulting strain confirmed the specificity of the edit performed and ruled out any undesired genome modification (Supplementary Figure S2 and Supplementary Tables S5 and S6) In sum, these results show that SURE editing enables gene platforms to be introduced into the target locus simultaneously to the deletion step, a feature that might be of special interest in genome streamlining processes.
Figure 6.

SURE editing-mediated insertion of gene platforms at a desired location. (A) Scheme depicting the chromosomal location of the region in which the targeted insertion of gene platforms was tested. A more detailed view of the target area is shown within the square. Genes encoded in this area are only present in the plus strand (blue arrows), whereas the gene identifiers associated to them are written in red or green, for essential and non-essential genes, respectively. Above the chromosome, the editing oligo and the selector plasmid transformed into the cells are depicted. HAs of the editing oligo (blue) have the sequence of the plus strand of the chromosome; the light-blue shadowed triangles indicate the genomic locations of the HA sequences, which are at a 5.5-kb distance to each other (B) Illustration depicting the main features included in the selector plasmid used for the targeted insertion of gene platforms (in this case, the mpn636 and mpn637 genes). Note that, for clarity, only the features important for the SURE editing process are indicated. (C) Scheme showing the chromosomal conformation of the modified area before the editing (WT genome) and after the editing with the plasmid inserted there (edited genome) or with the plasmid excised (edited + resolved genome). Note that after excision, the two essential genes cloned in the vector are maintained at the chromosome. Black arrows represent the oligos used for the PCR screening; the expected size for each situation is indicated above the dashed line connecting them. (D) Top: electrophoretic analysis of the PCR conducted in five different puromycin-resistant colonies (analysed colonies); a negative control and a WT sample are included as references. The ratio of edited clones is indicated based on the observed size of the PCR products. Bottom: electrophoretic analysis of the PCRs of five different colonies (analysed colonies) obtained after transforming one clone carrying the plasmid inserted at the edited locus with a suicide plasmid coding for Vcre. A negative control and a WT sample are included as references.
Adaptation of a cumate inducible system for Mycoplasma, and its use for generating an all-in-one SURE editing vector
We have shown that SURE editing is a highly versatile system that allows a wide variety of genome modifications to be carried out in a single step, with efficiencies always >50% of the screened colonies regardless of the type of modification attempted or the SSR used. Nonetheless, to obtain marker-free modifications and to excise the inserted vector, an additional transformation step with a suicide vector coding for SSR-B (i.e. Vcre recombinase) needs to be done. As the inclusion of an SSR-A coding gene into the sequence of the selector plasmid still resulted in a functional SURE editing system (Figures 4–6) we reasoned that it should be also possible to clone the coding sequence of an SSR-B. In this framework, the selector plasmid would carry all the elements required for a functional SURE editing technology (except for a SSAP); in other words, producing an on-demand, self-integrative and self-excisable plasmid. For this purpose, the SSR-A and SSR-B should act in a strictly temporary order to mediate integration and excision of the vector at will. Thus, their expression needs to be driven by two different inducible systems. Unfortunately, only a tetracycline-inducible system was available for Mycoplasmas (52,62); this system was used to drive the expression of the SSR-A upon addition of anhydrotetracyline (aTc). This forced us to adapt a new system based on cumate, in which the expression of SRR-B can be controlled in response of an external signal different from aTc. Based on this system we rationally designed different cumate responsive constructs for Mycoplasmas (see Materials and Methods section) and tested their performance driving the expression of a reporter gene (i.e. the coding sequence of venus fluorescent protein). The levels of venus fluorescence obtained with the three designs (dubbed Pcum1, Pcum2 and Pcum 2.1) (Supplementary Figure S1A) were tested across different inducer doses, and compared side by side with those produced by the only inducible system that has been previously reported for Mycoplasmas, based on the tetracycline repressor (62) (Supplementary Figure S1B). The leakiness (i.e. the ratio between the fluorescent signal in the absence of inducer, and that observed in the WT strain) was negligible for all systems except for the PCum1 system, which produced a fluorescent signal eight times higher than that observed in the control strain (Supplementary Figure S1C). In contrast, the inducibility (i.e. the fold-change in venus expression between the optimal induction condition and the uninduced state) was almost three times more pronounced in Pcum2.1 than in Ptet (Supplementary Figure S1D). Altogether, these results confirmed the development of a novel inducible system for M. pneumoniae based on Pcum2.1 design that clearly outperforms the previously available system.
With this novel inducible system at hand, we constructed an all-in-one selector plasmid for SURE editing that we termed pLoxPuroCreVcre (Figure 7A). We aimed to compare the performance of this selector plasmid with that observed for the selector vector that only codes for the puromycin resistance gene (i.e. pLoxPuro) or the one that additionally carries an SSR-A coding gene (i.e. pLoxPuroCre). Hence, we assessed in parallel the ability of the three plasmids to select the clones carrying the smallest (Δ90 bp) or the biggest (Δ30 kb) edits tested in this work. For this, equimolar amounts of the different selector plasmids were transformed together with the appropriate editing oligo into either M129-GP35-Cre (for pLoxPuro plasmid) or M129-GP35 (for pLoxPuroCre and pLoxPuroCreVcre plasmids). Regardless of the attempted modification, co-transformation of the editing oligo and pLoxPuroCre selector plasmid showed the highest number of puromycin-resistant colonies. In any case, all co-transformations resulted in an amount of puromycin-resistant colonies that was clearly higher that that obtained in their respective control transformations with only a selector plasmid (Supplementary Figure S8A). To confirm the correct identity of the obtained editing, we analysed five puromycin-resistant colonies of each cotransformation by PCR with oligos flanking the edited area. According to this analysis, pLoxPuro selector vector mediated the intended modification in four or five of the five analysed colonies for the Δ90 bp and Δ30 kb edits, respectively (Supplementary Figure S8B), whereas these percentages were 100% and 60% when pLoxPuroCre was used as selector vector (Supplementary Figure S8C). Although the use of pLoxPuroCreVcre as selector plasmid resulted in lower number of puromycin-resistant colonies as compared to pLoxPuro or PloxPuroCre, the percentage of analysed colonies carrying the intended modifications was as high as that observed with the pLoxPuro selector plasmid, of 100% for the Δ90 bp edit, and 80% for the Δ30 kb edit (Figure 7B). Next, three of the clones carrying the Δ30 kb edit selected with pLox66PuroCreVcre were grown in different combinations of puromycin and cumate to confirm the possibility of mediating vector excision without any additional round of transformation with a vector coding for Vcre. All clones were able to grow in all tested conditions except those involving the simultaneous presence of cumate and puromycin (Figure 7C). This is in line with the expected behaviour of the system, as cumate induction should lead to the expression of Vcre, which would excise the vector from the edited locus precluding the growth of the bacterium in puromycin. To further confirm this, we analysed by PCR the three clones grown in the presence of cumate, which gave PCR products with a size a compatible to the expected in case of vector excision (Figure 7D). Sequencing of these products confirmed the removal of the selector plasmid from the edited area (Supplementary Figure S8D). Altogether, these results confirmed the functionality of the all-in-one selector plasmid for SURE editing, producing markerless, genome-scale (up to 30 kb) edits in a single transformation step.
DISCUSSION
Mycoplasmas are interesting microorganisms for elucidating a minimal core machinery capable of sustaining autonomous growth; however, for the most part, they are recalcitrant to classic genome engineering approaches (47). This boosted the development of imaginative solutions, such as chemical synthesis (63,64) or in yeast modification of whole Mycoplasma genomes (65,66), for reintroduction of these tailored genomes into an acceptor cell using a process known as genome transplantation (67). Unfortunately, this approach is only available for a few species that are closely related to Mycoplasma capricolum, which is the only competent acceptor cell reported to date (68). In the specific case of M. pneumoniae, for which genome transplantation is not possible, there was a single report describing the generation a targeted gene knock-out in a seemingly unreliable genome editing protocol driven by inefficient host recombination machinery (69). We recently reported that GP35 SSAP can be used in recombineering protocols for this bacterium, using either oligos (23) or long-stretches of ssDNA (70) as a substrate for recombination. This allowed us to generate the first attenuated version of M. pneumoniae, which has been used as a live biotherapeutic product to fight S. aureus biofilms-associated infections (51). However, both engineering approaches show major limitations. In the case of oligo-recombineering, its frequency for ∼1 kb genetic changes is lower than that of Cas9 evaders, thus hampering the selection of edited cells (23). In contrast, long stretches of ssDNA facilitate direct selection of edited cells, as they can harbour antibiotic resistance genes. Nonetheless, their extended length limits their incorporation at the replication fork, and very few positive clones have been reported with this approach (70). Finally, GP35-mediated recombineering of long stretches of ssDNA has been employed to sequentially place lox sites at desired locations of M. pneumoniae chromosome and mediate a recombinase-mediated cassette exchange (RMCE) protocol. This approach requires the assembly of several customized DNA constructs and up to three different transformation steps, and it has failed to produce isolated, pure clones carrying the intended modifications (71).
In this work, we developed SURE editing, an efficient, simple and versatile engineering rationale that might be especially suitable for different genome engineering projects. For instance, using SURE editing, virtually all possible knockouts (KO) of a genome can be selected with a single selector plasmid. Hence, the generation of single-gene KO mutant libraries, such as the KEIO collection available for E. coli (72), seems to be a feasible endeavour with this method. Of note, the construction of the KEIO collection involved the generation of a recombinogenic PCR product substrate for each target gene, whereas the recombinogenic substrate in SURE editing is an editing oligonucleotide that can be obtained ready-to-use from different suppliers. Additionally, given that SURE editing is also capable of mediating targeted genome integration of gene platforms, it can be use in projects that require an increased copy number of a particular segment of DNA in a certain location. This might be of interest for instance when engineering the copy number of ribosomal RNA operons (rrn) in the chromosome. Although increasing rrn copies from the native seven copies to up to 10 did not show any effect in growth rate in E. coli (73), it seems that the rrn copy number strongly correlates with growth rate across different bacterial species (74). Therefore, altering rrn copy number at targeted positions in slow-dividing microorganisms, such as Mycoplasmas (which for the most part contain a single rrn copy) might be a feasible project with SURE editing. Lastly, as we showed in this study, SURE editing is particularly suited for genome streamlining protocols, given its ability to delete large chunks of DNA, and even to complement essential functions placed in the targeted area if required. Remarkably, genome minimization approaches undertaken in other bacterial species have revealed interesting emergent properties in the resulting strains, such as higher transformation efficiency, higher stability of heterologous DNA constructs, rewired metabolic networks and increased protein yields (75–77).
Several genome editing methods have been developed in the last years, and researchers can currently choose among different strategies depending on the final goal and the organism being engineered (Table 1). For instance, homologous recombination strategies relying on native recombination machinery can produce remarkable results in a wide variety of edits (i.e. deletions, insertions, replacements). In some cases, these homologous recombination strategies have been merged with RNA-guided programmable nucleases (i.e. Cas9) (78) or even DNA-guided programmable guided nucleases (i.e. NgAgo) (79) to create the desired edits. Also, the inclusion of different counterselection markers (80), such as sacB, thyA and I-SceI restriction site in the recombinogenic substrate, can led to the generation of scarless edits (57,81). On the other hand, there are other engineering methods that seem to operate independently of the host recombination machinery. For instance, when aiming for gene deletions, oligo-recombineering is a great alternative, as it does not require tailored DNA constructs to be made for each modification. However, when attempting large deletions, or when working with strains in which oligo-recombineering protocols are not highly efficient, the combination with Cas9 (23–28) or Cas12 (82)-mediated counterselection might be needed to boost its efficiency and to allow selection of edited cells at reasonable frequencies. Retron technology can produce recombinogenic substrates for SSAPs inside cells (83,84), achieving exceptional efficiencies for small edits (of more than 90% of screened cells), although its performance for larger deletions remains unexplored (84). Remarkably, retron-based editing has been demonstrated to work across different kingdoms of life (85). However, similar to oligo-recombineering protocols coupled to CRISPR counterselection, retron-based editing requires personalized DNA assemblies to be made for each modification. For large deletions, Cas3 might represent the preferred choice, as it can be programmed to cleave DNA at a precise location and promote proccessive degradation of the surrounding sequences in a bidirectional manner (86). Deletions ranging from 7 to 424 kb have been obtained with this method, but a repair template must be provided when aiming for precise deletions of a specific size. Also, this approach might be limited by the differential ability of each host to repair DSBs, as alternative end joining of microhomology regions seems to be involved in the resolution of the protocol (86).
Table 1.
Side by side comparison of different targeted genome editing methods across a set of desirable features for a given engineering system
| Genome editing method | Portability | Tailored constructs for each target | Direct selection of edited clones | Possible iterativity | Achievable edits | Reported scarless outcome |
|---|---|---|---|---|---|---|
| Plasmid or dsDNA classical homologous recombination | Limited to recombination-proficient strains | Yes | Yes | Yes | All | In some protocols (57) |
| Oligo-recombineering (16) (MAGE (22), pORTMAGE (97)) | Broad | No | No | Yes | Deletion, insertions only for few nucleotides | Yes |
| Oligo-recombineering coupled to CRISPR/Cas9 (23,25–27) (CRMAGE (24), CRAM (28)) | Broad | Yes | Yes, but influenced by Cas9 evaders | Yes | Deletion, insertions only for few nucleotides | Yes |
| Retrons(83,84) | In bacteria, only reported for E. coli, but probably broad as it also works in eukaryotes | Yes | No, but small deletions are obtained at exceptional efficiencies | Yes | Deletion, Insertions only for few nucleotides | Yes |
| Cas3 (86) | Broad but might be limited for the differential ability to repair DSBs | Yes | No | Yes | Only tested for deletions but the repair template has potential to mediate insertions | Yes |
| Transposon-associated CRISPR/Cas systems (CAST (87), INTEGRATE (88,89)) | So far reported in three species but theoretically broad | Yes | Yes | Yes | Insertion, but INTEGRATE also allows deletion (Cre/lox) | No |
| MobileII group introns (Targetron (90), Clostron (91), GETR (95)) | Quite broad | Yes | Yes | Yes | Insertion (limited to ∼1kb) GETR allows larger insertions and deletions (Cre/lox) | No |
| REXER (96) | Limited to species with efficient dsDNA recombineering protocols) | Yes | Yes | Yes | All | No |
| ORBIT (29) | Theoretically as broad as oligo-recombineering | No | Yes | No | All | No |
| SURE editing | Theoretically as broad as oligo-recombineering | No | Yes | Yes | All | Yes |
On the other hand, transposon-associated CRISPR/Cas systems might be an excellent choice for targeted gene insertions. Two different systems, termed CAST (87) and INTEGRATE (88,89), are available to mediate the insertion of the cargo sequence at variable distances downstream of the region recognized by the guide RNA, but the inverted repeat sequences associated to the Tn7-like transposon will remain at the edited area. Targeted insertion can also be achieved with engineered mobile group II introns (generally termed Targetrons (90) or Clostrons (91)), although with a much more limited cargo capacity (∼1 kb) (92) that might be occupied by an antibiotic resistance gene (93) or by a gene platform of interest if CRISPR/Cas9 is employed as counterselection for non-edited cells (94). To expand their possibilities, Targetrons have been combined with Cre/lox technology to enable large insertions and deletions, in multistep process termed genome editing via Targetrons and recombinases (GETR) (95). A similar approach was undertaken with INTEGRATE, thereby expanding the range of suitable modifications of the system (89). REXER is another interesting tool for mediating the insertion of large DNA sequences (96). REXER can catalyse not only insertions but also replacements, as it relies on recombineering dsDNA fragments rather than on integrative elements (such as Targetrons or INTEGRATE). However, while REXER is an extremely powerful method, it has only been tested in E. coli so far, and it involves the assembly of complex bacterial artificial chromosomes (BACs) that carry several personalized elements for each modification (such as homology regions, sgRNAs, etc).
Finally, ORBIT (29) uncouples the process of genome editing from the selection of edited cells thanks to the coordinated actions of an SSAP to mediate the intended modification, and an SSR to catalyse plasmid integration. ORBIT is a versatile method capable of mediating deletions as well as insertions and replacements, and it does not require the construction of personalized DNA constructs for each individual modification. SURE editing described in this work follows a similar engineering logic and hence compiles all the advantages of ORBIT while adding some extra features that solve certain limitations of the method. For instance, in ORBIT, markerless edits can be obtained only for gene deletions, as it uses the same SSR for plasmid insertion and excision; this preclude the removal of the antibiotic resistance gene if a gene platform must be introduced. Moreover, in both methods, the editing scars contain active RS for SSRs, which would preclude iterative rounds of genome editing. While this limitation remains unaddressed in ORBIT method, SURE editing offers two different approaches to overcome this drawback, based on the development of scarless editing and on the use of up to five different SSRs to expand the usability of the system. Finally, for SURE editing, we developed an all-in-one selector plasmid capable of mediating self-integration and excision on demand, thereby reducing the protocol steps required to obtain marker-free modifications to a single transformation step. Of note, both methods share a common logic based on the combination of oligo-recombineering and SSRs and have been demonstrated to be functional in bacteria genera that are phylogenetically quite distant, such as Mycobacterium and Mycoplasma. Hence, the work here describes an engineering rationale that not only represents a valuable addition to the limited toolbox available for Mycoplasmas, but might also provide a basis for developing a similar method in other bacteria, as it uses enzymes that work in many bacteria species.
DATA AVAILABILITY
Whole genome sequencing data are available at Array Express under accession E-MTAB-11600. All materials generated in this work are available upon request.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank the Genomics Unit at the CRG for assistance with the sequencing.
Contributor Information
Carlos Piñero-Lambea, Pulmobiotics ltd, Dr. Aiguader 88, Barcelona 08003, Spain.
Eva Garcia-Ramallo, Pulmobiotics ltd, Dr. Aiguader 88, Barcelona 08003, Spain.
Samuel Miravet-Verde, Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, Dr. Aiguader 88, Barcelona 08003, Spain.
Raul Burgos, Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, Dr. Aiguader 88, Barcelona 08003, Spain.
Margherita Scarpa, Pulmobiotics ltd, Dr. Aiguader 88, Barcelona 08003, Spain.
Luis Serrano, Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, Dr. Aiguader 88, Barcelona 08003, Spain; Universitat Pompeu Fabra (UPF), Barcelona 08002, Spain; ICREA, Pg. Lluís Companys 23, Barcelona 08010, Spain.
Maria Lluch-Senar, Pulmobiotics ltd, Dr. Aiguader 88, Barcelona 08003, Spain; Basic Sciences Department, Faculty of Medicine and Health Sciences Universitat Internacional de Catalunya, 08195 Sant Cugat del Vallès, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program [670216 (MYCOCHASSIS)]; Spanish Ministry of Economy, Industry and Competitiveness (MEIC) (to the EMBL partnership); Centro de Excelencia Severo Ochoa; CERCA Program from the Generalitat de Catalunya; European Union's Horizon 2020 Research; Innovation Program [634942 (MycoSynVac)]; LaCaixa Fundation [Livetherapeutics HR18-00058]; C.P.-L. acknowledges the support of ‘Programa Torres Quevedo’ grant [PTQ2020-011048] funded by MCIN/AEI/10.13039/501100011033; European Union ‘NextGenerationEU/PRTR’; M.L.-S. acknowledges the support from FEDER project from Instituto Carlos III (ISCIII, Acción Estratégica en Salud 2016) [CP16/00094].
Conflict of interest statement. None declared.
REFERENCES
- 1. Venter J.C., Cohen D.. The century of biology. N. Perspect. Q. 2014; 31:28–37. [Google Scholar]
- 2. Gleizer S., Ben-Nissan R., Bar-On Y.M., Antonovsky N., Noor E., Zohar Y., Jona G., Krieger E., Shamshoum M., Bar-Even A.et al.. Conversion of Escherichiacoli to generate all biomass carbon from CO2. Cell. 2019; 179:1255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ru J., Huo Y., Yang Y.. Microbial degradation and valorization of plastic wastes. Front. Microbiol. 2020; 11:442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Piñero-Lambea C., Ruano-Gallego D., Fernández L.Á.. Engineered bacteria as therapeutic agents. Curr. Opin. Biotechnol. 2015; 35:94–102. [DOI] [PubMed] [Google Scholar]
- 5. Adams B.L. The next generation of synthetic biology chassis: moving synthetic biology from the laboratory to the field. ACS Synth. Biol. 2016; 5:1328–1330. [DOI] [PubMed] [Google Scholar]
- 6. Blomfield I.C., Vaughn V., Rest R.F., Eisenstein B.I.. Allelic exchange in Escherichiacoli using the Bacillus subtilis sacB gene and a temperature-sensitive pSC101 replicon. Mol. Microbiol. 1991; 5:1447–1457. [DOI] [PubMed] [Google Scholar]
- 7. Jasin M., Schimmel P.. Deletion of an essential gene in Escherichiacoli by site-specific recombination with linear DNA fragments. J. Bacteriol. 1984; 159:783–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marx C.J., Lidstrom M.E.. Broad-host-range cre-lox system for antibiotic marker recycling in gram-negative bacteria. BioTechniques. 2002; 33:1062–1067. [DOI] [PubMed] [Google Scholar]
- 9. Murphy K.C. Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichiacoli. J. Bacteriol. 1998; 180:2063–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang Y., Buchholz F., Muyrers J.P., Stewart A.F.. A new logic for DNA engineering using recombination in Escherichiacoli. Nat. Genet. 1998; 20:123–128. [DOI] [PubMed] [Google Scholar]
- 11. Little J.W. An exonuclease induced by bacteriophage lambda. II. Nature of the enzymatic reaction. J. Biol. Chem. 1967; 242:679–686. [PubMed] [Google Scholar]
- 12. Clark A.J., Sandler S.J., Willis D.K., Chu C.C., Blanar M.A., Lovett S.T.. Genes of the RecE and RecF pathways of conjugational recombination in Escherichiacoli. Cold Spring Harb. Symp. Quant. Biol. 1984; 49:453–462. [DOI] [PubMed] [Google Scholar]
- 13. Li Z., Karakousis G., Chiu S.K., Reddy G., Radding C.M.. The beta protein of phage lambda promotes strand exchange. J. Mol. Biol. 1998; 276:733–744. [DOI] [PubMed] [Google Scholar]
- 14. Hall S.D., Kane M.F., Kolodner R.D.. Identification and characterization of the Escherichiacoli RecT protein, a protein encoded by the recE region that promotes renaturation of homologous single-stranded DNA. J. Bacteriol. 1993; 175:277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Datsenko K.A., Wanner B.L.. One-step inactivation of chromosomal genes in Escherichiacoli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ellis H.M., Yu D., DiTizio T., Court D.L.. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:6742–6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ricaurte D.E., Martínez-García E., Nyerges Á., Pál C., de Lorenzo V., Aparicio T.. A standardized workflow for surveying recombinases expands bacterial genome-editing capabilities. Microb. Biotechnol. 2018; 11:176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sun Z., Deng A., Hu T., Wu J., Sun Q., Bai H., Zhang G., Wen T.. A high-efficiency recombineering system with PCR-based ssDNA in Bacillussubtilis mediated by the native phage recombinase GP35. Appl. Microbiol. Biotechnol. 2015; 99:5151–5162. [DOI] [PubMed] [Google Scholar]
- 19. Filsinger G.T., Wannier T.M., Pedersen F.B., Lutz I.D., Zhang J., Stork D.A., Debnath A., Gozzi K., Kuchwara H., Volf V.et al.. Characterizing the portability of phage-encoded homologous recombination proteins. Nat. Chem. Biol. 2021; 17:394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wannier T.M., Ciaccia P.N., Ellington A.D., Filsinger G.T., Isaacs F.J., Javanmardi K., Jones M.A., Kunjapur A.M., Nyerges A., Pal C.et al.. Recombineering and MAGE. Nat Rev Methods Primers. 2021; 1:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wannier T.M., Nyerges A., Kuchwara H.M., Czikkely M., Balogh D., Filsinger G.T., Borders N.C., Gregg C.J., Lajoie M.J., Rios X.et al.. Improved bacterial recombineering by parallelized protein discovery. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:13689–13698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang H.H., Isaacs F.J., Carr P.A., Sun Z.Z., Xu G., Forest C.R., Church G.M.. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009; 460:894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Piñero Lambea C., Garcia-Ramallo E., Martinez S., Delgado J., Serrano L., Lluch-Senar M.. Mycoplasma pneumoniae genome editing based on oligo recombineering and cas9-mediated counterselection. ACS Synth. Biol. 2020; 9:1693–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ronda C., Pedersen L.E., Sommer M.O.A., Nielsen A.T.. CRMAGE: CRISPR optimized MAGE recombineering. Sci. Rep. 2016; 6:19452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aparicio T., de Lorenzo V., Martínez-García E.. CRISPR/Cas9-Based counterselection boosts recombineering efficiency in Pseudomonasputida. Biotechnol. J. 2018; 13:e1700161. [DOI] [PubMed] [Google Scholar]
- 26. Oh J.-H., van Pijkeren J.-P.. CRISPR-Cas9-assisted recombineering in Lactobacillusreuteri. Nucleic Acids Res. 2014; 42:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Corts A.D., Thomason L.C., Gill R.T., Gralnick J.A.. Efficient and precise genome editing in. ACS Synth. Biol. 2019; 8:1877–1889. [DOI] [PubMed] [Google Scholar]
- 28. Napolitano M.G., Landon M., Gregg C.J., Lajoie M.J., Govindarajan L., Mosberg J.A., Kuznetsov G., Goodman D.B., Vargas-Rodriguez O., Isaacs F.J.et al.. Emergent rules for codon choice elucidated by editing rare arginine codons in Escherichiacoli. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E5588–E5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Murphy K.C., Nelson S.J., Nambi S., Papavinasasundaram K., Baer C.E., Sassetti C.M.. ORBIT: a new paradigm for genetic engineering of mycobacterial chromosomes. Mbio. 2018; 9:e01467-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yus E., Maier T., Michalodimitrakis K., van Noort V., Yamada T., Chen W.-H., Wodke J.A.H., Güell M., Martínez S., Bourgeois R.et al.. Impact of genome reduction on bacterial metabolism and its regulation. Science. 2009; 326:1263–1268. [DOI] [PubMed] [Google Scholar]
- 31. Gibson D.G., Young L., Chuang R.-Y., Venter J.C., Hutchison C.A., Smith H.O.. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009; 6:343–345. [DOI] [PubMed] [Google Scholar]
- 32. Choi Y.J., Morel L., Le François T., Bourque D., Bourget L., Groleau D., Massie B., Míguez C.B.. Novel, versatile, and tightly regulated expression system for Escherichiacoli strains. Appl. Environ. Microbiol. 2010; 76:5058–5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Choi Y.J., Morel L., Bourque D., Mullick A., Massie B., Míguez C.B.. Bestowing inducibility on the cloned methanol dehydrogenase promoter (PmxaF) of Methylobacteriumextorquens by applying regulatory elements of Pseudomonasputida f1. Appl. Environ. Microbiol. 2006; 72:7723–7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kaczmarczyk A., Vorholt J.A., Francez-Charlot A.. Cumate-inducible gene expression system for sphingomonads and other alphaproteobacteria. Appl. Environ. Microbiol. 2013; 79:6795–6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Horbal L., Fedorenko V., Luzhetskyy A.. Novel and tightly regulated resorcinol and cumate-inducible expression systems for streptomyces and other actinobacteria. Appl. Microbiol. Biotechnol. 2014; 98:8641–8655. [DOI] [PubMed] [Google Scholar]
- 36. Seo S.-O., Schmidt-Dannert C.. Development of a synthetic cumate-inducible gene expression system for Bacillus. Appl. Microbiol. Biotechnol. 2019; 103:303–313. [DOI] [PubMed] [Google Scholar]
- 37. Mullick A., Xu Y., Warren R., Koutroumanis M., Guilbault C., Broussau S., Malenfant F., Bourget L., Lamoureux L., Lo R.et al.. The cumate gene-switch: a system for regulated expression in mammalian cells. BMC Biotechnol. 2006; 6:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Montero-Blay A., Miravet-Verde S., Lluch-Senar M., Piñero-Lambea C., Serrano L.. SynMyco transposon: engineering transposon vectors for efficient transformation of minimal genomes. DNA Res. 2019; 26:327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Le Grice S.F., Shih C.C., Whipple F., Sonenshein A.L.. Separation and analysis of the RNA polymerase binding sites of a complex Bacillussubtilis promoter. Mol. Gen. Genet. 1986; 204:229–236. [DOI] [PubMed] [Google Scholar]
- 40. Yus E., Yang J.-S., Sogues A., Serrano L.. A reporter system coupled with high-throughput sequencing unveils key bacterial transcription and translation determinants. Nat. Commun. 2017; 8:368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Langmead B., Wilks C., Antonescu V., Charles R.. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics. 2019; 35:421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.1000 Genome Project Data Processing Subgroup . The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Garrison E., Marth G.. Haplotype-based variant detection from short-read sequencing. 2012; arXiv doi:17 July 2012, preprint: not peer reviewedhttps://arxiv.org/abs/1207.3907.
- 45. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 46. Miravet-Verde S., Burgos R., Delgado J., Lluch-Senar M., Serrano L.. FASTQINS and ANUBIS: two bioinformatic tools to explore facts and artifacts in transposon sequencing and essentiality studies. Nucleic Acids Res. 2020; 48:e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Halbedel S., Stülke J.. Tools for the genetic analysis of Mycoplasma. Int. J. Med. Microbiol. 2007; 297:37–44. [DOI] [PubMed] [Google Scholar]
- 48. Sternberg N., Hamilton D.. Bacteriophage P1 site-specific recombination. I. Recombination between loxP sites. J. Mol. Biol. 1981; 150:467–486. [DOI] [PubMed] [Google Scholar]
- 49. Nagy A. Cre recombinase: the universal reagent for genome tailoring. Genesis. 2000; 26:99–109. [PubMed] [Google Scholar]
- 50. Suzuki E., Nakayama M.. VCre/VloxP and SCre/SloxP: new site-specific recombination systems for genome engineering. Nucleic Acids Res. 2011; 39:e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Garrido V., Piñero-Lambea C., Rodriguez-Arce I., Paetzold B., Ferrar T., Weber M., Garcia-Ramallo E., Gallo C., Collantes M., Peñuelas I.et al.. Engineering a genome-reduced bacterium to eliminate Staphylococcusaureus biofilms in vivo. Mol. Syst. Biol. 2021; 17:e10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mariscal A.M., González-González L., Querol E., Piñol J.. All-in-one construct for genome engineering using Cre-lox technology. DNA Res. 2016; 23:263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Beare P.A., Larson C.L., Gilk S.D., Heinzen R.A.. Two systems for targeted gene deletion in Coxiellaburnetii. Appl. Environ. Microbiol. 2012; 78:4580–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lluch-Senar M., Delgado J., Chen W.-H., Lloréns-Rico V., O’Reilly F.J., Wodke J.A., Unal E.B., Yus E., Martínez S., Nichols R.J.et al.. Defining a minimal cell: essentiality of small ORFs and ncRNAs in a genome-reduced bacterium. Mol. Syst. Biol. 2015; 11:780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Albert H., Dale E.C., Lee E., Ow D.W.. Site-specific integration of DNA into wild-type and mutant lox sites placed in the plant genome. Plant J. 1995; 7:649–659. [DOI] [PubMed] [Google Scholar]
- 56. Monteilhet C., Perrin A., Thierry A., Colleaux L., Dujon B.. Purification and characterization of the in vitro activity of I-Sce i, a novel and highly specific endonuclease encoded by a group i intron. Nucleic Acids Res. 1990; 18:1407–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pósfai G., Kolisnychenko V., Bereczki Z., Blattner F.R.. Markerless gene replacement in Escherichiacoli stimulated by a double-strand break in the chromosome. Nucleic Acids Res. 1999; 27:4409–4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sauer B., McDermott J.. DNA recombination with a heterospecific cre homolog identified from comparison of the pac-c1 regions of P1-related phages. Nucleic Acids Res. 2004; 32:6086–6095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Karimova M., Abi-Ghanem J., Berger N., Surendranath V., Pisabarro M.T., Buchholz F.. Vika/vox, a novel efficient and specific Cre/loxp-like site-specific recombination system. Nucleic Acids Res. 2013; 41:e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim A.I., Ghosh P., Aaron M.A., Bibb L.A., Jain S., Hatfull G.F.. Mycobacteriophage bxb1 integrates into the Mycobacteriumsmegmatis groEL1 gene. Mol. Microbiol. 2003; 50:463–473. [DOI] [PubMed] [Google Scholar]
- 61. Grindley N.D.F., Whiteson K.L., Rice P.A.. Mechanisms of site-specific recombination. Annu. Rev. Biochem. 2006; 75:567–605. [DOI] [PubMed] [Google Scholar]
- 62. Breton M., Sagné E., Duret S., Béven L., Citti C., Renaudin J.. First report of a tetracycline-inducible gene expression system for mollicutes. Microbiology (Reading). 2010; 156:198–205. [DOI] [PubMed] [Google Scholar]
- 63. Gibson D.G., Benders G.A., Andrews-Pfannkoch C., Denisova E.A., Baden-Tillson H., Zaveri J., Stockwell T.B., Brownley A., Thomas D.W., Algire M.A.et al.. Complete chemical synthesis, assembly, and cloning of a Mycoplasmagenitalium genome. Science. 2008; 319:1215–1220. [DOI] [PubMed] [Google Scholar]
- 64. Gibson D.G., Glass J.I., Lartigue C., Noskov V.N., Chuang R.-Y., Algire M.A., Benders G.A., Montague M.G., Ma L., Moodie M.M.et al.. Creation of a bacterial cell controlled by a chemically synthesized genome. Science. 2010; 329:52–56. [DOI] [PubMed] [Google Scholar]
- 65. Chandran S., Noskov V.N., Segall-Shapiro T.H., Ma L., Whiteis C., Lartigue C., Jores J., Vashee S., Chuang R.-Y.. TREC-IN: gene knock-in genetic tool for genomes cloned in yeast. BMC Genomics. 2014; 15:1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ruiz E., Talenton V., Dubrana M.-P., Guesdon G., Lluch-Senar M., Salin F., Sirand-Pugnet P., Arfi Y., Lartigue C.. CReasPy-Cloning: a method for simultaneous cloning and engineering of megabase-sized genomes in yeast using the CRISPR-Cas9 system. ACS Synth Biol. 2019; 8:2547–2557. [DOI] [PubMed] [Google Scholar]
- 67. Lartigue C., Glass J.I., Alperovich N., Pieper R., Parmar P.P., Hutchison C.A., Smith H.O., Venter J.C.. Genome transplantation in bacteria: changing one species to another. Science. 2007; 317:632–638. [DOI] [PubMed] [Google Scholar]
- 68. Labroussaa F., Lebaudy A., Baby V., Gourgues G., Matteau D., Vashee S., Sirand-Pugnet P., Rodrigue S., Lartigue C.. Impact of donor-recipient phylogenetic distance on bacterial genome transplantation. Nucleic Acids Res. 2016; 44:8501–8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Krishnakumar R., Assad-Garcia N., Benders G.A., Phan Q., Montague M.G., Glass J.I.. Targeted chromosomal knockouts in Mycoplasmapneumoniae. Appl. Environ. Microbiol. 2010; 76:5297–5299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Burgos R., Weber M., Martinez S., Lluch-Senar M., Serrano L.. Protein quality control and regulated proteolysis in the genome-reduced organism Mycoplasma pneumoniae. Mol. Syst. Biol. 2020; 16:e9530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Garcia-Morales L., Ruiz E., Gourgues G., Rideau F., Piñero-Lambea C., Lluch-Senar M., Blanchard A., Lartigue C.. A RAGE based strategy for the genome engineering of the human respiratory pathogen Mycoplasmapneumoniae. ACS Synth Biol. 2020; 9:2737–2748. [DOI] [PubMed] [Google Scholar]
- 72. Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K.A., Tomita M., Wanner B.L., Mori H.. Construction of Escherichiacoli K-12 in-frame, single-gene knockout mutants: the keio collection. Mol. Syst. Biol. 2006; 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gyorfy Z., Draskovits G., Vernyik V., Blattner F.F., Gaal T., Posfai G.. Engineered ribosomal RNA operon copy-number variants of E. coli reveal the evolutionary trade-offs shaping rRNA operon number. Nucleic Acids Res. 2015; 43:1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Roller B.R.K., Stoddard S.F., Schmidt T.M.. Exploiting rRNA operon copy number to investigate bacterial reproductive strategies. Nat Microbiol. 2016; 1:16160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Pósfai G., Plunkett G., Fehér T., Frisch D., Keil G.M., Umenhoffer K., Kolisnychenko V., Stahl B., Sharma S.S., de Arruda M.et al.. Emergent properties of reduced-genome Escherichiacoli. Science. 2006; 312:1044–1046. [DOI] [PubMed] [Google Scholar]
- 76. Komatsu M., Uchiyama T., Omura S., Cane D.E., Ikeda H.. Genome-minimized streptomyces host for the heterologous expression of secondary metabolism. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:2646–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Reuß D.R., Altenbuchner J., Mäder U., Rath H., Ischebeck T., Sappa P.K., Thürmer A., Guérin C., Nicolas P., Steil L.et al.. Large-scale reduction of the Bacillussubtilis genome: consequences for the transcriptional network, resource allocation, and metabolism. Genome Res. 2017; 27:289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. So Y., Park S.-Y., Park E.-H., Park S.-H., Kim E.-J., Pan J.-G., Choi S.-K.. A highly efficient CRISPR-Cas9-mediated large genomic deletion in Bacillussubtilis. Front. Microbiol. 2017; 8:1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Fu L., Xie C., Jin Z., Tu Z., Han L., Jin M., Xiang Y., Zhang A.. The prokaryotic argonaute proteins enhance homology sequence-directed recombination in bacteria. Nucleic Acids Res. 2019; 47:3568–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Khetrapal V., Mehershahi K., Rafee S., Chen S., Lim C.L., Chen S.L.. A set of powerful negative selection systems for unmodified enterobacteriaceae. Nucleic Acids Res. 2015; 43:e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hmelo L.R., Borlee B.R., Almblad H., Love M.E., Randall T.E., Tseng B.S., Lin C., Irie Y., Storek K.M., Yang J.J.et al.. Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange. Nat. Protoc. 2015; 10:1820–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yan M.-Y., Yan H.-Q., Ren G.-X., Zhao J.-P., Guo X.-P., Sun Y.-C.. CRISPR-Cas12a-Assisted recombineering in bacteria. Appl. Environ. Microbiol. 2017; 83:e00947-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Farzadfard F., Lu T.K.. Synthetic biology. Genomically encoded analog memory with precise in vivo DNA writing in living cell populations. Science. 2014; 346:1256272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Schubert M.G., Goodman D.B., Wannier T.M., Kaur D., Farzadfard F., Lu T.K., Shipman S.L., Church G.M.. High-throughput functional variant screens via in vivo production of single-stranded DNA. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2018181118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lopez S.C., Crawford K.D., Lear S.K., Bhattarai-Kline S., Shipman S.L.. Precise genome editing across kingdoms of life using retron-derived DNA. Nat. Chem. Biol. 2022; 18:199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Csörgő B., León L.M., Chau-Ly I.J., Vasquez-Rifo A., Berry J.D., Mahendra C., Crawford E.D., Lewis J.D., Bondy-Denomy J.. A compact cascade-cas3 system for targeted genome engineering. Nat. Methods. 2020; 17:1183–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Strecker J., Ladha A., Gardner Z., Schmid-Burgk J.L., Makarova K.S., Koonin E.V., Zhang F.. RNA-guided DNA insertion with CRISPR-associated transposases. Science. 2019; 365:48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Klompe S.E., Vo P.L.H., Halpin-Healy T.S., Sternberg S.H.. Transposon-encoded CRISPR-Cas systems direct RNA-guided DNA integration. Nature. 2019; 571:219–225. [DOI] [PubMed] [Google Scholar]
- 89. Vo P.L.H., Ronda C., Klompe S.E., Chen E.E., Acree C., Wang H.H., Sternberg S.H.. CRISPR RNA-guided integrases for high-efficiency, multiplexed bacterial genome engineering. Nat. Biotechnol. 2021; 39:480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Perutka J., Wang W., Goerlitz D., Lambowitz A.M.. Use of computer-designed group II introns to disrupt Escherichiacoli DExH/D-box protein and DNA helicase genes. J. Mol. Biol. 2004; 336:421–439. [DOI] [PubMed] [Google Scholar]
- 91. Heap J.T., Pennington O.J., Cartman S.T., Carter G.P., Minton N.P.. The clostron: a universal gene knock-out system for the genus Clostridium. J. Microbiol. Methods. 2007; 70:452–464. [DOI] [PubMed] [Google Scholar]
- 92. Plante I., Cousineau B.. Restriction for gene insertion within the Lactococcuslactis Ll.LtrB group II intron. RNA. 2006; 12:1980–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhong J., Karberg M., Lambowitz A.M.. Targeted and random bacterial gene disruption using a group II intron (targetron) vector containing a retrotransposition-activated selectable marker. Nucleic Acids Res. 2003; 31:1656–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Velázquez E., Lorenzo V.de, Al-Ramahi Y.. Recombination-independent genome editing through CRISPR/Cas9-enhanced targetron delivery. ACS Synth. Biol. 2019; 8:2186–2193. [DOI] [PubMed] [Google Scholar]
- 95. Enyeart P.J., Chirieleison S.M., Dao M.N., Perutka J., Quandt E.M., Yao J., Whitt J.T., Keatinge-Clay A.T., Lambowitz A.M., Ellington A.D.. Generalized bacterial genome editing using mobile group II introns and Cre-lox. Mol. Syst. Biol. 2013; 9:685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wang K., Fredens J., Brunner S.F., Kim S.H., Chia T., Chin J.W.. Defining synonymous codon compression schemes by genome recoding. Nature. 2016; 539:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Nyerges Á., Csörgő B., Nagy I., Bálint B., Bihari P., Lázár V., Apjok G., Umenhoffer K., Bogos B., Pósfai G.et al.. A highly precise and portable genome engineering method allows comparison of mutational effects across bacterial species. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:2502–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole genome sequencing data are available at Array Express under accession E-MTAB-11600. All materials generated in this work are available upon request.