


Abstract
tRNA molecules contain dense, abundant modifications that affect tRNA structure, stability, mRNA decoding and tsRNA formation. tRNA modifications and related enzymes are responsive to environmental cues and are associated with a range of physiological and pathological processes. However, there is a lack of resources that can be used to mine and analyse these dynamically changing tRNA modifications. In this study, we established tModBase (https://www.tmodbase.com/) for deciphering the landscape of tRNA modification profiles from epitranscriptome data. We analysed 103 datasets generated with second- and third-generation sequencing technologies and illustrated the misincorporation and termination signals of tRNA modification sites in ten species. We thus systematically demonstrate the modification profiles across different tissues/cell lines and summarize the characteristics of tRNA-associated human diseases. By integrating transcriptome data from 32 cancers, we developed novel tools for analysing the relationships between tRNA modifications and RNA modification enzymes, the expression of 1442 tRNA-derived small RNAs (tsRNAs), and 654 DNA variations. Our database will provide new insights into the features of tRNA modifications and the biological pathways in which they participate.
INTRODUCTION
Transfer RNA (tRNA) is a key molecule in the process of decoding genetic information. tRNA molecules contain dense, abundant chemical modifications and may represent a record of the ancient RNA world (1–5). Approximately 400 unique tRNA molecules can potentially be produced in human cells (6,7). On average, each tRNA carries 13 modification sites (8). Moreover, recent studies have shown that tRNA modifications are highly dynamic and show a wide range of responses to cellular metabolite levels and environmental stresses (9–16). Furthermore, these changes in tRNA modifications affect tRNA structure, codon recognition, and tRNA-derived small RNA (tsRNA) formation (17–24). A growing number of studies have found that alterations in tRNA modifications can cause serious diseases, referred to as RNA modopathies (25). These studies have led a new chapter in tRNA modification research, and future work will surely reveal the mechanisms of dynamic tRNA modifications.
There are several databases that collect tRNAs or modifications thereof. GtRNAdb (26) is the most comprehensive tRNA database, but it mainly collects the sequences and structures of tRNAs. Similarly, the T-psi-C database (27) focuses on the presentation of tRNA sequences and 3D structures. The Modomics database (28) provides comprehensive information concerning the chemical structures of modified ribonucleosides and their biosynthetic pathways. However, this database mainly includes early published results for individual tRNAs and lacks large-scale tRNA epitranscriptome data. RMBase is a database developed by our team based on high-throughput sequencing data to analyse RNA modifications (29). However, it collects data from genome-wide RNA modification detection techniques, mainly including m6A modifications on mRNAs. These databases are either relatively old or do not specifically focus on tRNA modifications, making it difficult to reveal the full landscape of the tRNA epitranscriptome.
Several epitranscriptomic technologies have been developed in recent years, which use special library construction strategies and identify tRNA modifications according to reverse transcription (RT) stop and misincorporation signatures (7,30–33). The most recent third-generation single-molecule sequencing strategy allows the direct detection of modifications on RNA molecules (34–38). However, current research on tRNA modopathies still faces many difficulties. First, each tRNA sequencing technology identifies tRNA modifications according to a different strategy. For example, QuantM-tRNA-Seq identifies modification sites based on the stop signal of tRNA fragments. mim-tRNA-seq (7) uses the RT misincorporation signature to identify tRNA modifications, while ARM-Seq (39) and DM-tRNA-seq (40) require the comparison of data obtained before and after AlkB treatment to demethylate common modifications in tRNAs. RBS-seq (41), developed based on bisulphite sequencing (42–45), utilizes chemical treatment to specifically identify m1A, Ψ and m5C. AQRNA-seq uses exonucleases to remove excess adapters to measure tRNA abundance and modification changes (31). The most recent development of Nanopore tRNA sequencing technology (34) allows the direct sequencing of tRNA molecules without reverse transcription steps, but it also produces a large number of miscalls. The differences in these sequencing technologies result in data including modification sites exhibiting completely different characteristics, making it difficult to compare data with each other. Second, the sequences of different tRNA isotypes are very similar, meaning that standard mapping strategies cannot precisely annotate the type of modifications at each site of each tRNA molecule. This can make it very difficult to identify the dynamic changes in these modifications at later stages. Finally, tRNA modifications are influenced by structural changes in tRNAs and differences in the levels of modification enzymes, and they determine the recognition of codons and the processing of tsRNAs (18–23). Current tRNA databases cannot store and display these complex factors simultaneously, thus limiting the subsequent resolution of their mechanisms and functions.
In summary, the detection of tRNA modifications requires specialized library construction strategies and bioinformatic analysis methods to ensure accuracy and high resolution. Based on the above research needs and problems, this study developed tModBase, a specialized resource for deciphering the landscape of tRNA modification profiles from epitranscriptome data.
MATERIALS AND METHODS
Data acquisition and download
For Homo sapiens (GRCh38/hg38), Mus musculus (GRCm39/mm39), Saccharomyces cerevisiae (S288c), Schizosaccharomyces pombe (972h-), Drosophila melanogaster (BDGP Rel. 6/dm6), Arabidopsis thaliana (TAIR10), Oryza sativa japonica group (Japanese rice IRGSP-1.0), Medicago truncatula A17 (MedtrA17_4.0), Mycobacterium bovis BCG (str. Pasteur 1173P2) and Escherichia coli (str. K-12 substr. MG1655), the mature tRNA sequences from GtRNAdb (26) were adapted, and identical sequences were merged at the isodecoder level. We used BLASTN to best match the dot-bracket structure and canonical structure from tRNAdb for each sequence in the tRNA reference library. For species with mitochondrial tRNA, we obtained mitochondrial tRNA reference sequences, dot-bracket structures and canonical structures from mitotRNAdb (46) or tRNAdb (46). The above steps are performed using the Python script.
The raw sequencing data were obtained from SRA files obtained with various sequencing technologies in the Gene Expression Omnibus (GEO) (47) database. The RNA-seq transcriptome profiling data of normal tumour samples from 32 cancer types were obtained from the TCGA database, and FPKM values were calculated using counts to represent the gene expression levels of the modification enzyme. RPM values were calculated to represent the expression levels of tRNA molecules. In addition, human (hg38) SNP data were obtained from the dbSNP database (48).
Annotation of known tRNA modifications and modification enzymes
Based on the available information on modifications for each species from the Modomics database modification were manually counted, and BLASTN (49) was used to align the modified sequences provided by Modomics with the compiled isodecoder sequence set. Modifications matching the tRNA molecules were screened from RMBase as complementary data. For human mitochondrial modifications, we collected more comprehensive annotation information from the literature (50).
tRNA quantification and identification of modification signals in epitranscriptome sequencing data
We collected sequence read data from 103 datasets of 11 high-throughput sequencing (HTS) technologies (Supplementary Table S1). These raw sequencing reads were subjected to quality control and sequence alignment, and the alignment results were quantified with the custom Python scripts.
DM-tRNA-seq
Before mapping, we used Trim_Galore! v0.6.7 for the quality control of all sequencing datasets from DM-tRNA-seq (40) with the following parameters:
-q 20 –phred33 –stringency 5 –length 15 -e 0.1
followed by alignment to the reference library using Bowtie2 (51) with the parameters:
D 20 -R 3 -N 1 -i S,1,0.50.
ARM-seq
We used the quality control tool Trim_Galore! v0.6.7 to trim reads with the following parameters:
-q 20 –phred33 –stringency 5 –length 15 -e 0.1
Followed by alignment to the reference library using Bowtie2 (51) with the following parameters:
–quiet –min-score G,1,8 –local -D 20 -R 3 -N 1 -L 10 -i S,1,0.5.
Multiple mapping reads were filtered out with the custom Python scripts.
YAMAT-seq
Since the original article for YAMAT-seq (30) did not specify quality control tools or parameters, we used Trim_Galore! v0.6.7 for the quality control of YAMAT-seq reads with the following parameters:
-q 20 –phred33 –stringency 5 -e 0.1
Since the maintenance of SHRiMP2, used in the YAMAT-seq article, had stopped, we used Bowtie2 (51) instead with the following parameters:
–quiet –min-score G,1,8 –local -D 20 -R 3 -N 1 -L 10 -i S,1,0.5.
hydro-tRNA-seq
We trimmed the hydro-tRNA-seq reads using Cutadapt v4.1 (52), and the 3′ adapter sequences of 4 replicates were as follows:
TCGTATGCCGTCCTCTGTTTG; TCACTTCGTGTCGTTCTGTGTGT; TCACTTCGTGTGCCGTCCTCTGTGTGTG; TCTAGTCGTATGCCGTCTTCTCTCTGTG
For sequence alignment, the default parameters of BWA-MEM were used.
QuantM-tRNA-seq
Commands for quality control and alignment were provided in the QuantM-tRNA-seq reference:
cutadapt –cut 2 -a CCAGTATCCAGTTGGAATT -g TCCAACTGGATACTGGN -e 0.2
bowtie2 –quiet –min-score G,1,8 –local -D 20 -R 3 -N 1 -L 10 -i S,1,0.5
mim-tRNA-seq
We used the pipeline provided by mim-tRNA-seq (7) for bioinformatics analysis and processed the results of the pipeline into a unified format through the custom script.
AQRNA-seq
In AQRNA-seq, fastxtoolkit was used for read quality control, but the adapter sequences were not given. Hence, we used Trim_Galore! v0.6.7 instead with the following parameters:
-q 20 –phred33 –stringency 5 –length 10 -e 0.1
We used BLAST for sequence alignment, and the parameters were as follows:
blast -perc identity 90 -word_size 9 -dust no -soft_masking false
BS-seq
We use the default parameters of Bismark (53) throughout the analysis of BS-seq data.
Pseudo-seq
The following command was used to perform quality control and sequence alignment as the Pseudo-seq article provided:
cutadapt -a ADAPTER_SEQ –overlap 3 –minimum-length 18 -o trimmed_reads.fastq.gz input_reads.fastq.gz.
bowtie2 –quiet –min-score G,1,8 –local -D 20 -R 3 -N 1 -L 10 -i S,1,0.5
The Pseudo-seq peak value was calculated for the paired + CMC/-CMC samples with the following formula:
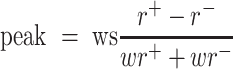 |
Here, ws is the window size, which is set to 10 because the reference tRNA sequences are small. In addition, r+ and r− are the numbers of reads terminating at the 3′ ends of the + CMC sample and -CMC sample, respectively. Moreover, wr+ and wr− are the numbers of all reads terminating within the windows for + CMC samples and -CMC samples, respectively. These calculations can be performed with the custom script.
m1A-seq
The supplementary file of the m1A-MAP article provided results including mismatch information at various positions in all the tRNA sequences, and we sorted the results into a unified format with the custom script.
Nanopore sequencing
After obtaining the original data (fast5 format) from Thomas et al. (34), we performed base calling using Guppy, which is recommended in the NanoProcess portion of the Master of Pores process. Then, we replaced U in the fastq format file with T. The obtained results were aligned with the reference tRNA sequences provided by the original article using Bowtie2 (51), filtered according to MAPQ scores greater than or equal to 1. Finally, the custom Python script was used to calculate the counts.
Differential expression analysis of tRNA and RNA modification enzymes
We collected all RNA-modifying enzymes responsible for tRNA modification from databases (28,29) and the literatures (54–56) and classified them into deamination, methylation, pseudouridylation, oxidation, hydroxylation, thiolation, acetylation, dihydrouridine modification, taurine modification, queuine modification, guanylylation and alkylation enzymes, for a total of 12 categories. In addition, we divided tRNA modification enzymes into two types, writers and erasers, according to their modification functions. To study the changes in modification enzymes in cancers, we downloaded data from >10 000 samples from 32 cancers available in the TCGA database (57). FPKM values were used to evaluate the expression of all genes based on the transcriptome sequencing data, and the expression levels of various modification enzymes were extracted from these data.
To compare the differences in tRNA expression between tumour and normal tissues, we first downloaded raw small RNA sequencing data from the TCGA database and then mapped them on mature tRNA sequences to quantify the tRNA expression levels in each sample. The results were next normalized using the expression levels of the modification enzymes with the following formula:
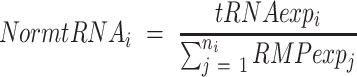 |
where, for a specific tRNA molecule i, the normalized value is the expression level of the tRNA divided by the sum of the expression levels of the modification enzymes (RNA modification Protein, RMP) corresponding to all modification sites (from j to ni) on that tRNA.
tsRNA level quantification and relationship analysis of modification sites
We previously established the tsRFinder tool for identifying tsRNA molecules from all small RNA samples in the TCGA database. Subsequently, we annotated the locations where the tsRNA molecules were generated on the tRNAs in detail in the identification results. The annotation results intersected with the modification sites on the tRNA molecules that we collected. Notably, studies have confirmed that the modification of tRNA has little effect on the identification of tsRNAs based on the TCGA library construction method (58).
Analysis of the relationship between RNA modification and DNA variation sites
To analyse the relationships between tRNA modification sites and known DNA variations, we downloaded the sites of known human DNA variations, including SNVs, insertions, deletions, and indels, from the dbSNP database. Then, the bedintersect tool (59) was used to identify the intersection between various DNA variants and all positions of tRNA modification sites in the reference tRNA sequences.
RESULTS
Dynamic interaction of tRNA modification positions and corresponding modification enzymes
A graph of the interaction between tRNA structure and modification is available on the tModBase home page (Figure 1). Users can intuitively check the modification type of each site in the consensus sequence of human cytoplasmic and mitochondrial tRNAs. For example, in human cytoplasmic tRNAs, position 34 is the site with the most abundant modification types, including a total of 20 modification types (Figure 1A). Additionally, this site corresponds to the wobble position of the mRNA codon, which affects the accuracy of codon recognition and the fidelity of translation. It can also be seen from the figure that whether the tRNA is cytoplasmic or mitochondrial, the most commonly affected modification site is pseudouridine (Ψ), and the bases with this modification are mainly distributed in the anticodon arm structure of the tRNA clover (Figure 1B, C).
Figure 1.

Dynamic interaction diagram of the modifications and modification enzyme types corresponding to each site in the tRNA consensus structure. (A) Modifications corresponding to each site in cytoplasmic tRNA, where the modification types at position 34 are highlighted. The type of modification corresponding to each site in cytoplasmic (B) and mitochondrial (C) tRNA molecules, with the modified sites affected by pseudouridine highlighted. (D) Map of possible tRNA sites of RNA-modifying enzymes, with the modification enzymes that affect the modification at position 34 of tRNA highlighted. The pencil icon represents an enzyme with writer function, and the eraser icon represents an enzyme with eraser function.
Different types of tRNA modifications are mediated by a variety of RNA modification enzymes (RNA modification proteins, RMPs). tModBase maps the modification types at each site to the corresponding modification enzymes and divides the modification enzymes into writers and erasers. For example, tRNA position 34 may be affected by 15 different writer enzymes (NSUN2, NSUN3, ADAT2, ADAT3, CTU1, CTU2, ELP3, MTO1, QTRT1, WDR6, PUS1, FTSJ1, ALKBH8, GTPBP3 and TRMU) and one eraser enzyme (ALKBH1) (Figure 1D). Users can easily search for modification enzymes based on the modification of interest and may search for the type and site of tRNA modification affected by a specific enzyme. In addition to the interactive diagram on the home page, users can access and filter more detailed information through three pages: tRNA, Modification, and Enzyme.
In conclusion, the currently known tRNA modification sites in humans and mice are well annotated in tModBase. However, we found that these annotated tRNAs represented only a small fraction of the total tRNA isotypes (Table 1). For example, the human genome contains a total of 432 tRNA loci, encoding 283 different tRNA isotypes, only 54 of which have well-annotated modification sites, accounting for 19% of all isotypes. Thus, the question arises of which sites on the remaining more than 80% of tRNA molecules are likely to contain annotations? We next mine through epitranscriptomic data.
Table 1.
List of the ratios of annotated modified tRNA molecules to the total number of tRNAs
| Species | # tRNA isotypes | # tRNA isotypes with annotated modification site | Percentage of known modified tRNAs (%) | # tRNA molecules with sequencing signals identified in epitranscriptomic data | Percentage of potential modified tRNAs identified by tModBase (%) |
|---|---|---|---|---|---|
| Homo sapiens | 283 | 54 | 19 | 274 | 97 |
| Mus musculus | 240 | 12 | 5 | 201 | 84 |
| Saccharomyces cerevisiae | 81 | 54 | 67 | 77 | 95 |
| Escherichia coli | 49 | 38 | 76 | 43 | 88 |
Mining tRNA modification signals from epitranscriptomic data
In recent years, a series of tRNA epitranscriptome sequencing technologies have been developed to obtain high-throughput tRNA modification signals. We collected and analysed 103 high-throughput sequencing datasets based on 11 library construction strategies for tRNA quantification and the identification of modification types from species including Homo sapiens, Mus musculus, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Drosophila melanogaster, Arabidopsis thaliana, Oryza sativa, Medicago truncatula, Solanum tuberosum, Mycobacterium tuberculosis variant bovis BCG and Escherichia coli (Supplementary Table S1). We developed corresponding analysis workflows for different library construction strategies (see Methods), and finally obtained the distribution of the modification signals (misincorporation or termination) in each tRNA molecule (Figure 2A). tModBase shows the overlapping relationships between the sites of different modification signals and the known modification sites in the molecule in detail. For example, according to the annotation, there is an m1A modification at the 9th site of mt-tRNA-His-GTG. Using mim-tRNA-seq technology (7), it can be observed that this site shows a strong A to T mismatch signal, which is determined by the properties of TGIRT RTase (60–62).
Figure 2.

Distribution of tRNA epitranscriptome sequencing signals displayed in tModBase. (A) Each row in the table represents the misincorporation signal detected in a tRNA isotype sequence. (B) The x-axis in the histogram represents the tRNA site in the consensus structure, and the y-axis represents the number of known modification types at that site. Each row of the heatmap represents a tRNA isotype, each column represents a site in the consensus structure of the tRNA, and the colour indicates the intensity of the misincorporation signal, such as the percentage of mismatches.
To better illustrate the common features of the distribution of modification signals on multiple tRNAs, we generated a heatmap of the distribution of modification signals of multiple tRNAs, corresponding to known modification sites, based on which the proportion and extent of the signal distribution on various tRNA molecules could be clearly displayed (Figure 2B).
Comparison of tRNA modification profiles between different tissues/cell lines
tModBase provides modification signal profiles recorded under various conditions, including profiles from three human cell lines (HEK293, K562 and iPSC) (Figure 3A), nine types of mouse tissues (heart, liver, cerebellum, cortex, medulla oblongata, spinal cord, tibialis, brain, and intestine) and different time points of T-cell activation in mice (naïve, 20 h after activation, 48 h after activation, 72 h after activation). In addition, tModBase provides the results of comparisons between the modification profiles of mouse tissues with or without microbial or virus infection (Figure 3B). Users can select tRNAs of interest to view the changes in the modification signals at various sites under different conditions.
Figure 3.

Modification profiles displayed in tModBase. Each row represents a condition: (A) different tissues of mouse and (B) the presence or absence of microorganisms, and each column represents a site on the tRNA molecule. The colour indicates the strength of the misincorporation signal, such as the percentage of mismatches.
Some sequencing methods (such as Pseudo-seq) require comparisons of datasets before and after chemical treatments (such as carbodiimide and CMC treatment). As a result, we calculated the rates of changes in the sequencing signals before and after treatment at each tRNA locus (Figure 4A). In addition, tModBase provides an analysis tool, Mod2Compare, for comparing any two epitranscriptome sequencing signals (Figure 4B). In Mod2Compare, the user can select any two datasets for comparison to obtain a downloadable table showing the misincorporation signal values detected in a common or unique part of the two datasets. In addition, mod2Compare provides an upload function, and the user can not only compare any two datasets contained in tModBase but can also upload a new dataset. With the two functions above, users can employ the first function to download the intersection or difference between any two datasets and then use the second function to reupload the results to a new dataset and compare it with a third dataset. Even more custom permutations can be performed by the user in a comparative manner.
Figure 4.

Comparison of modification profiles between different sequencing sets. (A) Distribution of misincorporation signal ratios between chemically treated and control groups. The x-axis represents each position of a tRNA molecule, and the y-axis represents the ratio of the misincorporation signal between chemically treated and control samples. (B) Scatter plot comparing the two datasets. The x- and y-axes represent the full range of misincorporation signal values in the two datasets selected by the user, with each point representing a site on a tRNA molecule.
Association between tRNA modification and tsRNA biogenesis
tRNA modifications affect the structure of the tRNA and the degree of resistance to nucleases at specific sites, which in turn triggers tsRNA processing (63). We summarized a list of the relationships between tRNA modifications and tsRNA processing positions reported in the literature, which lists tsRNA types, modification sites, modification enzymes, and the corresponding mechanisms of action. Our previous study revealed that tsRNAs are abnormally expressed in a variety of cancers, which is associated with patient survival and prognosis (64). To further clarify the relationship between tsRNA occurrence and tRNA modifications, we analysed the biogenesis sites of tsRNAs in 32 cancers and mapped them to known tRNA modification sites. Since modification changes are affected by modification enzymes, we also displayed the changes in various modification enzymes in 32 cancers on the Enzyme page. Additionally, we provide a graph comparing tRNA expression levels between tumour and normal tissues on the tRNA page. Researchers can screen modification enzymes and tsRNA molecules that undergo synergistic changes in tModBase, and then design experiments to deeply explore the mechanisms of abnormal tsRNA levels in cancer.
Diseases caused by tRNA modification
Disturbances in tRNA modification can cause serious diseases. However, the types of diseases identified thus far are still very limited. The relationships between reported tRNA modifications and four major disease types (mitochondrial disease, neurological disorder, cancer, and diabetes) are summarized in tModBase. The Mod2Disease page details disease names, affected genes, types of RNA modifications, and tRNA molecules. On this basis, we annotated whether the phenotype is caused by the influence of tRNA modification enzymes or the modifications themselves in detail and briefly summarized the corresponding pathogenic mechanisms (Supplementary Table S2). In addition, we analysed the relationships between tRNA modification sites and DNA variation sites. The tRNA molecules, modification sites, and modification types and DNA variation sites and variation types are annotated in detail. In the future, additional diseases caused by tRNA modification changes can be identified based on tModBase, and their pathogenic mechanisms can be further analysed.
DISCUSSION AND CONCLUSIONS
Unlike mRNA modifications, modifications on tRNA molecules are abundant and densely distributed. Traditional biochemical methods can identify the types of modifications present on tRNA molecules but cannot perform large-scale batch detection, let alone distinguish multiple tRNA isotypes. In recent years, a series of high-resolution experimental detection methods and bioinformatics analysis procedures focused on tRNA modifications and quantification have been developed. This study integrates and analyses currently reported tRNA epitranscriptome sequencing datasets and demonstrates the misincorporation and termination signals generated by these sequencing technologies in detail (Figure 5). Additional tRNA molecules likely to contain modifications were identified. Based on the identified misincorporation signals, users can design experiments to further discover new tRNA-modifying molecules and even rare types of modifications. To avoid the problem of false-positives caused by the use of a single dataset, tModBase provides the Mod2Compare tool to compare the differences in the identification signals of the two datasets. It is recommended that researchers use the result obtained from the intersection of multiple datasets as a more credible modification signal for validation.
Figure 5.

Major contents of tModBase.
Many modifications can cause misincorporation or termination in the RT process, causing the site to generate a detectable signal suggesting that the site is a possible modification site. However, relying on such a signal alone may produce a large number of false-positives, as unmodified bases may also cause misincorporation or termination during the RT process. This is one of the issues that tModBase is trying to address, that is, to provide cross-validation results for multiple datasets to increase accuracy. In fact, there may be multiple reasons for misincorporation or termination, including the library construction method, experimental technique, base type, modification type, tRNA structure, RT enzyme characteristics, or sequencing errors involved. To reflect this information comprehensively, we built a summary table on the position page that shows how many datasets contained a signal at a site, the base type at the site, whether previous studies reported the presence of modifications at the site, the reverse transcriptase used for sequencing, etc. Users can determine the confidence of a modification site based on these features. In the future, researchers can train machine learning models to make more accurate predictions of tRNA modification sites based on the above information provided by tModBase as features.
In addition, many modification types can lead to incomplete reverse transcription of tRNA and, thus, the lose of this part of the signal, which is also a very important issue. Fortunately, the tRNA sequencing technology collected in tModBase has been designed to address this issue during the library construction process. The basic idea includes three key points: (i) A 3′ adapter is ligated to tRNA 3′ ends. The adapter-ligated tRNA pools are used as templates for primer-dependent cDNA synthesis by TGIRT. (ii) The cDNA is circularized with CircLigase to provide a template for library construction by PCR. (iii) The previously ligated adapter is cleaved using a restriction endonuclease, thus linearizing the circularized RNA again while ensuring that both ends of the target are successfully ligated with the adapter sequences. Hence, the truncated cDNA resulting from the RT stop can also be detected in these libraries. We have summarized a table of the datasets used in tmodBase, describing the library construction strategy for the sequencing data and the reverse transcriptase (RTase) that is used. In the future, researchers can train machine learning models to make more accurate predictions of tRNA modification sites based on the above information provided by tModBase as features.
Using tModBase, researchers can intuitively observe the signal preferences at known modification sites and correlate them with the corresponding modification enzyme signatures. For example, TGIRT, a reverse transcriptase, can cause an RT misincorporation signal of A to T or G at m1A (determined by the structural properties of the enzyme itself) (62). By summarizing the properties of different reverse transcriptases, researchers can further apply them for the identification of tRNA modification types.
tRNA modifications change dynamically under different conditions, and earlier studies have detected the tRNA modification changes during cellular changes under stress conditions (imposed by temperature, chemical reagent treatment, etc.) using LC–MS/MS techniques (Supplementary Table S3) (9–11,13–15,65,66). Recent studies using HTS techniques have identified specific tRNA modifications in human and mouse cells that are significantly altered under pathogenic infection (67,68) and during T cell activation (32). This evidence implies that dynamic changes in tRNA modifications may be a general response of cells to stress conditions; however, the modifications that can be detected by LC–MS/MS methods cannot clarify the specific tRNA isotypes, and the data obtained from HTS are currently far from sufficient. tModBase will be updated continuously to collect and demonstrate the dynamic changes in tRNA modifications. In the future, we will continue to collect more dynamic modification data generated by HTS and LC–MS/MS methods and add them to tModBase. In addition, we provide a user-submitted data function that allows researchers to upload these data into tModBase if relevant work is published, and we believe that having such a dedicated resource will help advance the field.
The identification and quantification of tRNA modifications are ultimately aimed at deciphering their mechanisms and functions, but research in this area is just emerging (69). In addition to collecting published reports, tModBase also provides tools such as Enzyme and Mod2tsRNA to correlate tRNA modification with changes in modification enzyme and tsRNA. In the future, researchers can integrate and analyse multiple factors to establish a regulatory network of tRNA modifications, RNA modification enzymes, and tsRNA levels to understand the molecular mechanisms that affect cell function and fate.
Compared with other tRNA and RNA modification databases focused mainly on tRNA sequences or modifications of other RNA molecules, tModBase has some distinct, important advantages (Figure 5). The advances in tModBase are as follows: (i) tModBase records 27 types of modifications, including m7G, t6A, Ψ and m1A, at each site in human and mouse tRNA molecules. (ii) We collected 103 datasets from 11 types of high-throughput tRNA sequencing technologies (Supplementary Table S1) and customized the data according to the applied library construction strategies. The signal of each sequencing technology and its dynamics under different conditions are demonstrated. In addition, tModBase provides tools for cross-sectional comparison between different sequencing results. (iii) tModBase matches the modification at each tRNA site to the expression of the enzyme that mediates that modification and shows the dynamics of these modification enzymes in 32 types of cancers. (iv) tModBase shows the overlap of tRNA modification sites with DNA variations, including SNVs, insertions, and deletions. Users can explore the variation sites corresponding to the modification sites in any tRNA and the associated diseases. (v) tModBase demonstrates the relationship between tRNA modification sites and the production positions of tRNA-derived small RNAs (tsRNAs). We provide the Mod2tsRNA tool to analyse the expression levels of tsRNA near tRNA modification sites, which helped to reveal the relationships between tRNA modifications and tsRNA production mechanisms. In addition, tModBase demonstrates the relationships between known tRNA modification sites and tsRNA processing from the published literature. (vi) tModBase includes four types of human diseases caused by aberrant tRNA modifications, including mitochondrial diseases, neurological disorders, cancers, and diabetes, and their associated tRNA molecules, modification types, and modification enzymes. (vii) The analysis results and display charts of all high-throughput sequencing data in tModBase are available for download so that users can conduct in-depth analysis and research based on tModBase.
DATA AVAILABILITY
tModBase is freely available at https://www.tmodbase.com/.
Supplementary Material
ACKNOWLEDGEMENTS
The results shown here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. We gratefully acknowledge contributions from the TCGA Research Network and its TCGA Pan-Cancer Analysis Working Group. We sincerely thank Guanzheng Luo and Zhang Zhang of Sun Yat-sen University for their valuable comments during the article revision process.
Contributor Information
Hao-Tian Lei, MOE Key Laboratory of Gene Function and Regulation, State Key Laboratory of Biocontrol, School of Life Sciences, Sun Yat-sen University, Guangzhou 510275, P.R. China.
Zhang-Hao Wang, Division of Biosciences, University College London, London WC1E 6BT, UK.
Bin Li, MOE Key Laboratory of Gene Function and Regulation, State Key Laboratory of Biocontrol, School of Life Sciences, Sun Yat-sen University, Guangzhou 510275, P.R. China.
Yang Sun, MOE Key Laboratory of Gene Function and Regulation, State Key Laboratory of Biocontrol, School of Life Sciences, Sun Yat-sen University, Guangzhou 510275, P.R. China.
Shi-Qiang Mei, MOE Key Laboratory of Gene Function and Regulation, State Key Laboratory of Biocontrol, School of Life Sciences, Sun Yat-sen University, Guangzhou 510275, P.R. China.
Jian-Hua Yang, MOE Key Laboratory of Gene Function and Regulation, State Key Laboratory of Biocontrol, School of Life Sciences, Sun Yat-sen University, Guangzhou 510275, P.R. China.
Liang-Hu Qu, MOE Key Laboratory of Gene Function and Regulation, State Key Laboratory of Biocontrol, School of Life Sciences, Sun Yat-sen University, Guangzhou 510275, P.R. China.
Ling-Ling Zheng, MOE Key Laboratory of Gene Function and Regulation, State Key Laboratory of Biocontrol, School of Life Sciences, Sun Yat-sen University, Guangzhou 510275, P.R. China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key R&D Program of China [2022YFC3400400]; National Natural Science Foundation of China [32270604, 31900903]; Guangdong Province [2021A1515010542, 2022A1515011321]; Guangdong Province Key Laboratory of Computational Science; Guangdong Province Computational Science Innovative Research Team (in part). Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Rios A.C., Tor Y.. On the origin of the canonical nucleobases: an assessment of selection pressures across chemical and early biological evolution. Isr. J. Chem. 2013; 53:469–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schneider C., Becker S., Okamura H., Crisp A., Amatov T., Stadlmeier M., Carell T.. Noncanonical RNA nucleosides as molecular fossils of an early earth-generation by prebiotic methylations and carbamoylations. Angew. Chem. Int. Ed. 2018; 57:5943–5946. [DOI] [PubMed] [Google Scholar]
- 3. Müller F., Escobar L., Xu F., Węgrzyn E., Nainytė M., Amatov T., Chan C.Y., Pichler A., Carell T.. A prebiotically plausible scenario of an RNA–peptide world. Nature. 2022; 605:279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Macé K., Gillet R.. Origins of tmRNA: the missing link in the birth of protein synthesis?. Nucleic Acids Res. 2016; 44:8041–8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de Farias S.T., Jose M.V.. Transfer RNA: the molecular demiurge in the origin of biological systems. Prog. Biophys. Mol. Biol. 2020; 153:28–34. [DOI] [PubMed] [Google Scholar]
- 6. Goodenbour J.M., Pan T.. Diversity of tRNA genes in eukaryotes. Nucleic Acids Res. 2006; 34:6137–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Behrens A., Rodschinka G., Nedialkova D.D.. High-resolution quantitative profiling of tRNA abundance and modification status in eukaryotes by mim-tRNAseq. Mol. Cell. 2021; 81:1802–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pan T. Modifications and functional genomics of human transfer RNA. Cell Res. 2018; 28:395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Begley U., Dyavaiah M., Patil A., Rooney J.P., Direnzo D., Young C.M., Conklin D.S., Zitomer R.S., Begley T.J.. Trm9-Catalyzed tRNA modifications link translation to the DNA damage response. Mol. Cell. 2007; 28:860–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chan C.T.Y., Pang Y.L.J., Deng W., Babu I.R., Dyavaiah M., Begley T.J., Dedon P.C.. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat. Commun. 2012; 3:937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chan C.T.Y., Dyavaiah M., Demott M.S., Taghizadeh K., Dedon P.C., Begley T.J.. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 2010; 6:e1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Patil A., Dyavaiah M., Joseph F., Rooney J.P., Chan C.T.Y., Dedon P.C., Begley T.J.. Increased tRNA modification and gene-specific codon usage regulate cell cycle progression during the DNA damage response. Cell Cycle. 2012; 11:3656–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chionh Y.H., McBee M., Babu I.R., Hia F., Lin W., Zhao W., Cao J., Dziergowska A., Malkiewicz A., Begley T.J.et al.. tRNA-mediated codon-biased translation in mycobacterial hypoxic persistence. Nat. Commun. 2016; 7:13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chan C.T.Y., Deng W., Li F., Demott M.S., Babu I.R., Begley T.J., Dedon P.C.. Highly predictive reprogramming of tRNA modifications is linked to selective expression of codon-biased genes. Chem. Res. Toxicol. 2015; 28:978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Noon K.R., Guymon R., Crain P.F., McCloskey J.A., Thomm M., Lim J., Cavicchioli R.. Influence of temperature on tRNA modification in Archaea: Methanococcoides burtonii (Optimum growth temperature [Topt], 23°C) and Stetteria hydrogenophi. J. Bacteriol. 2003; 185:5483–5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dixit S., Kessler A.C., Henderson J., Pan X., Zhao R., D’Almeida GabrielS., Kulkarni S., Rubio Mary, Anne T., Hegedűsová E.et al.. Dynamic queuosine changes in tRNA couple nutrient levels to codon choice in trypanosoma brucei. Nucleic Acids Res. 2021; 49:12986–12999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang W., Foo M., Eren A.M., Pan T.. tRNA modification dynamics from individual organisms to metaepitranscriptomics of microbiomes. Mol. Cell. 2022; 82:891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jackman J.E., Montange R.K., Malik H.S., Phizicky E.M.. Identification of the yeast gene encoding the tRNA m1G methyltransferase responsible for modification at position 9. RNA. 2003; 9:574–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schmidt P.G., Sierzputowska-Gracz H., Agris P.F.. Internal motions in yeast phenylalanine transfer RNA from carbon-13 NMR relaxation rates of modified base methyl groups: a model-free approach. Biochemistry. 1987; 26:8529–8534. [DOI] [PubMed] [Google Scholar]
- 20. Fleming B.A., Blango M.G., Rousek A.A., Kincannon W.M., Tran A., Lewis AdamJ., Russell ColinW., Zhou Q., Baird L.M., Barber AmeliaE.et al.. A tRNA modifying enzyme as a tunable regulatory nexus for bacterial stress responses and virulence. Nucleic Acids Res. 2022; 50:7570–7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cantara W.A., Bilbille Y., Kim J., Kaiser R., Leszczynska G., Malkiewicz A., Agris P.F.. Modifications modulate anticodon loop dynamics and codon recognition of E. coli tRNA(Arg1,2). J. Mol. Biol. 2012; 416:579–597. [DOI] [PubMed] [Google Scholar]
- 22. Pereira M., Ribeiro D.R., Pinheiro M.M., Ferreira M., Kellner S., Soares A.R.. m5U54 tRNA hypomodification by lack of TRMT2A drives the generation of tRNA-Derived small RNAs. Int. J. Mol. Sci. 2021; 22:2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schaefer M., Pollex T., Hanna K., Tuorto F., Meusburger M., Helm M., Lyko F.. RNA methylation by dnmt2 protects transfer RNAs against stress-induced cleavage. Genes & Development. 2010; 24:1590–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Torres A.G., Reina O., Stephan-Otto Attolini C., Ribas De Pouplana L.. Differential expression of human tRNA genes drives the abundance of tRNA-derived fragments. Proc. Natl Acad. Sci. U.S.A. 2019; 116:8451–8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Suzuki T. The expanding world of tRNA modifications and their disease relevance. Nat. Rev. Mol. Cell Biol. 2021; 22:375–392. [DOI] [PubMed] [Google Scholar]
- 26. Chan P.P., Lowe T.M.. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016; 44:D184–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sajek M.P., Woźniak T., Sprinzl M., Jaruzelska J., Barciszewski J.. T-psi-C: user friendly database of tRNA sequences and structures. Nucleic Acids Res. 2019; 48:D256–D260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Boccaletto P., Stefaniak F., Ray A., Cappannini A., Mukherjee S., Purta E., Kurkowska M., Shirvanizadeh N., Destefanis E., Groza P.et al.. MODOMICS: a database of RNA modification pathways. 2021 update. Nucleic Acids Res. 2022; 50:D231–D235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xuan J.-J., Sun W.-J., Lin P.-H., Zhou K.-R., Liu S., Zheng L.-L., Qu L.-H., Yang J.-H.. RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2017; 46:D327–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shigematsu M., Honda S., Loher P., Telonis A.G., Rigoutsos I., Kirino Y.. YAMAT-seq: an efficient method for high-throughput sequencing of mature transfer RNAs. Nucleic Acids Res. 2017; 45:e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hu J.F., Yim D., Ma D., Huber S.M., Davis N., Bacusmo J.M., Vermeulen S., Zhou J., Begley T.J., Demott M.S.et al.. Quantitative mapping of the cellular small RNA landscape with AQRNA-seq. Nat. Biotechnol. 2021; 39:978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rak R., Polonsky M., Eizenberg-Magar I., Mo Y., Sakaguchi Y., Mizrahi O., Nachshon A., Reich-Zeliger S., Stern-Ginossar N., Dahan O.et al.. Dynamic changes in tRNA modifications and abundance during T cell activation. Proc. Natl Acad. Sci. U.S.A. 2021; 118:e2106556118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carlile T.M., Rojas-Duran M.F., Zinshteyn B., Shin H., Bartoli K.M., Gilbert W.V.. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature. 2014; 515:143–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thomas N.K., Poodari V.C., Jain M., Olsen H.E., Akeson M., Abu-Shumays R.L.. Direct nanopore sequencing of individual full length tRNA strands. ACS Nano. 2021; 15:16642–16653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yuan X., Su Y., Zhang X., Turkel S., Shi S., Wang X., Choi E.-J., Wu W., Liu H., Viner R.et al.. MLC seq: de novo sequencing of full-length tRNA isoforms by mass ladder complementation. 2021; bioRxiv doi:23 May 2021, preprint: not peer reviewed 10.1101/2021.05.22.445286. [DOI]
- 36. Workman R.E., Tang A.D., Tang P.S., Jain M., Tyson J.R., Razaghi R., Zuzarte P.C., Gilpatrick T., Payne A., Quick J.et al.. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat. Methods. 2019; 16:1297–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garalde D.R., Snell E.A., Jachimowicz D., Sipos B., Lloyd J.H., Bruce M., Pantic N., Admassu T., James P., Warland A.et al.. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods. 2018; 15:201–206. [DOI] [PubMed] [Google Scholar]
- 38. Jain M., Fiddes I.T., Miga K.H., Olsen H.E., Paten B., Akeson M.. Improved data analysis for the MinION nanopore sequencer. Nat. Methods. 2015; 12:351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cozen A.E., Quartley E., Holmes A.D., Hrabeta-Robinson E., Phizicky E.M., Lowe T.M.. ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat. Methods. 2015; 12:879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zheng G., Qin Y., Clark W.C., Dai Q., Yi C., He C., Lambowitz A.M., Pan T.. Efficient and quantitative high-throughput tRNA sequencing. Nat. Methods. 2015; 12:835–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Khoddami V., Yerra A., Mosbruger T.L., Fleming A.M., Burrows C.J., Cairns B.R.. Transcriptome-wide profiling of multiple RNA modifications simultaneously at single-base resolution. Proc. Natl Acad. Sci. U.S.A. 2019; 116:6784–6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Squires J.E., Patel H.R., Nousch M., Sibbritt T., Humphreys D.T., Parker B.J., Suter C.M., Preiss T.. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012; 40:5023–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schaefer M., Pollex T., Hanna K., Lyko F.. RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res. 2008; 37:e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gu W., Hurto R.L., Hopper A.K., Grayhack E.J., Phizicky E.M.. Depletion of Saccharomyces cerevisiae tRNAHis Guanylyltransferase Thg1p leads to uncharged tRNAHis with additional m5C. Mol. Cell. Biol. 2005; 25:8191–8201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tuorto F., Liebers R., Musch T., Schaefer M., Hofmann S., Kellner S., Frye M., Helm M., Stoecklin G., Lyko F.. RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat. Struct. Mol. Biol. 2012; 19:900–905. [DOI] [PubMed] [Google Scholar]
- 46. Jühling F., Mörl M., Hartmann R.K., Sprinzl M., Stadler P.F., Pütz J.. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2008; 37:D159–D162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Edgar R. Gene expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002; 30:207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Smigielski E.M. dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res. 2000; 28:352–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Boratyn G.M., Camacho C., Cooper P.S., Coulouris G., Fong A., Ma N., Madden T.L., Matten W.T., McGinnis S.D., Merezhuk Y.et al.. BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 2013; 41:W29–W33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Suzuki T., Yashiro Y., Kikuchi I., Ishigami Y., Saito H., Matsuzawa I., Okada S., Mito M., Iwasaki S., Ma D.et al.. Complete chemical structures of human mitochondrial tRNAs. Nat. Commun. 2020; 11:4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kechin A., Boyarskikh U., Kel A., Filipenko M.. cutPrimers: a new tool for accurate cutting of primers from reads of targeted next generation sequencing. J. Comput. Biol. 2017; 24:1138–1143. [DOI] [PubMed] [Google Scholar]
- 53. Krueger F., Andrews S.R.. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011; 27:1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. de Crécy-Lagard V., Boccaletto P., Mangleburg C.G., Sharma P., Lowe T.M., Leidel S.A., Bujnicki J.M.. Matching tRNA modifications in humans to their known and predicted enzymes. Nucleic Acids Res. 2019; 47:2143–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pereira M., Francisco S., Varanda A., Santos M., Santos M., Soares A.. Impact of tRNA modifications and tRNA-Modifying enzymes on proteostasis and human disease. Int. J. Mol. Sci. 2018; 19:3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Suzuki T. The expanding world of tRNA modifications and their disease relevance. Nat. Rev. Mol. Cell Biol. 2021; 22:375–392. [DOI] [PubMed] [Google Scholar]
- 57. Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R.M., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M.. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013; 45:1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Telonis A.G., Loher P., Magee R., Pliatsika V., Londin E., Kirino Y., Rigoutsos I.. tRNA fragments show intertwining with mRNAs of specific repeat content and have links to disparities. Cancer Res. 2019; 79:3034–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shurtleff M.J., Yao J., Qin Y., Nottingham R.M., Temoche-Diaz M.M., Schekman R., Lambowitz A.M.. Broad role for YBX1 in defining the small noncoding RNA composition of exosomes. Proc. Natl Acad. Sci. U.S.A. 2017; 114:E8987–E8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Reinsborough C.W., Ipas H., Abell N.S., Nottingham R.M., Yao J., Devanathan S.K., Shelton S.B., Lambowitz A.M., Xhemalçe B.. BCDIN3D regulates tRNAHis 3′ fragment processing. PLoS Genet. 2019; 15:e1008273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li X., Xiong X., Zhang M., Wang K., Chen Y., Zhou J., Mao Y., Lv J., Yi D., Chen X.W.et al.. Base-Resolution mapping reveals distinct m(1)A methylome in Nuclear- and Mitochondrial-Encoded transcripts. Mol. Cell. 2017; 68:993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chen Q., Zhang X., Shi J., Yan M., Zhou T.. Origins and evolving functionalities of tRNA-derived small RNAs. Trends Biochem. Sci. 2021; 46:790–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang J.-H., Chen W.-X., Mei S.-Q., Yang Y.-D., Yang J.-H., Qu L.-H., Zheng L.-L.. tsRFun: a comprehensive platform for decoding human tsRNA expression, functions and prognostic value by high-throughput small RNA-Seq and CLIP-Seq data. Nucleic Acids Res. 2022; 50:D421–D431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Endres L., Begley U., Clark R., Gu C., Dziergowska A., Małkiewicz A., Melendez J.A., Dedon P.C., Begley T.J.. Alkbh8 regulates selenocysteine-protein expression to protect against reactive oxygen species damage. PLoS One. 2015; 10:e0131335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Alings F., Sarin L.P., Fufezan C., Drexler H.C.A., Leidel S.A.. An evolutionary approach uncovers a diverse response of tRNA 2-thiolation to elevated temperatures in yeast. RNA. 2015; 21:202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jungfleisch J., Böttcher R., Talló-Parra M., Pérez-Vilaró G., Merits A., Novoa E.M., Díez J.. CHIKV infection reprograms codon optimality to favor viral RNA translation by altering the tRNA epitranscriptome. Nat. Commun. 2022; 13:4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tucker J.M., Schaller A.M., Willis I., Glaunsinger B.A.. Alteration of the premature tRNA landscape by gammaherpesvirus infection. Mbio. 2020; 11:e02664-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Phizicky E.M., Hopper A.K.. tRNA biology charges to the front. Genes Dev. 2010; 24:1832–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
tModBase is freely available at https://www.tmodbase.com/.