


Abstract
Quick growth restart after upon encountering favourable environmental conditions is a major fitness contributor in natural environment. It is widely assumed that the time required to restart growth after nutritional upshift is determined by how long it takes for cells to synthesize enough ribosomes to produce the proteins required to reinitiate growth. Here we show that a reduction in the capacity to synthesize ribosomes by reducing number of ribosomal RNA (rRNA) operons (rrn) causes a longer transition from stationary phase to growth of Escherichia coli primarily due to high mortality rates. Cell death results from DNA replication blockage and massive DNA breakage at the sites of the remaining rrn operons that become overloaded with RNA polymerases (RNAPs). Mortality rates and growth restart duration can be reduced by preventing R-loop formation and improving DNA repair capacity. The same molecular mechanisms determine the duration of the recovery phase after ribosome-damaging stresses, such as antibiotics, exposure to bile salts or high temperature. Our study therefore suggests that a major function of rrn operon multiplicity is to ensure that individual rrn operons are not saturated by RNAPs, which can result in catastrophic chromosome replication failure and cell death during adaptation to environmental fluctuations.
Graphical Abstract
Graphical Abstract.
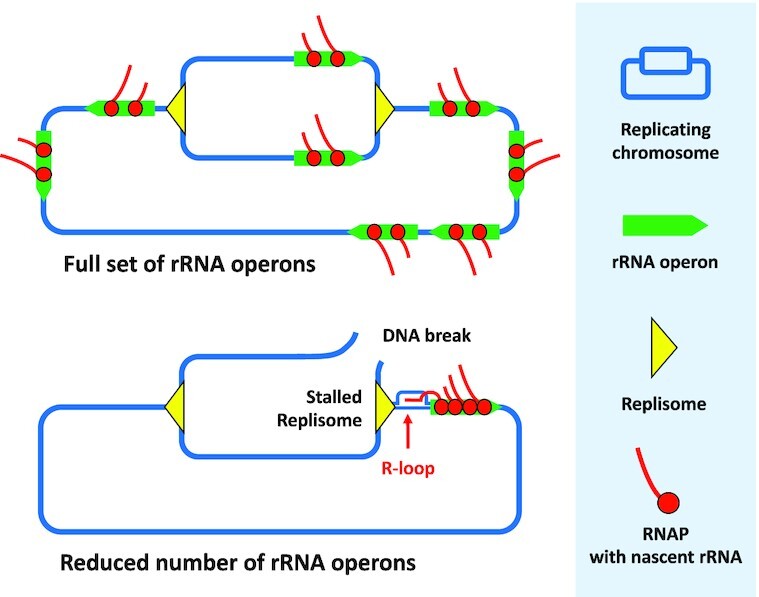
rRNA operon multiplicity assures genome stability by decreasing occurrence of deleterious replication-transcription conflicts during adaptation to environmental fluctuations.
INTRODUCTION
The capability for rapid growth and reproduction when resources are abundant is an essential component of the evolutionary success of microbes in constantly fluctuating natural environments. Different mechanisms determine growth and reproduction rates, but protein synthesis plays a paramount role. Because it is the most energetically expensive process in the cell, resource abundance is indispensable for efficient and extensive protein synthesis. Indeed, as much as 70% of energy resources are devoted to the translation machinery and protein synthesis in rapidly growing Escherichia coli cells (1). In cells growing in rich medium, ribosomes constitute up to 50% of dry mass, ribosomal proteins represent about 20–25% of all proteins, ribosomal RNA (rRNA) synthesis represents about 73% of the total RNA synthesis and requires 68% of all transcribing RNA polymerases (RNAPs) (2–6). Consequently, cells must constantly adapt the rate of ribosome production to resource availability. This is primarily achieved by regulating rRNA synthesis, to which the synthesis of ribosomal proteins is adjusted (7,8).
The genes encoding rRNAs in bacteria are organized in rRNA operons (rrn). In E. coli all rrn operons have the same structure: P1 and P2 promoters-16S rRNA-spacer tRNA(s)-23S rRNA-5S rRNA-distal tRNA(s) (9). Strong rrn operon promoters ensure that the transcription rate per rRNA gene is much higher than transcription rates for protein-coding genes (10). Nevertheless, for most bacteria, a single rrn operon is not sufficient to ensure fast growth and reproduction. Because the functional product of rrn operons is RNA and not protein, and a translation amplification step is therefore not possible, the best solution to this problem is to increase the number of the rrn operons per genome. Indeed, the number of rrn operons per bacterial genome, which varies from 1 to 21, is correlated with the maximal growth rates of different species (11–13).
The capacity for rapid growth is not the sole purpose of high rrn operon copy number per genome. For example, it is possible to delete a few rrn operons in both E. coli and Bacillus subtilis, which have 7 and 10 operons, respectively, without having a major effect on cellular ribosome concentration or growth rate (14,15). This is due to increased transcription of the remaining rrn copies through the derepression of a feedback regulatory mechanism. Reducing the number of rrn operons in E. coli results in an increased number of RNA polymerases (RNAP) per remaining rRNA gene through enhanced promoter initiation frequency, accompanied by an increased RNAP elongation rate (14). Therefore, in wild-type E. coli, 7 rrn operons are not fully saturated with the RNAPs even at highest growth rates. It was previously shown that the full set of rrn operons is required for rapid adaptation to favourable environmental conditions, such as nutrient upshift or temperature change (16,17). This was attributed primarily to a difficulty in rapidly making enough ribosomes for optimal growth in the new conditions, because the demand on the remaining rrn operons is quickly saturated. However, introduction of additional chromosomal and plasmid-borne rrn operons in E. coli increased lag phase and doubling times both under stable and fluctuating growth conditions (18,19). Therefore, the E. coli rrn operon copy number seems to have been optimised during evolution to ensure survival and maximise fitness in fluctuating natural environments.
The rrn operons in E. coli and other bacteria are oriented in such a way that they are transcribed in the same orientation as chromosome replication (20), to reduce the possibility of deleterious head-on collisions between the DNA replication and RNA transcription machineries (21). However, it was reported that rrn operons are nonetheless hotspots for transcription-dependent DNA replication blockage in ∼ 5–10% of cells in rapidly growing B. subtilis populations (22), suggesting that highly transcribed rrn operons might be obstacles for DNA replication progression in vivo. Because DNA replication blockages can result in DNA breakage and cell death, they must be avoided or rapidly resolved. We hypothesized that the large number of rrn operons per genome, which decreases the density of transcribing RNAPs per operon, may reduce the probability of DNA replication blockages.
To test this hypothesis, we examined the impact of varying the number of rrn operons in the E. coli genome on cell growth, DNA replication, genome stability and mortality rates. We found that decreasing number of rrn operons increases the lag-time before cell populations can resume growth upon exiting from the stationary phase mostly due to high mortality rates. The primary cause of cell death is DNA breakage resulting from transcription-dependent DNA replication blockages at the sites of the remaining overexpressed rrn operons. We also observed transcription-replication conflicts in cells that have a full set of rrn operons during recovery from stresses that lead to ribosome degradation, and that these conflicts can be avoided by increasing number of rrn operons. We conclude that rrn operon multiplicity, which ensures that individual rrn operons are not saturated by RNAP, is a guarantor of genome stability.
MATERIAL AND METHODS
Strains, media, culture conditions and chemicals
Bacterial strains and plasmids used in this study are described in Table 1. All strains are derivatives of the E. coli K12 MG1655 strain, which has a standard number of seven rrn operons per genome that is referred as wild-type (WT). All mutant derivatives were constructed by P1vir transduction of the alleles from either the Keio collection (23) or from our laboratory collection. Pre-cultures for all experiments were grown at 30°C because it was previously shown that mutants that have problems with DNA replication, such as holD, suffer less at 30°C (24). In addition, E. coli cells growing at low temperature have lower base-pair substitutions rates (25), which should reduce the probability that suppressor mutations will arise in strains having reduced number of rrn operons. Experiments were performed using Luria-Bertani (LB) medium (Difco) or M9 Minimal salts (Serva) supplemented with 0.4% glucose. When needed, medium was supplemented with antibiotics (Sigma-Aldrich) at the following concentrations: ampicillin (Amp) 100 μg ml−1, chloramphenicol (Cm) 30 μg ml−1, kanamycin (Kan) 100 μg ml−1, and tetracycline (Tet) 12.5 μg ml−1. Medium was supplemented with IPTG (Sigma-Aldrich) at the concentrations: 0.01 mM and 0.1 mM, to induce Ptac-uvsW and pACYC184 Ptrc-rnh, respectively.
Table 1.
Bacterial strains and plasmids
| Name | E. coli Stains | Genotype | Sources |
|---|---|---|---|
| SF1 | MG1655 : 7 rrn (WT) | rph-1λ– | |
| SF2 | 1 rrnC | rph-1λ–Δ(rrsH-aspU)794::FRT Δ(rrfG-rrsG)791::FRT Δ(rrfF-rrsD)793::FRT Δ(rrsA-rrfA)792::FRT Δ(rrsB-rrfB)790::FRT Δ(rrsE-rrfE)789::FRT [ptRNA67] = p15a aadA | (33) |
| SF3 | 1 rrnE | rph-1λ–Δ(rrsH-aspU)794::FRT Δ(rrfG-rrsG)791::FRT Δ(rrfD-rrsD)793::FRT Δ(rrsC-trpT)795::FRT Δ(rrsA-rrfA)792::FRT Δ(rrsB-rrfB)790::FRT [ptRNA67] = p15a aadA | (33) |
| SF4 | 2 rrnC rrnD | SF2 but rrnD+ | This study |
| SF5 | 2 rrnC rrnB | rph-1λ–Δ(rrsH-aspU)794::FRT Δ(rrfG-rrsG)791::FRT Δ(rrfF-rrsD)793::FRT Δ(rrsA-rrfA)792::FRTΔ(rrsE-rrfE)789::FRT [ptRNA67] = p15a aadA | (33) |
| SF6 | 1 rrnD | SF4 but Δ(rrsC-trpT)795::FRT | This study |
| SF7 | 3 rrn | rph-1λ–Δ(rrfG-rrsG)791::FRT Δ(rrfF-rrsD)793::FRT Δ(rrsA-rrfA)792::FRT Δ(rrsB-rrfB)790::FRT | (33) |
| SF8 | 4 rrn | rph-1λ–Δ(rrfG-rrsG)791::FRTΔ(rrsA-rrfA)792::FRT Δ(rrsB-rrfB)790::FRT | (33) |
| SF9 | 5 rrn | rph-1λ–Δ(rrsE-rrfE)789::FRT, Δ(rrfG-rrsG)791::FRT | (33) |
| SF10 | 6 rrn | rph-1λ–Δ(rrsE-rrfE)785(::FRT) | (33) |
| SF11 | 8 rrn | rph-1λ–+ rrnB(araC)71267 | (18) |
| SF12 | 9 rrn | rph-1λ–+ rrnB (araC)71267, rrnB(yibF)3761992 | (18) |
| SF13 | 10 rrn | rph-1λ–+ rrnB(araC)71267, rrnB(yibF)3761992, rrnB (mocA) 3015910 | (18) |
| SF14 | dnaA(Sx) | as SF1 but dnaA(Sx) 721 zib::Tn10 | Thisstudy,(72) |
| SF15 | 1 rrnC dnaA(Sx) | as SF2 but dnaA(Sx) 721 zib::Tn10 | This study, (72) |
| SF16 | ΔdinG | as SF1 but ΔdinG:: kan | This studya |
| SF17 | 1 rrnC ΔdinG | as SF2 but ΔdinG:: kan | This studya |
| SF18 | ΔgreA | as SF1 but ΔgreA:: kan | This studya |
| SF19 | 1 rrnC ΔgreA | as SF2 but ΔgreA :: kan | This studya |
| SF20 | ΔhflX | as SF1 but dnaA(Sx) 721 zib::Tn10 | This study |
| SF21 | 1rrnC ΔhflX | as SF2 but dnaA(Sx) 721 zib::Tn10 | This study |
| SF22 | lexA1 (Ind–) | as SF1 but lexA1 malB::Tn9 | This study |
| SF23 | 1 rrnC lexA1 (Ind–) | as SF2 but lexA1 malB::Tn9 | This study |
| SF24 | lexA (Def) ΔsulA | as SF1 but lexA:: kanΔsulA | This studya |
| SF25 | 1 rrnC lexA (Def) ΔsulA | as SF2 but lexA:: kan ΔsulA | This studya |
| SF26 | Δmfd | as SF1 but Δmfd :: kan | This studya |
| SF27 | 1 rrnC Δmfd | as SF2 but Δmfd :: kan | This studya |
| SF28 | Δmfd rnh + | as SF26 + pACYC184-rnh | This study |
| SF29 | 1 rrnC Δmfd rnh + | as SF27 + pACYC184-rnh | This study |
| SF30 | Δmfd uvsW + | as SF26 but Δ(argF-lac)U169 Ptac-uvsW-amp | This study |
| SF31 | 1 rrnC Δmfd uvsW + | as SF27 but Δ(argF-lac)U169 Ptac-uvsW-amp | This study |
| SF32 | nusG F165A | as SF1 but nusGF165A:: kan | Thisstudy,(52) |
| SF33 | 1 rrnC nusGF165A | as SF2 but nusGF165A:: kan | This study, (52) |
| SF34 | nusG F165A rpoC* | as SF32 but rpoCΔ215–220 thiC::tn10 | Thisstudy, (73) |
| SF35 | nusG F165A rnh+ | as SF32 + pACYC184-rnh | This study |
| SF36 | ΔrapA | as SF1 but ΔrapA :: kan | This studya |
| SF37 | 1 rrnC ΔrapA | as SF2 but ΔrapA :: kan | This studya |
| SF38 | ΔrecA | as SF1 but ΔrecA :: kan | This studya |
| SF39 | 1 rrnC ΔrecA | as SF2 but ΔrecA :: kan | This studya |
| SF40 | ΔrecA rnh+ | as SF38 + pACYC184-rnh | This study |
| SF41 | 1 rrnC ΔrecA rnh+ | as SF39 + pACYC184-rnh | This study |
| SF42 | ΔrecA uvsW + | as SF38 butΔ(argF-lac)U169 Ptac-uvsW-amp | This study |
| SF43 | 1 rrnC ΔrecA uvsW + | as SF39 butΔ(argF-lac)U169 Ptac-uvsW-amp | This study |
| SF44 | ΔrecB | as SF1 but ΔrecB :: kan | This studya |
| SF45 | 1 rrnC ΔrecB | as SF2 but ΔrecB :: kan | This studya |
| SF46 | ΔrecF | as SF1 but ΔrecF :: kan | This studya |
| SF47 | 1 rrnC ΔrecF | as SF2 but ΔrecF :: kan | This studya |
| SF48 | ΔrecQ | as SF1 but ΔrecQ :: kan | This studya |
| SF49 | 1 rrnC ΔrecQ | as SF2 but ΔrecQ :: kan | This studya |
| SF50 | Δrep | as SF1 but Δrep :: kan | This studya |
| SF51 | 1 rrnC Δrep | as SF2 but Δrep :: kan | This studya |
| SF52 | rnh+ | as SF1 + pACYC184-rnh | This study |
| SF53 | 1 rrnC rnh+ | as SF2 + pACYC184-rnh | This study |
| SF54 | ΔrnhA | as SF1 but ΔrnhA :: kan | This studya |
| SF55 | 1 rrnC ΔrnhA | as SF2 but ΔrnhA :: kan | This studya |
| SF56 | ΔrnhA uvsW + | as SF54 but Δ(argF-lac)U169 Ptac-uvsW-amp | This study |
| SF57 | 1 rrnC ΔrnhA uvsW + | as SF55 but Δ(argF-lac)U169 Ptac-uvsW-amp | This study |
| SF58 | rpoC* | as SF1 but rpoCΔ215–220 thiC::tn10 | This study |
| SF59 | 1 rrnC rpoC* | as SF2 but rpoCΔ215–220 thiC::tn10 | This study |
| SF60 | ΔruvA | as SF1 but ΔruvA :: kan | This studya |
| SF61 | 1 rrnC ΔruvA | as SF2 but ΔruvA :: kan | This studya |
| SF62 | ΔuvrD | as SF1 but ΔuvrD:: kan | This studya |
| SF63 | 1 rrnC ΔuvrD | as SF2 but ΔuvrD:: kan | This studya |
| SF64 | uvsW + | as SF1 but Δ(argF-lac)U169 Ptac- uvsW -amp | Thisstudy, (74) |
| SF65 | 1 rrnC uvsW + | as SF2 but Δ(argF-lac)U169 Ptac- uvsW -amp | This study |
| SF66 | 1 rrn P1rrnB- gfp | as SF2 but attB::P1rrnB- gfpASV | Thisstudy, (27) |
| SF67 | 2 rrn P1rrnB- gfp | as SF5 but attB:: P1rrnB- gfpASV | This study |
| SF68 | 3 rrn P1rrnB- gfp | as SF7 but attB:: P1rrnB- gfpASV | This study |
| SF69 | 4 rrn P1rrnB- gfp | as SF8 but attB:: P1rrnB- gfpASV | This study |
| SF70 | 5 rrn P1rrnB- gfp | as SF9 but attB:: P1rrnB- gfpASV | This study |
| SF71 | 6 rrn P1rrnB- gfp | as SF10 but attB:: P1rrnB- gfpASV | This study |
| SF72 | 7 rrn P1rrnB- gfp | as SF1 but attB:: P1rrnB- gfpASV | This study |
| SF73 | 8 rrn P1rrnB- gfp | as SF11 but attB:: P1rrnB- gfpASV | This study |
| SF74 | 9 rrn P1rrnB- gfp | as SF12 but attB:: P1rrnB- gfpASV | This study |
| SF75 | 10 rrn P1rrnB- gfp | as SF13 but attB::P1rrnB- gfpASV | This study |
| SF76 | 7 rrn P1rrnB- gfp rpoC* | as SF72 but rpoCΔ215–220 thiC::tn10 | |
| SF77 | 1 rrn P1rrnB- gfp rpoC* | as SF66 but rpoCΔ215–220 thiC::tn10 | This study |
| SF78 | 1 rrn PrecA-yfp | as SF2 but intC::PrecA-yfp-cm | This study |
| SF79 | 2 rrn PrecA-yfp | as SF5 but intC::PrecA-yfp-cm | This study |
| SF80 | 3 rrn PrecA-yfp | as SF7 but intC::PrecA-yfp-cm | This study |
| SF81 | 4 rrn PrecA-yfp | as SF8 but intC::PrecA-yfp-cm | This study |
| SF82 | 5 rrn PrecA-yfp | as SF9 but intC::PrecA-yfp-cm | This study |
| SF83 | 6 rrn PrecA-yfp | as SF10 but intC::PrecA-yfp-cm | This study |
| SF84 | 7 rrn PrecA-yfp | as SF1 but intC::PrecA-yfp-cm | This study |
| SF85 | 8 rrn PrecA-yfp | as SF11 but intC::PrecA-yfp-cm | This study |
| SF86 | 9 rrn PrecA-yfp | as SF12 but intC::PrecA-yfp-cm | This study |
| SF87 | 10 rrn PrecA-yfp | as SF13 but intC::PrecA-yfp-cm | This study |
| SF88 | 1 rrn PrecA-yfp lexA1 (Ind–) | as SF78 but SF22 | This study |
| SF89 | 7 rrn PrecA-yfp lexA1 (Ind–) | as SF84 but SF22 | This study |
| SF90 | 1 rrn PrpsT-gfp | as SF2 but SF101 | This study |
| SF91 | 7 rrn PrpsT-gfp | as SF1 but SF101 | This study |
| SF92 | MG1655 Pbla-gfp | as SF1 but galK::Pbla-gfp-cm | Lab collection |
| SF93 | 1 rrn Pbla-gfp | as SF2 but SF92 | This study |
| SF94 | 7 rrn Pbla-gfp | as SF1 but SF92 | This study |
| SF95 | 1 rrn pK4-16 | as SF2 but SF102 | This study |
| SF96 | 7 rrn pK4-16 | as SF1 but SF102 | This study |
| Name | B.subtilis Stains | Genotype | Sources |
| SF97 | 10 rrn | trpC2 | (75) |
| SF98 | 1 rrn | trpC2 ΔrrnHG1 ΔrrnO1 ΔrrnD1 ΔrrnE1 ΔrrnB2 ΔrrnI2 ΔrrnW2 ΔrrnJ1::cat (Δ9 rrnA+) | (75) |
| Plasmids | |||
| SF99 | DH5alpha pCP20 | Δ(argF-lac)169, phi80dlacZ58 (M15), glnV44(AS), λ–, rfbD1, gyrA96 (NalR), recA1, endA1, spoT1, thi-1, hsd | (76) |
| SF100 | P rnh+ | [pEM001] = pACYC184-rnh | (66) |
| SF101 | P rpsT | Promoter specific GFP - kan | (26) |
| SF102 | pK4-16 | pSC101 ori, rrnB - kan | (33) |
aDerivative strains were constructed using P1 transduction of alleles from Keio collection (Baba et al., 2006).
Determination of growth curves
Overnight cultures were diluted 1/1,000 and dispensed in 96-well plates. The volume of the cultures in each well was 150 μl of LB medium and 20 μl of mineral oil. It has been previously found that mineral oil does not affect the bacterial growth rates and does not increase the expression of genes that are induced under anaerobic conditions (26). During growth at 37°C with shaking, OD600nm was measured every 2 min over 15 h of growth using a Tecan Infinite M200 PRO microplate reader. Lag time was defined as a time required after dilution to reach the first point of intersection of an extrapolation of the linear portion of the growth curve (semi-log plot) with the growth curve itself. The doubling time was defined as the time required for the bacterial population to increase OD600nm from 0.1 to 0.2.
Measuring expression of fluorescent protein-based transcription reporters
Overnight cultures were diluted 1/1,000 and dispensed in 96-well plates. The volume of the cultures in each well was 150 μl of LB medium and 20 μl of mineral oil. During growth at 37°C, OD600nm and fluorescence was measured every 2 min over 15 h of growth using a Tecan Infinite M200 PRO microplate reader. For GFP-based reporters, excitation at 480 nm and emission at 510 nm was used for analysis. For the YFP-based reporter, excitation at 490 nm and emission at 530 nm was used for analysis.
Microfluidics experiments
Cells from overnight cultures were concentrated 10 × by centrifugation, after which they were loaded into a microfluidic mother machine device (27) by diffusion. The mother machine microfluidic device consists of separate growth channels, which are closed on one end and open to the media channel on other. The ‘mother’ cell is retained at the closed end, while sister and daughter cells are pushed along the growth channel towards the media channel until they are washed away.
Fresh LB medium was infused with a syringe pump at 1 ml/h. For each experiment, images were acquired for 5 h at 5 min intervals using Metamorph software and a Nikon Eclipse Ti fluorescence microscope equipped with a motorized stage and a CCD camera (Photometrics CoolSnap HQ2). To extract information about the cells from the images, each channel containing cells was cut and pasted on a new image and aligned from left to right according to the acquisition time. The resulting image contained cell segmentation information and was analysed by ImageJ. The cell lineage was determined by visualizing the arrows connecting the cells. The division time of one cell was defined as the time between the birth of one cell and its first division.
DNA content analysis
DNA content quantification was performed as previously described (28). Overnight cultures were diluted 1/1,000 in fresh LB medium and incubated at 37°C with agitation until OD600nm reached 0.05. Cephalexin (10 μg ml−1) was then added at time 0 to block cell division. Rifampicin (150 μg ml−1) was added either simultaneously with cephalexin at time 0 or after 40 min of incubation with cephalexin. Rifampicin inhibits initiation of replication but allows ongoing replication rounds to finish, thus enabling estimation of the number of chromosome equivalents. Cells were incubated simultaneously with both antibiotics at 37°C for an additional 90 min. Cells were washed with PBS before being fixed with 100% cold MeOH for 1 h. Fixed cells were washed with PBS and the DNA was stained with DAPI (4′,6-diamidino-2-phenylindole, 0.3 μg ml−1). For each time point, a minimum of 50,000 cells was analysed using Beckman Coulter Gallios flow cytometer with a maximum of 2,000 events counted per second. DAPI was excited with a UV laser at 355 nm and read at 450 nm. The number of the peaks of histograms showing amount of DNA in these cells corresponds to the number of fully replicated chromosomes and reflects the number of origins present in the cell at the time the drugs were added. Number of genome equivalents (N) was calculated using stationary phase cells (1N) as a reference.
Quantification of DNA damage
DNA damage was quantified using the Promega Fluorometric DeadEndTM Fluorometric TUNEL system. TUNEL is an acronym for terminal deoxynucleotidyl transferase biotin-dUTP nick end labelling. Overnight cultures were diluted 1/1,000 in fresh LB medium and incubated at 37°C with agitation until they reached the OD600nm 0.05. 5 × 106 cells were then centrifuged at 5,000 rpm for 10 min at 4°C, washed twice with cold PBS, and resuspended in 0.5 ml of PBS. Cells were fixed with 5 ml 1% MeOH-free formaldehyde on ice for 20 min then washed twice with cold PBS, and permeabilized with 0.2% Triton X-100 solution for 5 min at room temperature. Cells were then washed twice with PBS and stained according to the manufacturer's recommendations. Green fluorescence was measured using Gallios flow cytometer (Beckman Coulter). A minimum of 50,000 cells was analysed for each time point with a maximum of 2,000 events counted per second.
Total RNA quantification
Total RNA quantification was performed as previously described (29). Briefly, overnight cultures were diluted 1/1,000 in fresh LB medium and incubated at 37°C with agitation until they reached OD600nm 0.05 for the lag phase and 0.7 for the exponential phase. For each growth phase, 1 ml of culture was centrifuged 2 min at 13.000 rpm. The supernatant was removed and pellet was resuspended in 140 μl of RNA extraction solution (18 mM of EDTA, 0.025% of SDS, 1% of 2-mercaptoethanol and 95% of formamide) and kept in dry ice. Then, cells were vortexed 10 min at maximal speed and incubated 10 min at 80°C. Cells were centrifuged 10 min at 4°C and supernatant was transferred to a clean tube. RNA was quantified by measuring OD260nm with nanodrop (RNA extraction solution was used as a blank).
Quantification of 70S ribosome concentrations
The 70S ribosome concentration quantification was performed as previously described (30). Briefly, overnight cultures were diluted 1/1,000 in fresh LB medium and incubated at 37°C with agitation until they reached OD600nm 0.05 for the lag phase and 0.7 for exponential phase. For each growth phase, an aliquot of culture was removed for serial dilutions for determined viable cell count. The rest of cells was centrifuged 10 min at 4°C and pellet was resuspended in 20 ml of Buffer A (20 mM Tris pH7.5, 0.2 M of NH4Cl and 10 mM of MgCl2) + 6 mM β-mercaptoethanol. Cells were then centrifuged at 6,000 rpm for 10 min at 4°C and pellets stored over night at −20°C if necessary. The pellet was resuspended in 1.7 ml in buffer A + 6 mM β-mercaptoethanol + 4 μl DNase I (10 mg/ml). Cells were broken twice in a chilled French press (900psi, medium pressure) and then transferred to a clean tube for centrifugation at 13,000 rpm at 4°C for 30 min. The supernatant was transferred to a TLA 100.3 tube for ultracentrifugation (80,000 rpm at 4°C for 40 min). The pellet was resuspended in 500 μl Buffer A + 6 mM β-mercaptoethanol and vortexed for 20 min in cold-room. Once the solution was homogenous, OD260nm was measured with a Nanodrop using buffer A as a blank. The concentration of 70S ribosomes was calculated with an extinction coefficient of 24 pmol/OD260nm unit: OD260nm x 24 x (6.02 × 1011) / viable count, where 6.02 × 1011 is Avogadro's number (molecules per pmole).
Determination of mutation rates
To determine rate of appearance of rifampicin-resistant (RifR) or nalidixic acid-resistant (NalR) mutants, overnight cultures were diluted 1/10,000 in fresh LB medium and incubated overnight at 37°C with shaking at 150 rpm. The next day, 5 ml of fresh LB medium was inoculated with 20–100 bacterial cells from overnight cultures, so that there were no pre-existing antibiotic-resistant mutants in the starting inoculum. Cultures were incubated for 20 h at 37°C with shaking at 150 rpm. Subsequently, appropriate dilutions were plated on LB agar plates and on selective media to determine total number of colony forming units (CFUs): LB was supplemented with 100 μg ml−1 of rifampicin or 20 μg ml−1 of nalidixic acid to determine number of the antibiotic resistant mutants. Plates were incubated 24 h at 37°C. Mutation rates and their respective confidence intervals were estimated by applying the Ma–Sandri–Sarkar Maximum Likelihood Estimation Method using the Fluctuation Analysis Calculator (31) (https://lianglab.brocku.ca/FALCOR/).
Detection of the RNA:DNA hybrids by immunoblotting with S9.6 monoclonal antibody
Overnight cultures were diluted 1/1,000 in fresh LB medium in flasks and incubated at 37°C with agitation until they reached OD600nm 0.05. Total nucleic acids were extracted using a Wizard Genomic DNA kit (Promega). Next, 1 μg of nucleic acids from each sample was spotted on Hybond-N + nylon membrane (Amersham pharmacia biotech) using a slot blot apparatus with vacuum suction. Membranes were then UV-crosslinked (0.12 J/m2), blocked in 5% milk/TBST (Tris buffer saline + 0.5% Tween 20 – Bio-RAD), and incubated overnight at 4°C. Antibody S9.6 (Merck) (1 μg) was added in 1% milk/TBST for 2 h at 4°C. Blots were washed 3 times with TBST and secondary antibody (1:10,000 goat anti-mouse HRP) was added for 1 h at RT. Blots were washed 3 times with TBS followed by reaction with enzyme-conjugated anti-mouse secondary antibody and detection with a chemiluminesence kit (Clarity ACL Bio-Rad). For loading controls of the different nucleic acid samples, the membrane was washed in distilled water, and transferred in 5% acetic acid with shaking for 15 min, and then stained with 0.05% methylene blue in 0.5 M sodium acetate buffer (pH5.2). As control for the S9.6 antibody specificity, we used ΔrnhA mutant and an addition of RNaseH (Invitrogen) (Supplementary Figure S5BC). For dot blot quantification, we used the « blot analysis » function in the ImageJ software (version 1.52a). The method consists of subtracting the background and measuring the integrated density of each point.
Quantification DNA degradation around rrn loci by qPCR analysis
Overnight cultures were diluted 1/1,000 in fresh LB medium in flasks and incubated at 37°C with agitation until they reached OD600nm 0.05. Total nucleic acids were extracted using a Wizard Genomic DNA kit (Promega). Quantitative PCR were performed using the Qiagen kit QuantiFast® SYBR® Green PCR in a LigthCycler® 480 instrument (Roche Molecular Systems, Inc) with specific oligonucleotides. Fold change are presented as relative values using the Ct approach with the specific control locus. For this experiment, primers that allow amplification of two 0.5 kb-long regions, which are located 2 kb upstream and downstream of a rrn operon in WT strains and in different strains having only one rrn operon were used. For each sample, a set of primers allowing amplification of the control locus located 250 kb upstream of the rrn copy was used.
Primers for rrnC±, upstream amplification: For 5′ CGCACAACAGTTAGCTGCACA 3′, Rev 5′ ACGGATTGGCGCTCATTTCATA 3′; downstream: For 5′ TCTGACCCAGTCAGCAGTGAG 3′, Rev 5′ CGATCAAACCTGCCTCATGCTG 3′. upstream amplification −250 kb: For 5′ GGGCAGAACTTCCCGGTACTC 3′, Rev 5′ AGGCTGATATCCATCCGCGAA 3′
Primers for rrnE±, upstream amplification: For 5′ AACAACGTCGTCCAGTCCGC 3′, Rev 5′ AGGTCGAAACGGGTTGCAAG 3′; downstream: For 5′ GTCAGGAGACGCTGCCTTCA 3′, Rev 5′ CTTAAGCATTGCCATGACCGC 3′. upstream amplification −250 kb: For 5′ GGACAAGCAGCACTCTGATGT 3′, Rev 5′ GCAGTTTCGCCGGGTTAG 3′
Primers for rrnD±, upstream amplification: For 5′AAATCGGTCAGCGCGTAATG 3′, Rev 5′ GCTGGGTCTGGTTACCCT 3′; downstream: For 5′ ACCTGCTAACGCGATGATTA 3′, rev 5′ CCGACGTTGGCTCATCAAA 3′. upstream amplification −250 kb: For 5′ CGTTGATTCCGGGAAAGGGG 3′. rev 5′ TCGCACCGACAATGTAGTTGC 3′
Primers for rpsT loci, upstream amplification: For 5′ GCAGCAGCGCGCCTTTTTCC 3′, Rev 5′ ACGTTAACGGCCCAATCTCG 3′; downstream: For 5′ CGCTCGAATCTGTCCGGTAA 3′, rev 5′ TTTCACTGCAAGGCGTCACG 3′. upstream amplification −250 kb: For 5′ ATGTTGAAATCCCCCCTGTT 3′. rev 5′ GTTCAATGGTCAGCGCCGTT 3′
Double strand DNA breaks detection using Gam-GFP reporter
Double-strand DNA breaks were scored by quantifying Gam-GFP fluorescent foci as described previously (32). Overnight cultures were diluted 1/1,000 in fresh LB medium and incubated in flask at 37°C with agitation until they reached OD600nm 0.05. Cells were washed with PBS and inoculated onto a solid matrix of minimal medium agarose in microscope cavity slides. Cells were examined with a 100 × objective and images were acquired using Metamorph software and a Nikon Eclipse Ti fluorescence microscope equipped with a motorized stage and a CCD camera (Photometrics CoolSnap HQ2).
Viability assay
Overnight cultures were diluted 1/1,000 in fresh LB medium and incubated at 37°C with agitation at 150 rpm. At different time points, aliquots of cells were transferred to clean tubes and centrifuged at 13,000 rpm at 4°C for 10 min. Next, the pellet was resuspended in PBS with 1 μM propidium iodide (PI). After 15 min of incubation at room temperature in the dark, cells were washed in 10–2 M MgSO4 then analysed using Gallios flow cytometer (Beckman Coulter). PI florescence was measured with a 488 nm excitation laser and 620 nm emission filter. Cells killed by incubation at 90°C for 5 min were used as a positive control. A minimum of 50,000 cells was analysed for each time point with a maximum of 2,000 events counted per second.
Determining duration of the recovery phase after exposure to different stress conditions
Overnight cultures were diluted 1/1,000 in fresh LB medium and incubated in flask at 37°C with agitation until they reached OD600nm 0.7. Next, cells were diluted 1:100 in fresh LB and grown again without and with different stresses: 30 min at 50°C; 5 min with 1 μg ml−1 of colistin; 60 min with 5% of sodium deoxycholate; 60 min with 1M of NaCl. The survival of different strains for a given stress was not different according to the CFU count (see Supplementary Tables S4-S5). After the stress, cells were washed three times in LB or M9 media and dispensed in 96-well plates. The volume of the cultures in each well was 150 μl and 20 μl of mineral oil. OD600nm was then measured every 2 min over 15 h of growth using a Tecan Infinite M200 PRO microplate reader.
Competition of strains in flask cultures
Overnight cultures of the WT strain and strains with different rrn operon numbers were diluted 1/1,000 in fresh LB medium, mixed at 1/1 (v/v) ratio to a final volume of 10 ml in 125 ml Erlenmeyer flasks incubated overnight with shaking at 37°C. Next day, 1μl of the overnight culture was transferred into 10 ml LB fresh medium and grown again. Competition cycles were repeated for three days. To track progress, samples were taken from overnight cultures after each competition cycle, i.e. overnight cultures. Appropriate dilutions of these overnight cultures were spread on LB plates to determine a total number of CFUs. The competing strains were differentiated based on different fluorescence, i.e. they carried different fluorescent reporters: GFP and RFP. To verify that the florescence reporters have no impact on competition, the WT strains carrying GFP and RFP fluorescent reporters were also put in competition. Colony fluorescence was detected using Illumatool Tunable Lighting System. Fitness differences were estimated by calculating the competitive index (Day 0 CFU (mutant/WT) / Days 1–2 or 3 CFU (mutant/WT)).
Statistical analysis
All analyses were done using GraphPad Prism 8 (GraphPad Software, Inc., La Jolla, CA).
RESULTS
rrn operon multiplicity modulates duration of the lag phase and growth rates
We first investigated the impact of rrn operon multiplicity on growth of the E. coli MG1655 strain in rich LB medium (Supplementary Figure S1). This strain, which has 7 rrn operons per genome, is referred to as wild type (WT) in the text (Supplementary Figure S2A). The growth of strains with 8, 6 or 5 rrn operons was not significantly different from WT (Supplementary Figures S1AB and S1A1-2, Supplementary Table S1). However, strains with 10, 9, 4, 3, 2 or 1 rrn operons all had significantly longer lag-phases, i.e. a delay before the population of cells resumes growth after exiting from the stationary phase, and longer doubling times than WT.
The growth defects of strains having 1, 2 or 3 rrn operons were not due to the absence of tRNA genes, which were parts of deleted rrn operons, because they were complemented with a plasmid expressing the 6 missing tRNA genes (33). As a control, we also introduced a plasmid carrying the rrnB operon into a strain having only the rrnC operon and the tRNA plasmid, and observed that it eliminated both the lag phase and doubling time difference with the WT (Supplementary Figure S1B45, S46). The growth defects of strains with 1 or 2 rrn operons were not specific for the remaining operons because strains having different sets of remaining rrn operons had very similar phenotypes (Supplementary Figures S1A5-S6, Supplementary Table S2). In minimal medium, a decrease in the number of rrn operons below 5 also increased lag phase and reduced exponential growth rate (Supplementary Figures S1A3-S4, Supplementary Table S1), while an increase in the number of rrn operons above 7 did not significantly affect growth.
Next, we quantified total RNA content in different strains in exponential and lag phases (Supplementary Figure S1CD). The total amount of RNA is a good proxy for the amount of rRNA because it represents most of the total cellular RNA. While RNA amounts in strains having from 5 to 10 rrn operons were not significantly different, they were significantly lower in strains having fewer than 6 rrn operons during lag phase and fewer than 5 rrn copies during exponential growth. A strong negative correlation was observed between the cellular RNA amount in the lag or exponential phase cells, respectively, and the lag phase duration or the growth rate (Supplementary Figure S2BC).
We also monitored the impact of rrn operon deletions on the expression of the remaining operons during lag and growth phases (Figure 1E-F and Supplementary Figure S2D), by using a fusion of the rrnB operon promoter P1 to the gene coding for an unstable green fluorescent protein (34,35). While the levels of reporter expression were not significantly different between strains having between 5 and 10 rrn operons, they increased with the decrease in number of rrn operons below 5. Importantly this increase relative to WT was much more pronounced during lag phase than during exponential growth. We also observed a positive correlation between the duration of lag phase and the peak level of the rrnB operon promoter P1 expression (Supplementary Figure S2E).
Figure 1.

Growth, RNA content, P1rrnB expression and DNA replication of cells having different rrn operon copy numbers growing in LB medium. (A) Lag time and (B) doubling time, calculated from the growth curves (see Supplementary Figures S1A1, S3). Total RNA content in (C) lag phase and (D) exponential phase cells. (A-C) Each dot represents the mean value (± SD) from three independent experiments. Asterisks show significant difference compared to the WT (t-test: *P < 0.05, **P < 0.01, ****P < 0.0001). (E-F) P1rrnB-gfp expression during first 5 h of growth. Each data point represents the mean value (± SD) from three independent experiments. (G) Lag phase cells were treated simultaneously with rifampicin and cephalexin (first row; time 0), or 40 min with cephalexin only, and then also with rifampicin (second row). DNA content was measured after 90 min of incubation with both antibiotics. Number of genome equivalents (N) was calculated using stationary phase, 1N cells, as a reference. The results are representative of three independent experiments.
Reduction of rrn operon perturbs DNA replication
We investigated whether the reduction of the number of rrn operons impacted chromosome replication during lag phase, using a rifampicin/cephalexin run-out DNA replication assay (28) (Figure 1G). The treatment of cells with rifampicin and cephalexin inhibits new rounds of replication initiation and cell division, respectively, but allows ongoing DNA replication to finish. DNA is stained with DAPI and analysed by flow cytometer. The number of peaks showing the cellular DNA amounts corresponds to the number of fully replicated chromosomes per cell and reflects the number of replication origins present in the cell at the time the drugs were added. The distribution of cellular DNA content in discrete narrow peaks indicated that the vast majority of the cells having 7 and 6 rrn operons completed replication of chromosomes. However, the distribution of cellular DNA contents in peaks with connected wide bases indicated that chromosome replication was not completed in most of the cells having 1 to 4 rrn operons.
Decreasing rrn operon multiplicity increases genome instability
The blockage of the DNA replication is known to induce SOS response in bacteria (36). This genotoxic stress response regulon is composed of more than 40 genes, most of which code for DNA damage processing functions, including the recA gene. RecA protein is necessary for SOS induction and for homologous recombination. Using a PrecA-yfp reporter, we showed that the SOS response was much more strongly induced in populations of cells having fewer than 5 rrn operons compared to WT cells during lag phase (Figure 2A). Importantly, the peak level of SOS induction in different strains correlated positively with the duration of lag phase (Supplementary Figure S2G), and with the rrnB operon promoter P1 expression (Supplementary Figure S2F).
Figure 2.

SOS induction, DNA repair, DNA damage, and mortality rates of cells growing in LB medium. (A) Expression of the SOS induction reporter PrecA-yfp in cells having different rrn operon copy numbers. Each data point represents the mean value (± SD) from three independent experiments. (B) Duration of the lag phase of different mutant derivatives of rrnC+ strain having different capacities to repair DNA lesions calculated from growth curves (see Supplementary Figure S1B). Each dot represents the mean value (± SD) from three independent experiments. Asterisks show significant differences compared to the strain with 1 rrn. (C) DNA damage detected in the lag phase cells using TUNEL assay and flow cytometer. Each bar represents the mean value (± SD) from three independent experiments. Asterisks show significant differences between strains. (D) Detection of double-strand breaks. Lag phase cells carrying a Gam-GFP reporter fusion were analysed using a fluorescence microscope. Inserted photographs show Gam-GFP foci in the cells having 1 rrn operon. Each bar represents the mean value (± SD) from three independent experiments. Total of 1,329 and 1,606 cells having 7 and 1 rrn were analysed, respectively. Asterisks show significant differences compared to WT. (E) Cell death measured during growth in the LB medium in populations of 7 rrn and 1 rrn strains (upper panel). Dead cells were detected using propidium iodide (PI) staining and flow cytometer (lower panel). Each result represents the mean value (± SD) from three independent experiments. (F-I) Growth of individual cells in the ‘mother’ machine microfluidic device in LB medium. Each panel represents growth of an individual mother cell. (F) 99% of WT cells with 7 rrn operons start growing and dividing after short lag phase. (G) 70% of cells with 1 rrn operon start growing and dividing after longer lag phase. (H) 10% of cells with 1 rrn operon undergoing transient crisis, i.e. filamentation, after which it resumes growth. (I) 20% of cells with 1 rrn operon increase in size, divide a few times and then die. The presented results are representative of three independent experiments. t-test: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
We performed two control experiments to confirm that the difference in a PrecA-yfp induction between WT and strains having reduced number of rrn operons was due to genuine SOS induction and not due to increase in the number of RNAPs available for transcription from other promoters. For this, we first showed that the introduction of the lexA(Ind–) allele, which prevents SOS induction, essentially extinguished the florescence signal in a 1 rrn operon strain carrying a PrecA-yfp reporter (Fig S2H). Second, we introduced a constitutive Pbla-gfp reporter and observed no difference in florescence signal between strains having 1 rrn operon and WT (Fig S2I).
To characterize the molecular mechanisms responsible for the observed phenotypes, we decided to use strain having only 1 rrn operon, rrnC, referred as rrnC+ in the text. We first examined the impact of SOS induction on growth, by introducing the lexA(Def), which derepresses SOS induction, and lexA1(Ind–) alleles (37). The lexA(Def) and lexA1(Ind–) alleles considerably shortened and increased the lag phase of the rrnC+ strain, respectively, while they had no impact on growth of the WT (Figure 2B and Supplementary Figures S1B1-S4, Supplementary Table S3). Thus, the ability to mount an SOS response plays an important role in determining the lag time of cells lacking rrn operons, suggesting that an inability to make sufficient ribosomes may not be the only problem in these strains.
Because the SOS response is triggered by the accumulation of persistent single-strand DNA (ssDNA), as a result of processing of DNA replication blockages and DNA lesions, we inquired whether rrnC+cells had an increased quantity of DNA breaks during lag phase. Using a terminal deoxynucleotidyl transferase dUTP nick end-labelling (TUNEL) assay, we showed that 0.3% of WT and 44.6% of rrnC+cells had a positive TUNEL signal (Figure 2C). To further confirm that rrnC+cells have more DNA breaks than WT, we deleted the recA, recB, recF and ruvA genes, encoding different enzymes involved in homologous recombination (38). Deletions of recA, recB and ruvA genes increased the lag phase of a WT to a small degree, but the increase was considerably greater for the rrnC+strain (Figure 2B and Supplementary Figures S1B5-S12, Supplementary Table S3). The absence of RecF, which participates in the repair of the single-strand gaps, had no effect in either strain. Because RecB is a component of the RecBCD complex that processes double strand breaks (DSB), we investigated whether DSBs are generated in the genomes of the rrnC+cells by using bacteriophage Mu double-strand DNA end-binding protein Gam fused to GFP (32). 1.4% of WT and 47.4% of rrnC+cells had at least one Gam-GFP focus (Figure 2D).
Because it is known that SOS induction increases mutagenesis, we quantified the rate of appearance of rifampicin (RifR) or nalidixic acid (NalR) resistant mutants in the different strains. The rate of appearance of RifR and NalR mutants was 3.8-fold and 10.7-fold higher, respectively, in rrnC+compared to WT (Supplementary Figure S4). The increase in mutation rates was SOS-dependent because it disappeared in a lexA1 (Ind–) strain. DNA Pol IV, encoded by SOS-controlled dinB gene, was responsible for the most of the increased mutagenesis. This further confirmed that the rrnC+strain has increased incidence of DNA replication blockage, because Pol IV has access to the replication fork only when replicative DNA polymerase Pol III is blocked (39).
Low rrn operon multiplicity increases mortality rates
Because extensive DNA damage can result in cell death, we asked if the longer lag phase of cells with low rrn operons multiplicity could be the result of increased mortality rates. Cell viability was assessed using propidium iodide (PI) staining followed by flow cytometry (Figure 2E). PI enters dead cells with permeabilized membranes and binds to DNA by intercalating between the bases, but it cannot enter live cells with intact membranes (40). Stationary phase cultures of both WT and rrnC+ strains contained about 0.01% dead cells. During lag phase, the fraction of dead cells increased 2-fold in WT and 4,600-fold in rrnC+populations (Figure 2E). Thus, while the population of WT cells experience a modest increase in mortality rates during lag-phase, the population of rrnC+ cells suffers a massive loss of viability. The rrnC+ culture starts growing only when the problem leading to cell death is somehow resolved in the surviving cells. This clearly shows that it is not possible to quantify an impact of mortality rates on population growth using only optical density and CFU-based measurements (Figure 2E and Supplementary Figure S2L).
Next, we evaluated growth of individual WT and rrnC+cells in a microfluidic ‘mother machine’ device (41). rrnC+cells had on average a 5.25-fold longer lag phase, i.e. time to first division after coming out of overnight stationary phase, compared to WT cells (Supplementary Figures S2FG and S3A). After lag phase, 99% of WT mother cells, but only 70% of rrnC+mother cells, started cycles of growing and dividing, with a two-fold longer division time for rrnC+cells compared to WT (Supplementary Figures S2FG). About 10% of rrnC+cells started filamenting, i.e. cell length increased by more than 2-fold, before resuming normal division and growth cycles (Figure 2H and Supplementary Figure S3B). However, about 20% of rrnC+cells started growing, some of which divided 1 or 2 times, and then stopped growing, presumably because they died (Figure 2I and Supplementary Figure S3B). Importantly, when one cell died, the sister cell continued growing and dividing in around 80% of cases, while in around 20% of cases, both sister cells died (Supplementary Figure S3C). Both filamentation and cell death occurred during first 5 divisions of the ‘mother’ cell (Supplementary Figure S3D). These experiments confirm that reduction of the number of the rrn operons results in longer lag phase and higher mortality rates of individual cells.
DNA breaks are caused by transcription-replication conflicts
Because DNA replication and transcription occurs simultaneously on a common DNA template in bacteria, and because the DNA replication machinery moves about 10 to 20-times faster than elongating RNAP, frequent collisions are unavoidable but efficiently dealt with in WT cells (21). We hypothesized that replication-transcription conflicts might increase in strains carrying rrn operon deletions to a level that cannot be efficiently prevented and/or processed by available cellular machinery.
We tested this hypothesis by reducing transcription of rrn operons with the introduction of the rpoC* (Δ215–220) allele (42). RpoC* alters the kinetic properties of RNAP, such that it requires higher concentrations of initiator NTP for efficient transcription initiation from the rrn P1 promoter in vitro. Since these concentrations are not attained in vivo, the net result is a reduction in transcription initiation at rrn promoters. We anticipated that this phenotype would give a competitive edge to DNA replication. The rpoC* allele indeed significantly decreased lag phase, mortality rates, DNA replication problems, SOS induction, the amount of DNA damage, mutagenesis and rrnB operon promoter P1 expression in the rrnC+strain (Figure 3A-F, Supplementary Figures S1B13-S14, S2JK and Supplementary Table S3). The rpoC* allele also significantly decreased the lag phase in rrnE+ and rrnD+ strains, showing that observed effect is not specific for the rrnC operon (Supplementary Figures S1A7-S8, Supplementary Table S2). Importantly, the rpoC* allele did not impact the ribosome concentration in the rrnC+cells (Figure 3G), indicating that the improvement in general cell health, and in particular the shortening of the lag phase, is not due to increased ribosome production.
Figure 3.

Impact of RNAP modulators on growth and mortality rates, SOS induction, DNA damage, mutagenesis and ribosome content of cells growing in LB medium. (A) Duration of the lag phase calculated from growth curves for different mutant strains (see Supplementary Figure S1B). Each dot represents the mean value (± SD) from three independent experiments. Asterisks show significant differences compared to the 1 rrn strain (t-test ****P < 0.0001). (B) Impact of the rpoC* allele on death of 1 rrn cells measured during growth in the LB medium. Dead cells were detected using propidium iodide (PI) staining and flow cytometer. (C) Impact of the rpoC* allele on DNA replication in 7 rrn and 1 rrn cells. Lag phase cells were treated simultaneously with rifampicin and cephalexin (first column), or 40 min with cephalexin only, and then also with rifampicin (second column). DNA content was measured after 90 min of incubation with both antibiotics. The results are representative of three independent experiments. (D) Impact of the rpoC* allele on the SOS induction measured using the PrecA-yfp reporter in 1 rrn cells. (E) Impact of RNAP modulators on DNA damage in 7 rrn and 1 rrn cells. DNA damage was quantified using TUNEL-based detection kit. Each bar represents the mean value (± SD) from three independent experiments. Asterisks show significant differences compared to the 1 rrn strain (t-test: *P < 0.05, **P < 0.01). (F) Impact of the rpoC* allele on rates of the appearance of nalidixic acid (left) and rifampicin resistant (right) mutants in populations of 7 rrn and 1 rrn cells. Each bar represents median with 95% confidence interval from at least five independent experiments each with five independent cultures. Asterisks show significant differences (Wilcoxon-test: ****P < 0.0001). (G) Concentration of 70S ribosomes in cells having 1 and 7 rrn operons and their rpoC* derivatives in lag and exponential growth phase cells. Each bar represents the mean value (± SD) from three independent experiments. Asterisks show significant differences (t-test ****P < 0.0001).
We additionally reduced the probability of replication-transcription conflicts by decreasing the frequency of DNA replication initiation using the dnaA(Sx) allele (43). As anticipated, this allele reduced the lag phase of the rrnC+ strain, while it did not affect the WT (Figure 3A and Supplementary Figures S1B15-S16, Supplementary Table S3).
Lastly, we asked whether RNAP stalling contributes to increased lag phase in cells with fewer rrn operons. We did this by deleting the mfd, rapA and greA genes (44). The Mfd protein displaces RNAPs stalled by DNA lesions, RapA recycles RNAPs blocked by DNA supercoiling and GreA releases stalled RNAPs by stimulating backtracking and mRNA shortening (44). The deletion of each of these genes increased the lag phase of the rrnC+strain but not of the WT (Figure 3A and Supplementary Figures S1B17-S22, Supplementary Table S3), indicating that RNAP stalling contributes to increased lag phase in cells with one rrn operon. Indeed, inactivation of the mfd gene in the rrnC+strain also increased the fraction of cells having DNA breaks detected by TUNEL assay (Figure 3E). Inactivation of the mfd gene additionally increased the lag phase in rrnE+ and rrnD+ strains significantly, indicating that observed effect is not specific for the rrnC operon (Supplementary Figures S1A9-S10, Supplementary Table S2).
Reduction of rrn operon copy number increases R-loop formation
R-loops are nucleic acid structures where the nascent RNA invades the template DNA strand outside of the transcription bubble, leaving the displaced coding DNA single stranded. Highly expressed gene loci have been associated with the generation of R-loops, possibly because their formation is favoured by the negative supercoiling that occurs behind actively transcribing RNAPs (45–47). R-loops can provoke DNA replication fork collapse, DNA breakage and increased mutation rates (21). We tested whether the reduction in the rrn operon copy number resulted in increased generation of R-loops in lag phase using the S9.6 antibody, which specifically detects DNA:RNA hybrids (Figure 4A). The amount of DNA:RNA hybrids was similar in the WT and in strains having 6 and 5 rrn operons. However, it increased significantly as the number of rrn operons decreased below 5. A large quantity of DNA:RNA hybrids was also observed in cells having only the rrnE or rrnD operons (Supplementary Figure S5A), indicating that accumulation of the DNA:RNA hybrids is not specific for the rrnC+ strain. We also observed high mortality rates and an increased amount of DNA:RNA hybrids in the B. subtilis mutant cells having only 1 rrn operon compared to the WT during lag phase (Supplementary Figure S5H-I), suggesting that this phenomenon is not restricted to E. coli.
Figure 4.
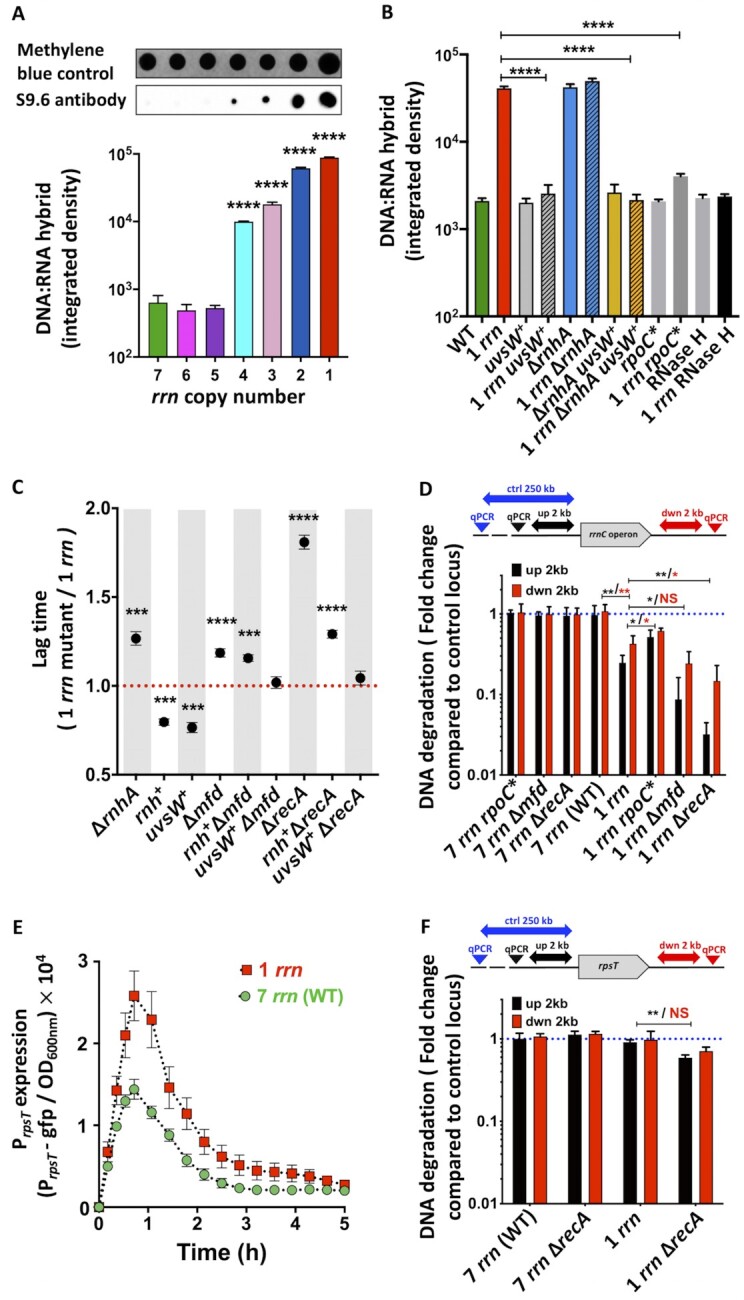
R-loops, DNA degradation and growth of mutants with different capacities to deal with stalled RNAPs and R-loops. (A) Detection of the DNA:RNA hybrids in lag phase cells having different rrn operon copy numbers using S9.6 antibody and dot-blot analysis. First row: Staining with methylene blue was used to estimate total amount of nucleic acids. Bar graphs represent quantification of the dot blots (second row). Each bar represents the mean value (± SD) of three independent experiments. Asterisks show significant differences compared to the WT. (B) Detection of the DNA:RNA hybrids in mutants with different capacities to deal with stalled RNAPs and R-loops during lag phase using S9.6 antibody and dot-blot analysis. (+) marks the overproduction of uvsW gene. Bar graphs represent quantification of the dot blots. Each bar represents the mean value (± SD) of three independent experiments. Asterisks show significant differences compared to the 1 rrn strain. (C) Duration of the lag times of mutants with different capacities to deal with stalled RNAPs and R-loops calculated from growth curves (see Supplementary Figure S1B). Each dot represents the mean value (± SD) of three independent experiments. (+) marks the overproduction of rnh and uvsW genes. Asterisks show significant differences compared to the 1 rrn strain. (D) Quantification of DNA degradation around the rrnC operon in strains having 1 (rrnC) and 7 rrn operons and their recA, rpoC* and mfd mutant derivatives during lag phase. DNA degradation was quantified using qPCR. Primers allowing amplification of the regions that are located 2 kb upstream and downstream of the rrnC operon and primers allowing amplification of the control locus located 250 kb upstream of the rrnC operon were used. Each bar represents the mean values (± SD) of three independent experiments. Asterisks show significant differences. (E) Expression of the PrpsT-gfp reporter in 7 rrn and 1 rrn strains. Each data point represents the mean value (± SD) from three independent experiments. (F) Quantification of DNA degradation around the rpsT gene in strains having 1 (rrnC) and 7 rrn operons and their recA mutant derivatives during lag phase. DNA degradation was quantified using qPCR. Primers allowing amplification of the regions that are located 2 kb upstream and downstream of the rpsT gene and primers allowing amplification of the control locus located 250 kb upstream of the rpsT gene were used. Each bar represents the mean values (± SD) of three independent experiments. Asterisks show significant differences according to t-test: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We further confirmed presence of DNA:RNA hybrids by modulating the expression of genes coding for enzymes that destroy these hybrids (Figure 4B and Supplementary Figure S5B-C). Deletion of the rnhA gene coding for RNase HI, which degrades the RNA component of an RNA:DNA hybrid (48,49) increased the lag phase of the rrnC+strain (Supplementary Figures S1B23-S24, Supplementary Table S3). In contrast, the overproduction of RNase HI reduced the lag phase not only of the rrnC+strain, but also the rrnC+ recA, and rrnC+ mfd strains, while growth of the WT, recA and mfd strains was not impacted (Figure 4C and Supplementary Figure S1B25-S30, Supplementary Table S3). Similarly, overproduction of the bacteriophage T4 UvsW helicase, which unwinds DNA:RNA hybrids (50,51), reduced the lag phase of the rrnC+, rrnC+ rnhA, rrnC+ recA, and rrnC+ mfd strains, without impacting growth of the WT, rnhA, recA and mfd strains (Supplementary Figures S1B31-S38, Supplementary Table S3). The overproduction of the UvsW protein reduced the amount of DNA:RNA hybrids detected using S9.6 antibody in the rnhA, rrnC+, and rrnC+ rnhA strains to the WT level (Figure 4BSupplementary Figure S5B-C). Therefore, the increased lag times of strains carrying fewer rrn operons are correlated with an inability to efficiently resolve R-loops.
rrn operons are DSB-prone genomic loci
The increased requirement for functional homologous recombination, as well as the TUNEL and Gam-GFP assays, showed increased DSBs in strains carrying fewer rrn operons. We asked whether these DSBs occurred randomly or at certain genomic hotspots? The involvement of the replication-transcription conflicts in the generation of DSBs, suggested two non-exclusive possibilities: (i) a reduced ribosome pool could result in transcription-translation uncoupling and RNAP stalling at highly expressed protein-coding gene loci; and (ii) the higher RNAP occupancy of the remaining rrn operons could promote higher levels of R-loop formation and DSBs at these loci.
We tested the first hypothesis by introducing the nusG F165A allele. NusG binds to a ribosomal protein S10 and acts as an adapter between RNA polymerase and the 30S ribosomal subunit. The F165A mutation weakens the NusG:S10 interaction, leading to transcription-translation uncoupling (52). The nusG F165A mutant allele had a longer lag phase than the WT (Supplementary Figure S1B39, Supplementary Table S3) that was shortened by introduction of the rpoC* allele and by the overproduction of RNase HI, indicating that the longer lag phase is due to RNAP blockage and R-loop formation (Supplementary Figures S1B41-S42, Supplementary Table S3). However, the nusG F165A allele increased lag phase of the rrnC+ strain to the same extent as the WT (Supplementary Figure S1B40, Supplementary Table S3), suggesting that this effect is independent of rrn copy number. Next, we deleted the hflX gene, which codes for a GTPase that rescues stalled ribosomes by separating the two ribosomal subunits and allowing them to dissociate from the mRNA (53). Deletion of hflX did not change growth patterns of either the WT or rrnC+strain (Supplementary Figures S1B43-S44, Supplementary Table S3). Therefore, transcription-translation uncoupling at sites of protein-coding genes is not the major contributor to the increase in lag phase in strains with reduced rrn multiplicity.
We tested second hypothesis by examining the appearance of chromosomal gaps at the unique rrn locus in the rrnC+ strain and its recA knockout derivative. The ends of DSBs are processed by helicase-nuclease activity of the RecBCD protein complex, which loads the RecA protein on the generated single-strand DNA to initiate DSB repair (38). In a recA mutant, RecBCD continues degrading DNA, thus leaving large gaps that can be detected by qPCR analysis. We used primers that allow amplification of two 0.5 kb-long regions that are located 2 kb upstream and downstream of the rrnC operon and a set of primers allowing amplification of a control locus located 250 kb bases upstream of the rrnC operon (Figure 4D). There was no significant difference in the amplification of regions flanking the rrnC operon or the control locus in WT, rpoC*, mfd and recA strains. However, the regions flanking the rrnC operon were significantly less amplified than the control locus in the rrnC+ compared to WT strain. The introduction of the rpoC* allele and deletion of the mfd gene, decreased and increased DNA degradation in the rrnC+ strain, respectively. The strongest increase in DNA degradation was observed when recA gene was inactivated in the rrnC+ strain, which indicates that DSBs were generated at the rrnC operon locus.
Disintegration of stalled replication forks would be expected to result in the degradation of DNA upstream of fork, whereas the downstream DNA should be less affected. However, we observed massive DNA degradation both upstream and downstream of the single remaining rrnC operon that is a major transcription-replication collision site. This might suggest the full destruction of the replication fork, similar to observed previously for ligase-deficient death (54). Only upon inactivation of the recA gene in the rrnC+ strain, the upstream sequences suffered greater levels of degradation than those downstream of the rrnC operon. This phenomenon was not peculiar to the genomic region surrounding rrnC, because the same results were observed at the rrnD and rrnE loci in recA derivatives of the strains having only one of these operons (Supplementary Figure S5D-E). Inactivation of the mfd gene significantly increased degradation of the sequences upstream but not those downstream of the rrnC operon in rrnC+ strain.
To further confirm that the over-transcribed rrn operons are preferential DSBs hotspots, we investigated whether there was also increased DNA degradation around another gene that was overproduced during the lag phase in the rrnC+ compared to WT strain. For this, we choose the rpsT gene that codes for S20 ribosomal protein (Figure 4E). There was no significant difference in the amplification of regions flanking the rpsT gene or the control locus in WT and rrnC+ strain (Figure 4F). Inactivation of the recA gene slightly increased the degradation of the DNA upstream of rpsT in the rrnC+ strain compared to the WT strain, but did not have a significant effect downstream. Thus, the remaining over-transcribed rrn operon is an elevated DSB hotspot in 1 rrn cells compared to other sites.
R-loop processing capacity impacts growth restart after ribosome-damaging stresses
We wondered if replication-transcription conflicts, observed in strains having reduced number of the rrn operons upon exit from stationary phase, would also impact growth restart upon ribosome-damaging treatment of WT cells. Recovery after ribosome destruction is expected to require high-level transcription of rrn operons to rapidly restore the full translational capacity required for rapid growth.
We measured the duration of the recovery phase after exposure to 4 different stresses (Supplementary Table S4), all of which cause degradation of stable RNA, albeit to different levels (Figure 5 and Supplementary Figure S1C and S5F). (1) Colistin damages membranes and allows entry of the periplasmic endoribonuclease RNase I, encoded by rna gene, into the cytoplasm (55). By quantifying total RNA content in WT and rna mutant cells, we confirmed that colistin treatment induces extensive RNase I-dependent RNA degradation (Figure 5A). In agreement with our hypothesis, inactivation of the rna gene significantly decreased the duration of the post-stress recovery phase after colistin treatment (Figure 5B and Supplementary Figures S1C1-S2). (2) High salt concentrations induce osmotic shock leading to membrane damage and RNase I release (56), which explains why deletion of the rna gene significantly decreased the duration of the recovery phase after salt shock (Supplementary Figure S1C5). High salt concentrations also impact DNA supercoiling, thus blocking RNAP, which can be recycled by the RapA protein (44). Accordingly, the ΔrapA mutant showed longer recovery times than WT after salt stress (Supplementary Figure S1C5). (3) Exposure to high temperature (e.g. 50°C) damages ribosomes and induces extensive rRNA degradation (57) (Supplementary Figure S5F). Surprisingly, deletion of the rna gene did not impact the duration of recovery after heat-shock, suggesting that it either (i) does not damage membranes and/or that it denatures RNase I, or (ii) allows other RNase(s) to degrade rRNA, by unfolding rRNA or damaging ribosomal proteins. (4) Bile salts (e.g. sodium deoxycholate) decrease membrane integrity (58) and induced the degradation of stable RNA (Supplementary Figure S5F). Similar to heat-shock, deletion of the rna gene did not impact the duration of recovery after treatment with sodium deoxycholate (Supplementary Figure S1C4), suggesting that some other RNase is responsible for ribosomal RNA turnover under these conditions.
Figure 5.

RNA degradation, DNA:RNA hybrids and growth resumption after colistin treatment. (A) Total RNA content in different strains after colistin treatment, as well as in untreated controls. Each bar represents the mean values (± SD) of three independent experiments. Asterisks show significant differences compared to the WT. (B and E) Duration of the recovery time after colistin treatment of different strains that was calculated from growth curves (see Supplementary Figure S1C). (+) marks the overproduction of rnh and uvsW genes. Each dot represents the mean value (± SD) of three independent experiments. Asterisks show significant differences compared to the WT. (C) Detection of DNA:RNA hybrids during recovery of the WT strain after colistin treatment using S9.6 antibody. Bars represent quantification of the dot blot analysis. Asterisks show significant difference compared to the untreated WT. (D) DNA damage quantification in WT and its mutant derivatives after colistin treatment using TUNEL-based detection kit.). (+) marks the overproduction of uvsW gene. Each bar represents the mean value (± SD) of three independent experiments. Asterisks show significant differences. Asterisks show significant differences according to t-test: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Consistent with what we observed for the rrnC+ strain, overexpression of the rnhA or uvsW genes, or introduction of the rpoC* allele, reduced the recovery phase of strains with the full rrn complement after all 4 stress treatments, while inactivation of the mfd gene increased the length of the lag (Figure 5B and Supplementary Figures S1C6-S15). This indicates that all 4 stresses cause increased replication-transcription conflicts and the generation of R-loops, resulting in longer recovery phases. Deletion of the hflX and rapA genes had no effect on the duration of the recovery phase after colistin or heat shock treatments (Figure 5B and Supplementary Figures S1C2-S3). Because HflX affects ribosomes stalled at protein coding loci, the absence of an effect of hflX gene deletion on the duration of the recovery phase for these two stress conditions is consistent with replication-transcription conflicts primarily occurring at rrn operon loci, as in the rrnC+ strain, while the lack of effect of the rapA deletion suggests that these conflicts are not caused by increased DNA superhelicity. We confirmed that the amount of DNA:RNA hybrids increased considerably during recovery after treatment with colistin and heat-shock, using S9.6 antibodies (Figure 5C and Supplementary Figure S5G). Interestingly, deletion of hflX and rapA genes did increase the duration of the recovery phase after sodium deoxycholate and osmotic shock (Supplementary Figures S1C4-S5), suggesting that these conditions may also cause RNAP stalling at protein coding loci and that this stalling is potentially linked to increased DNA superhelicity.
Deletion of the recA gene significantly increased the duration of the recovery phase after treatments with all 4 stressors (Figure 5B and Supplementary Figure S1C 16–20). Overproduction of the UvsW helicase decreased the impact of the recA gene deletion on recovery phase for all treatments, albeit to different extents (Figure 5B and Supplementary Figure S1C 16–20). Finally, by using TUNEL assay, we found that the recA gene deletion and the overproduction of the UvsW helicase in recA mutant, respectively increased and reduced the amount of DNA damage after colistin treatment (Figure 5D). These results support our hypothesis that the growth restart after ribosome destructive treatments is delayed by transcription replication conflicts that cause DNA breakage, similar to the delay of growth restart after stationary phase in mutants with reduced numbers of rrn operons.
rrn operon multiplicity impacts growth restart after ribosome-damaging stress
While decreasing number of the rrn operons per genome results in an increased number of RNAPs per rrn operon (14), the opposite was observed upon introduction of a multicopy plasmid carrying rrn operon in the WT (59). For this reason, we hypothesized that increasing number of rrn operons above seven might reduce probability that replication-transcription conflicts arise during the post-stress recovery period.
We first evaluated how increasing rrn copy number above WT level impacted growth in LB without any exogenous stress in a single growth cycle and in 3-day batch culture competition experiments. Strains with more than 8 rrn operons had significantly longer doubling times and lag phases (Supplementary Figure S1AB, Supplementary Table S1), and lower fitness than WT (Supplementary Figure S6). Next, we exposed strains pre-grown in LB to different stress conditions and measured the duration of the recovery phase (Figure 5E and Supplementary Figures S1C21-S30, Supplementary Table S5). Strains with more and less rrn operons had respectively shorter and longer recovery phases than WT, after all treatments, albeit to different degrees. The impact of rrn copy number variation was strongest after heat shock and colistin treatments, and somewhat less pronounced after osmotic shock and sodium deoxycholate treatments. We obtained qualitatively similar results in minimal M9 medium, i.e. higher-than-WT numbers of rrn operons shortened the duration of the recovery phase after exposure to heat shock, colistin, and sodium deoxycholate, but the number of extra operons required to see this effect varied. For example, the recovery after osmotic shock was improved only for the strain having 10 rrn operons (Supplementary Figures S1C26-S30, Supplementary Table S5).
DISCUSSION
The number of rrn operons per bacterial genome, which varies widely between different bacterial species, is generally correlated with maximal growth rates (11,12,17,60). However, it has been observed that a complete set of rrn operons exceeds requirements for the maximal growth rates for different species (14,15). This ‘excess’ capacity is considered to be required for rapid adjustment to improved environmental conditions, such as nutrient upshifts and more favourable temperatures, because it ensures that cells can more rapidly attain higher translational capacity (16,17). However, our study shows that a major role of ‘excess’ rrn operon multiplicity is also to ensure that individual rrn operons are not saturated by RNAPs, which can result in catastrophic failures in genome replication and cell death (Figure 6).
Figure 6.

Molecular mechanisms involved in the processing of causes and consequences of replication/transcription conflicts. (A) Modulation of the lag phase duration of the strain having 1 rrn operon (rrnC) relative to WT having 7 rrn operons (0%) by different genetic modifications tested in this study (+/-%). (B) Model showing how transition from stationary to growth phase in cells with reduced numbers of rrn operons and recovery from ribosome damaging treatments in cells with full complement of rrn operons, cause crowding and stalling of RNAPs, R-loop formation, replicon stalling and DNA breakage, most probably upon arrival of the next replicon, at the rrn locus. Proteins involved in the processing of causes and consequences of these replication/transcription conflicts are indicated.
Our conclusion is based on the observation that the long transition from stationary to growth phase in E. coli strains with low numbers of rrn operons per chromosome is largely due to high mortality rates (Figure 2E). During lag phase, these cells initiate DNA replication like WT cells, but they have severe problems with DNA replication elongation, as well as increased DNA breakage, SOS induction and mutagenesis (Figures 1G, 2A-D and 3F). The primary cause of these deleterious events is a blockage of the DNA replication machinery by stalled RNAPs and formation of R-loops at the sites of the remaining over-transcribed rrn operons (Figure 6B). The RNAP stalling in cells with one rrn operon was revealed by increased lag phase of Δmfd, ΔrapA or ΔgreA mutants (Figure 3A and Supplementary Figures S1B17-S22, Supplementary Table S3). The survival and recovery of cells with one rrn operon requires a functional homologous recombination machinery (Figure 2B). That a functional homologous recombination machinery is necessary for the elimination of the deleterious consequences of persistent R-loops was previously shown by the synthetic lethality of a rnhA null mutation with the inactivation of recA, recB and polA genes (61).
The reduction of lag phase by reducing R-loop formation or by improving DNA repair capacity accounted for the major portion of the effect previously thought to be primarily due to insufficient translational capacity (Figure 6A). For example, introducing the rpoC* allele, or resolving DNA:RNA hybrids by overproducing the UvsW helicase, reduced the duration of the lag phase of the rrnC+strain relative to WT by 72% and 63%, respectively (Figures 3A, 4C and 6A). Importantly, the rpoC* allele reduced the length of the lag phase, without a concomitant increase in the number of ribosomes (Figure 3G). Although we cannot exclude that the rpoC* allele rescues growth of the single rrnC+ strain by improving the quality of ribosome synthesis, our model is that the rpoC* allele reduces replication-transcription conflicts by reducing transcription initiation.
Increasing the cellular capacity to deal with DNA damage by constitutively derepressing the SOS regulon reduced the duration of the lag phase of the rrnC+strain relative to WT by 87% (Figures 2B and 6A). The importance of DNA repair was also confirmed by observation that a lack of RecA, which plays a crucial role both in the SOS induction and homologous recombination, increased the duration of the lag phase of the rrnC+strain by 137% (Figures 2B and 6A). Remarkably, overproduction of the UvsW helicase totally abolished the difference in lag phases between the ΔrecA rrnC+and ΔrecA strains (Figures 4C and 6A). Therefore, the time required to synthesize the cellular components necessary for regrowth would appear to have little impact, relative to the resolution of DNA damage, on the longer lag phase of cells with reduced numbers of rrn operons.
Our observations raised following questions: (i) Why are rrn operons such hotspots for DSBs? (ii) Why does the replication catastrophe happen at this particular growth phase? We suspect the answer to the first question lies in the known link between high-level transcription and R-loop formation. With fewer rrn operons to make rRNA, cells load as many RNAPs as possible onto the remaining rrn operons (increasing from an average of 53 RNAPs/operon in WT cells to 71 RNAPs/operon in a 3 rrn strain, for example (14). This, coupled with the G/C-rich and G-quadruplex formation prone nature of rrn operons, which also favour R-loop formation (62,63) likely explains why the sole remaining rrn operon in the 1 rrn strain is such a hot-spot for DSBs.
The fact that rrn operons are hotspots for replication-transcription conflicts is also supported by observation that in absence of the RNase H, which degrades the RNA in DNA:RNA hybrids, DNA replication blockage at rrn operons increases considerably (64). It was also reported that the absence of DNA topoisomerase I increases occurrence of R-loops within the E. coli rrnB rRNA operon, while RNase H overproduction decreases it (65–67). rRNA genes were also shown to be a hot-spots for R-loop formation in absence of RNase H1 or DNA topoisomerase 1 in yeast and human cells (68,69). This means that R-loops are frequently formed in rRNA genes/operons under normal conditions but that R-loop processing enzymes are able to eliminate most of them.
To answer the second question, the number of rrn operons per cell and not per chromosome should be taken into account. Upon exit from stationary phase, E. coli cells start replicating the chromosome from the origin of replication (70). When cells grow slowly, cell division starts after completion of chromosome duplication and before a new round of DNA replication is initiated. When cells grow fast, a second (and sometimes third) replication initiation event occurs before cell division. Therefore, the ori/ter ratio is 2 and 4 in slowly and rapidly growing cells, respectively. Consequently, the copy number of the rrn operons that are close the ori region, progressively increases during the transition from the stationary to lag and exponential growth phases. For example, in E. coli the number of the rrn operons per cell increases from 7 in non-growing cells to as many as 38 during maximal growth, with those closest to the origin accounting for the major proportion (14). The number of rrn operons per cell is most limiting during lag phase before chromosome multiplication increases the copy number. The tendency to over-transcribe a limited number of rrn operons likely explains why replication-transcription conflicts occur mostly during the exit from stationary phase. Cells that survive long enough and replicate enough chromosomes will eventually have sufficient rrn operons per cell to avoid RNAP stalling. Importantly, we observed that low rrn operon multiplicity increased mortality rates and R-loops amounts also in B. subtilis cells during lag phase, suggesting this is a widespread phenomenon (Supplementary Figure S5HI).
Our study shows that rrn operon multiplicity, which ensures that individual rrn operons are not saturated by RNAPs during environmental fluctuations, can be considered as a genome stability insurance policy. We propose that this may be an important component of the selective pressure, which determines optimal lower limit of the rrn operon copy number for a given bacterial species. The question is what may determine the upper limit of rrn operon copy number? The full set of 7 rrn operons in E. coli, was clearly not sufficient to diminish probability of the RNAP stalling and R-loop formation during the recovery from stress conditions that cause ribosome degradation (Figure 5B). We showed that higher-than-normal number of rrn operons per chromosome, i.e. above 7 in E. coli, can significantly reduce length of the recovery phase following such stresses (Figure 5E and Supplementary Figure S1C). So, the question is why E. coli does not have more rrn operons?
The answer may come from the observation that the extra copies of rrn operons increased the lag phase and doubling time of unstressed E. coli cells growing in rich medium, which corroborates previously published observations (Supplementary Figure S6) (18,19). It was also reported that E. coli cells with 8, 9 and 10 rrn operons are significantly larger than WT cells (18). This may result from deleterious perturbations of the repartition of RNAPs between rrn operons and other genes. Another nonexclusive possibility is that additional rrn operons are a genomic liability, because recombination between such repeated DNA sequences can result in chromosomal rearrangements, many of which can be deleterious or even lethal if they cause large deletions (71). Therefore, it can be hypothesized that the observed trade-offs determine the upper limit of rrn operon copy number for a given species in its natural habitat.
DATA AVAILABILITY
We comply with the Minimal Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines. Other details, including the sequences of primers used in the quantitative PCR, are supplied in Materials and Methods section of this manuscript.
Supplementary Material
ACKNOWLEDGEMENTS
The authors want to thank M. Gottesman (Columbia University, New York, NY, USA), J. Gowrishankar (Centre for DNA Fingerprinting and Diagnostics, Hyderabad, India), Z. Györfy (University of Szeged, Hungary), I. Iost (ARNA Laboratory, Bordeaux, France), S. Lovett (Brandeis University, Waltham, MA, USA), and B. Michel (Institut de Biologie Intégrative de la Cellule, Gif sur Yvette, France) for kindly providing bacterial strains and plasmids. Clara Delaroque for qPCR technical help.
Notes
Present address: Tanja Dapa, Instituto Gulbenkian de Ciência, 2780-156 Oeiras, Portugal.
Contributor Information
Sebastien Fleurier, Department of Infection, Immunity and Inflammation, Institut Cochin, Inserm U1016, CNRS UMR8104, Université de Paris, 75014 Paris, France.
Tanja Dapa, Department of Infection, Immunity and Inflammation, Institut Cochin, Inserm U1016, CNRS UMR8104, Université de Paris, 75014 Paris, France.
Olivier Tenaillon, INSERM U1137, Université de Paris, 75018 Paris, France.
Ciarán Condon, Institut de Biologie Physico-Chimique, CNRS UMR8261, Université de Paris, 75005 Paris, France.
Ivan Matic, Department of Infection, Immunity and Inflammation, Institut Cochin, Inserm U1016, CNRS UMR8104, Université de Paris, 75014 Paris, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
IM is supported by Labex ‘Who am I?’ Idex ANR-11-IDEX-0005-01 / ANR-11-LABX-0071-Idex-Sorbonne Paris, by ANR-18-CE35-007-02 and ANR-20-PAMR-0002 grants. CC is funded by ANR grant ARNr-QC and Labex Dynamo.
Conflict of interest statement. None declared.
REFERENCES
- 1. Russell J.B., Cook G.M.. Energetics of bacterial growth: balance of anabolic and catabolic reactions. Microbiol. Rev. 1995; 59:48–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Condon C., Squires C., Squires C.L.. Control of rRNA transcription in Escherichia coli. Microbiol. Rev. 1995; 59:623–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dennis P.P., Bremer H.. Modulation of chemical composition and other parameters of the cell at different exponential growth rates. EcoSal Plus. 2008; 3: 10.1128/ecosal.5.2.3. [DOI] [PubMed] [Google Scholar]
- 4. Gagarinova A., Stewart G., Samanfar B., Phanse S., White C.A., Aoki H., Deineko V., Beloglazova N., Yakunin A.F., Golshani A.et al.. Systematic genetic screens reveal the dynamic global functional organization of the bacterial translation machinery. Cell Rep. 2016; 17:904–916. [DOI] [PubMed] [Google Scholar]
- 5. Klumpp S., Hwa T.. Growth-rate-dependent partitioning of RNA polymerases in bacteria. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:20245–20250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schmidt A., Kochanowski K., Vedelaar S., Ahrné E., Volkmer B., Callipo L., Knoops K., Bauer M., Aebersold R., Heinemann M.. The quantitative and condition-dependent Escherichia coli proteome. Nat. Biotechnol. 2016; 34:104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ehrenberg M., Bremer H., Dennis P.P.. Medium-dependent control of the bacterial growth rate. Biochimie. 2013; 95:643–658. [DOI] [PubMed] [Google Scholar]
- 8. Jinks-Robertson S., Nomura M.. Neidhardt F.C., Ingraham J.L., Low K.B., Magasanik B., Schaechter M., Umbarger H.E.. Ribosomes and tRNA. Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology. 1987; Washington DC: American Society for Microbiology; 1358–1385. [Google Scholar]
- 9. Brosius J., Dull T.J., Sleeter D.D., Noller H.F.. Gene organization and primary structure of a ribosomal RNA operon from Escherichia coli. J. Mol. Biol. 1981; 148:107–127. [DOI] [PubMed] [Google Scholar]
- 10. Klumpp S., Hwa T.. Traffic patrol in the transcription of ribosomal RNA. RNA Biology. 2009; 6:392–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Couturier E., Rocha E.P.C.. Replication-associated gene dosage effects shape the genomes of fast-growing bacteria but only for transcription and translation genes: gene dosage effects and genome organisation. Mol. Microbiol. 2006; 59:1506–1518. [DOI] [PubMed] [Google Scholar]
- 12. Roller B.R.K., Stoddard S.F., Schmidt T.M.. Exploiting rRNA operon copy number to investigate bacterial reproductive strategies. Nat Microbiol. 2016; 1:16160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stoddard S.F., Smith B.J., Hein R., Roller B.R.K., Schmidt T.M.. rrnDB: improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res. 2015; 43:D593–D598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Condon C., French S., Squires C., Squires C.L.. Depletion of functional ribosomal RNA operons in Escherichia coli causes increased expression of the remaining intact copies. EMBO J. 1993; 12:4305–4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yano K., Wada T., Suzuki S., Tagami K., Matsumoto T., Shiwa Y., Ishige T., Kawaguchi Y., Masuda K., Akanuma G.et al.. Multiple rRNA operons are essential for efficient cell growth and sporulation as well as outgrowth in Bacillus subtilis. Microbiology. 2013; 159:2225–2236. [DOI] [PubMed] [Google Scholar]
- 16. Condon C., Liveris D., Squires C., Schwartz I., Squires C.L.. rRNA operon multiplicity in Escherichia coli and the physiological implications of rrn inactivation. J. Bacteriol. 1995; 177:4152–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Klappenbach J.A., Dunbar J.M., Schmidt T.M.. rRNA operon copy number reflects ecological strategies of bacteria. Appl. Environ. Microbiol. 2000; 66:1328–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gyorfy Z., Draskovits G., Vernyik V., Blattner F.F., Gaal T., Posfai G.. Engineered ribosomal RNA operon copy-number variants of E. coli reveal the evolutionary trade-offs shaping rRNA operon number. Nucleic Acids Res. 2015; 43:1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cabrera J.E., Jin D.J.. Active transcription of rRNA operons is a driving force for the distribution of RNA polymerase in bacteria: effect of extrachromosomal copies of rrnB on the in vivo localization of RNA polymerase. JB. 2006; 188:4007–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rocha E.P. Order and disorder in bacterial genomes. Curr. Opin. Microbiol. 2004; 7:519–527. [DOI] [PubMed] [Google Scholar]
- 21. Merrikh H., Zhang Y., Grossman A.D., Wang J.D.. Replication–transcription conflicts in bacteria. Nat. Rev. Microbiol. 2012; 10:449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Merrikh H., Machón C., Grainger W.H., Grossman A.D., Soultanas P.. Co-directional replication–transcription conflicts lead to replication restart. Nature. 2011; 470:554–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K.A., Tomita M., Wanner B.L., Mori H.. Construction of Escherichiacoli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006; 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Viguera E., Petranovic M., Zahradka D., Germain K., Ehrlich D.S., Michel B.. Lethality of bypass polymerases in Escherichia coli cells with a defective clamp loader complex of DNA polymerase III: lethality of bypass polymerases in a clamp loader mutant. Mol. Microbiol. 2003; 50:193–204. [DOI] [PubMed] [Google Scholar]
- 25. Foster P.L., Niccum B.A., Popodi E., Townes J.P., Lee H., MohammedIsmail W., Tang H.. Determinants of base-pair substitution patterns revealed by whole-genome sequencing of DNA mismatch repair defective Escherichiacoli. Genetics. 2018; 209:1029–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zaslaver A., Bren A., Ronen M., Itzkovitz S., Kikoin I., Shavit S., Liebermeister W., Surette M.G., Alon U.. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat. Methods. 2006; 3:623–628. [DOI] [PubMed] [Google Scholar]
- 27. Mathieu A., Fleurier S., Frénoy A., Dairou J., Bredeche M.-F., Sanchez-Vizuete P., Song X., Matic I.. Discovery and function of a general core hormetic stress response in E. coli induced by sublethal concentrations of antibiotics. Cell Rep. 2016; 17:46–57. [DOI] [PubMed] [Google Scholar]
- 28. Boye E., Løbner-Olesen A.. Bacterial growth control studied by flow cytometry. Res. Microbiol. 1991; 142:131–135. [DOI] [PubMed] [Google Scholar]
- 29. Stead M.B., Agrawal A., Bowden K.E., Nasir R., Mohanty B.K., Meagher R.B., Kushner S.R.. RNA snap TM: a rapid, quantitative and inexpensive, method for isolating total RNA from bacteria. Nucleic Acids Res. 2012; 40:e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liveris D., Klotsky R.A., Schwartz I.. Growth rate regulation of translation initiation factor IF3 biosynthesis in Escherichia coli. J. Bacteriol. 1991; 173:3888–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hall B.M., Ma C.-X., Liang P., Singh K.K.. Fluctuation analysis calculator: a web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics. 2009; 25:1564–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shee C., Cox B.D., Gu F., Luengas E.M., Joshi M.C., Chiu L.-Y., Magnan D., Halliday J.A., Frisch R.L., Gibson J.L.et al.. Engineered proteins detect spontaneous DNA breakage in human and bacterial cells. Elife. 2013; 2:e01222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Quan S., Skovgaard O., McLaughlin R.E., Buurman E.T., Squires C.L.. Markerless Escherichiacoli rrn deletion strains for genetic determination of ribosomal binding sites. G3. 2015; 5:2555–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bartlett M.S., Gourse R.L.. Growth rate-dependent control of the rrnB P1 core promoter in Escherichia coli. J. Bacteriol. 1994; 176:5560–5564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shah D., Zhang Z., Khodursky A., Kaldalu N., Kurg K., Lewis K.. [No title found]. BMC Microbiol. 2006; 6:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cohen S.E., Foti J.J., Simmons L.A., Walker G.C.. The SOS regulatory network. EcoSal Plus. 2008; 3: 10.1128/ecosalplus.5.4.3. [DOI] [PubMed] [Google Scholar]
- 37. Mount D.W., Low K.B., Edmiston S.J.. Dominant mutations (lex) in Escherichia coli K-12 which affect radiation sensitivity and frequency of ultraviolet lght-induced mutations. J. Bacteriol. 1972; 112:886–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bell J.C., Kowalczykowski S.C.. Mechanics and single-molecule interrogation of DNA recombination. Annu. Rev. Biochem. 2016; 85:193–226. [DOI] [PubMed] [Google Scholar]
- 39. Jarosz D.F., Beuning P.J., Cohen S.E., Walker G.C.. Y-family DNA polymerases in Escherichia coli. Trends Microbiol. 2007; 15:70–77. [DOI] [PubMed] [Google Scholar]
- 40. Sgorbati S., Barbesti S., Citterio S., Bestetti G., DeVecchi R.. Characterization of number, DNA content, viability and cell size of bacteria from natural environments using DAPI/PI dual staining and flow cytometry. Minerva Biotecnologica. 1996; 8:9–15. [Google Scholar]
- 41. Potvin-Trottier L., Luro S., Paulsson J.. Microfluidics and single-cell microscopy to study stochastic processes in bacteria. Curr. Opin. Microbiol. 2018; 43:186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bartlett M.S., Gaal T., Ross W., Gourse R.L.. RNA polymerase mutants that destabilize RNA polymerase-promoter complexes alter NTP-sensing by rrn P1 promoters. J. Mol. Biol. 1998; 279:331–345. [DOI] [PubMed] [Google Scholar]
- 43. Ginés-Candelaria E., Blinkova A., Walker J.R.. Mutations in Escherichia coli dnaA which suppress a dnaX(Ts) polymerization mutation and are dominant when located in the chromosomal allele and recessive on plasmids. J. Bacteriol. 1995; 177:705–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Borukhov S., Lee J., Laptenko O.. Bacterial transcription elongation factors: new insights into molecular mechanism of action: Bacterial transcription elongation factors: Gre, NusA and Mfd. Mol. Microbiol. 2004; 55:1315–1324. [DOI] [PubMed] [Google Scholar]
- 45. Chan Y.A., Aristizabal M.J., Lu P.Y.T., Luo Z., Hamza A., Kobor M.S., Stirling P.C., Hieter P.. Genome-wide profiling of yeast DNA:RNA hybrid prone sites with DRIP-chip. PLos Genet. 2014; 10:e1004288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stork C.T., Bocek M., Crossley M.P., Sollier J., Sanz L.A., Chédin F., Swigut T., Cimprich K.A.. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. Elife. 2016; 5:e17548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wahba L., Costantino L., Tan F.J., Zimmer A., Koshland d. S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes Dev. 2016; 30:1327–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Crooke S.T., Lemonidis K.M., Neilson L., Griffey R., Lesnik E.A., Monia B.P.. Kinetic characteristics of Escherichia coli RNase H1: cleavage of various antisense oligonucleotide-RNA duplexes. Biochem. J. 1995; 312:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hogrefe H.H., Hogrefe R.I., Walder R.Y., Walder J.A.. Kinetic analysis of Escherichia coli RNase H using DNA-RNA-DNA/DNA substrates. J. Biol. Chem. 1990; 265:5561–5566. [PubMed] [Google Scholar]
- 50. Carles-Kinch K., George J.W., Kreuzer K.N.. Bacteriophage T4 UvsW protein is a helicase involved in recombination, repair and the regulation of DNA replication origins. EMBO J. 1997; 16:4142–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dudas K.C., Kreuzer K.N.. UvsW protein regulates bacteriophage T4 origin-dependent replication by unwinding R-Loops. Mol. Cell. Biol. 2001; 21:2706–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Saxena S., Myka K.K., Washburn R., Costantino N., Court D.L., Gottesman M.E.. Escherichia coli transcription factor NusG binds to 70S ribosomes: NusG binding to ribosomes couples transcription with translation. Mol. Microbiol. 2018; 108:495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang Y., Mandava C.S., Cao W., Li X., Zhang D., Li N., Zhang Y., Zhang X., Qin Y., Mi K.et al.. HflX is a ribosome-splitting factor rescuing stalled ribosomes under stress conditions. Nat. Struct. Mol. Biol. 2015; 22:906–913. [DOI] [PubMed] [Google Scholar]
- 54. Kouzminova E.A., Kuzminov A.. Chromosome demise in the wake of ligase-deficient replication. Mol. Microbiol. 2012; 84:1079–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Deutscher M.P. Degradation of stable RNA in bacteria. J. Biol. Chem. 2003; 278:45041–45044. [DOI] [PubMed] [Google Scholar]
- 56. Abrell J.W. Ribonuclease I released from Escherichia coli by osmotic shock. Arch. Biochem. Biophys. 1971; 142:693–700. [DOI] [PubMed] [Google Scholar]
- 57. Tolker-Nielsen T., Molin S.. Role of ribosome degradation in the death of heat-stressed Salmonellatyphimurium. FEMS Microbiol. Lett. 1996; 142:155–160. [DOI] [PubMed] [Google Scholar]
- 58. Merritt M.E., Donaldson J.R.. Effect of bile salts on the DNA and membrane integrity of enteric bacteria. J. Med. Microbiol. 2009; 58:1533–1541. [DOI] [PubMed] [Google Scholar]
- 59. Voulgaris J., French S., Gourse R.L., Squires C., Squires C.L.. Increased rrn gene dosage causes intermittent transcription of rRNA in Escherichia coli. J. Bacteriol. 1999; 181:4170–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Acinas S.G., Marcelino L.A., Klepac-Ceraj V., Polz M.F.. Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. JB. 2004; 186:2629–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kogoma T., Hong X., Cadwell G., Barnard K., Asai T.. Requirement of homologous recombination functions for viability of the Escherichia coli cell that lacks RNase HI and exonuclease V activities. Biochimie. 1993; 75:89–99. [DOI] [PubMed] [Google Scholar]
- 62. Bartas M., Čutová M., Brázda V., Kaura P., Šťastný J., Kolomazník J., Coufal J., Goswami P., Červeň J., Pečinka P.. The presence and localization of G-Quadruplex forming sequences in the domain of bacteria. Molecules. 2019; 24:1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brambati A., Zardoni L., Nardini E., Pellicioli A., Liberi G.. The dark side of RNA:DNA hybrids. Mutation Research/Reviews in Mutation Research. 2020; 784:108300. [DOI] [PubMed] [Google Scholar]
- 64. Maduike N.Z., Tehranchi A.K., Wang J.D., Kreuzer K.N.. Replication of the Escherichiacoli chromosome in RNase HI-deficient cells: multiple initiation regions and fork dynamics: replication in RNase HI-deficient Escherichiacoli. Mol. Microbiol. 2014; 91:39–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hraiky C., Raymond M.-A., Drolet M.. RNase H overproduction corrects a defect at the level of transcription elongation during rRNA synthesis in the absence of DNA Topoisomerase I in Escherichiacoli. J. Biol. Chem. 2000; 275:11257–11263. [DOI] [PubMed] [Google Scholar]
- 66. Massé E., Phoenix P., Drolet M.. DNA topoisomerases regulate R-loop formation during transcription of the rrnB operon in Escherichiacoli. J. Biol. Chem. 1997; 272:12816–12823. [DOI] [PubMed] [Google Scholar]
- 67. Vydzhak O., Luke B., Schindler N.. Non-coding RNAs at the Eukaryotic rDNA Locus: RNA–DNA hybrids and beyond. J. Mol. Biol. 2020; 432:4287–4304. [DOI] [PubMed] [Google Scholar]
- 68. El Hage A., French S.L., Beyer A.L., Tollervey D.. Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev. 2010; 24:1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shen W., Sun H., De Hoyos C.L., Bailey J.K., Liang X., Crooke S.T.. Dynamic nucleoplasmic and nucleolar localization of mammalian RNase H1 in response to RNAP I transcriptional R-loops. Nucleic Acids Res. 2017; 45:10672–10692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Skarstad K., Katayama T.. Regulating DNA replication in bacteria. Cold Spring Harb. Perspect. Biol. 2013; 5:a012922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hill C.W. Large genomic sequence repetitions in bacteria: lessons from rRNA operons and Rhs elements. Res. Microbiol. 1999; 150:665–674. [DOI] [PubMed] [Google Scholar]
- 72. Sutera V.A., Lovett S.T.. The role of replication initiation control in promoting survival of replication fork damage. Mol. Microbiol. 2006; 60:229–239. [DOI] [PubMed] [Google Scholar]
- 73. Boubakri H., de Septenville A.L., Viguera E., Michel B.. The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J. 2010; 29:145–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Krishna Leela J., Syeda A.H., Anupama K., Gowrishankar J.. Rho-dependent transcription termination is essential to prevent excessive genome-wide R-loops in Escherichia coli. Proc. Natl. Acad. Sci. 2013; 110:258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nanamiya H., Sato M., Masuda K., Sato M., Wada T., Suzuki S., Natori Y., Katano M., Akanuma G., Kawamura F.. Bacillus subtilis mutants harbouring a single copy of the rRNA operon exhibit severe defects in growth and sporulation. Microbiology. 2010; 156:2944–2952. [DOI] [PubMed] [Google Scholar]
- 76. Datsenko K.A., Wanner B.L.. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. 2000; 97:6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We comply with the Minimal Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines. Other details, including the sequences of primers used in the quantitative PCR, are supplied in Materials and Methods section of this manuscript.