


Abstract
AI-driven protein structure prediction, most notably AlphaFold2 (AF2) opens new frontiers for almost all fields of structural biology. As traditional structure prediction methods for transmembrane proteins were both complicated and error prone, AF2 is a great help to the community. Complementing the relatively meager number of experimental structures, AF2 provides 3D predictions for thousands of new alpha-helical membrane proteins. However, the lack of reliable structural templates and the fact that AF2 was not trained to handle phase boundaries also necessitates a delicate assessment of structural correctness. In our new database, Transmembrane AlphaFold database (TmAlphaFold database), we apply TMDET, a simple geometry-based method to visualize the likeliest position of the membrane plane. In addition, we calculate several parameters to evaluate the location of the protein into the membrane. This also allows TmAlphaFold database to show whether the predicted 3D structure is realistic or not. The TmAlphaFold database is available at https://tmalphafold.ttk.hu/.
INTRODUCTION
The folding problem (i.e. correct prediction of the 3D structure of any protein based on their amino acid sequence alone) has been a central unresolved question for many decades in the 20th and the early 21st century. However, as our knowledge gradually expanded with the availability of more and more experimentally determined structures, new methods became available. Although we still cannot predict the structure of every possible polymer based on their monomers alone, the geometry of natural proteins can now be reasonably guessed using homology-based approaches. The application of machine learning and state-of-the-art neural nets and language models resulted in the creation of the AlphaFold2 algorithm (1), which demonstrates an unprecedented breakthrough in protein structure prediction.
Nowhere is structure prediction more welcome, than in protein groups where structural studies are cumbersome and difficult, such as membrane-embedded or transmembrane (TM) proteins. The functions of transmembrane proteins are diverse, and the majority of current therapeutic drug targets are membrane proteins. Although they constitute around 20–30% of the proteome of different organisms (2,3), experimental 3D structures for these molecules are underrepresented about tenfold compared to non-TM proteins (4), because structural studies on them are difficult and expensive. The lipid bilayer divides the molecular environment of a TM protein into three different phases (two aqueous and one lipid-filled intramembrane compartment) that the very same protein will experience, while also intersecting phase boundaries. Proper embedding of natural proteins requires a multi-step folding process (5). Membrane proteins are also radically different from their cytosolic or secreted counterparts in that they interface the outer and inner aqueous phase with a hydrophilic, but the lipid-filled membrane compartment with a hydrophobic surface at the same time. Finally, there are a lot of geometric restraints associated with the way a protein chain can traverse the membrane. The overwhelming majority of TM proteins contain highly hydrophobic alpha-helical segments, in a preferentially perpendicular orientation that roughly match the height of the complete membrane. With a relative scarcity of complete, 3D experimental templates, these physico-chemical restraints are difficult to follow by currently available, machine learning-based prediction methods (6–8).
For experimentally determined membrane protein structures, there are already two methods (TMDET (9) and PPM (10)) that can reconstruct the position of the original membrane plane for alpha-helical transmembrane proteins. Using these algorithms on purely prediction-based protein structures is fairly straightforward, and they also provide a simple geometric approach for the assessment of model quality. We can also compare these theoretical models with low-resolution global topology predictions (6–8). The latter algorithms are fairly accurate and provide an additional basis for quality assessment. We are well aware that other TM proteins also exist, such as pore-forming beta-barrels, with many examples consisting of multiple chains. As the latter lack clearly defined hydrophilic interior and hydrophobic exterior sides, they cannot be confidently identified using monomeric AF2 structures only. Beta barrels and some multimeric pore-forming toxins were therefore not included in the presented TmAlphaFold database, only genuine alpha-helical TM proteins. TmAlphaFold database not only provides an open-access resource to visualize the likely orientation of 215 844 alpha-helical TM proteins regarding the membrane plane, but also displays any structural conflicts the AlphaFold2 models might have.
DATA RESOURCES AND METHODOLOGY
Resources
The AlphaFold Protein Structure Database (AFDB) (11) was used as a source of AlphaFold2 predicted structures. We ran the CCTOP (6), TOPCONS2 (7) consensus methods and DeepTMHMM (8) algorithms on our cluster to discriminate between TM and non-TM proteins. We used CCTOP to predict the topology of TM proteins, however, we replaced the older version of signal peptide prediction with SignalP6 (12). Structures were sliced into fragments based on their Predicted Alignment Error matrix using the Agglomerative Clustering function of the Sklearn python package (https://scikit-learn.org/). Helical residues were defined using DSSP (13). The TMDET algorithm (9) was used to detect the membrane plane in the AlphaFold structures (and in their fragments).
Technical details
The web page of TmAlphaFold database was written in PHP using the Laravel framework with Livewire package, which makes working with user interactions more manageable. To visualize topology data over amino acid sequences, protvista packages were utilized from EBI web components GitHub repository (https://github.com/ebi-webcomponents/nightingale), while 3D structures were visualized using a locally modified version of Mol* (14). The modified version can show the membrane as two planes around the investigated TM protein using the results of TMDET. Sequences and topology data as well as information about protein similarities provided by BLAST (15) are stored in a MySQL database. Data can be downloaded either for each protein separately or for whole genomes as gzipped packages. For programmable access to the database, an API is also provided.
RESULTS
Data processing
We used a combination of three state-of-the-art prediction methods to select TM proteins from all protein structures deposited into AFDB. Our internal tests showed that CCTOP has the highest sensitivity, however, the intersection of the three prediction methods produced better specificity (Supplementary Tables 1 and 2). Therefore, we accepted a protein as TM if either (i) all three prediction methods detected a TM segment, or (ii) the manually curated cases where CCTOP predicted a TM segment, but TOPCONS2 or DeepTMHMM did not find membrane region(s).
In the TmAlphaFold database we reconstructed the membrane bilayer using the coordinates in the PDB files and Predicted Aligned Error files. After removing the signal peptide, we used agglomerative clustering to slice the structures into potential domains (the threshold was set to 7). We utilized the TMDET algorithm to detect the membrane bilayer in the full structure and the fragments as well. However, note that residues with low pLDDT were omitted (the default threshold was set to 70, for helical residues to 50). In most cases, the results for the fragments and the full structure were in full agreement. In case they contradicted, we created an assembly from the fragments where only compatible ones were kept (we checked if the rotations of the fragments regarding the membrane plane were similar).
By the end, we obtained multiple individual fragments, the assembled structure and the original full structure, together with the membrane plane definitions. We found that out of these three approaches, selecting the one with the most TM segments provided the most realistic model. This is intuitively easy to understand, considering that the most common error of AF2 was to randomly position non-TM segments into the membrane plane (confusing TMDET), while the most characteristic error of fragmentation was to inadvertently split TM domains. Sometimes, however, this strategy did not yield a positive outcome, as TMDET could not detect the membrane, or the structure had a lot of geometric errors (see Quality assessment). In such cases, we masked every residue that was not predicted as TM by CCTOP, and made an attempt to find the membrane plane in the resulting structure. For a more detailed description of the algorithm see Supplementary Material or visit the webpage of TmAlphaFold database.
Quality assessment
Our data processing pipeline found and reconstructed the membrane bilayer in most cases (depending on the quality of the AlphaFold2 predicted structure), however, several geometric (or topological) errors may still arise in the structures. We defined ten commonly seen problems and evaluated all the predicted structures to assess their quality. These scores are related to the quality of structures: comparing the Root Mean Square Deviation between the experimental and predicted structures (Supplementary Table 3), the higher ratio of ‘Excellent’ quality level was assigned to proteins with lower RMSD (Supplementary Figure S1). We found that signal peptides, short helical segments and low-reliability segments (or in another interpretation, intrinsically disordered regions) often fell into the membrane bilayer. The latter can produce more complicated problems, as they can also work as a linker and turn the direction of the polypeptide chain, so an ordered domain penetrates the membrane. Sometimes we detected additional membrane segments in the fragments, however, they fall outside from the membrane plane in the final result. We also compared the structure and the membrane plane to the CCTOP topology prediction result, to check if all segments were found (or if there are extra helical segments in the membrane). Using these flags, we categorize each structure as excellent, good, fair, poor or failed. Notably, these quality checks do not necessarily mean that the predicted structure is erroneous, it is up to the user to judge the structure based on the evidence. For a detailed list of all quality flags see Supplementary Material or the webpage.
Membrane orientation and quality assessment of membrane proteins from AF
The TmAlphaFold database contains 215 844 TM proteins, and in 203 077 (94.09%) the membrane bilayer is also reconstructed. In the majority of cases, the quality of the structure is excellent (45.16%) or good (21.51%). In a lower proportion of proteins, the quality was fair (25.08%) or poor (2.21%). In 12 767 proteins (6.05%), the membrane bilayer could not be reconstructed. Regarding all the sequences from the 16 model organisms, 32 global health organisms and SwissProt database, ∼22% of the proteins contained at least one TM segment. The most abundant class is bitopic proteins (32.42%), which are often receptors or responsible for cell adhesion. 7TM proteins are also abundant (8.38%), especially in mammalians where the majority of them belonging to the class of GPCRs.
Webpage of TmAlphaFold database
The home page of the TmAlphaFold database provides an easily accessible user interface to inspect the structures together with the reconstructed membrane plane and the detected errors. Users can search for proteins using their UniProt ID, UniProt Accession, Gene name or protein name. The results can be filtered based on the source organism, the quality of the structure (see Quality assessment), CCTOP topology prediction evidence level and the number of TM segments. Summary information about the protein structures is also available on the search results page.
On the protein page five panels are available. The information panel shows general information about the protein: protein name, organism, subcellular localization from UniProt (16) and a hyperlink to the AFDB. Furthermore, Q value determined by TMDET, evidence and reliability levels from CCTOP are all displayed. On the TM panel, the proposed topography of the protein is shown (considering TMDET result): correct TM helices are marked with yellow (re-entrant loops are orange), false positive ones are red and false negatives are blue. For a quick comparison, the CCTOP prediction result, and—if available—related structures from PDBTM (4) are also shown. Last, but not least, fragments generated by agglomerative clustering from PAE matrices (see Methods) and their membrane segments are also displayed. The ‘3D’ tab shows the AlphaFold2 structure together with the reconstructed membrane plane. Geometric errors detected from multiple quality steps are also marked (with the same color-coding as on the topography panel). On the evaluation panel, the results of quality checks are indicated. On the download tab, we provide the rotated structure (so the Z axis is perpendicular to the membrane plane), the TMDET result file, the evaluation result and the CCTOP prediction results. On the download page, all data bundled by proteomes can also be downloaded as a single compressed file.
The statistics page can be used to quickly access basic statistics of TmAlphaFold database (number of entries categorized by evaluation results, by evidence, by the number of transmembrane segments and by species). Users can also generate custom charts using any combination of the following information: quality of the structure, organisms, number of TM segments and CCTOP evidence level. Multiple charts can be added to a single page, and the link can be saved, bookmarked for later use, or shared.
DISCUSSION
Case studies
The strength of TmAlphaFold database is the various filtering techniques, that can help to highlight the potential membrane domain. Without these steps traditional algorithms designed to find the membrane plane would fail, as other segments often penetrate the lipid bilayer. However, establishing the membrane plane is by no means straightforward in many cases, as the following examples show:
ASAH_HUMAN (Q9NR71) is a ceramidase anchored into the plasma membrane. This bitopic, type II transmembrane protein has a disordered region that connects the ceramidase domain with the TM segment. AF2 correctly predicts each segment (considering the observation that low pLDDT values correlate with intrinsic disorder (17)), however, the orientation of these segments relative to each other is problematic, as the flexible linker turns back the polypeptide chain and places the domain next to the TM region. In this case, by slicing the structure into fragments based on the Predicted Alignment Error matrix, the membrane region can be correctly identified (Figure 1A).
Figure 1.
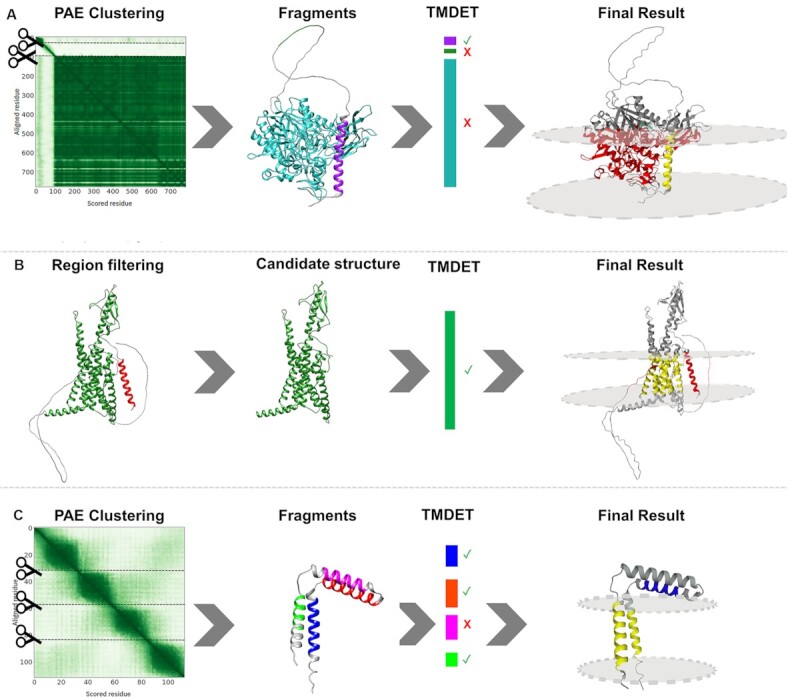
(A) ASAH_HUMAN (Q9NR71) Left to right: The structure was sliced into potential domains based on the Predicted Alignment Error matrix; The structure fell apart into three fragments: 2 domains (purple and cyan) and a flexible linker (green); TMDET detected the membrane plane in the helical domain (purple); The final result shows the TM helix (yellow), however, the other globular fragment is not compatible with the membrane plane (red). (B) PTH2R_HUMAN (P49190) Left to right: signal peptide (red) and low reliability regions (grey) are detected; These regions might penetrate the membrane, therefore they are masked out; TMDET detects the membrane plane in the structure; The final result shows the TM domain (yellow), while masked out segments embedded in the membrane are highlighted (red). (C) TM14C_HUMAN (Q9P0S9) Left to right: The structure was sliced into potential domains based on the Predicted Alignment Error matrix; The structure fell apart into four helical fragments (blue, orange, purple and green); TMDET detected membrane helices in three fragments; The membrane plane cannot be positioned in a way that all three helices are included. Green and blue fragments are in the membrane plane (yellow), the extra TM segment is highlighted (blue). Notably, in comparison with the experimental structure (2los_A), the orientation of the N-terminal TM helix is wrong, one TM segment is erroneously predicted to lie parallelly to the membrane plane.
PTH2R_HUMAN (P49190) is a GPCR-superfamily hormone receptor with seven transmembrane helices. The protein also has an N-terminal signal peptide that is cleaved in the mature protein, although AF2 folds this stretch into the membrane domain. The C-terminal segment is predicted to be disordered (18), however, this region is also (erroneously) positioned inside the proposed membrane layer according to AF2. By filtering these segments out, the correct membrane bilayer can be reconstructed (19) (Figure 1B).
TM14C_HUMAN (Q9P0S9) is a transmembrane protein, probably involved in heme biosynthesis. AF2 predicts 4 alpha-helix in the protein, two pairs from which both could define the membrane plane. Considering the NMR structure of the protein (20), AF2 correctly identifies all secondary structure, however, their orientation relative to each other is wrong—the middle TM helix is predicted parallel to the membrane next to the interface helix, and therefore the orientation of the N-terminal TM helix compared to the C-terminal one is wrong. NMR structures were not used to train AF2, yet it was shown that DeepMind's algorithm is still accurate on them (21)—with a few exceptions (Figure 1C).
Limitations
When constructing the TmAlphaFold database, we heavily relied on the TMDET software to detect the membrane plane. TMDET is also routinely used for maintaining the PDBTM database. However, it has a very high sensitivity with modest specificity, thus manual curation is also required. While the weekly update for PDB can be handled easily, the high number of structures from AFDB, that need to be checked is several orders of magnitude higher. Therefore, we decided to use sequence-based filtering of TM proteins. TM filtering capacity and specificity of such methods is high, yielding only a handful of false positives on our benchmarking test. However, when processing around 1 million sequences the number of false positive predictions is expected to be in the range of thousands. A few days before the manuscript submission, a new language model-based TM protein filtering and topology prediction algorithm was released (TMbed) (22). Although it can handle a huge number of sequences in a relatively short period of time, this will only be implemented in the next version of the database.
One limitation of our method (and in general, AFDB), is that it does not consider the quaternary structure of proteins. A high fraction of membrane proteins forms oligomer structures, however, by predicting them in monomer form both TMDET and even the AlphaFold2 algorithm might fail. Furthermore, when detecting the membrane plane, TMDET calculates the solvent accessible surface areas, which might be different for monomer and multimer versions of the proteins. Another problem arises from dynamically folded structures, such as pore-forming toxins. Unfortunately, some proteins have more than one fundamentally different, stable 3D structures, where one is cytoplasmic and the other one is membrane inserted. In these cases, AlphaFold2 tends to predict the soluble, non-membrane form. Therefore, we generally avoided the inclusion of these proteins as TM, even if a structurally unmodeled TM form might exist.
AlphaFold2 often aims to produce compact 3D structures, that may lead to fold additional elements to the membrane domain. For example, OR1L1_HUMAN (Q8NH94) is an olfactory receptor, with seven TM regions. AF2 places an extra helical segment next to the transmembrane segments (Supplementary Figure S2a). Since there is no geometrical violation, this is not detected during the quality check as a problem. Notably, according to UniProt it is uncertain that the initiating Methionine can be found at position 1 or 51 (https://www.uniprot.org/uniprotkb/Q8NH94/entry). Taking this initiation site ambiguity into account, for the shorter protein, AF2 would probably predict a correct structure.
In some other cases AlphaFold2 does not prefer the compact structures, most notably in all-alpha structures. We found several examples, when two or more transmembrane alpha helices are modeled as one long helix. The most spectacular is RCF1_YEAST (Q03713) (Supplementary Figure S2b). This is a mitochondrial protein with an experimentally determined NMR structure (5nv8) that shows five TM helices, bringing another example where structural problems raises when no x-ray structure is available. Although correctly recognizing an alpha-helix rich structure, AF2 only positions two TM helices into positions that overlap reasonably with that of the experimental structure. The rest are left as dangling outside the membrane plane, and hence they cannot be recognized as being TM by the TMDET algorithm. Non-compact structures may be also produced when there is a high fraction of low pLDDT (probably disordered) residues in the structure. E9AGZ2_LEIIN (E9AGZ2) is probably a type I TM protein from Leishmania pathogen, where all topology prediction algorithms agree in the TM segment around the 220–240 segment (Supplementary Figure S2c). The protein has a very highly disordered extracellular part with potential amyloid-forming segments judged based on the sequence. AF2 cannot really handle the structure, long disordered regions connecting alpha-helical segments that are loosely placed within the membrane plane.
Naturally, AF2 is not equipped to deal with structures that has no reliable experimental template for (divergent domains or understudied protein structures). As an example, CLCL1_HUMAN (Q8IZS7) encodes a fast-evolving immune receptor lectin (23) with orthologs found in most other mammals: however, due to its unexpectedly high rate of amino acid exchange, AF2 fails to confidently predict its initial segment as a signal-anchor (TM segment). This example illustrates that in the case of divergent sequences, AF2 can also be unreliable at finding TM helices (Supplemantary Figure S2d).
We are well aware that even predominantly alpha-helical membrane proteins can sometimes have non-helical segments also inserted into the membrane. Most of these segments are just re-entrant loops, but fully transiting segments are not unheard of. They most commonly occur at the core of multi-pass TM proteins, where - shielded from the environment—arbitrary folds can occur, including even beta-sheets. ABCB_ASPFU (Q4WT65) is an ATP-binding cassette transporter, providing an example where most of the TM domain is folded correctly, except one TM helix (902–922)—according to the Predicted Alignment Error matrix this region could not be reliably aligned to the rest of the TM domain—our fragmentation also handles it as a separate entity (Supplementary Figure S2e). Notably, OmegaFold, a newly developed alignment-free folding method (https://github.com/HeliXonProtein/OmegaFold) predicts this region as a regular alpha-helix. This example suggests that the alignment sometimes struggles on TM regions, advocating the necessity of membrane protein specific alignment algorithms or the utilization of alignment free algorithms. Further examples, where TM regions were predicted to have non-helical secondary structure include the Escherichia coli urea permease URAA_ECOLI (P0AGM7, Supplementary Figure S2f), the cyanobacterial bicarbonate transporter BICA_SYNY3 (Q55415, Supplementary Figure S2g) or the human voltage sensor prestin S26A5_HUMAN (P58743, Supplementary Figure S2h) (24–26). These rare, unusual TM segments (that sometimes might represent genuine, experimental structures) will be labeled as ‘non-transmembrane segment in membrane’ under the current algorithm, thus critical assessment is advised for all users.
Finally, AF2 is not equipped to deal with structures it has barely any template for, such as stand-alone TM segments (this is a recurring problem for single-pass membrane proteins). Therefore, clashes with intra- or extracellular domains or weakly predicted helices are very common for single-pass TM proteins. This problem is compounded for TM segments with an unusual amino acid composition (such as Gly- or Ala- rich TM segments), where even the geometry prediction of AF2 can fail, giving non-helical conformations. Thus, even if the segment in question is unequivocally identified as a TM segment (and not a signal peptide) by other methods, it might still not be detectable on the 3D folded structure. On the other hand, these issues are encountered with multi-pass TM proteins less commonly.
Comparison with other resources
During the construction of our database, we noted that for many of the TM proteins, predicted 3D structures are also available from Membranome 3.0 (27). This database only focuses on single-pass membrane proteins but processes them very differently from our methods. Unlike Membranome 3.0, we did not aim to ‘repair’ the AF2-predicted membrane proteins by fragmenting and reorienting them. TmAlphaFold database only applies the membrane segment prediction on intact, original AF2 output structures, and indicate slight errors without the explicit aim of ‘repairing’ faulty geometries. This process can be dangerous if the protein is not proven to be a membrane protein with all certainty. On the other hand, our resource is aimed to include all TM proteins, hence providing a much higher coverage for proteomes than Membranome 3.0.
Our method found 87% of the 5758 TM helices that were listed in Membranome 3.0, reflecting a good agreement between the two databases. A few positive cases showed the presence of more than one TM helices and multi-pass topology for the protein. When our algorithm was not able to find the TM helix described in Membranome 3.0, it was not possible to define the membrane plane. One of the most significant reasons for failed TM identifications is the overprediction of signal peptides, hindered the erroneous detection of non-cleavable signal anchors (e.g. in a lot of Golgi-resident glycosyl transferases).
FURTHER DIRECTIONS
The current version of the TmAlphaFold database is based on the v3 release of the AFDB. We plan to regularly update the database upon changes in AFDB. During the development of the current version, a new release (v4) of AFDB became available, increasing the number of structures from ∼1 million to ∼200 million (https://github.com/deepmind/alphafold). Although this change is not yet reflected in the current version of TmAlphaFold database, we plan to keep adding structures to precisely reflect the membrane world of predicted AF2 structures.
DATA AVAILABILITY
The TmAlphaFold database is available at https://tmalphafold.ttk.hu/.
Supplementary Material
Contributor Information
Laszlo Dobson, Protein Bioinformatics Research Group, Institute of Enzymology, Research Centre for Natural Sciences, Magyar Tudósok körútja 2, H-1117 Budapest, Hungary; Structural and Computational Biology Unit, European Molecular Biology Laboratory, Meyerhofstraße 1, 69117 Heidelberg, Germany.
Levente I Szekeres, Protein Bioinformatics Research Group, Institute of Enzymology, Research Centre for Natural Sciences, Magyar Tudósok körútja 2, H-1117 Budapest, Hungary.
Csongor Gerdán, Protein Bioinformatics Research Group, Institute of Enzymology, Research Centre for Natural Sciences, Magyar Tudósok körútja 2, H-1117 Budapest, Hungary.
Tamás Langó, Protein Bioinformatics Research Group, Institute of Enzymology, Research Centre for Natural Sciences, Magyar Tudósok körútja 2, H-1117 Budapest, Hungary.
András Zeke, Protein Bioinformatics Research Group, Institute of Enzymology, Research Centre for Natural Sciences, Magyar Tudósok körútja 2, H-1117 Budapest, Hungary.
Gábor E Tusnády, Protein Bioinformatics Research Group, Institute of Enzymology, Research Centre for Natural Sciences, Magyar Tudósok körútja 2, H-1117 Budapest, Hungary.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 101028908; Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund [K132522]. Funding for open access charge: Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund [K132522].
Conflict of interest statement. None declared.
REFERENCES
- 1. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A.et al.. Highly accurate protein structure prediction with AlphaFold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Käll L., Sonnhammer E.L.L.. Reliability of transmembrane predictions in whole-genome data. FEBS Lett. 2002; 532:415–418. [DOI] [PubMed] [Google Scholar]
- 3. Dobson L., Reményi I., Tusnády G.E.. The human transmembrane proteome. Biol. Direct. 2015; 10:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kozma D., Simon I., Tusnády G.E.. PDBTM: protein data bank of transmembrane proteins after 8 years. Nucleic Acids Res. 2013; 41:D524–D529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bowie J.U. Solving the membrane protein folding problem. Nature. 2005; 438:581–589. [DOI] [PubMed] [Google Scholar]
- 6. Dobson L., Reményi I., Tusnády G.E.. CCTOP: a consensus constrained TOPology prediction web server. Nucleic Acids Res. 2015; 43:W408–W412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tsirigos K.D., Peters C., Shu N., Käll L., Elofsson A.. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 2015; 43:W401–W407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hallgren J., Tsirigos K.D., Pedersen M.D., Armenteros J.J.A., Marcatili P., Nielsen H., Krogh A., Winther O.. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. 2022; bioRxiv doi:10 April 2022, preprint: not peer reviewed 10.1101/2022.04.08.487609. [DOI]
- 9. Tusnády G.E., Dosztányi Z., Simon I.. TMDET: web server for detecting transmembrane regions of proteins by using their 3D coordinates. Bioinformatics. 2005; 21:1276–1277. [DOI] [PubMed] [Google Scholar]
- 10. Lomize M.A., Pogozheva I.D., Joo H., Mosberg H.I., Lomize A.L.. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 2012; 40:D370–D376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Varadi M., Anyango S., Deshpande M., Nair S., Natassia C., Yordanova G., Yuan D., Stroe O., Wood G., Laydon A.et al.. AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022; 50:D439–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Teufel F., Almagro Armenteros J.J., Johansen A.R., Gíslason M.H., Pihl S.I., Tsirigos K.D., Winther O., Brunak S., von Heijne G., Nielsen H.. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022; 40:1023–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Frishman D., Argos P.. Knowledge-based protein secondary structure assignment. Proteins. 1995; 23:566–579. [DOI] [PubMed] [Google Scholar]
- 14. Sehnal D., Bittrich S., Deshpande M., Svobodová R., Berka K., Bazgier V., Velankar S., Burley S.K., Koča J., Rose A.S.. Mol* viewer: modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021; 49:W431–W437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 16. UniProt Consortium UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019; 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ruff K.M., Pappu R.V.. AlphaFold and implications for intrinsically disordered proteins. J. Mol. Biol. 2021; 433:167208. [DOI] [PubMed] [Google Scholar]
- 18. Mészáros B., Erdos G., Dosztányi Z.. IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018; 46:W329–W337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang X., Cheng X., Zhao L., Wang Y., Ye C., Zou X., Dai A., Cong Z., Chen J., Zhou Q.et al.. Molecular insights into differentiated ligand recognition of the human parathyroid hormone receptor 2. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2101279118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klammt C., Maslennikov I., Bayrhuber M., Eichmann C., Vajpai N., Chiu E.J.C., Blain K.Y., Esquivies L., Kwon J.H.J., Balana B.et al.. Facile backbone structure determination of human membrane proteins by NMR spectroscopy. Nat. Methods. 2012; 9:834–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fowler N.J., Williamson M.P.. The accuracy of protein structures in solution determined by AlphaFold and NMR. Structure. 2022; 30:925–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bernhofer M., Rost B.. TMbed: transmembrane proteins predicted through language model embeddings. BMC Bioinformatics. 2022; 23:326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ryan E.J., Marshall A.J., Magaletti D., Floyd H., Draves K.E., Olson N.E., Clark E.A.. Dendritic cell-associated lectin-1: a novel dendritic cell-associated, C-type lectin-like molecule enhances T cell secretion of IL-4. J. Immunol. 2002; 169:5638–5648. [DOI] [PubMed] [Google Scholar]
- 24. Lu F., Li S., Jiang Y., Jiang J., Fan H., Lu G., Deng D., Dang S., Zhang X., Wang J.et al.. Structure and mechanism of the uracil transporter UraA. Nature. 2011; 472:243–246. [DOI] [PubMed] [Google Scholar]
- 25. Wang C., Sun B., Zhang X., Huang X., Zhang M., Guo H., Chen X., Huang F., Chen T., Mi H.et al.. Structural mechanism of the active bicarbonate transporter from cyanobacteria. Nat. Plants. 2019; 5:1184–1193. [DOI] [PubMed] [Google Scholar]
- 26. Ge J., Elferich J., Dehghani-Ghahnaviyeh S., Zhao Z., Meadows M., von Gersdorff H., Tajkhorshid E., Gouaux E.. Molecular mechanism of prestin electromotive signal amplification. Cell. 2021; 184:4669–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lomize A.L., Schnitzer K.A., Todd S.C., Cherepanov S., Outeiral C., Deane C.M., Pogozheva I.D.. Membranome 3.0: database of single-pass membrane proteins with AlphaFold models. Protein Sci. 2022; 31:e4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The TmAlphaFold database is available at https://tmalphafold.ttk.hu/.