


Abstract
The extracellular matrix (ECM) is a complex assembly of proteins that constitutes the scaffold organizing cells, tissues, and organs. Over the past decade, mass-spectrometry-based proteomics has become the method of choice to profile the composition of the ECM, or the matrisome, of tissues. To assist non-specialists with the reuse of ECM proteomic datasets, we released MatrisomeDB (https://matrisomedb.org) in 2020. Here, we report the expansion of the database to include 25 new curated studies on the ECM of 24 new tissues in addition to datasets on tissues previously included, more than doubling the size of the original database and achieving near-complete coverage of the in-silico predicted matrisome. We further enhanced data visualization by maps of peptides and post-translational-modifications detected onto domain-based representations and 3D structures of ECM proteins. We also referenced external resources to facilitate the design of targeted mass spectrometry assays. Last, we implemented an abstract-mining tool that generates an enrichment word cloud from abstracts of studies in which a queried protein is found with higher confidence and higher abundance relative to other studies in MatrisomeDB.
Graphical Abstract
Graphical Abstract.
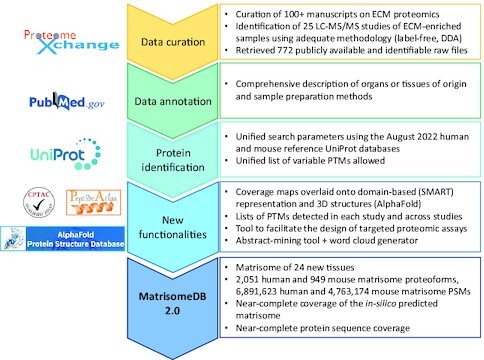
Graphical abstract depicts the main steps of the workflow developed to expand the content and functionalities of MatrisomeDB.
INTRODUCTION
The extracellular matrix (ECM) is a complex assembly of proteins forming the scaffold supporting cell and tissue organization (1,2). In addition to its structural roles, the ECM provides biochemical and mechanical signals that orchestrate a plethora of cellular processes ranging from proliferation and survival, to stemness, differentiation, and migration. ECM remodeling is integral to physiological events, including embryonic development (3), scarring (4) and aging (5,6). An imbalance in the ECM, caused either by mutations in ECM genes or by alterations in ECM metabolism (excessive accumulation or, on the contrary, destruction), is a hallmark of many diseases such as cancer and fibrosis (7–10). The ECM thus constitutes an immense reservoir of disease biomarkers and therapeutic targets that has remained, until recently, largely under-explored.
The insolubility of ECM proteins, mediated by their extensive post-translational modifications (PTMs) and incorporation in higher-order complexes, hindered, for a long time, their comprehensive biochemical analysis. In addition, the lack of framework to define the parts list of the ECM has limited big data annotation and further prevented the application of high-throughput methods to profile the composition of the ECM. Over the past decade, different approaches relying on sequential protein extraction or decellularization have been developed to achieve enrichment of ECM proteins (11–14). Moreover, different protein solubilization and digestion methods have been devised to make the ECM amenable to bottom-up mass spectrometric analysis. Last, we have defined the ‘matrisome’ as the compendium of genes encoding proteins that can contribute to form an ECM (15–17). Combined, these developments have contributed to make mass-spectrometry the state-of-the-art method to characterize the protein composition of the ECM (18,19). With the broad adoption by scientists and publishers of Findable, Accessible, Interoperable and Reusable (FAIR) guiding principles (20), datasets from existing ECM proteomic studies are now largely available from public repositories. However, they are difficult to utilize without specific expertise. To improve the accessibility of this rich source of data, we released in 2020, MatrisomeDB, the ECM protein knowledge database (21). In its original version, MatrisomeDB included datasets from 17 studies on the ECM of 15 normal tissue types, six cancer types and other diseases including vascular defects and lung and liver fibroses. Since its release in 2020, MatrisomeDB has received nearly 123,000 visits (Figure 1) from researchers based all over the world, and its online availability averaged 97.02%.
Figure 1.

MatrisomeDB usage. The color code indicates, for each country, the number of visits for the January 2020–September 2022 period. Red dots depict the cities from which connections to MatrisomeDB were established.
The landscape of ECM proteomic research has developed considerably over the past three years, prompting us to update MatrisomeDB. Here, we report on four major advances: (i) the horizontal expansion of MatrisomeDB 2.0 which now includes compositional information on the ECM of 24 new tissues and organs, two new cancer types, and datasets reporting changes in the ECM during aging, during different stages of disease progression, or during chemotherapeutic treatment (Table 1); (ii) the addition of modules to visualize peptide mapping on schematic representation of the domain-based organization of ECM proteins and on 3D-structures predicted by AlphaFold; (iii) cross-referencing to external resources to advance the use of targeted mass-spectrometry to study ECM proteins and (iv) an abstract-mining tool to support the generation of novel hypotheses to advance our understanding of the roles of the ECM in health and disease.)
Table 1.
List of studies included in MatrisomeDB 2.0
| Physiological System | Sample Description | Sample Preparation | Dataset identifier | Reference |
|---|---|---|---|---|
| Human tissues | ||||
| Cardiovascular System | Decellularized human umbilical artery | Sequential extraction with CHAPS and SDS; Tryptic digestion | PXD020187 | Mallis et al., 2020 (35) |
| Digestive System | Human gastric antrum adenocarcinoma, distant mucosa, and adjacent mucosa | Decellularization with SDS and DNase; Deglycosylation (PNGaseF) followed by digestion with Lys-C and trypsin | PXD029782 | Moreira et al., 2022 (37) |
| Digestive System | Human liver metastases from colorectal tumors and adjacent normal liver | ECM enrichment using sequential compartmental protein extraction; Protein solubilization in urea; Deglycosylation (PNGaseF) followed by tryptic digestion | PXD010252 | Yuzhalin et al., 2018 (54) |
| Integumentary System | Human dermis samples from normal skin, scar tissue, and keloids | Extraction with NaCl (pre-decellularization) and GuHCl, followed by deglycosylation and tryptic digestion | PXD015057 | Barallobre-Barreiro et al., 2019 (38) |
| Integumentary System | Human dermis samples from young and elderly patients | Extraction with GuHCl; Chemical digestion with cyanogen bromide (CNBr) followed by tryptic digestion | PXD015982 | McCabe et al., 2020 (39) |
| Musculoskeletal System | Human nucleus pulposus and annulus fibrosus from young and aged intervertebral discs | Solubilization with GuHCl and urea; Digestion with trypsin and Lys-C | PXD018193 | Tam et al., 2020 (43) |
| Reproductive System | Human Fallopian Tube | ECM enrichment using sequential compartmental protein extraction; Deglycosylation (PNGaseF) followed by digestion with Lys-C and trypsin | PXD023707 | Renner et al., 2021 (41) |
| Reproductive System | Normal human omentum and human omental metastases of high-grade serous ovarian cancer, before and after chemotherapy | ECM enrichment using sequential compartmental protein extraction; Urea solubilization; Deglycosylation (PNGaseF) followed by tryptic digestion | PXD004060 | Pearce et al., 2018 (53) |
| Reproductive System | Human ovarian tissue | Extraction with either RapiGest (RP), ProteaseMax (PM), and 3273 (also known as MaSDeS or n-dodecyl β-D-maltoside); Digestion with Lys-C and trypsin | PXD020823 | Ouni et al., 2020 (42) |
| ECM of human tumor xenografts growing in mice | ||||
| Digestive System | Wild-type (BxPC3) and K-Ras-mutant (AsPC1) human pancreatic adenocarcinoma xenografts grown in mice | ECM enrichment using sequential compartmental protein extraction; Deglycosylation (PNGaseF) followed by digestion with Lys-C and trypsin | MSV000082639 | Tian et al., 2019 (51) |
| Reproductive System | Human triple-negative breast cancer xenograft (MDA-MB-231) metastases to mouse brain, lung, liver, and bone and matched normal tissues: murine lung, liver, and bone | ECM enrichment using sequential compartmental protein extraction; Deglycosylation (PNGaseF) followed by digestion with Lys-C and trypsin | MSV000084023 | Hebert et al., 2020 (52) |
| ECM produced by human cells in vitro | ||||
| Musculoskeletal System | ECM produced by human dental pulp stem cells | Decellularization with Triton-X100 and NH4OH and sequential extraction protocol; Protein solubilization in urea; Deglycosylation (PNGaseF) followed by digestion in Lys-C and trypsin | PXD018951 | Nowwarote et al., 2022 (57) |
| Respiratory System | Decellularized ECM produced by human lung fibroblasts | Decellularization with Triton X-100 and NH4OH; Protein solubilization in urea; Tryptic digest; deglycosylated (TFMS or NEB Deglycosylation Mix II) and non-deglycosylated samples analyzed | PXD007700 | Lansky et al., 2019 (58) |
| Urinary System | ECM of human induced pluripotent stem cell-derived kidney organoids | Decellularization with Triton-X100 and ECM enrichment using sequential compartmental protein extraction; Solubilization in SDS followed by tryptic digestion | PXD025838 | Morais et al., 2022 (59) |
| Mouse tissues | ||||
| Cardiovascular System | Murine aorta from wild-type and Adamts5Δcat KO mice treated with angiotensin II | Protein extraction and solubilization with SDS and GuHCl; Deglycosylation (PNGaseF) followed by tryptic digestion | PXD009410 | Fava et al., 2018 (36) |
| Digestive System | Murine normal liver; xenografts: human colorectal tumor (MC38) metastases to mouse liver | Decellularization with NH4OH; Protein solubilization in urea; Deglycosylation (PNGaseF) followed by tryptic digestion | PXD013350 | Yuzhalin et al., 2019 (55) |
| Integumentary System | Murine back skin tissue | Comparative analysis of different ECM protein preparation methods: 1) ECM enrichment using sequential compartmental protein extraction; 2) Extraction using SDS, Triton-X-100 and GuHCl, 3) Enzymatic decellularization using phospholiase A2 and DOC, 4) Quantitative detergent solubility profiling (QDSP); Deglycoslyation (PNGaseF) followed by digestion with Lys-C and trypsin | PXD025842 | Dussoyer et al., 2021 (40) |
| Musculoskeletal System | Murine proximal femoral epiphysis cartilage from control Col2a1-Cre mouse | Decellularization using DOC; Digestion with Lys-C and trypsin | PXD027109 | Bubb et al., 2021 (44) |
| Musculoskeletal System | Murine adult (12mo) and elderly (24mo) gastrocnemius muscle | Protein solubilization in urea-thiourea and GuHCl; Tryptic digestion | PXD027895 | Lofaro et al., 2021 (45) |
| Musculoskeletal System | Murine soleus muscle, tendon, and myotendinous junction | Comparative analysis of ECM extraction methods: 1) GuHCl extraction followed by tryptic digestion; 2) ECM enrichment using sequential compartmental protein extraction followed by deglycosylation (chondroitinase ABC) and digestion with Lys-C and trypsin | MSV000085253 | Jacobson et al., 2020 (46) |
| Musculoskeletal System | Murine femoral and tibia knee cartilage from control mouse | Protein extraction with GuHCl and solubilization in urea; Deglycosylation (chondroitinase ABC), followed by digestion with Lys-C and trypsin | PXD019374 | Georgieva et al., 2020 (47) |
| Musculoskeletal System | Murine lumbar and tail annulus fibrosus and nucleus pulposus intervertebral disc | Soluble fraction from GuHCl extraction resuspended in urea; Digestion with Lys-C and trypsin | PXD006671 | Kudelko et al., 2021 (48) |
| Nervous and Sensory Systems | Murine brain lysate | Samples lysed with SDS and Triton X-100 followed by acetone precipitation; Deglycosylation (chondroitinase ABC or heparin lyase I, II, and III) and digestion with Lys-C and trypsin | PXD017513 | Sethi et al., 2020 (50) |
| Nervous and Sensory Systems | Murine cerebral cortex, lateral sub-ependymal zone, olfactory bulb | Quantitative detergent solubility profiling (QDSP); Digestion with Lys-C and trypsin | PXD016632 | Kjell et al., 2020 (49) |
| Respiratory System | Murine lung from young and elderly mice | Quantitative detergent solubility profiling (QDSP); Digestion with Lys-C and trypsin | PXD012307 | Angelidis et al., 2019 (56) |
DATABASE INFRASTRUCTURE
Literature curation and criteria for dataset inclusion
ProteomeXchange (22), a database that consolidates publicly available proteomic datasets from partner repositories such as PRIDE (23) and MassIVE (https://massive.ucsd.edu), was primarily searched for the term ‘extracellular matrix.’ We also searched the repository for related terms (e.g. ‘matrisome’, ‘ECM’) to ensure we captured all potentially relevant datasets. Data derived from human (Homo sapiens) and murine (Mus musculus) tissues or cells were retained. Corresponding publications were then subjected to a preliminary screen to determine their relevance to MatrisomeDB. Additional papers compiled via PubMed alerts including the keywords ‘extracellular matrix’ and ‘proteomics’, or ‘ECM’ and ‘proteomics’, or ‘matrisome’ were also manually curated. Studies deemed relevant were then screened for the availability of data in .raw format in a public repository. Importantly, we also screened for the availability of correspondence files allowing the assignment of raw files to a detailed sample description. The methods and supplemental methods sections of the papers that passed the preliminary screening steps were manually curated for adherence to the following eligibility criteria:
Methods were assessed to determine whether and how samples were fractionated to enrich for ECM proteins. Studies were included if they comprised an ECM-enrichment (sequential compartmental extraction) or decellularization step (e.g. deoxycholate, NH4OH, SDS). A second stipulation imposed that ECM-enriched samples be extracted in conditions allowing ECM protein solubilization (e.g. guanidine hydrochloride, GuHCl or a high concentration of urea). Samples generated with no ECM enrichment steps were eliminated. Because of their large size, ECM proteins are difficult to resolve by SDS-PAGE, we thus also excluded studies using SDS-PAGE to fractionate proteins. Note that studies reporting the profiling of both soluble and insoluble protein fractions were kept in their entirety and were labeled as such (Supplementary Table S1).
Bottom-up mass spectrometry studies where peptides were generated via enzymatic cleavage (Lys-C, trypsin; Table 1).
Only raw data acquired via LC–MS/MS on an OrbiTrap-type instrument were included. Furthermore, we only included files generated using a data-dependent acquisition (DDA) mode.
This extensive manual curation process ensures that datasets included in MatrisomeDB were obtained using overall similar experimental pipelines, allowing for inter-study comparison.
Last, we resorted to personal communication to obtain additional information regarding the sample preparation method and file descriptions when these were not publicly available, or to resolve any discrepancies. If we were unable to obtain sufficient information on a given study, we did not retain it.
Mass spectrometry database search and processing
All raw data files were converted and searched using uniform parameters and against the same and most recent reference proteome databases (UniProt 08/2022). The reference databases used contain, in total, 101,761 human proteoforms and 63,668 mouse proteoforms. Common contaminating proteins were also appended to the reference databases (24). The raw data were searched by the MSFragger search engine (25) using a closed search method with ±20 ppm as precursor mass tolerance. We allowed carbamido-methylation for all studies as fixed post-translational modification. We also allowed for the following variable post-translational modifications: deamidation/citrullination R[0.9840], methionine oxidation M[15.9949], serine, threonine, and tyrosine phosphorylation S[79.9663], T[79.9663], Y[79.9663], acetylation n[42.0106], the ECM-specific proline and lysine hydroxylations P[15.9949], K[15.9949]. The enzyme used for in-silico digestion is trypsin with two allowed miscleavages. The search results were then filtered with 1% false discovery rate (FDR) at the peptide-spectra-match (PSM), peptide and protein levels. Importantly, through uniform search parameters and FDR filters level, a unified search allows for data harmonization and for inter-study comparison.
NEW DATABASE FUNCTIONALITIES
Peptide and post-translational modification (PTM) mapping to domain-based representation of ECM proteins
The previous release of MatrisomeDB allowed the visualization of sequence coverage on the primary amino-acid sequences of proteins. In this new release, we enhanced the sequence coverage visualization and provided an overlay of peptides detected on the schematic organization depicting the domain-based organization of ECM proteins using the Simple Modular Architecture Research Tool (SMART,26). As a result, an interactive domain coverage and a PTMs annotation plot are generated by a custom backend Python script, for each protein query. At the top: a grey bar chart represents the sequence coverage of the queried protein binned by groups of 10 amino acids. In the center portion of the page, users are provided with a schematic domain-based organization of the queried protein. All PTMs detected are mapped at the bottom of the domain-based representation. For clarity, domains containing a large number of PTMs are merged but all identified PTMs including types and positions are made available in a separate downloadable table available via the ‘Open PTM occurrence table’ link located above the graph (Figure 4B inset and Supplementary Table S2B). Users can also download data on domain coverage and PTM positioning with regards to domains via links provided above the coverage plots (Figure 4B, Supplementary Table S2A and C, respectively). Tools located in the upper right corner allow users to hover over the domain representation to show the domains’ names, start and end positions, and domains’ sequence coverage. The interactive displays of both the domain coverage/PTMs graph and PTM table on the web interface are enabled by a custom Python script powered by Bokeh and available at https://github.com/Matrisome/MatrisomeDB2. Data are provided for the protein of interest both in a given sample type and across the complete MatrisomeDB dataset.
Figure 4.

New functionalities implemented to enhance data visualization and generate new hypotheses. Using as an example protein–glutamine gamma-glutamyltransferase 2 (TGM2, UniProt P21980) in pancreatic ductal adenocarcinoma xenograft (BxPC3) samples, we illustrate the novel functionalities of the database. (A) External references: MatrisomeDB 2.0 provide links to (i) the Simple Modular Architecture Research Tool (SMART) providing users with a graphic representation of the domain-based organization of the queried protein, (ii) if available, to the validated CPTAC assay(s) for targeted mass spectrometry experiments and (iii) to the Peptide Atlas page of the queried protein. (B) Sequence coverage and PTM mapping onto the domain-based representation of TGM2 retrieved from SMART. The control panel in the upper right corner allows for an interactive experience (red arrow). Black arrows point to links that will generate tables including domain-level percentage of sequence coverage (Supplementary Table S2A), list of PTMs (inset and Supplementary Table S2B) and, list of PTMs with respect to domain positioning (Supplementary Table S2C). (C) Peptide and PTM mapping onto 3D-predicted model of TGM2 by AlphaFold using SCV. Sequence coverage is depicted in red. Regions of TGM2 not covered by any peptide in the pancreatic ductal adenocarcinoma xenograft (BxPC3) samples are depicted in gray. In this example, we selected to display methionine, proline, and lysine oxidations in light blue, acetylations in pink, deamidations in orange, and serine, threonine and tyrosine phosphorylations in dark blue. These colors are customizable using the right-hand panel (red arrow). Blue arrow points to the AlphaFold Protein Structure Database page of the queried protein.
Peptide and post-translational modification (PTM) mapping to 3D structures or models of ECM proteins from the AlphaFold database
In addition to the 1D coverage maps overlaid to primary amino-acid sequences (MatrisomeDB 1.0) and domain-based depiction of matrisome proteins (see below), we have leveraged AlphaFold's validated or predicted structures of proteins (27,28) to provide interactive 3D coverage of matrisome proteins included in MatrisomeDB 2.0. This new feature is available to users from the protein-coverage map page, by clicking on ‘Open sample data (or global data) in SCV’. User-customizable 3D interactive sequence coverage/PTMs visualization is enabled by connecting MatrisomeDB with Sequence Coverage Visualizer (SCV; (29)), a web app we recently developed to visualize 3D sequence coverage. For proteins with known structures, the AlphaFold versions were used. These structures are essentially identical to the corresponding Protein Data Bank (PDB) structures (30) as they were used for training the AlphaFold model. For unknown structures, AlphaFold-predicted models were used (27,28). All 3D sequence coverage HTML pages of queried proteins were pre-generated by the static SCV scripts served on MatrisomeDB server.
Cross-referencing to external resources to facilitate the design of targeted MS experiments
Targeted mass spectrometry approaches such as selected or multiple reaction monitoring (SRM/MRM) allow the quantification of the abundance of a given protein, or sets of proteins in complex samples, with high accuracy (31), yet these methodologies have not yet been broadly applied to ECM research. This is partly due to the fact that these approaches rely on prior knowledge on proteins of interest and the characterization of proteotypic peptides that allow the design and validation of SRM or MRM assays (31). One of the goals of the National Cancer Institute's Clinical Proteomic Tumor Analysis Consortium (CPTAC) is to devise and validate proteotypic peptides amenable to targeted mass-spectrometry assays (31,32). Validated assays are then made publicly available via the CPTAC assay portal (33). We retrieved the CPTAC assay database (2022.04.17 version) that included 3,006 peptides and identified 336 peptides matching 192 matrisome proteins. If available, a link pointing to the CPTAC page will be displayed at the top of the page reached after clicking on the MatrisomeDB ‘Description’ column of the main search output table. For matrisome proteins for which no CPTAC-validated assays are available, we are providing link to the Peptide Atlas (34) that lists predicted highly observable peptides that can be leveraged by users for the design of targeted MS experiments.
Abstract-enrichment word cloud
In MatrisomeDB 1.0, we provided hyperlinks to the primary research publications reporting queried proteins. To provide better reference visualization in MatrisomeDB 2.0, we added an enrichment word cloud. Upon user's query for a protein, the top three projects for which the queried protein has the highest normalized spectral abundance factor (NSAF) and confidence score are first selected. Then the enrichment index of each word in the abstracts of these three projects is computed by the following formula, after removing of a list of pre-defined common words:
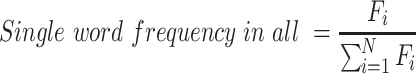 |
 |
 |
where 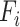 indicates the word frequency of the
indicates the word frequency of the 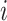 th word from the entire abstracts pool and
th word from the entire abstracts pool and 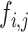 indicates the frequency of the same word in the abstracts pool of the selected projects, in which the queried protein has the highest abundance and confidence. The enrichment index of each word is then computed as the relative difference of the normalized word frequency between selected abstracts and all abstracts. Enrichment is visualized by font size, so that larger words correspond to more enriched terms representing a high correlation with the queried protein. A Custom Python script was used to generate the enrichment word cloud and is available at https://github.com/Matrisome/MatrisomeDB2.
indicates the frequency of the same word in the abstracts pool of the selected projects, in which the queried protein has the highest abundance and confidence. The enrichment index of each word is then computed as the relative difference of the normalized word frequency between selected abstracts and all abstracts. Enrichment is visualized by font size, so that larger words correspond to more enriched terms representing a high correlation with the queried protein. A Custom Python script was used to generate the enrichment word cloud and is available at https://github.com/Matrisome/MatrisomeDB2.
Dataset submission page
One of the bottlenecks to the horizontal expansion of MatrisomeDB was the lack of information allowing us to match raw files to the samples they relate to. To overcome this, we have built a new submission interface, allowing researchers to submit their datasets for consideration to MatrisomeDB. This interface will require researchers to provide a link to raw mass spectrometry data, but also detailed sample descriptions, including tissue of origin, information on the ECM enrichment method employed, and protein and/or peptide fractionation method, as well as a list linking each raw file to the sample it corresponds to. Examples of detailed sample descriptions are provided in Supplementary Table S1, column H. We are aiming to update MatrisomeDB periodically, with datasets added in batches, with formal documented new releases.
RESULTS AND DISCUSSION
Horizontal expansion
Upon completion of the curation process, we added 772 raw files from the datasets of 25 studies (Table 1 and Supplementary Table S1). This significantly expands the content of the database which includes new healthy tissues, such as the ECM of umbilical arteries (35) and the aorta (36), the stomach (37), the skin, including healthy skin from different anatomical regions and patients of different ages, but also scars and keloids (38–40), the Fallopian tubes (41) and ovaries (42), different musculoskeletal structures (43–48), and different regions of the brain (49,50). It also includes two cancer types not included before: pancreatic ductal adenocarcinoma (51) and gastric antrum adenocarcinoma (37), and data on the ECM of metastases arising from mammary tumors growing in different distant organs (52) and from metastases of ovarian tumors growing in the omentum treated or not with chemotherapy (53). MatrisomeDB 2.0 also includes two new studies on the ECM of hepatic metastasis from colorectal tumors (54,55) and an additional dataset on the ECM of aging lung (56). Last, we included three studies reporting the composition of the ECM produced in vitro by dental pulp stem cells (57), lung fibroblasts (58) and kidney organoids (59). Importantly, this release includes, for the first time, data on the ECM of aging tissues (skin, lung, intervertebral discs). MatrisomeDB 2.0 now includes 2,051 human and 949 mouse matrisome proteoforms, 6,891,623 human and 4,763,174 mouse matrisome-protein-derived peptide-to-spectrum matches (PSMs).
The expansion of MatrisomeDB results in near complete coverage of the matrisome and of matrisome protein sequences
Using de-novo sequence analysis, we predicted that there are 1,027 genes in the human genome encoding ECM and ECM-associated proteins (15,17). We further divided this in-silico predicted matrisome into two main divisions: (i) structural components of the ECM or the ‘core matrisome’ including ECM glycoproteins, collagens, and proteoglycans, which are highly insoluble in nature and (ii) matrisome-associated proteins including ECM-affiliated proteins, ECM regulators, and secreted factors known or expected to bind to structural components of the ECM. The aggregation of the datasets from MatrisomeDB 2.0 resulted in the detection of almost all the proteins of the matrisome, a significant improvement over the aggregated data from MatrisomeDB 1.0 (Figure 2A). Namely, in MatrisomeDB 1.0, we had experimental of evidence for 89.4% of the core matrisome and only 59.4% for matrisome-associated proteins. These numbers are now 98.3% and 97.3%, respectively. The experimental coverage of ECM-affiliated proteins (+38.6%) and secreted factors (+51.2%) benefited the most from the inclusion of additional datasets to MatrisomeDB (Figure 1B and 1C). This is likely explained by fact that we not only included new tissues, but we also included protein samples of different solubility in this new release of the database.
Figure 2.

The horizontal expansion of MatrisomeDB results in near complete coverage of the matrisome and of matrisome protein sequences. (A) Bar graph represents, for each matrisome category, the percentage of the in-silico-predicted human matrisome supported by experimental evidence in MatrisomeDB 2.0. Lower bars (lighter shades) indicate coverage in MatrisomeDB 1.0, upper bars (darker shades) indicate the coverage achieved in MatrisomeDB 2.0. (B) Bar graph represents, for each matrisome category, the average protein sequence coverage (%) in MatrisomeDB 1.0 (lighter shades) and in MatrisomeDB 2.0 (darker shades). (C) Histograms represent, for each matrisome category, the frequency of average protein sequence coverage (%) between MatrisomeDB 1.0 (lighter shades) and MatrisomeDB 2.0 (darker shades).
The horizontal expansion of MatrisomeDB also resulted in achieving near-complete protein sequence coverage for proteins across all matrisome categories (93.2% on average). This represents a significant improvement over the 24.6% average sequence coverage reached upon aggregation of MatrisomeDB 1.0 datasets (Figure 2B). This significant improvement has the potential to uncover novel facets of ECM proteins (detection of isoforms arising from splice variants, single amino-acid variants) and support novel hypotheses regarding the roles of these proteins in pathophysiological contexts (60,61).
Updated functionalities of the main search page
The search functionality of MatrisomeDB remains unchanged and a search's primary output will be a set of heat maps depicting the confidence score and abundance with which proteins are identified in studies, see MatrisomeDB 1.0 (21). We enhanced the primary output by providing an enrichment word cloud generated upon query depicting terms enriched in abstracts of studies in which the protein of interest is detected with higher confidence and higher abundance. In the example presented in Figure 3, we searched for ‘TGM2’, the gene symbol of the protein-glutamine gamma-glutamyltransferase 2, across all datasets of MatrisomeDB 2.0. The resulting cloud points to an enrichment of a set of terms including ‘PDAC’ (for pancreatic ductal adenocarcinoma), ‘pancreatitis’, ‘MC38’ (a metastatic colorectal cancer cell line), ‘metastases’, and ‘airway’, that can point users to possible roles for TGM2 in these processes. Note that the content of the cloud will change if users further narrow the analysis to a given species or set of samples.
Figure 3.

Abstract-mining tool. Searching for ‘TGM2’, the protein-glutamine gamma-glutamyl transferase 2 across the entire MatrisomeDB 2.0 dataset returns an enrichment word cloud generated from the abstracts of studies in which a queried protein is found with higher confidence and higher abundance relative to other studies in MatrisomeDB. Enriched words across abstracts appear in larger font. Users can right-click (or Control-click on Mac devices) on the word cloud to save the image.
Cross-referencing to external resources to design targeted mass spectrometry assays for accurate ECM protein quantification in complex samples
Upon selecting a protein of interest, users land on a page providing a set of links pointing to external resources. These include a link to the NCI CPTAC assay portal, for proteins for which SRM/MRM assays have been validated, and a link to the Peptide Atlas, which users can then query to design their own targeted MS assay (Figure 4A).
Peptide and PTM mapping on 2D domain-based representation of matrisome proteins
Using as an example the human form of protein-glutamine gamma-glutamyltransferase 2 (TGM2, UniProt P21890) detected in pancreatic ductal adenocarcinoma xenograft (BxPC3) samples, we illustrate here additional functionalities implemented in MatrisomeDB 2.0. Users can now visualize the mapping of peptides and PTMs overlaid onto the domain-based representation of TGM2 (Figure 4B). The grey histogram located above the domain-based depiction represents the percentage of sequence coverage. users can retrieve information regarding the percentage of sequence coverage for each domain by clicking on the ‘Download domain coverage csv’ link (Supplementary Table S2A). A set of controls located in the upper right corner allows users to zoom in or out, hover over the domains to get additional information, and to save the image (Figure 4B, red arrow). By selecting ‘Open sample PTM occurrence table’ users will be taken to a page listing the types of PTMs detected and their position (Figure 4B, inset) and will be given the option to download the table in a .csv format (Supplementary Table S2B). The ‘Download domain PTM coverage csv’ link generates a table listing the position of PTMs detected with respect to domain positioning (Supplementary Table S2C). Note that not all PTMs are listed in this table since some may fall outside defined protein domains.
Peptide and PTM mapping on Alpha-Fold predicted 3D structures of matrisome proteins
In our example, users can now visualize the mapping of peptides and PTMs overlaid onto the AlphaFold-predicted 3D structure of TGM2 (Figure 4C). To do so, users can select the option ‘Open sample data in SCV’ located at the top of the domain-based representation of the protein queried (Figure 4B). A set of controls located in the upper right corner allow users to change the colors of the PTMs and covered sequence (Figure 4C, red arrow), to rotate the 3D structure, and to take a screenshot of the image (Figure 4C). To allow users to evaluate the confidence of AlphaFold-predicted structure, we are also including a link to the AlphaFold Protein Structure Database page of the queried protein (Figure 4C, blue arrow; in the present example, the link will point to https://alphafold.ebi.ac.uk/entry/P21980). In a future release of MatrisomeDB, we will enhance this feature by providing, when available, 3D peptide and PTM mapping overlaid onto experimentally validated structures of matrisome proteins retrieved from the Protein Data Bank (30).
Of note, users can visualize peptide and PTM mapping on 2D and 3D representations of matrisome proteins for a given sample type (as shown in Figure 4 for TGM2 in pancreatic ductal adenocarcinoma xenograft samples) or for all datasets across MatrisomeDB (‘global’ view).
FUTURE DEVELOPMENTS
With this new deployment, we more than doubled the size of MatrisomeDB, expanding its content to nearly 40 different broad tissue types and including new types of samples such as aging tissues and organoids. Anticipating that mass-spectrometry-based proteomics will remain the state-of-the-art approach to profile, in an unbiased manner, the composition of tissues, we will plan a future release to further expand the content of the database to tissues or diseases not represented yet.
In addition, beyond the standard approach of using label-free samples and a data-dependent acquisition mode, proteomics can be conducted using labeled samples, for example to more accurately evaluate protein abundance or multiplex the analysis (62), or using data-independent acquisition (63–65). These alternate modalities are not yet broadly applied to study the ECM, but if they became more broadly adopted, we would envision devising a pipeline to include such studies to MatrisomeDB.
In order to further facilitate the mining of MatrisomeDB data, we will work towards building connections with other ECM-related databases including MatrixDB which reports ECM protein–protein and ECM protein–glycan interactions (66), MatriNet which reports gene interaction networks within the ECM (67), and basement membraneBASE which focuses on a subset of matrisome proteins forming the specialized ECM basement membrane and provides an atlas of basement membrane components across tissues and pathophysiological processes (68).
Over the past decade, ECM-focused proteomics has become a powerful tool that allows unbiased characterization of the ECM protein signature of healthy or diseased tissues, making the discovery of potential prognostic biomarkers and treatment targets attainable. It is our hope to contribute to this effort by facilitating the re-use of existing proteomic datasets and demonstrating how data obtained by querying MatrisomeDB can be a powerful hypothesis-generating tool.
CITING MATRISOME DB
For a general citation of MatrisomeDB, researchers should cite this publication. In addition, the following citation format is suggested when referring to specific data obtained from MatrisomeDB: https://matrisomedb.org.
DATA AVAILABILITY
All raw mass spectrometry datasets were retrieved from public repositories (see Table 1, for identifiers). All codes are available at https://github.com/Matrisome/MatrisomeDB2.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank all the members of the Naba and Gao laboratories for helpful discussions. The authors would also like to thank the authors of studies included in MatrisomeDB who have kindly accepted to assist with the data curation, and providing, when needed, additional information on sample identification and annotation.
Contributor Information
Xinhao Shao, Department of Pharmaceutical Sciences, University of Illinois at Chicago, Chicago, IL 60612, USA.
Clarissa D Gomez, Department of Physiology and Biophysics, University of Illinois at Chicago, Chicago, IL 60612, USA.
Nandini Kapoor, Department of Physiology and Biophysics, University of Illinois at Chicago, Chicago, IL 60612, USA.
James M Considine, Department of Physiology and Biophysics, University of Illinois at Chicago, Chicago, IL 60612, USA.
Christopher Grams, Department of Pharmaceutical Sciences, University of Illinois at Chicago, Chicago, IL 60612, USA.
Yu (Tom) Gao, Department of Pharmaceutical Sciences, University of Illinois at Chicago, Chicago, IL 60612, USA; University of Illinois Cancer Center, Chicago, IL 60612, USA.
Alexandra Naba, Department of Physiology and Biophysics, University of Illinois at Chicago, Chicago, IL 60612, USA; University of Illinois Cancer Center, Chicago, IL 60612, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [1U01HG012680-01 to A.N. and Y.G., 1R21CA261642-01A1 to A.N. and Y.G., 5R35GM133416 to Y.G.]; University of Illinois Cancer Center [2020-PP-07 to A.N. and Y.G.]; C.G. is supported by an award from the LAS Undergraduate Research Initiative (LASURI) at UIC; Summer Research Opportunities Program (SROP); Graduate Pathways to Success Program (GPS) at UIC. Funding for open access charge: University of Illinois Cancer Center (to A.N. and Y.G.).
Conflict of interest statement. A.N. has sponsored research agreements with Boehringer-Ingelheim in support of work independent from the present study. X.S., C.G., J.C., N.K., and Y.G. declare no competing interests.
REFERENCES
- 1. Hynes R.O., Yamada K.M.. Extracellular Matrix Biology. Cold Spring Harbor Perspectives in Biology. 2012; NY: Cold Spring Harbor Laboratory Press, Cold Spring Harbor. [Google Scholar]
- 2. Karamanos N.K., Theocharis A.D., Piperigkou Z., Manou D., Passi A., Skandalis S.S., Vynios D.H., Orian-Rousseau V., Ricard-Blum S., Schmelzer C.E.H.et al.. A guide to the composition and functions of the extracellular matrix. FEBS J. 2021; 288:6850–6912. [DOI] [PubMed] [Google Scholar]
- 3. Walma D.A.C., Yamada K.M.. The extracellular matrix in development. Development. 2020; 147:dev175596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moretti L., Stalfort J., Barker T.H., Abebayehu D. The interplay of fibroblasts, the extracellular matrix, and inflammation in scar formation. J. Biol. Chem. 2022; 298:101530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marino G.E., Weeraratna A.T.. A glitch in the matrix: age-dependent changes in the extracellular matrix facilitate common sites of metastasis. Aging Cancer. 2020; 1:19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ewald C.Y. The matrisome during aging and longevity: a systems-level approach toward defining matreotypes promoting healthy aging. Gerontology. 2020; 66:266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Karamanos N.K., Theocharis A.D., Neill T., Iozzo R.V.. Matrix modeling and remodeling: a biological interplay regulating tissue homeostasis and diseases. Matrix Biol. 2019; 75–76:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Theocharis A.D., Manou D., Karamanos N.K.. The extracellular matrix as a multitasking player in disease. FEBS J. 2019; 286:2830–2869. [DOI] [PubMed] [Google Scholar]
- 9. Socovich A.M., Naba A.. The cancer matrisome: from comprehensive characterization to biomarker discovery. Semin. Cell Dev. Biol. 2019; 89:157–166. [DOI] [PubMed] [Google Scholar]
- 10. Lamandé S.R., Bateman J.F.. Genetic disorders of the extracellular matrix. Anat Rec (Hoboken). 2020; 303:1527–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McCabe M.C., Schmitt L.R., Hill R.C., Dzieciatkowska M., Maslanka M., Daamen W.F., van Kuppevelt T.H., Hof D.J., Hansen K.C.. Evaluation and refinement of sample preparation methods for extracellular matrix proteome coverage. Mol. Cell. Proteomics. 2021; 20:100079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Taha I.N., Naba A.. Exploring the extracellular matrix in health and disease using proteomics. Essays Biochem. 2019; 63:417–432. [DOI] [PubMed] [Google Scholar]
- 13. Randles M., Lennon R.. Applying proteomics to investigate extracellular matrix in health and disease. Curr. Top. Membr. 2015; 76:171–196. [DOI] [PubMed] [Google Scholar]
- 14. Krasny L., Huang P.H.. Advances in the proteomic profiling of the matrisome and adhesome. Expert Rev. Proteomics. 2021; 18:781–794. [DOI] [PubMed] [Google Scholar]
- 15. Naba A., Clauser K.R., Hoersch S., Liu H., Carr S.A., Hynes R.O.. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell. Proteomics. 2012; 11:M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Naba A., Hoersch S., Hynes R.O.. Towards definition of an ECM parts list: an advance on GO categories. Matrix Biol. 2012; 31:371–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gebauer J.M., Naba A.. Ricard-Blum S. The matrisome of model organisms: from in-silico prediction to big-data annotation. Extracellular Matrix Omics. 2020; Cham: Springer International Publishing; 17–42.Biology of Extracellular Matrix. [Google Scholar]
- 18. Naba A., Ricard-Blum S.. Ricard-Blum S. The extracellular matrix goes -omics: resources and tools. Extracellular Matrix Omics. 2020; Cham: Springer International Publishing; 1–16.Biology of Extracellular Matrix. [Google Scholar]
- 19. Naba A., Clauser K.R., Ding H., Whittaker C.A., Carr S.A., Hynes R.O.. The extracellular matrix: tools and insights for the “omics” era. Matrix Biol. 2016; 49:10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wilkinson M.D., Dumontier M., Aalbersberg Ij.J., Appleton G., Axton M., Baak A., Blomberg N., Boiten J.-W., da Silva Santos L.B., Bourne P.E.et al.. The FAIR guiding principles for scientific data management and stewardship. Sci. Data. 2016; 3:160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shao X., Taha I.N., Clauser K.R., Gao Y., Naba A.. MatrisomeDB: the ECM-protein knowledge database. Nucleic Acids Res. 2020; 48:D1136–D1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Deutsch E.W., Bandeira N., Sharma V., Perez-Riverol Y., Carver J.J., Kundu D.J., García-Seisdedos D., Jarnuczak A.F., Hewapathirana S., Pullman B.S.et al.. The proteomexchange consortium in 2020: enabling ‘big data’ approaches in proteomics. Nucleic Acids Res. 2020; 48:D1145–D1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Perez-Riverol Y., Bai J., Bandla C., García-Seisdedos D., Hewapathirana S., Kamatchinathan S., Kundu D.J., Prakash A., Frericks-Zipper A., Eisenacher M.et al.. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022; 50:D543–D552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mellacheruvu D., Wright Z., Couzens A.L., Lambert J.-P., St-Denis N., Li T., Miteva Y.V., Hauri S., Sardiu M.E., Low T.Y.et al.. The CRAPome: a contaminant repository for affinity purification mass spectrometry data. Nat. Methods. 2013; 10:730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kong A.T., Leprevost F.V., Avtonomov D.M., Mellacheruvu D., Nesvizhskii A.I.. MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods. 2017; 14:513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Letunic I., Khedkar S., Bork P.. SMART: recent updates, new developments and status in 2020. Nucleic Acids Res. 2021; 49:D458–D460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tunyasuvunakool K., Adler J., Wu Z., Green T., Zielinski M., Žídek A., Bridgland A., Cowie A., Meyer C., Laydon A.et al.. Highly accurate protein structure prediction for the human proteome. Nature. 2021; 596:590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A.et al.. Highly accurate protein structure prediction with alphafold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shao X., Grams C., Gao Y.. Sequence coverage visualizer: a web application for protein sequence coverage 3D visualization. 2022; bioRxiv doi:13 January 2022, preprint: not peer reviewed 10.1101/2022.01.12.476109. [DOI] [PMC free article] [PubMed]
- 30. consortium wwPDB Protein data bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019; 47:D520–D528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Carr S.A., Abbatiello S.E., Ackermann B.L., Borchers C., Domon B., Deutsch E.W., Grant R.P., Hoofnagle A.N., Hüttenhain R., Koomen J.M.et al.. Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol. Cell. Proteomics. 2014; 13:907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Whiteaker J.R., Halusa G.N., Hoofnagle A.N., Sharma V., MacLean B., Yan P., Wrobel J.A., Kennedy J., Mani D.R., Zimmerman L.J.et al.. CPTAC assay portal: a repository of targeted proteomic assays. Nat. Methods. 2014; 11:703–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Whiteaker J.R., Halusa G.N., Hoofnagle A.N., Sharma V., MacLean B., Yan P., Wrobel J.A., Kennedy J., Mani D.R., Zimmerman L.J.et al.. Using the CPTAC assay portal to identify and implement highly characterized targeted proteomics assays. Methods Mol. Biol. 2016; 1410:223–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Desiere F., Deutsch E.W., King N.L., Nesvizhskii A.I., Mallick P., Eng J., Chen S., Eddes J., Loevenich S.N., Aebersold R.. The peptideatlas project. Nucleic Acids Res. 2006; 34:D655–D658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mallis P., Sokolis D.P., Makridakis M., Zoidakis J., Velentzas A.D., Katsimpoulas M., Vlahou A., Kostakis A., Stavropoulos-Giokas C., Michalopoulos E.. Insights into biomechanical and proteomic characteristics of small diameter vascular grafts utilizing the human umbilical artery. Biomedicines. 2020; 8:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fava M., Barallobre-Barreiro J., Mayr U., Lu R., Didangelos A., Baig F., Lynch M., Catibog N., Joshi A., Barwari T.et al.. Role of ADAMTS-5 in aortic dilatation and extracellular matrix remodeling. ATVB. 2018; 38:1537–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moreira A.M., Ferreira R.M., Carneiro P., Figueiredo J., Osório H., Barbosa J., Preto J., Pinto-do-Ó P., Carneiro F., Seruca R.. Proteomic identification of a gastric tumor ECM signature associated with cancer progression. Front. Mol. Biosci. 2022; 9:818552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barallobre-Barreiro J., Woods E., Bell R.E., Easton J.A., Hobbs C., Eager M., Baig F., Ross A.M., Mallipeddi R., Powell B.et al.. Cartilage-like composition of keloid scar extracellular matrix suggests fibroblast mis-differentiation in disease. Matrix Biol. Plus. 2019; 4:100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McCabe M.C., Hill R.C., Calderone K., Cui Y., Yan Y., Quan T., Fisher G.J., Hansen K.C.. Alterations in extracellular matrix composition during aging and photoaging of the skin. Matrix Biol. Plus. 2020; 8:100041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dussoyer M., Page A., Delolme F., Rousselle P., Nyström A., Moali C.. Comparison of extracellular matrix enrichment protocols for the improved characterization of the skin matrisome by mass spectrometry. J. Proteomics. 2021; 251:104397. [DOI] [PubMed] [Google Scholar]
- 41. Renner C., Gomez C., Visetsouk M.R., Taha I., Khan A., McGregor S.M., Weisman P., Naba A., Masters K.S., Kreeger P.K.. Multi-modal profiling of the extracellular matrix of human fallopian tubes and serous tubal intraepithelial carcinomas. J. Histochem. Cytochem. 2022; 70:151–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ouni E., Ruys S.P.D., Dolmans M.-M., Herinckx G., Vertommen D., Amorim C.A.. Divide-and-Conquer matrisome protein (DC-MaP) strategy: an MS-Friendly approach to proteomic matrisome characterization. Int. J. Mol. Sci. 2020; 21:E9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tam V., Chen P., Yee A., Solis N., Klein T., Kudelko M., Sharma R., Chan W.C., Overall C.M., Haglund L.et al.. DIPPER, a spatiotemporal proteomics atlas of human intervertebral discs for exploring ageing and degeneration dynamics. Elife. 2020; 9:e64940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bubb K., Holzer T., Nolte J.L., Krüger M., Wilson R., Schlötzer-Schrehardt U., Brinckmann J., Altmüller J., Aszodi A., Fleischhauer L.et al.. Mitochondrial respiratory chain function promotes extracellular matrix integrity in cartilage. J. Biol. Chem. 2021; 297:101224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lofaro F.D., Cisterna B., Lacavalla M.A., Boschi F., Malatesta M., Quaglino D., Zancanaro C., Boraldi F.. Age-related changes in the matrisome of the mouse skeletal muscle. Int. J. Mol. Sci. 2021; 22:10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jacobson K.R., Lipp S., Acuna A., Leng Y., Bu Y., Calve S.. Comparative analysis of the extracellular matrix proteome across the myotendinous junction. J. Proteome Res. 2020; 19:3955–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Georgieva V.S., Etich J., Bluhm B., Zhu M., Frie C., Wilson R., Zaucke F., Bateman J., Brachvogel B.. Ablation of the miRNA cluster 24 has profound effects on extracellular matrix protein abundance in cartilage. Int. J. Mol. Sci. 2020; 21:4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kudelko M., Chen P., Tam V., Zhang Y., Kong O.-Y., Sharma R., Au T.Y.K., To M.K.-T., Cheah K.S.E., Chan W.C.W.et al.. PRIMUS: comprehensive proteomics of mouse intervertebral discs that inform novel biology and relevance to human disease modelling. Matrix Biol. Plus. 2021; 12:100082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kjell J., Götz M.. Filling the gaps – a call for comprehensive analysis of extracellular matrix of the glial scar in region- and injury-specific contexts. Front. Cell Neurosci. 2020; 14:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sethi M.K., Downs M., Zaia J.. Serial in-solution digestion protocol for mass spectrometry-based glycomics and proteomics analysis. Mol Omics. 2020; 16:364–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tian C., Clauser K.R., Öhlund D., Rickelt S., Huang Y., Gupta M., Mani D.R., Carr S.A., Tuveson D.A., Hynes R.O.. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:19609–19618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hebert J.D., Myers S.A., Naba A., Abbruzzese G., Lamar J.M., Carr S.A., Hynes R.O.. Proteomic profiling of the ECM of xenograft breast cancer metastases in different organs reveals distinct metastatic niches. Cancer Res. 2020; 80:1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pearce O.M.T., Delaine-Smith R.M., Maniati E., Nichols S., Wang J., Böhm S., Rajeeve V., Ullah D., Chakravarty P., Jones R.R.et al.. Deconstruction of a metastatic tumor microenvironment reveals a common matrix response in human cancers. Cancer Discov. 2018; 8:304–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yuzhalin A.E., Gordon-Weeks A.N., Tognoli M.L., Jones K., Markelc B., Konietzny R., Fischer R., Muth A., O’Neill E., Thompson P.R.et al.. Colorectal cancer liver metastatic growth depends on PAD4-driven citrullination of the extracellular matrix. Nat. Commun. 2018; 9:4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yuzhalin A.E., Lim S.Y., Gordon-Weeks A.N., Fischer R., Kessler B.M., Yu D., Muschel R.J.. Proteomics analysis of the matrisome from MC38 experimental mouse liver metastases. Am. J. Physiol. Gastrointest. 2019; 317:G625–G639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Angelidis I., Simon L.M., Fernandez I.E., Strunz M., Mayr C.H., Greiffo F.R., Tsitsiridis G., Ansari M., Graf E., Strom T.-M.et al.. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat. Commun. 2019; 10:963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nowwarote N., Petit S., Ferre F.C., Dingli F., Laigle V., Loew D., Osathanon T., Fournier B.P.J.. Extracellular matrix derived from dental pulp stem cells promotes mineralization. Front. Bioeng. Biotechnol. 2022; 9:740712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lansky Z., Mutsafi Y., Houben L., Ilani T., Armony G., Wolf S.G., Fass D. 3D mapping of native extracellular matrix reveals cellular responses to the microenvironment. JSBX. 2019; 1:100002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Morais M.R.P.T., Tian P., Lawless C., Murtuza-Baker S., Hopkinson L., Woods S., Mironov A., Long D.A., Gale D.P., Zorn T.M.T.et al.. Kidney organoids recapitulate human basement membrane assembly in health and disease. Elife. 2022; 11:e73486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rekad Z., Izzi V., Lamba R., Ciais D., Van Obberghen-Schilling E.. The alternative matrisome: alternative splicing of ECM proteins in development, homeostasis and tumor progression. Matrix Biol. 2022; 111:26–52. [DOI] [PubMed] [Google Scholar]
- 61. Izzi V., Davis M.N., Naba A.. Pan-Cancer analysis of the genomic alterations and mutations of the matrisome. Cancers. 2020; 12:2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zecha J., Satpathy S., Kanashova T., Avanessian S.C., Kane M.H., Clauser K.R., Mertins P., Carr S.A., Kuster B.. TMT labeling for the masses: a robust and cost-efficient, in-solution labeling approach. Mol. Cell. Proteomics. 2019; 18:1468–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sajic T., Liu Y., Aebersold R.. Using data-independent, high-resolution mass spectrometry in protein biomarker research: perspectives and clinical applications. Proteomics Clin. Appl. 2015; 9:307–321. [DOI] [PubMed] [Google Scholar]
- 64. Ludwig C., Gillet L., Rosenberger G., Amon S., Collins B.C., Aebersold R.. Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol. Syst. Biol. 2018; 14:e8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shi T., Song E., Nie S., Rodland K.D., Liu T., Qian W.-J., Smith R.D.. Advances in targeted proteomics and applications to biomedical research. Proteomics. 2016; 16:2160–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Clerc O., Deniaud M., Vallet S.D., Naba A., Rivet A., Perez S., Thierry-Mieg N., Ricard-Blum S.. MatrixDB: integration of new data with a focus on glycosaminoglycan interactions. Nucleic Acids Res. 2019; 47:D376–D381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kontio J., Soñora V.R., Pesola V., Lamba R., Dittmann A., Navarro A.D., Koivunen J., Pihlajaniemi T., Izzi V.. Analysis of extracellular matrix network dynamics in cancer using the matrinet database. Matrix Biol. 2022; 110:141–150. [DOI] [PubMed] [Google Scholar]
- 68. Jayadev R., Morais M.R.P.T., Ellingford J.M., Srinivasan S., Naylor R.W., Lawless C., Li A.S., Ingham J.F., Hastie E., Chi Q.et al.. A basement membrane discovery pipeline uncovers network complexity, regulators, and human disease associations. Sci. Adv. 2022; 8:eabn2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw mass spectrometry datasets were retrieved from public repositories (see Table 1, for identifiers). All codes are available at https://github.com/Matrisome/MatrisomeDB2.