

Abstract
Background
Olutasidenib (FT-2102) is a highly potent, orally bioavailable, brain-penetrant and selective inhibitor of mutant isocitrate dehydrogenase 1 (IDH1). The aim of the study was to determine the safety and clinical activity of olutasidenib in patients with relapsed/refractory gliomas harboring an IDH1R132X mutation.
Methods
This was an open-label, multicenter, nonrandomized, phase Ib/II clinical trial. Eligible patients (≥18 years) had histologically confirmed IDH1R132X-mutated glioma that relapsed or progressed on or following standard therapy and had measurable disease. Patients received olutasidenib, 150 mg orally twice daily (BID) in continuous 28-day cycles. The primary endpoints were dose-limiting toxicities (DLTs) (cycle 1) and safety in phase I and objective response rate using the Modified Response Assessment in Neuro-Oncology criteria in phase II.
Results
Twenty-six patients were enrolled and followed for a median 15.1 months (7.3‒19.4). No DLTs were observed in the single-agent glioma cohort and the pharmacokinetic relationship supported olutasidenib 150 mg BID as the recommended phase II dose. In the response-evaluable population, disease control rate (objective response plus stable disease) was 48%. Two (8%) patients demonstrated a best response of partial response and eight (32%) had stable disease for at least 4 months. Grade 3‒4 adverse events (≥10%) included alanine aminotransferase increased and aspartate aminotransferase increased (three [12%], each).
Conclusions
Olutasidenib 150 mg BID was well tolerated in patients with relapsed/refractory gliomas harboring an IDH1R132X mutation and demonstrated preliminary evidence of clinical activity in this heavily pretreated population.
Keywords: brain penetration, glioma, IDH1, mutant, olutasidenib
Key Points.
Olutasidenib is a potent, brain-penetrant, selective inhibitor of mutant IDH1
Olutasidenib was well tolerated in patients with relapsed/refractory gliomas
The disease control rate (objective response plus stable disease) was 48%
Importance of the Study.
Olutasidenib is a brain-penetrant, selective inhibitor of mutant IDH1. In this study, olutasidenib 150 mg twice daily was well tolerated in patients with relapsed or refractory gliomas harboring an IDH1 mutation. Olutasidenib plasma concentrations, at levels predicted to safely provide benefit, were maintained over the duration of treatment, and measurement of olutasidenib in cerebrospinal fluid provided evidence of brain penetration. Olutasidenib demonstrated preliminary evidence of clinical activity in heavily pretreated patients with progressive/recurrent, enhancing, high-grade and low-grade gliomas. Magnetic resonance images for two patients with a partial response provided evidence of an ≥80% reduction in tumor burden (ie, MRI cross-sectional tumor area) and 32% (8/25) patients had stable disease for >4 months.
Gliomas represent the most common malignant primary brain tumors in adults and pose ongoing challenges in terms of mortality associated with the disease and morbidity associated with available treatment options.1–3 Despite current standard of care treatments, which include surgery, radiotherapy, and chemotherapy, these tumors inexorably recur, progress in grade, and patients eventually succumb to their disease.4 In addition to high mortality, late radiation-induced cognitive decline, a delayed complication of radiation therapy, can affect up to half of adult patients with brain tumors at least 6 months after radiotherapy.2 Particularly for patients with lower-grade gliomas (LGG), who are expected to live a relatively long life, cognitive impairment of this nature can present ongoing challenges in terms of quality of life, employment, financial burden, and independence.5
More than 70% of patients with LGG (World Health Organization [WHO] grades II/III) and approximately 5‒7% of patients with glioblastomas harbor mutations in the gene encoding for the isocitrate dehydrogenase protein 1 (IDH1).6–8 IDH1 catalyzes the conversion of isocitrate to alpha-ketoglutarate (α-KG) and, in the process, generates NADPH from cofactor NADP+ to increase antioxidant protection.9,10 α-KG-dependent enzymes use α-KG as a substrate to maintain normal energy metabolism in the cytosol and to facilitate gene expression accommodating of cellular differentiation via the regulation of DNA and histone methylation patterns in the nucleus.11,12 In gliomagenesis, IDH1 mutations alter the function of IDH via neoenzymatic gain-of-function activity.12 This drives the conversion of α-KG to the structurally similar oncometabolite R-2-hydroxyglutamate (R-2-HG) and consumes NADPH.13
Supraphysiological concentrations of R-2-HG, a competitive inhibitor of α-KG-dependent enzymes, disrupts cellular metabolism, resulting in transcription-altering hypermethylation, and inhibition of cellular differentiation.14,15IDH mutations can predispose cells to malignant transformation and augment tumor progression.16,17 In preclinical models, mIDH1 has been shown to promote glioma tumorigenesis in concert with other co-operative molecular alterations.18IDH1 mutations at codon 132 are heterozygous and result in dysfunctional IDH1 proteins when a common arginine residue in the catalytic site is substituted by a single histidine, serine, leucine, glycine, or cysteine residue (R132X).19 Of note, gliomas are known to have a high percentage of R132H mutations, and available data indicate that patients harboring non-R132H mutations have a more favorable prognosis than patients with R132H mutations.20,21 It is also important to note that in addition to molecular characteristics such as IDH1 mutation status, tumor grading remains an important predictor of clinical outcome.22 Based on the pivotal role that IDH1 plays, through its effector 2-HG, in glioma initiation, cellular metabolism, epigenetic modulation, redox regulation, and DNA repair, targeted inhibition of mutant IDH1 becomes an attractive therapeutic strategy for maximal 2-HG depletion.23 Of note, the safety and clinical activity of ivosidenib, an inhibitor of mutant IDH1 and vorasidenib, a dual inhibitor of mutant IDH1/2, were recently evaluated in phase I studies in patients with mutant IDH gliomas.24,25 Both drugs showed a favorable safety profile, with reversible dose-limiting toxicities (DLTs) of elevated transaminases for vorasidenib at doses ≥ 100 mg. In patients with non-enhancing gliomas, ivosidenib showed prolonged disease control and reduced tumor growth and vorasidenib showed preliminary antitumor activity. For both studies, no patients with enhancing gliomas had a confirmed response, though prolonged stable disease was observed in an important number of patients.
Olutasidenib (FT-2102) is a highly potent, orally bioavailable, brain-penetrant, and selective inhibitor of mutant IDH1.26 Olutasidenib was designed to reduce R-2-HG and revert pathologic epigenetic modifications that impair cellular differentiation to restore regulatory enzyme function. In an ongoing phase I/II study, olutasidenib, as monotherapy or in combination with azacitidine, has been shown to be well tolerated and induce durable complete remissions in a subset of patients with high-risk relapsed/refractory acute myeloid leukemia (AML) and has shown preliminary evidence of clinical activity in patients with myelodysplastic syndrome (MDS).27–30
Here, we report on the phase Ib/II study evaluating the safety, tolerability, pharmacokinetics (PK), and clinical activity of olutasidenib monotherapy in a cohort of patients with relapsed or refractory gliomas harboring IDH1 mutations.31
Methods
Study Design and Participants
This was an open-label, multicenter, nonrandomized, phase Ib/II, basket study of advanced solid malignancies including gliomas assessed in parallel groups across 26 global sites in North America, Europe, and Asia-Pacific. Here, we report the results of the patients who received olutasidenib monotherapy in the glioma cohort.
Screening for patient eligibility was performed ≤30 days from the first dose of study drug. In phase Ib of this study, treatment was initiated in a safety lead-in period over a 28-day cycle during which dose-limiting toxicities (DLTs) in patients receiving olutasidenib were monitored using a standard 3 + 3 design. The planned starting dose of olutasidenib was 150 mg twice daily, equivalent to the recommended phase II dose (RP2D) for patients with acute myeloid leukemia or myelodysplastic syndrome in a phase I/II hematological study.27
In phase II, an optimal Simon’s two-stage design was used to assess efficacy and confirm the overall safety of olutasidenib. Eight patients with relapsed or refractory glioma were to be enrolled and treated with the RP2D for olutasidenib in stage I, and if ≥1 responses were observed, an additional 15 patients could be enrolled and treated until disease progression without clinical benefit or unacceptable toxicity (Figure 1).
Fig. 1.

Trial profile. DLT, dose-limiting toxicity; IDH1R132X, isocitrate dehydrogenase 1 mutation.
Patients considered for this study were aged ≥18 years, had histologically confirmed IDH1R132X mutant glioma that relapsed or progressed on or following standard therapy, measurable disease, and a life expectancy of ≥4 months (see Supplementary Material for additional detail regarding glioma-specific inclusion criteria). Patients had to be recovered to National Cancer Institute Common Terminology Criteria for Adverse Event (CTCAE; version 4.03) grade ≤ 2 or baseline toxicity (excluding alopecia) since prior therapy, have adequate hepatic, renal, bone marrow, and cardiac function, and an Eastern Cooperative Oncology Group (ECOG) performance status of 0–2. There were no eligibility restrictions for tumor grade at baseline. All prior antitumor treatments were allowed, except for prior IDH1 inhibitor treatment; the minimum elapsed time from last dose of prior treatment to first dose of olutasidenib was specified by the study protocol. Per protocol, patients were excluded if they had prior radiation therapy within 4 weeks of the first dose of study treatment. Enrolment of patients who had failed bevacizumab was allowed per protocol. Patients who underwent a previous solid organ or hematopoietic cell transplant were excluded.
All patients gave written informed consent before screening and enrolment and the study was conducted in accordance with the principles of the Declaration of Helsinki and approved by the institutional ethical review boards or local ethics committees of participating institutions. Additional study details are available in the protocol.
Procedures
Patients with relapsed or refractory gliomas harboring IDH1R132X mutations received olutasidenib orally, as a 150-mg capsule twice daily, in continuous 28-day cycles. Capsules were received at least 2-hours postprandially or at least 30 min before a meal.
DLTs were assessed during the safety lead-in period in phase I using the CTCAE version 4.03. DLTs were events unrelated to underlying disease and considered related to olutasidenib. Any adverse events (AEs) that met the criteria for a DLT but occurred after cycle 1 or during stage I of phase II were also considered. DLTs comprised clinically relevant grade ≥3 nonhematologic laboratory findings with clinical sequelae or requiring treatment, and all grade ≥3 toxicities (excluding grade 3 nausea, vomiting, or rash lasting <72 hours with optimal medical management), as well as grade 4 neutropenia lasting >7 days and grade ≥3 thrombocytopenia and grade ≥3 febrile neutropenia.
Safety monitoring of AEs, including AEs of special interest, and serious AEs occurred from the time of signed informed consent through 28 days after the last dose of olutasidenib. AEs were graded for severity according to the CTCAE 4.03 and investigators assessed their relatedness to olutasidenib. Treatment-emergent adverse events (TEAEs) were summarized using the Medical Dictionary for Regulatory Activities (MedDRA; version 21.0). Other key safety assessments included clinical laboratory measurements (blood chemistry and hematology tests done prior to dosing on dosing days), additional liver function tests (days 8 and 22 [±2] of cycle 2 and beyond), ECOG performance status, and physical exams. PK assessments are described in the Supplementary Material.
For the assessment of antitumor activity, investigators evaluated best response in accordance with the modified Response Assessment in Neuro-Oncology (RANO) 2017 criteria and RANO-LGG.32,33 Tumor responses were assessed using contrast-enhanced magnetic resonance imaging (MRI). Efficacy assessments were on day 1 (±7) of cycles 3, 5, 7, 9, and 12, and every three cycles thereafter. All responses were confirmed with a follow-up MRI obtained at least 4 weeks after the initial partial response.
For exploratory assessments, central blinded volumetric assessments of tumor size changes (ie, tumor burden) utilized MRI and included the analysis of T2 or fluid attenuated inversion recovery (FLAIR) tumor volume, and post-gadolinium T1-weighted volumes (ie, nonenhancing and enhancing).
Survival follow-up assessments occurred every 3 months, after documented disease progression or the start of a new anticancer therapy, for up to 2 years following the first dose of olutasidenib or up to 1 year after the last dose, whichever was longer.
Outcomes
The primary endpoints were DLTs in the safety lead-in period, AEs, and safety laboratory values in phase Ib and objective response rate (ORR) per assessment criteria in phase II. ORR was defined as the proportion of patients with a best response of complete response, partial response, or for LGGs, a minor response. Secondary endpoints were AEs, laboratory values (phase II), PK values derived from olutasidenib in plasma or cerebrospinal fluid as available, ORR (phase Ib), and time-to-event endpoints (phase Ib/II) including progression-free survival (PFS), time to progression, time to response, duration of response, and overall survival.
Statistical Analysis
The data cutoff date for this analysis was December 3, 2020. The Simon’s two-stage design employed used a one-sided alpha of 0.025, power of 80%, a null hypothesis of 5% ORR, assuming the true ORR was 25%. In stage II, the null hypothesis of 5% ORR was to be rejected if ≥4 responses were achieved out of a total of 23 patients (17%). The null hypothesis rate of 5% was chosen assuming the possibility of a placebo response rate of 5% based on potential for variability in imaging and historical control response rates.34 The 25% ORR alternative hypothesis rate was selected to be superior to the rate of temozolomide in recurrent GBM patients and allowed for a reasonably small sample size in this signal-seeking study.35,36
Analysis sets utilized in this study were: the DLT-evaluable population, participants who had a DLT during cycle 1 or completed ≥75% of the prescribed cycle 1 dose in the safety lead-in period; the safety population, all patients who received any amount of olutasidenib during the study; the response-evaluable population, all patients with measurable disease who received any amount of olutasidenib and had ≥1 post-baseline response assessment, or discontinued treatment owing to disease progression within 8 [+2] weeks of first olutasidenib dose; and the PK population, patients from stage I who received ≥1 dose of olutasidenib and for whom ≥1 primary PK parameter was calculable.
ORR was evaluated at the one-sided, 0.025 level of significance. Post-hoc analyses summarized disease control rate (objective response plus stable disease) using descriptive statistics. Time-to-event data were summarized as the median (interquartile range [IQR]) using Kaplan–Meier methodology with associated two-sided 95% confidence intervals (CI) and percentage of censored observations. All other data were analyzed using SAS statistical software (version 9.3) to calculate descriptive statistics.
The study was registered with ClinicalTrials.gov, NCT03684811 and the European Clinical Trials database, Eudra-CT, number 2018-001796-21.
Results
Between November 7, 2018 and February 3, 2020, 26 patients with glioma were screened for this study. All 26 patients comprised the safety analysis set, three (12%) patients were in the DLT-evaluable analysis set, 25 (96%) were in the response-evaluable analysis set, and 13 (50%) made up the PK analysis set. Five (19%) patients remained on treatment at the data cutoff date.
Baseline characteristics in the safety population are summarized (Table 1). The median age of patients was 45 (IQR 40‒49) years, the majority were male (17 [65%]), and White (25 [96%]). At study enrolment, most patients (22 [85%]) had tumors classified as WHO grade III/IV: grade III (15 [58%]) and grade IV (seven [27%]). The most common types of IDH1-mutated glioma were anaplastic astrocytoma (11 [42%] patients) and glioblastoma (seven [27%]). Most patients (22 [85%]) had the IDH1R132H mutation subtype (Table 1). At study enrolment, tumors were enhancing in 23 (88%) patients. Overall, patients had experienced disease recurrence a median 3.4 months (IQR 1.3‒37.4) since last recurrence. Patients had received a median of 2 (IQR 1‒3) prior regimens with 11 (42%) patients receiving ≥3 prior regimens. All patients had prior radiation therapy (seven with prior re-radiation), all had at least one surgery prior to enrolment, including five patients who had surgery/biopsy immediately before enrolment (<2 months) with no therapy from the time of the surgical procedure to the time of enrolment, and 23 (88%) had prior chemotherapy. Patients most commonly received prior: temozolomide (23 [88%] patients), lomustine (nine [35%]), procarbazine (five [19%]), and bevacizumab (four [15%]).
Table 1.
Baseline Characteristics
| Characteristics*,† | Olutasidenib (N = 26) |
|---|---|
| Age in years | |
| Median (IQR) | 45 (40‒49) |
| By category | |
| 18–40 | 7 (27%) |
| 41–59 | 16 (62%) |
| ≥60 | 3 (12%) |
| Sex‡ | |
| Female | 9 (35%) |
| Male | 17 (65%) |
| Race | |
| White | 25 (96%) |
| Not reported | 1 (4%) |
| Ethnicity | |
| Hispanic | 2 (8%) |
| Non-Hispanic | 22 (85%) |
| Not reported | 2 (8%) |
| ECOG performance status | |
| 0 | 10 (38%) |
| 1 | 13 (50%) |
| 2 | 3 (12%) |
| Grade at initial diagnosis | |
| II | 7 (27%) |
| III | 14 (54%) |
| IV | 3 (12%) |
| Unknown/NA | 2 (8%) |
| IDH1 mutation subtype | |
| R132H | 22 (85%) |
| R132L | 2 (8%) |
| R132C | 1 (4%) |
| R132G | 1 (4%) |
| Baseline steroid use | |
| Yes | 5 (19%) |
| No | 21 (81%) |
| Median (IQR) years since initial diagnosis | 6 (4-11) |
| Median (IQR) years since last recurrence | 0.3 (0.1-3.1) |
| Prior therapies** | |
| Surgery only | 2 (8%) |
| Systemic therapy and surgery | 24 (92%) |
| Radiation | 26 (100%) |
| Median (IQR) number of prior surgeries | 2 (1‒2) |
| Median (IQR) years since last surgery | 1.8 (0.5–2.7) |
| Median (IQR) years since last radiation | 1.9 (0.9–6.4) |
| Median (IQR) number prior regimens§ | 2 (1‒3) |
| Number of prior regimens§ | |
| 1 | 7 (27%) |
| 2 | 6 (23%) |
| 3 | 7 (27%) |
| >3 | 4 (15%) |
| Glioma diagnosis | |
| Diffuse astrocytoma, IDH mutant | 1 (4%) |
| Anaplastic astrocytoma, IDH mutant*** | 12 (46%) |
| Oligodendroglioma, IDH mutant | 4 (15%) |
| Anaplastic oligodendroglioma, IDH mutant | 2 (8%) |
| Glioblastoma, IDH mutant | 7 (27%) |
| Tumor type | |
| Enhancing | 23 (88%) |
| Non-enhancing | 3 (12%) |
| Glioma grade at study enrolment (per WHO)**** | |
| II | 4 (15%) |
| III | 15 (58%) |
| IV | 7 (27%) |
*Data are n (%) and median (IQR), unless shown otherwise.
†Percentages may not sum to 100% due to rounding.
‡Sex (as assigned at birth).
§ n = 24.
**All prior antitumor treatments were allowed, except for prior IDH1 inhibitor treatment; the minimum elapsed time from last dose of prior treatment to first dose of olutasidenib was specified by the study protocol.
***Includes one patient with a diagnosis of “anaplastic astrocytoma, NOS”; patient was confirmed to harbor an R132H IDH1 mutation.
****Includes updated tumor grade information for 5 patients with surgery with ~2 months of enrolment.
ECOG, Eastern Cooperative Oncology Group; IQR, interquartile range; NA, not applicable, NOS, not otherwise specified; STD, standard deviation; WHO, World Health Organization.
The median follow-up time was 15.1 months (IQR 7.3‒19.4). The overall median duration of olutasidenib treatment was 4.2 months (1.5–15.2); up to 21 (28-day) cycles of olutasidenib were received by five patients in the safety analysis set. Mean compliance was 100% (standard deviation 1) during cycle 1 and 99% (3) overall. Protocol-defined disease progression was the most common primary reason for treatment discontinuation (20/26 [77%] patients) and death was the most common reason for study discontinuation (13 [50%] patients; Figure 1).
In the phase Ib safety lead-in period, the first three patients who received 150 mg of olutasidenib twice daily experienced no DLTs. In view of available PK data (Supplementary Table 1 and Figure 1), and supportive safety, PK, and pharmacodynamic data in patients with acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS),37 the initially selected olutasidenib dosing regimen was determined to be tolerable and safe, and was chosen as the RP2D for further consideration in patients with glioma in this study.
TEAEs were reported for all patients (26 [100%]), with treatment-related events in 23 (88%) patients. Serious AEs (SAEs) were reported for 11 (42%) patients, with treatment-related serious events in three (12%) patients. The most common (>25%) TEAEs were: nausea (14/26 [54%] patients), fatigue (13 [50%]), alanine aminotransferase (ALT) increased, diarrhea, and headache (eight [31%] each), and constipation and fall (seven [27%] each) (Table 2 and Supplementary Table 2). Most events were mild or moderate in severity and did not result in treatment modification or discontinuation. Grade 3‒4 TEAEs (irrespective of causality) were reported for 11 (42%) patients. Grade 3 events included ALT increased and aspartate aminotransferase (AST) increased, each in three (12%) patients, hemiparesis in two (8%), and all other events were reported in one (4%) patient each (Table 2 and Supplementary Table 3). One (4%) patient had a grade 4 event of treatment-related acute hepatitis. Two (8%) patients had fatal disease progression not related to treatment within 28 days after the last dose of olutasidenib. The most common (>20%) treatment-related AEs were nausea (ten [38%]), ALT increased (eight [31%]), and fatigue (seven [27%]); treatment-related grade 3 events reported in more than one patient were ALT and AST increased (three [12%] each) (Supplementary Table 4). As noted above, SAEs were reported in 11 (42%) patients, with treatment-related SAEs in three (12%) patients (one [4%] patient each with grade 4 acute hepatitis or grade 3 platelet count decreased, and one [4%] patient with both nausea and vomiting, each of grade 3 severity). Olutasidenib was withdrawn for the one patient with grade 4 acute hepatitis which resolved within 3 weeks with supportive treatment. Four (15%) patients had dose reductions that were treatment-related (three [12%] for ALT increased and one [4%] for hypophosphatemia) and five (19%) patients had treatment-related dose interruptions.
Table 2.
Summary of All Grades and Grades 3‒4 Treatment-emergent Adverse Events Reported for ≥10% of Patients (All Grades) in the Safety Population
| Preferred term*,† | Any grade N = 26 |
Grades 3–4§ N = 26 |
|---|---|---|
| Nausea | 14 (54%) | 1 (4%) |
| Fatigue | 13 (50%) | 0 |
| Alanine aminotransferase increased | 8 (31%) | 3 (12%) |
| Diarrhea | 8 (31%) | 0 |
| Headache | 8 (31%) | 1 (4%) |
| Constipation | 7 (27%) | 0 |
| Fall | 7 (27%) | 0 |
| Aspartate aminotransferase increased | 5 (19%) | 3 (12%) |
| Dysgeusia | 5 (19%) | 0 |
| Seizure | 5 (19%) | 0 |
| Vomiting | 5 (19%) | 1 (4%) |
| Dizziness | 4 (15%) | 0 |
| Hypertension | 4 (15%) | 0 |
| Insomnia | 4 (15%) | 0 |
| Platelet count decreased | 4 (15%) | 1 (4%) |
| Upper respiratory tract infection | 4 (15%) | 0 |
| Aphasia | 3 (12%) | 0 |
| Confusional state | 3 (12%) | 0 |
| Decreased appetite | 3 (12%) | 0 |
| Dyspepsia | 3 (12%) | 0 |
| Epistaxis | 3 (12%) | 0 |
| Hemiparesis | 3 (12%) | 2 (8%) |
| Hypophosphatemia | 3 (12%) | 1 (4%) |
| Muscular weakness | 3 (12%) | 0 |
| Paresthesia | 3 (12%) | 0 |
Data are n (%).
*Safety analysis set was defined as all patients who received any amount of study drug of olutasidenib.
†Per protocol, disease progression was not considered an adverse event when assessed by the Investigator to be unrelated to olutasidenib.
§All listed grade 3‒4 events were grade 3 and considered related to study drug except for headache and hemiparesis.
The majority of abnormal laboratory values were grades 1-2 and not clinically significant. One (4%) patient with acute hepatitis had concurrent grade 4 elevations in ALT and total bilirubin which decreased to grade 1 and grade 2, respectively, and resolved in 3 weeks. Mean changes from baseline in vital signs were also not of clinical significance across cycles through to end of treatment and the safety follow-up visit.
Steady state PK parameters for olutasidenib were achieved by cycle 1, day 8 and systemic exposure remained consistent for the duration of treatment (Supplementary Figure 1). On day 1 of cycle 2, the mean maximum plasma concentration at steady state was 2832 (CV%, 31.0) ng/mL and the mean area under the curve was 19,500 h·ng/mL (Supplementary Table 1). In cerebrospinal fluid (CSF) samples collected from two patients, the measured olutasidenib concentration ranged from 22.5 to 31.8 ng/mL and the unbound brain partition coefficients (Kpuu) were 0.79 and 0.54, respectively (Supplementary Table 5).
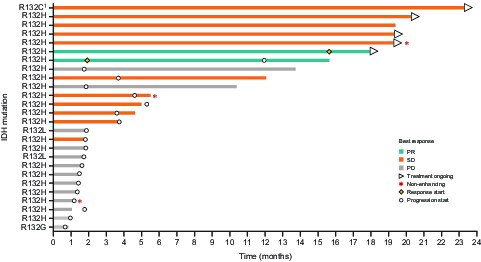
Following exposure to olutasidenib, 2/25 (8%) patients in the response-evaluable set (95% CI 1.0‒26.0%) demonstrated an objective response (both best responses of partial response) (Table 3 and Figure 2A), which did not meet the primary endpoint for activity. The partial responses per RANO were closely aligned with the independent central volumetric assessments (Figure 2B). The median time to response was 8.8 months (IQR 1.9‒15.6). Of the two responders, one patient’s duration of ongoing response was 2.3 months as of the data cut-off and the duration of response for the second patient was 10.0 months. The best overall responses per RANO for individual patients are shown in conjunction with duration of olutasidenib treatment (Supplementary Figure 2). Both responders had enhancing tumors at baseline; one was grade III and the other was a grade IV glioblastoma. Molecular profiling of these patients at the time of initial diagnosis demonstrated IDH1 R132H and TP53 mutations. One of these tumors also showed ATRX loss (Supplementary Table 6) and both tumors tested negative for CDKN2 loss.
Table 3.
Tumor burden assessed using RANO (2D) and volumetry (3D) in the response-evaluable analysis set*
| Investigator-assessed responses using RANO criteria | n/N | ORR | 95% CI |
|---|---|---|---|
| Objective response rate (ORR) | 2/25 | 8% | 1.0‒26.0% |
| Best overall response | |||
| Partial response (PR) | 2/25 | 8% | 1.0‒26.0% |
| Stable disease (SD)† | 10/25 | 40% | |
| Progressive disease (PD)‡ | 13/25 | 52% | |
| Clinical benefit rate (CR+PR+SD) | 12/25 | 48% | |
| Disease control rate (PR+SD) | 12/25 | 48% | |
| SD >4 months | 8/25 | 32% | |
| Blinded independent central review using volumetry | n/N§ | % | |
| ≥50% decrease | 4/24 | 17% | |
| >25% decrease but <50% decrease | 2/24 | 8% | |
| ≤25% decrease and ≤25% increase | 7/24 | 29% | |
| >25% increase | 11/24 | 46% |
*Response-evaluable analysis set was defined as all patients with measurable disease who received any amount of olutasidenib and had ≥1 post-baseline response assessment, or discontinued treatment owing to disease progression within 8 (+2) weeks of first olutasidenib dose.
†Two patients had non-enhancing tumors.
‡One patient had a non-enhancing tumor.
§One patient was non-evaluable for volumetric assessment.
2D=two-dimensional; 3D=three-dimensional; CI=confidence interval; RANO=Response Assessment in Neuro-Oncology.
Fig. 2.

Best response per RANO criteria and percentage change from baseline following olutasidenib therapy. (A) Investigator assessment using RANO and (B) volumetric assessments of tumor burden by central review. Color-coding of best response is provided according to RANO evaluation for both panels. Changes exceeding 100% are reported as 100%. BICR, blinded independent central review; PD, progressive disease; PR, partial response; RANO, Response Assessment in Neuro-Oncology; SD, stable disease.
Data showing tumor shrinkage with olutasidenib monotherapy are shown for the above two patients (Supplementary Figure 3). Target lesions identified using baseline brain MRIs were followed on study treatment; areas of cystic change and/or necrosis on T2/FLAIR for volumetric analysis were included in the estimate of total lesion volume and nonenhancing tumor volume on post-gadolinium T1-weighted images. Each set of MRIs is accompanied by longitudinal data depicting the percentage change in tumor burden by treatment cycle. The overall reductions in tumor burden reached or exceeded 80% over the course of treatment with olutasidenib for these patients.
The disease control rate (objective response plus stable disease) was 48% (12/25 patients), with a median duration of disease control of 8.6 (95% CI 5.8‒14.8) months. Ten (40%) response-evaluable patients had a best overall response of stable disease, with a median duration of 5.0 (95% CI 4.2‒14.6) months; eight of ten (80%) had stable disease for >4 months. Four of the ten (40%) patients who had stable disease achieved a reduction in the sum of products of the diameters <50% which did not qualify for partial response. Upon completion of the blinded independent central review, no target lesion was identified for one of 25 (4%) patients. Four out of 24 (17%) patients with central blinded volumetric assessments exhibited a ≥50% decrease in tumor burden after receiving olutasidenib, and an additional five patients had a reduction in tumor volume of <50% (Figure 2, Table 2).
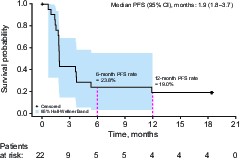
As of December 3, 2020, with five (19%) patients ongoing, 19 PFS events had occurred with a median PFS of 1.9 months (95% CI 1.8‒4.6) and a median time to progression of 1.9 months (95% CI 1.8‒4.6) in the response-evaluable population (Supplementary Figure 4A). For patients with low-grade gliomas (n = 4), the median PFS was 16.9 (95% CI −0.9 to 27.1) months. Six- and 12-month PFS rates were 25.0% (95% CI 10.2‒43.1%) and 20.8% (7.6‒38.5%), respectively (Supplementary Figure 4A). At the time of analysis, 13 deaths (50%) had occurred. Median overall survival was 17.2 months (95% CI 11.5–not estimable) (Supplementary Figure 4B). Six- and 12-month survival rates were 80.8% (95% CI 59.8‒91.5%) and 67.9% (45.7‒82.6%), respectively. The PFS of patients with enhancing gliomas who received olutasidenib monotherapy is also shown (Supplementary Figure 5).
Discussion
Despite genomic and metabolomic advances in glioma over the last 15 years, therapeutic improvements have translated to minimal clinical benefit and glioma patients continue to have a poor clinical outcome with high morbidity, including cognitive decline, and high mortality. Therefore, effective and less toxic treatments for glioma remain an unmet medical need.38 In this study, we demonstrated that olutasidenib monotherapy, at the RP2D of 150 mg twice daily, was well tolerated in patients with glioma with no DLTs reported during the safety lead-in period of the study. At the RP2D, steady state olutasidenib plasma concentrations were above the preclinical minimum considered effective, yet below the concentration predicted to pose a QT prolongation risk (Supplementary Figure 1). Olutasidenib measured in CSF samples from two patients confirmed brain penetration at levels expected to be clinically meaningful. Although the study failed to meet the primary efficacy endpoint of ORR, olutasidenib monotherapy provided preliminary evidence of clinical activity, with a disease control rate of 48% in heavily pretreated patients with high-grade glioma.
In this study of patients with recurrent/relapsing glioma, olutasidenib monotherapy provided encouraging preliminary evidence of clinical activity, and a 12-month PFS rate of 20.8%. Contrasted with the recently published data of vorasidenib and ivosidenib in patients with enhancing glioma where objective responses were not observed,24,25 two patients with enhancing, high-grade glioma (one anaplastic astrocytoma and one glioblastoma) treated with olutasidenib in this study had a durable partial response (>80% tumor reduction). In the recent study of ivosidenib in patients with advanced IDH1-mutated gliomas, SD was reported in 45.2% of patients with enhancing disease with a median PFS of 1.4 months. For patients with nonenhancing gliomas receiving ivosidenib, the ORR was 2.9% (1 partial response), with 85.7% patients achieving SD, and a median PFS of 13.6 months.25 Similarly, in the study of vorasidenib in patients with mutant IDH1/2 glioma, SD was reported in 56.7% of patients with enhancing disease with a median PFS of 3.6 months, while for patients with nonenhancing disease, 72.7% of patients had a best response of SD and the median PFS was 36.8 months.24 While it is difficult to compare across studies with different patient populations, in our analysis of patients with predominantly enhancing IDH1-mutant gliomas treated with olutasidenib, we observed a best response of SD in 40% of patients with a median PFS of 1.9 months. The low number of patients with nonenhancing disease in this study limits our ability to draw any meaningful conclusions about olutasidenib activity in patients with nonenhancing tumors, and additional studies are warranted. Importantly, by central blinded volumetric assessments, 4/24 (17%) patients exhibited a ≥50% decrease in tumor burden after receiving olutasidenib, with an additional five patients having a reduction in tumor volume <50%, raising the question if additional volumetric assessments may be needed to better evaluate treatment response in IDH-mutant gliomas. Of note, most patients in the study (85%) had canonical R132H mutations so it is difficult to draw any meaningful conclusions about potential differences in outcomes across R132H and non-R132H patients. Both patients with a partial response had R132H mutations, for the four patients with non-R132H mutations, one (nonevaluable) patient with R132C had a best response of stable disease with >23 months on olutasidenib therapy while the two patients with R132L and the one patient with R132G had disease progression after approximately 1–2 months of treatment.
It has been suggested that the lack of single-agent antitumor efficacy of vorasidenib in patients with enhancing gliomas may be due to the presence of additional genomic alterations in these tumors that can bypass the need for the mIDH enzyme for tumor maintenance.24 As our protocol did not mandate tumor biopsy nor genomic sequencing immediately preceding study enrolment, we are unable to further address this question. For future studies, mandatory genomic analyses at time of enrolment may help to shed light on the concept that the efficacy of single-agent mIDH inhibitors may be linked to the need for the mIDH enzyme for tumor maintenance, based on the presence or absence of additional genomic alterations, rather than on the enhancing component in imaging, or tumor grading.
Overall, our preliminary data are promising considering that olutasidenib was well tolerated as a single agent and demonstrated antitumor activity and disease control in a population with relapsed or refractory, heavily pretreated, and predominantly enhancing IDH1-mutated tumors. These data support further exploration of olutasidenib combination trials in the upfront or recurrent setting for IDH1-mutant high-grade gliomas.
Limitations of our study include the relatively small number of patients as well as the absence of updated genomic sequence at enrolment and on-treatment/end-of-treatment biopsies that would be addressing some of our pending questions. Although the preliminary CSF data for two patients are encouraging, additional confirmatory analyses would be beneficial. Genomic sequencing at enrolment may help to identify additional driver mutations that these tumors may add during the course of the disease and would further our understanding of the role of IDH mutations in the recurrent, advance setting. An on-treatment/end-of-treatment biopsy may confirm the biological effect of the drug, identify a potential mechanism of resistance, and allow for the prediction of useful combinatory approaches. Furthermore, patients were not stratified by tumor grade or enhancing status at baseline; therefore limiting our ability to draw any conclusions about olutasidenib activity in patients with low-grade gliomas or patients with non-enhancing tumors; these patient populations will need to be included in future studies.
We conclude that clinically relevant olutasidenib plasma concentrations were reached at the RP2D of 150 mg twice daily, a dose sufficient to penetrate the blood‒brain barrier yet observed to be tolerable with no unexpected safety concerns. This phase Ib/II study in patients with relapsed or refractory and predominantly enhancing gliomas harboring IDH1R132X mutations provides preliminary evidence of the clinical activity and prolonged disease control that can be achieved with oral olutasidenib in this heavily pretreated population.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors wish to thank the patients and their families, the investigators, and all site personnel who participated in this study. Medical writing assistance was provided by Brooke Harrison (Forma Therapeutics, Inc.). Medical writing assistance was also provided by Sue Reinwald (Engage Scientific Solutions) and was funded by Forma Therapeutics, Inc.
Contributor Information
Macarena I de la Fuente, Sylvester Comprehensive Cancer Center and Department of Neurology, University of Miami, Miami, Florida, USA.
Howard Colman, Huntsman Cancer Institute, University of Utah, Salt Lake City, Utah, USA.
Mark Rosenthal, Peter MacCallum Cancer Centre Melbourne , Victoria, Australia.
Brian A Van Tine, Washington University in St. Louis School of Medicine, St. Louis, Missouri, USA.
Danijela Levacic, Baylor and Scott White Vasicek Cancer Center, Baylor University Temple, Temple, Texas, USA.
Tobias Walbert, Henry Ford Cancer Institute, Henry Ford Health System and Wayne State University, Detroit, Michigan, USA.
Hui K Gan, Olivia Newton-John Cancer Wellness and Research Centre Austin Hospital, Heidelberg, Victoria, Australia.
Maria Vieito, Vall d’Hebron Institute of Oncology, Barcelona, Spain.
Mohammed M Milhem, Holden Comprehensive Cancer Center, University of Iowa, Iowa City, Iowa, USA.
Kathryn Lipford, Forma Therapeutics, Inc., Watertown, Massachusetts, USA.
Sanjeev Forsyth, Forma Therapeutics, Inc., Watertown, Massachusetts, USA.
Sylvie M Guichard, Forma Therapeutics, Inc., Watertown, Massachusetts, USA.
Yelena Mikhailov, Forma Therapeutics, Inc., Watertown, Massachusetts, USA.
Alexander Sedkov, Forma Therapeutics, Inc., Watertown, Massachusetts, USA.
Julie Brevard, Forma Therapeutics, Inc., Watertown, Massachusetts, USA.
Patrick F Kelly, Forma Therapeutics, Inc., Watertown, Massachusetts, USA.
Hesham Mohamed, Forma Therapeutics, Inc., Watertown, Massachusetts, USA.
Varun Monga, Holden Comprehensive Cancer Center, University of Iowa, Iowa City, Iowa, USA.
Funding
This study was funded by Forma Therapeutics, Inc., Watertown, MA, USA.
Conflict of interest statement. MID reports funding to her institution to conduct this clinical trial and support for attending an advisory board meeting from Forma Therapeutics as well as participation as an advisory board member for Agios Pharmaceuticals and Forma Therapeutics. HC reports advisory board/consulting fees from Forma Therapeutics, Best Doctors/Teladoc, Orbus Therapeutics, Adastra Pharmaceuticals, Bristol-Myers Squibb (BMS), Deciphera Pharmaceuticals, Private Health, Beyer, AbbVie, NewLink Genetics Corporation, and Karyopharm Therapeutics as well as support for attending advisory board meetings and/or travel from Forma Therapeutics (outside the submitted work), Orbus Therapeutics, Adastra Pharmaceuticals, Deciphera, Bayer, AbbVie, and Karyopharm Therapeutics. MR and DL report no conflicts of interest. BV reports research grants from Pfizer, Merck, TRACON Pharmaceuticals, and GSK, royalties or licenses from Accuronix Therapeutics for Sigma-2 Receptor Ligands and Therapeutic Uses Thereof (006766) and Modular Platform for Targeted Therapeutic Delivery (006755), Sigma-2 Receptor Ligand Drug Conjugates as Antitumor Compounds, Methods of Synthesis and Uses Thereof (014229), consulting fees from ADRx and Ayala Pharmaceuticals, consulting/advisor fees from Cytokinetics, Inc., and Bayer, payment or honoraria from Adaptimmune Ltd., and GSK for speaker bureaus, and from Bionest Partners for a televised interview and Intellisphere LLC for a virtual workshop, payment from Hinshaw & Culbertson LLP, Rodney Law, and the CRICO Risk Management Foundation for expert legal testimony and payment from Tracey & Fox Law Firm for expert deposition, support for attending meetings and/or travel from GSK and Adaptimmune Ltd., participation on advisory boards for Adaptimmune Ltd., Apexigen Inc., Daiichi Sankyo, Deciphera Pharmaceuticals, Inc., Epizyme, GSK, Novartis, and Eli Lilly, as well as a leadership or a fiduciary role for Polaris as a board member. TW reports payment or honoraria from AstraZeneca and Orbus Therapeutics as well as participation on a Data Safety Monitoring Board or Advisory Board for Novocure. HKG reports consulting fees for BMS, payment or honoraria from Eisai and Merck Serono for speaker bureaus, and receipt of laboratory reagents from AbbVie. MV reports consulting fees from Roche and Debioparm. MM reports consulting fees from Syneos Health, Exicure, Inc., Novartis, Immunocore, BioNTech, BluePrint Medicines Corporation, Amgen, Array BioPharma Inc., and Trieza. KL, SF, SG, YM, AS, JB, and PFK are employees of Forma Therapeutics and shareholders of stocks or stock options from Forma Therapeutics. HM is a prior employee of Forma Therapeutics and a shareholder of stocks or stock options from Forma Therapeutics. VM reports funding to his institution to conduct the clinical trial, support for medical writing, and honoraria for an advisory board meeting from Forma Therapeutics all in relation to the present manuscript, a foundation grant, Rising Tide Foundation Clinical Cancer Research, to conduct a trial and a grant to his institution from Prelude Therapeutics to fund a clinical trial, payment or honoraria from Gunderson Medical Foundation for a Grand Round presentation, support for travel and/or attending meetings from Forma Therapeutics, Deciphera Pharmaceuticals, and GlaxoSmithKline (GSK) and advisory board participation is reported for Forma Therapeutics and Astex Pharmaceuticals.
Authorship statement. All authors contributed to study conceptualization, the methodology and investigation, data collection, and project administration. AS and JB were responsible for the formal analysis. MID and VM verified the data reported in the manuscript. All authors had access to and interpreted the data, participated in writing the original draft, revised subsequent drafts for important intellectual content, and provided final approval of the submitted version.
References
- 1. Lapointe S, Perry A, Butowski NA. Primary brain tumours in adults. The Lancet 2018;392(10145):432–446. [DOI] [PubMed] [Google Scholar]
- 2. Cramer CK, Cummings TL, Andrews RN, et al. Treatment of radiation-induced cognitive decline in adult brain tumor patients. Curr Treat Options Oncol. 2019;20(5):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang J, Yu J, Tu L, et al. Isocitrate dehydrogenase mutations in glioma: from basic discovery to therapeutics development. Front Oncol. 2019;9(506). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stylli SS. Novel treatment strategies for glioblastoma. Cancers 2020;12(2883):28831–28811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brown PD, Buckner JC, Uhm JH, Shaw EG. The neurocognitive effects of radiation in adult low-grade glioma patients. Neuro Oncol 2003;5(3):161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell 2013;155(2):462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Myung JK, Cho HJ, Park CK, et al. IDH1 mutation of gliomas with long-term survival analysis. Oncol Rep. 2012;28(5):1639–1644. [DOI] [PubMed] [Google Scholar]
- 9. Reitman ZJ, Yan H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J Natl Cancer Inst. 2010;102(13):932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fernandez-Marcos PJ, Nóbrega-Pereira S. NADPH: new oxygen for the ROS theory of aging. Oncotarget 2016;7(32):50814–50815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Raineri S, Mellor J. IDH1: linking metabolism and epigenetics. Front Genet. 2018;9:493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu A, Hou C, Chen H, Zong X, Zong P. Genetics and epigenetics of glioblastoma: applications and overall incidence of IDH1 mutation. Front Oncol. 2016;6(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009;462(7274):739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011;19(1):17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012;483(7390):474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cohen AL, Holmen SL, Colman H. IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep. 2013;13(5):345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jezek P. 2-Hydroxyglutarate in cancer cells. Antioxid Redox Signal. 2020;33(13):903–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Philip B, Yu DX, Silvis MR, et al. Mutant IDH1 promotes glioma formation in vivo. Cell Rep 2018;23(5):1553–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rakheja D, Medeiros LJ, Bevan S, Chen W. The emerging role of d-2-hydroxyglutarate as an oncometabolite in hematolymphoid and central nervous system neoplasms. Front Oncol. 2013;3:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Franceschi E, De Biase D, Di Nunno V, et al. IDH1 non-canonical mutations and survival in patients with glioma. Diagnostics (Basel) 2021;11(2):342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tesileanu CMS, Vallentgoed WR, Sanson M, et al. Non-IDH1-R132H IDH1/2 mutations are associated with increased DNA methylation and improved survival in astrocytomas, compared to IDH1-R132H mutations. Acta Neuropathol. 2021;141(6):945–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ng SC, de la Monte SM, Conboy GL, Karns LR, Fishman MC. Cloning of human GAP-43: growth association and ischemic resurgence. Neuron. 1988;1(2):133–139. [DOI] [PubMed] [Google Scholar]
- 23. Molenaar RJ, Maciejewski JP, Wilmink JW, van Noorden CJF. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene 2018;37(15):1949–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mellinghoff IK, Penas-Prado M, Peters KB, et al. Vorasidenib, a dual Inhibitor of mutant IDH1/2, in recurrent or progressive glioma; results of a first-in-human phase I trial. Clin Cancer Res. 2021;27(16):4491–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mellinghoff IK, Ellingson BM, Touat M, et al. Ivosidenib in isocitrate dehydrogenase 1-mutated advanced glioma. J Clin Oncol. 2020;38(29):3398–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Caravella JA, Lin J, Diebold RB, et al. Structure-based design and identification of FT-2102 (olutasidenib), a potent mutant-selective IDH1 inhibitor. J Med Chem. 2020;63(4):1612–1623. [DOI] [PubMed] [Google Scholar]
- 27. Watts JM, Baer MR, Yang J, et al. Olutasidenib (FT-2102), an IDH1m inhibitor as a single agent or in combination with azacitidine, induces deep clinical responses with mutation clearance in patients with acute myeloid leukemia treated in a phase 1 dose escalation and expansion study. Blood 2019;134(Supplement 1):231–231. [Google Scholar]
- 28. Cortes JE, Wang ES, Watts JM, et al. Olutasidenib (FT-2102) induces rapid remissions in patients with IDH1-mutant myelodysplastic syndrome: results of phase 1/2 single agent treatment and combination with azacitidine. Blood 2019;134(Supplement 1):674–674. [Google Scholar]
- 29. De Botton S, Yee KW, Recher C, et al. Effect of olutasidenib (FT-2102) on complete remissions in patients with relapsed/refractory (R/R) mIDH1 acute myeloid leukemia (AML): results from a planned interim analysis of a phase 2 clinical trial. J Clin Oncol. 2021;39. [Google Scholar]
- 30. Cortes JE, Esteve J, Bajel A, et al. Olutasidenib (FT-2102) in combination with azacitidine induces durable complete remissions in patients with mIDH1 acute myeloid leukemia. Blood 2021;138(Supplement 1):698–698. [Google Scholar]
- 31. NCT03684811. A study of FT 2102 in participants with advanced solid tumors and gliomas with an IDH1 mutation. 2020; https://www.clinicaltrials.gov/ct2/show/NCT03684811. Accessed October 17, 2020.
- 32. Ellingson BM, Wen PY, Cloughesy TF. Modified criteria for radiographic response assessment in glioblastoma clinical trials. Neurotherapeutics 2017;14(2):307–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van den Bent MJ, Wefel JS, Schiff D, et al. Response assessment in neuro-oncology (a report of the RANO group): assessment of outcome in trials of diffuse low-grade gliomas. Lancet Oncol. 2011;12(6):583–593. [DOI] [PubMed] [Google Scholar]
- 34. Wong ET, Hess KR, Gleason MJ, et al. Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol. 1999;17(8):2572–2578. [DOI] [PubMed] [Google Scholar]
- 35. Chen C, Xu T, Lu Y, Chen J, Wu S. The efficacy of temozolomide for recurrent glioblastoma multiforme. Eur J Neurol. 2013;20(2):223–230. [DOI] [PubMed] [Google Scholar]
- 36. Huang J, Chaudhary R, Cohen AL, et al. A multicenter phase II study of temozolomide plus disulfiram and copper for recurrent temozolomide-resistant glioblastoma. J Neurooncol. 2019;142(3):537–544. [DOI] [PubMed] [Google Scholar]
- 37. Watts JM, Baer MR, Lee S, et al. A phase 1 dose escalation study of the IDH1m inhibitor, FT-2102, in patients with acute myeloid leukemia or myelodysplastic syndrome. J Clin Oncol. 2018;36(15 (Supplement):7009. [Google Scholar]
- 38. Shergalis A, BankheadA, 3rd, Luesakul U, Muangsin N, Neamati N. Current challenges and opportunities in treating glioblastoma. Pharmacol Rev. 2018;70(3):412–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.