


Abstract
Understanding gene expression patterns across different human cell types is crucial for investigating mechanisms of cell type differentiation, disease occurrence and progression. The recent development of single-cell RNA-seq (scRNA-seq) technologies significantly boosted the characterization of cell type heterogeneities in different human tissues. However, the huge number of datasets in the public domain also posed challenges in data integration and reuse. We present Human Universal Single Cell Hub (HUSCH, http://husch.comp-genomics.org), an atlas-scale curated database that integrates single-cell transcriptomic profiles of nearly 3 million cells from 185 high-quality human scRNA-seq datasets from 45 different tissues. All the data in HUSCH were uniformly processed and annotated with a standard workflow. In the single dataset module, HUSCH provides interactive gene expression visualization, differentially expressed genes, functional analyses, transcription regulators and cell–cell interaction analyses for each cell type cluster. Besides, HUSCH integrated different datasets in the single tissue module and performs data integration, batch correction, and cell type harmonization. This allows a comprehensive visualization and analysis of gene expression within each tissue based on single-cell datasets from multiple sources and platforms. HUSCH is a flexible and comprehensive data portal that enables searching, visualizing, analyzing, and downloading single-cell gene expression for the human tissue atlas.
INTRODUCTION
The human body is composed of various tissues and cells. Characterizing the expression patterns of different cells is crucial for probing cellular functions and molecular events in development and disease (1,2). Single-cell RNA-seq has proven to be a powerful technology to investigate the heterogeneity of human cell types in various tissues (3–5). Several human single-cell atlas projects, such as JingleBells (6), SCPortalen (7), PanglaoDB (8), scHCL (9) and Tabula Sapiens (10) have been developed in the past years and greatly promote our understanding of cellular heterogeneity in human tissues. However, there are still many limitations of the current human scRNA-seq atlas project. First, most of the above projects or databases only collect the scRNA-seq dataset or markers without processing them, and a lack of uniform data processing and harmonization will greatly affect data reuse (11,12). Second, several recent projects, such as the scHCL and Tabula Sapiens, provide uniformly processed datasets and online visualization functions. They highly rely on single technology platforms (Microwell-seq for the scHCL, 10X-Genomics and Smart-seq2 for Tabula Sapiens), which might have limited coverage on different cell types due to the bias in cell type capturing efficiency of different technologies (13). Finally, none of these projects or databases provided advanced analysis functions such as functional analyses, cell–cell interactions (CCI), and transcriptional regulator analyses, which are important to understanding the function of novel cell types and their association with normal and disease phenotypes (12,14). Therefore, a comprehensive database for human cell tissue expression atlas is urgently required.
Here, we present the Human Universal Single Cell Hub (HUSCH), a scRNA-seq database for visualizing and analyzing human gene expression across different tissues. The HUSCH database contains 185 datasets from 45 human tissues covering 7 different platforms. All the datasets were uniformly processed and annotated. For each dataset, HUSCH provides detailed cell type annotation, expression visualization, marker gene identification, functional analyses, transcription factor and cell–cell interaction analyses. HUSCH also integrates different datasets within the same tissue to enable cross datasets analyses. Finally, HUSCH provided an automatic online cell type annotation function with the curated reference.
MATERIALS AND METHODS
Data collection and curation
We collected human tissue scRNA-seq datasets from several public databases, including GEO (15), the single-cell portal from the broad institute, ArrayExpress from EMBL-EBI (16), the human cell atlas data portal (17), and the 10X Genomics website. For GEO and ArrayExpress datasets, we have built a text-mining-based workflow to automatically crawl and download scRNA-seq datasets. The keywords used for searching data include ‘single-cell RNA sequencing’, ‘scRNAseq’, ‘single cell’ or related technology terms such as ‘CellRanger’, ‘Seurat’ and ‘10X Genomics’. The collected records were then manually cleaned and only those relevant to scRNA-seq were kept. Since HUSCH is aimed at normal scRNA-seq datasets from human tissues, datasets from in vitro cultures and other species were removed and for the disease datasets, only the normal cells were included in the HUSCH database. We also collected the original cell annotations, if provided by paper, and corresponding cell type-specific gene markers for better annotating the datasets from different sources. Meta-information such as tissue type, genome assembly version, sequencing platform, sequencing type, cell number, sample resource, related publications, and donor information are also parsed. After filtering low-quality datasets and datasets with cell numbers <1000, the HUSCH database contains 185 high-quality single-cell datasets across 45 different tissues (Supplementary Table S1). All the downstream analyses were based on the expression matrix of the raw count (if available), TPM, or FPKM for each dataset. The source codes for collecting and processing scRNA-seq datasets are deposited in the GitHub repository (https://github.com/wanglabtongji/HUSCH).
Data pre-processing
The data pre-processing steps of HUSCH could be separated into two major parts, single dataset pre-processing and multiple datasets integration within the same tissue. For single-cell datasets processing, the scRNA-seq datasets were first cleaned to an expression matrix with columns as cells and rows as genes, other meta-information was cleaned as a meta table with rows as cells and columns as meta-information. Then the expression matrix and meta matrix were processed automatically using an analyses workflow based on MAESTRO v1.1.0 (18). The workflow will perform quality control, dimension reduction, unsupervised clustering, batch effect removal, differential expression (DE), initial cell type annotation based on DE genes, gene set enrichment analyses (GSEA)(19), transcriptional regulators and cell–cell interaction analyses (Supplementary Figure S1). For multiple datasets processing within the same tissue, all the processed datasets from that tissue will be integrated using R package harmony v1.0 (20). To confirm the integration results and harmonize inconsistent annotations between different datasets, we first classified the cell types into four major lineage levels including immune, stromal, endothelial, and tissue-specific cells, a similar definition used in the Tabula Sapiens data portal (10). If a single dataset showed inconsistent cell type annotations at the major lineage level with other datasets for the same cluster, the cell type annotations will be curated by voting. The integration steps will be repeated until all four major lineages were consistent (Supplementary Figure S2). After harmonization, if there are still a few cell types from a single dataset that might be mixed with the wrong lineage, these cell types will be removed from the tissue-integrated results but are still kept on the individual dataset page.
Quality control
Datasets containing <1000 cells were not included in the HUSCH database. Two metrics were used to assess the quality of cells: the number of detected genes per cell and the number of total counts (UMI) per cell (library size). The low-quality cells with a library size of <1000 or detected gene numbers of <500 were removed from the downstream analyses.
Clustering and differential gene analyses
For each dataset, the HUSCH preprocessing workflow identified the top 2000 variable features and perform PCA for dimension reduction, KNN and Louvain algorithm for unsupervised clustering (21,22). The number of PCs and the resolution for graph-based clustering were adjusted according to the number of cells per dataset. The uniform manifold approximation and projection (UMAP) were used to visualize the gene expression, and the Wilcoxon rank-sum test was used to identify the DE genes between different clusters based on the logFC > 0.25 and FDR < 1E–5.
Batch effect removal
Datasets from various donors or samples are usually impacted by batch effects in the majority of datasets. To systematically assess the batch effects, each dataset was quantified using a metric based on information entropy and the Euclidean distance between cell coordinates in the UMAP graph. A system's complexity can be reflected in its information entropy, and a greater entropy number indicates that different batches of cells are mixed more uniformly. The entropy was calculated using
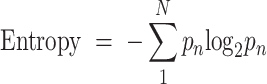 |
where 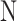 is the number of batches and
is the number of batches and 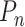 denotes the percentage of the 30 cells in the neighborhood that belong to a certain batch. We regard the result of the maximum over the median of all entropy values as a criterion for whether or not we need to remove the batch effect. If
denotes the percentage of the 30 cells in the neighborhood that belong to a certain batch. We regard the result of the maximum over the median of all entropy values as a criterion for whether or not we need to remove the batch effect. If 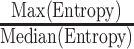 is too large, it indicates that the majority of the entropy values are scattered on the smaller side, which means the surrounding cells are not evenly distributed among different batches for the majority of the cells. After testing, we set 4 as the threshold and remove batch effects using conventional correlation analysis (CCA) in Seurat v4.0.4 for datasets above the threshold (23).
is too large, it indicates that the majority of the entropy values are scattered on the smaller side, which means the surrounding cells are not evenly distributed among different batches for the majority of the cells. After testing, we set 4 as the threshold and remove batch effects using conventional correlation analysis (CCA) in Seurat v4.0.4 for datasets above the threshold (23).
Cell type annotation
The cell types in HUSCH datasets are defined as major-lineage level and minor-lineage level. As described in the data pre-processing section, the major-lineage level includes immune, stromal (including fibroblast, pericyte, myofibroblast, and muscle cell), endothelial, and tissue-specific cells (including epithelial and other tissue-specific cell types). The minor-lineage level was the original cell type annotation level. Datasets with original cell type annotation will be annotated directly using the original label. For datasets without original cell type annotation provided by the original study, HUSCH will automatically annotate each cluster using cell type-specific marker genes (Supplementary Table S2). Briefly, cell type marker genes were collected from the original studies or the public resources and curated manually. Then HUSCH will apply automatic cell type annotation using the cell type scoring function in MAESTRO based on the DE genes for each cluster. After the automatic annotation, we manually corrected all of the annotated cell types by checking cell type marker genes’ expression levels. The cluster will be assigned as the cell types that showed the highest score using its corresponding markers. However, the two different annotation strategies will lead to inconsistent cell type names. Then HUSCH will unify the cell type names based on the standard cell type names from Cell Ontology (24) (Supplementary Table S3).
Functional enrichment analysis
To better understand the function of different cell types and clusters, we performed gene set enrichment analyses (GSEA) using the top 200 DE genes of each cluster. The genes were ranked based on the fold change from the differential expression analyses. We collected 236 gene sets for GSEA in total, including 186 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, and 50 hallmark pathways from the Molecular Signatures Database (MSigDB v7.1; 34). Significant up-regulated, and down-regulated pathways (FDR ≤ 0.05) in each cluster were identified and visualized to enable the functional enrichment analyses between different clusters. The GSEA analyses were achieved using GSEA v4.0.3 for Linux and the heatmaps were generated using the ComplexHeatmap R package v1.99.5 (25).
Transcription regulator prediction
The transcriptional regulators (TR) of each cell type cluster were inferred using LISA v2.2.2 (26). For each cluster, the top 500 positive differentially expressed genes are used as input as a query gene list, and 8000 random sample background genes were used to control the bias. Other parameters are set as default for the TR analyses. After getting the TR enrichment from different cell types, we normalized the TR enrichment P-values from LISA using log z-score transformation and generated the TR enrichment heatmaps to better visualize the cell type-specific TRs.
Cell-cell interaction analysis
The cell–cell interaction (CCI) analyses were performed using CellChat v. 1.4.0 (27) with the CellChatDB.human database. The aggregated CCI network was calculated by counting the number of links or summarizing the communication probability between different cell types, which is visualized using a circle plot with the netVisual_circle function. For each cell type, the significant in and out CCIs were also visualized in HUSCH using the netVisual_bubble function. All the CCIs with a P-value < 0.01 were visualized for a certain cell type.
Online automatic cell type annotation
With the comprehensive human cell type atlas, HUSCH also provides an online cell type annotation function using a deep learning-based framework SELINA (28). Briefly, the annotated datasets from HUSCH were trained as a reference. The SELINA algorithm includes three major steps, cell type balancing for rare cell types using SMOTE(29) techniques, a multi-adversarial domain adaptation network for correcting batch effect between different platforms, and an autoencoder for removing batch effect between query and reference datasets. The users could either upload the single-cell expression matrix or cluster averaged expression matrix, HUSCH will run SELINA in the background and return the cell type annotation by email. For jobs with single-cell level annotation, HUSCH requires the users to input the gene symbol types, PC number, and clustering resolutions for pre-processing the scRNA-seq dataset online. No data processing parameters are required for cluster-level annotation.
Web portal for the database
We developed the HUSCH web portal to show the analysis results in a user-friendly manner based on the uniformly processed scRNA-seq datasets. From the web portal, all of the processed and annotated datasets can be searched, viewed, and downloaded. The interfaces of HUSCH were designed with Vue.js, and all the interactive functions of HUSCH, such as online expression visualization and cell type prediction were implemented with python. HUSCH is deployed with the Apache2 HTTP server and is publicly accessible at http://husch.comp-genomics.org without any registration or login requirements.
RESULTS
Dataset summary
The HUSCH database includes the single-cell transcriptome atlas of 2 865 305 cells from 185 high-quality human datasets, covering 45 different tissue types and 7 platforms (Figure 1). The majority of the HUSCH datasets were generated using 10X-Genomics and Microwell-seq platforms, which are the two most widely used commercialized scRNA-seq platforms. On average, each dataset contains ∼15 000 cells and ∼18 000 genes. Among the 45 tissues, blood is the tissue with the most abundant dataset, with >20 datasets and nearly 500K cells. The lung, prostate, eye and adipose tissues have >150K cells, and the large-Intestine, bone marrow, lung, and skin tissues have >10 datasets, indicating these tissues are more investigated or are easier to perform scRNA-seq experiments. Most of the tissues in HUSCH include more than two datasets, except for some small tissues such as the fallopian tube and the common bile duct, which only have one dataset for each tissue.
Figure 1.

Statistics of HUSCH datasets. HUSCH includes 185 high-quality single-cell datasets, covering nearly 3 million cells across 45 different human tissues. The left figure shows the total number of cells for each tissue, and the right figure shows the total number of datasets for each tissue.
Single dataset exploration in HUSCH
The dataset module of HUSCH includes sample information, clustering results, cell type annotations, cell type statistics, and marker genes, downstream functional analyses for 185 datasets (Figure 2). There are mainly three major functions of the dataset module, single dataset exploration, single tissue exploration, and data download. We first used datasets HU_0322_Blood_GSE159929 and HU_0104_Airway_GSE102580 as examples to illustrate how to explore a single dataset in the dataset module.
Figure 2.

Overview of the HUSCH workflow and functions. The datasets in the HUSCH database were automatically parsed from multiple different public databases. All datasets were then processed using a uniform workflow based on MAESTRO, including quality control, batch effect removal, cell clustering, differential expression analysis, cell type and tissue level integration. For each dataset, HUSCH displayed relevant study information, including tissue type, assembly, platform, the number of patients and cells, and related study information. In the Dataset Exploration module, HUSCH provides three main functions: single-dataset exploration, single-tissue exploration, and data download. HUSCH also allows online cell type annotation using the curated expression atlas as a reference.
Cell type composition and gene expression visualization
If the users select a certain tissue, all the datasets of that tissue will be listed as a table, which shows the basic information of each dataset including publication date and PMID, data source, sample stage, sequencing platforms, sequencing technology, and cell numbers (Figure 3A, Supplementary Figure S3A). For a certain dataset at the Overview tab, HUSCH will display the cell clustering UMAP, cell type annotation UMAP, cell type statistics, and top DE genes of that cell type (Figure 3B, Supplementary Figure S3B). HUSCH also allows users to select different cell types and rank the DE genes by P-value or log2FC. In the Gene tab, HUSCH provides an online gene expression visualization function for multiple genes using UMAP. Besides, HUSCH also allows visualizing the gene expression using a violin plot at cell type or cluster level (Figure 3C, Supplementary Figure S3C). For the blood dataset, we visualized the expression of CD14, CD8A, and CD19 using a violin plot, and the results demonstrate the clear cell type-specific expression of these genes with CD14 highly enriched in monocytes, CD8A enriched in CD8 T-cells, and CD19 enriched in B-cells as well as plasmacytoid dendritic cells (Figure 3C).
Figure 3.

Expression visualization of a single dataset in HUSCH. (A) Datasets table of the blood tissue. Users could click on a certain dataset and enter the single dataset exploration page. (B) The overview tab of the HU_0322_Blood_GSE159929 dataset. Two UMAP plots with cells colored by cluster ID (left) and cell type (right) are displayed at the top of the tab. The bottom table below shows DE genes in each cell type cluster. (C) The gene tab of the single dataset exploration page. The expression of genes of interest can be visualized at single-cell and cell type resolution. The upper figures showed the expression visualization in single-cell resolution by UMAP, and the bottom violin plot visualizes the expression at the cell type level.
Advanced analysis
HUSCH also provides several advanced analysis results for each dataset including GSEA, transcription regulator identification, and cell–cell interaction analyses.
In the GSEA tab, the pre-calculated GSEA results are available for the users to investigate the potential function of different cell types. We gathered 236 gene sets from MSigDB, including KEGG and hallmark to annotate the function of each cell type. The differentially expressed genes in each cluster will be used to identify the enriched up or down-regulated pathways, which will be displayed using heatmaps (Figure 4A, Supplementary Figure S3D). For example, the B-cell receptor signaling pathway and intestinal immune network for IgA production are highly enriched in the B-cells of the blood dataset. Besides, the two B-cell clusters also showed slight differences in functions, indicating they might be different sub-types of B-cells (Figure 4A).
Figure 4.

Advanced analyses of a single dataset in HUSCH. (A) GSEA results of a single dataset. The enriched up- or down-regulated hallmark pathways in each cluster are visualized in heatmaps. (B) CCI results of a single dataset. The left top heatmap displays the number of interactions between different cell types, the right top circle plot displays the number of interactions between a certain cell type and other cell types. The bottom dot plot displays significant CCIs (P-value < 0.01) of selected cell types. Left, the selected cell type as receiver cells, right, the selected cell type as sender cells. (C) The left one is heatmap of significant enriched transcriptional regulators predicted by LISA for each cell type cluster, the right one is dotplot of the rank for regulators predicted in cluster CD4T_C0.
All the cell–cell interactions in HUSCH were generated by CellChat. In the cell–cell interaction tab, HUSCH summarizes the number of significant CCIs between different cell type clusters using a heatmap. Besides, for a single-cell cluster, HUSCH will also display the number of interactions of this cluster with all other clusters using a circle plot (Figure 4B, Supplementary Figure S3E). Finally, HUSCH will display the significant ligand–receptor pairs for each cell type cluster using a dot plot, whether this cell type work as sender cells or receiver cells. In the blood example dataset, we can see that the CD8T_C3 cluster interacts with other antigen presentation cells like B and pDCs using MHC-II and CD8A/B interactions. Also, the CD8T_C3 cluster could work as a sender cell to modulate the functions of Monocyte_C5 through CD99 and ANXA1-dependent interactions (Figure 4B).
Finally, in the TF tab, HUSCH provides transcription regulator (TR) predictions for each cell type cluster (Figure 4C, Supplementary Figure S3F). The TRs predicted using the LISA algorithm could reflect the lineage and differentiation status of a certain cell type. For example, both BCL6 and FOXO1 are well-known functional regulators in B-cells (30), SPI1 is important for characterizing monocyte identity, and GATA3, as well as T, is important in T-cell functions (Figure 4C) (31). These analyses suggest that the predicted TRs using scRNA-seq could accurately reflect its cell type lineages and may be used to discover other novel regulators for uncharacterized cell types.
Single tissue exploration in HUSCH
The integration of different datasets within one tissue will not only include a wider range of covered cell types but also amend potential cell type annotation errors from a single dataset. We then integrated datasets from different patients, sources, and platforms within the same tissue using harmony, and generated the integrated datasets for each tissue for visualizing gene expression across different datasets (Figure 5, Supplementary Figure S4). We take blood and nose as examples of the tissue integration result. Integrating 21 blood datasets significantly enlarged the covered cell types (Figure 5A). In addition, when we visualized the gene expression using heatmaps, all of the included datasets show high expression of CD14 in monocytes, indicating the high consistency between different blood datasets (Figure 5B).
Figure 5.

Tissue level expression visualization in HUSCH. (A) The cell type annotation of integrated blood tissue. (B) Heatmap showing the expression of CD14 across different blood datasets in HUSCH. The color indicates the expression level of the gene. (C) The cell type annotation of integrated bladder tissue, the UMAP was plotted for four major lineages, including tissue-specific, immune, endothelial and stromal. (D) Heatmap summarizing the number of significant CCIs between different cell type clusters. (E) Heatmap of significant enriched transcriptional regulators predicted by LISA for each cell type cluster.
For complexed tissues like the bladder, we first separated different cell types into four major lineages, including immune, stromal (fibroblast, fibsmo, and smooth muscle cell), endothelial, and tissue-specific (epithelia and other unclassified cell types). This major lineage separation enables HUSCH to curate the cell type consistency in complex tissues (Figure 5C).
Advanced analysis
For each integrated tissue, HUSCH also provides various exploration results, such as GSEA, transcription regulator identification, and evaluations of cell–cell interactions, using the same methods with single dataset analyses. We use the integrated blood dataset as an example. In the cell–cell interaction tab, myeloid clusters show apparently more interactions with other cell types (Figure 5D). In the TF tab, SPI1, which is a well-known functional regulator in monocyte, was predicted to be highly enriched in mono/macrophage (Figure 5E). These analyses show the reliability and wide utility of the integrated HUSCH datasets.
Online automatic cell type annotations
Although HUSCH provides a comprehensive expression atlas of human scRNA-seq data. Considering the rapid accumulation of scRNA-seq data in the public domain, HUSCH also offers functions for users to annotate their own scRNA-seq data using HUSCH data as a reference (Supplementary Figure S5). Users could choose from uploading scRNA-seq data either at the single-cell or cluster resolution, and HUSCH will process and annotate the scRNA-seq data using the cell type labels transferred from the reference data in the database, and send the annotated result to users by email (Methods).
DISCUSSION
Single-cell RNA-seq has the ability to identify rare cell types in tissues with unprecedented accuracy and speed, making it an indispensable tool for investigating cellular heterogeneities across different species. To understand the complicated cell type compositions and expression heterogeneity in the human body, many scRNA-seq datasets have been produced. However, there is still a lack of a well-curated, consistently processed, and annotated data gateway for large-scale data reuse. Here, we present HUSCH, a comprehensive database providing a user-friendly web resource for interactive gene expression visualization of cellular differences across various human tissues at the single-cell level. HUSCH has a number of benefits over the available single-cell resources. Firstly, HUSCH includes the single-cell transcriptome atlas of around 3 million cells from 185 high-quality human normal tissue datasets, covering 45 tissue types, 270 cell types and 7 platforms, different scRNA-seq datasets were uniformly processed, annotated, and batched corrected, which removes the barriers for data re-use. Secondly, for biologists who want to fully comprehend and research human biology at a cellular level, the integrated datasets in HUSCH will be a tremendous resource for visualizing gene expression conveniently without processing data by themselves. Finally, HUSCH offers a wealth of functions for users to dig into the data, understanding the CCIs and crucial TFs that may drive cell type differentiation and functions. We will continue to incorporate new datasets as well as novel functions to enhance the HUSCH database in the future.
DATA AVAILABILITY
The codes used for data processing in the HUSCH database are deposited in the GitHub repository at https://github.com/wanglabtongji/HUSCH.
The expression matrix, sample meta-information, differential expression gene list, transcription factors, and cell–cell interactions displayed in the HUSCH database could be directly downloaded from http://husch.comp-genomics.org/.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the Bioinformatics Supercomputer Center of Tongji University for offering computing resources. The authors acknowledge the authors from published studies to share their single-cell RNA-seq data on human clinical samples.
Contributor Information
Xiaoying Shi, Key Laboratory of Spine and Spinal Cord Injury Repair and Regeneration of Ministry of Education, Department of Orthopedics, Tongji Hospital, School of Life Science and Technology, Tongji University, Tongji, 200092, China; Frontier Science Center for Stem Cells, School of Life Sciences and Technology, Tongji University, Shanghai 200092, China.
Zhiguang Yu, State Key Laboratory for Conservation and Utilization of Subtropical Agro-bioresources, College of Life Science and Technology, Guangxi University, Guangxi 530004, China.
Pengfei Ren, Key Laboratory of Spine and Spinal Cord Injury Repair and Regeneration of Ministry of Education, Department of Orthopedics, Tongji Hospital, School of Life Science and Technology, Tongji University, Tongji, 200092, China; Frontier Science Center for Stem Cells, School of Life Sciences and Technology, Tongji University, Shanghai 200092, China.
Xin Dong, Key Laboratory of Spine and Spinal Cord Injury Repair and Regeneration of Ministry of Education, Department of Orthopedics, Tongji Hospital, School of Life Science and Technology, Tongji University, Tongji, 200092, China; Frontier Science Center for Stem Cells, School of Life Sciences and Technology, Tongji University, Shanghai 200092, China.
Xuanxin Ding, Key Laboratory of Spine and Spinal Cord Injury Repair and Regeneration of Ministry of Education, Department of Orthopedics, Tongji Hospital, School of Life Science and Technology, Tongji University, Tongji, 200092, China; Frontier Science Center for Stem Cells, School of Life Sciences and Technology, Tongji University, Shanghai 200092, China.
Jiaming Song, State Key Laboratory for Conservation and Utilization of Subtropical Agro-bioresources, College of Life Science and Technology, Guangxi University, Guangxi 530004, China.
Jing Zhang, Research Center for Translational Medicine, Shanghai East Hospital, School of Life Science and Technology, Tongji University, Shanghai, China.
Taiwen Li, State Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, Research Unit of Oral Carcinogenesis and Management, Chinese Academy of Medical Sciences, West China Hospital of Stomatology, Sichuan University, Chengdu 610041, China.
Chenfei Wang, Key Laboratory of Spine and Spinal Cord Injury Repair and Regeneration of Ministry of Education, Department of Orthopedics, Tongji Hospital, School of Life Science and Technology, Tongji University, Tongji, 200092, China; Frontier Science Center for Stem Cells, School of Life Sciences and Technology, Tongji University, Shanghai 200092, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [32222026 and 32170660 to C.W., 81972551 to T.L.]; Shanghai Rising Star Program [21QA1408200 to C.W.]; Natural Science Foundation of Shanghai [21ZR1467600 to C.W.]; Natural Science Foundation of Sichuan Province [2022NSFSC0054 to T.L.]; Young Elite Scientist Sponsorship Program by CAST [2021QNRC001 to T.L.]. Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Wagner A., Regev A., Yosef N.. Revealing the vectors of cellular identity with single-cell genomics. Nat. Biotechnol. 2016; 34:1145–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tanay A., Regev A.. Scaling single-cell genomics from phenomenology to mechanism. Nature. 2017; 541:331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Papalexi E., Satija R.. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 2018; 18:35–45. [DOI] [PubMed] [Google Scholar]
- 4. Stubbington M.J.T., Rozenblatt-Rosen O., Regev A., Teichmann S.A.. Single-cell transcriptomics to explore the immune system in health and disease. Science. 2017; 358:58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pijuan-Sala B., Guibentif C., Gottgens B.. Single-cell transcriptional profiling: a window into embryonic cell type specification. Nat. Rev. Mol. Cell Biol. 2018; 19:399–412. [DOI] [PubMed] [Google Scholar]
- 6. Ner-Gaon H., Melchior A., Golan N., Ben-Haim Y., Shay T.. JingleBells: a repository of immune-related single-cell RNA-Sequencing datasets. J. Immunol. 2017; 198:3375–3379. [DOI] [PubMed] [Google Scholar]
- 7. Abugessaisa I., Noguchi S., Bottcher M., Hasegawa A., Kouno T., Kato S., Tada Y., Ura H., Abe K., Shin J.W.et al.. SCPortalen: human and mouse single-cell centric database. Nucleic Acids Res. 2018; 46:D781–D787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Franzen O., Gan L.M., Bjorkegren J.L.M.. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database (Oxford). 2019; 2019:baz046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Han X., Zhou Z., Fei L., Sun H., Wang R., Chen Y., Chen H., Wang J., Tang H., Ge W.et al.. Construction of a human cell landscape at single-cell level. Nature. 2020; 581:303–309. [DOI] [PubMed] [Google Scholar]
- 10. Tabula Sapiens C., Jones R.C., Karkanias J., Krasnow M.A., Pisco A.O., Quake S.R., Salzman J., Yosef N., Bulthaup B., Brown P.et al.. The tabula sapiens: a multiple-organ, single-cell transcriptomic atlas of humans. Science. 2022; 376:eabl4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stuart T., Satija R.. Integrative single-cell analysis. Nat. Rev. Genet. 2019; 20:257–272. [DOI] [PubMed] [Google Scholar]
- 12. Elmentaite R., Dominguez Conde C., Yang L., Teichmann S.A.. Single-cell atlases: shared and tissue-specific cell types across human organs. Nat. Rev. Genet. 2022; 23:395–410. [DOI] [PubMed] [Google Scholar]
- 13. Argelaguet R., Cuomo A.S.E., Stegle O., Marioni J.C.. Computational principles and challenges in single-cell data integration. Nat. Biotechnol. 2021; 39:1202–1215. [DOI] [PubMed] [Google Scholar]
- 14. Armingol E., Officer A., Harismendy O., Lewis N.E.. Deciphering cell–cell interactions and communication from gene expression. Nat. Rev. Genet. 2021; 22:71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M.et al.. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013; 41:D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Athar A., Fullgrabe A., George N., Iqbal H., Huerta L., Ali A., Snow C., Fonseca N.A., Petryszak R., Papatheodorou I.et al.. ArrayExpress update - from bulk to single-cell expression data. Nucleic Acids Res. 2019; 47:D711–D715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Regev A., Teichmann S.A., Lander E.S., Amit I., Benoist C., Birney E., Bodenmiller B., Campbell P., Carninci P., Clatworthy M.et al.. The human cell atlas. Elife. 2017; 6:e27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang C., Sun D., Huang X., Wan C., Li Z., Han Y., Qin Q., Fan J., Qiu X., Xie Y.et al.. Integrative analyses of single-cell transcriptome and regulome using MAESTRO. Genome Biol. 2020; 21:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S.et al.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S.. Fast, sensitive and accurate integration of single-cell data with harmony. Nat. Methods. 2019; 16:1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Butler A., Hoffman P., Smibert P., Papalexi E., Satija R.. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018; 36:411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu C., Su Z.. Identification of cell types from single-cell transcriptomes using a novel clustering method. Bioinformatics. 2015; 31:1974–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M.et al.. Integrated analysis of multimodal single-cell data. Cell. 2021; 184:3573–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Osumi-Sutherland D., Xu C., Keays M., Levine A.P., Kharchenko P.V., Regev A., Lein E., Teichmann S.A.. Cell type ontologies of the human cell atlas. Nat. Cell Biol. 2021; 23:1129–1135. [DOI] [PubMed] [Google Scholar]
- 25. Gu Z., Eils R., Schlesner M.. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016; 32:2847–2849. [DOI] [PubMed] [Google Scholar]
- 26. Qin Q., Fan J., Zheng R., Wan C., Mei S., Wu Q., Sun H., Brown M., Zhang J., Meyer C.A.et al.. Lisa: inferring transcriptional regulators through integrative modeling of public chromatin accessibility and chip-seq data. Genome Biol. 2020; 21:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jin S., Guerrero-Juarez C.F., Zhang L., Chang I., Ramos R., Kuan C.H., Myung P., Plikus M.V., Nie Q.. Inference and analysis of cell–cell communication using cellchat. Nat. Commun. 2021; 12:1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ren P., Shi X., Dong X., Yu Z., Ding X., Wang J., Sun L., Yan Y., Hu J., Zhang P.et al.. SELINA: single-cell assignment using multiple-adversarial domain adaptation network with Large-scale references. 2022; bioRxiv doi:17 January 2022, preprint: not peer reviewed 10.1101/2022.01.14.476306. [DOI] [PMC free article] [PubMed]
- 29. Chawla NV, Bowyer KW, Hall LO, Kegelmeyer WP. SMOTE: synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002; 16:321–357. [Google Scholar]
- 30. Boller S., Grosschedl R.. The regulatory network of B-cell differentiation: a focused view of early B-cell factor 1 function. Immunol. Rev. 2014; 261:102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Novershtern N., Subramanian A., Lawton L.N., Mak R.H., Haining W.N., McConkey M.E., Habib N., Yosef N., Chang C.Y., Shay T.et al.. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011; 144:296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The codes used for data processing in the HUSCH database are deposited in the GitHub repository at https://github.com/wanglabtongji/HUSCH.
The expression matrix, sample meta-information, differential expression gene list, transcription factors, and cell–cell interactions displayed in the HUSCH database could be directly downloaded from http://husch.comp-genomics.org/.