


Abstract
The 5′ and 3′ untranslated regions of eukaryotic mRNAs (UTRs) play crucial roles in the post-transcriptional regulation of gene expression through the modulation of nucleo-cytoplasmic mRNA transport, translation efficiency, subcellular localization, and message stability. Since 1996, we have developed and maintained UTRdb, a specialized database of UTR sequences. Here we present UTRdb 2.0, a major update of UTRdb featuring an extensive collection of eukaryotic 5′ and 3′ UTR sequences, including over 26 million entries from over 6 million genes and 573 species, enriched with a curated set of functional annotations. Annotations include CAGE tags and polyA signals to label the completeness of 5′ and 3′UTRs, respectively. In addition, uORFs and IRES are annotated in 5′UTRs as well as experimentally validated miRNA targets in 3′UTRs. Further annotations include evolutionarily conserved blocks, Rfam motifs, ADAR-mediated RNA editing events, and m6A modifications. A web interface allowing a flexible selection and retrieval of specific subsets of UTRs, selected according to a combination of criteria, has been implemented which also provides comprehensive download facilities. UTRdb 2.0 is accessible at http://utrdb.cloud.ba.infn.it/utrdb/
INTRODUCTION
Even after over two decades since the completion of the first draft assembly of the human genome, and the compilation of a complete catalog of all the human genes, we are still far from a detailed appreciation of the molecular mechanisms that regulate the spatio-temporal expression of protein coding genes and their translation into their ‘final’ gene products. While technological advances such as the massive, parallel sequencing of RNAs have enabled the quantitative and precise measure of RNA expression levels in a wide range of human cells and tissues (1), several independent lines of evidence confirmed that mRNAs and proteins levels are only partly correlated, and in general mRNA levels alone are not predictive of the concentration or activity of proteins in vivo (2–4). Several biological mechanisms that regulate post-transcriptional mRNAs stability and translation are at the base of this observation. For example, the nucleo-cytoplasmic transport of the newly synthesized RNAs (5), their stability (6), translation efficiency (7) and subcellular localization (5) are regulated at the post-transcriptional level. In protein-coding mRNAs, such regulation is mainly mediated by cis-acting elements located in the 5′ and 3′ untranslated regions (5′UTRs, 3′UTRs) also through the interaction with complementary RNAs (e.g. miRNAs) or enzymatically driven chemical modifications (e.g. m6A) or editing processes (e.g. A to I). Evolutionarily free from the constraint of encoding proteins, UTRs may modulate gene expression through various functional elements which have been characterized and experimentally validated in numerous mRNAs (8,9). These elements may correspond to short oligonucleotide tracts whose biological activity relies on both their sequence and secondary structures (10). For example, some specific sequence motifs function as target sites for RNA binding proteins, modification or editing enzymes or interact directly with the translational machinery.
The 5′UTRs may contain structural motifs and upstream Open Reading Frames (uORFs) that regulate translation efficiency or promote cap-independent translation in specific physiological conditions (11). uORFs, are found in nearly half of human 5′UTR mRNA transcripts (12), and can suppress translation through various mechanisms, including translation reinitiation, ribosome leaky scanning or the functional activity of the uORF-encoded peptide. 3′UTRs may contain, as well, sequence and structural motifs modulating mRNA stability, translational efficiency and subcellular localization in several ways. In particular, their degenerate complementary interaction with miRNAs has been associated with translation downregulation (13).
Additionally, alternative cleavage and polyadenylation (APA), a widespread mechanism to generate mRNA isoforms with alternative 3′ UTRs, has been implicated in the modulation of protein abundance, as well as in the regulation of mRNA localization and the spatial organization of protein synthesis (14). Finally, all the regulatory mechanisms described above can be further modulated by specific RNA editing or chemical modifications.
Although UTRs comprise only <1% of the human genome, about 3.7% of all currently known human genetic variants are located in UTRs, suggesting that they might represent a hotspot of genetic variation in human protein coding genes, a consideration that further emphasizes the importance of performing/unraveling accurate functional annotation of UTR elements (15). Indeed, GWAS investigations identified several genetic variants in UTR regions. These variants, potentially associated with complex disease risks, may affect gene expression, thus playing a critical functional role, but are usually more tricky to functionally assess with respect to variants affecting coding sequences. Moreover, 599 variants (0.5% of the total number) in the UTRs of protein coding genes are known to be implicated in the pathogenesis of human disorders according to the ClinVar database (16).
Therefore, the availability of an extensive collection of highly curated annotations of functional elements in human UTRs could represent an invaluable resource for a better understanding of gene expression regulation and its role in health and disease conditions. Importantly, similar considerations could be applied to several model and non-model organisms, for which a reference genome assembly and annotation is currently available. At present >500 complete genomes are available through the dedicated resources curated by EMBL/Ensembl (17). Accurate and comprehensive annotations of UTR regions could provide a useful resource for advancing functional genomics also in these species, and for the execution of complex and detailed comparative genomic analyses.
To address these issues, we have developed a new version of UTRdb (18), a collection of 5′ and 3′UTR sequences derived from eukaryotic mRNA collected in the Ensembl (17) and Ensembl Genomes (19) databases with over 500 different species represented.
UTRdb is based on a gene and transcript-centric annotation model, which facilitates the full integration with other specialized databases and resources, and, at the same time, permits the compilation of detailed reports providing comprehensive information on alternative 5′ and 3′UTRs. UTRdb entries boast a comprehensive collection of annotations, including: regulatory elements collected in Rfam (20), conserved elements in PhastCons (21), ADAR-mediated RNA editing (22) and m6A modifications (23). In addition, upstream open reading frames, complemented with mass spectrometry proteomic data (when available) (24), and Internal Ribosome entry sites (25) are provided for 5′ UTRs, while experimentally validated miRNA targets are reported for 3′UTRs (26,27). Finally, to provide information concerning the completeness of 5′ and 3′ UTR sequences and/or alternative TSS/TTS, UTRdb integrates a large collection of CAGE experiments (28) and provides annotations of experimentally validated poly-A signals (29).
We believe that by providing a comprehensive and annotation-rich collection of annotations UTRdb may be regarded as a highly useful resource for investigations on the regulatory roles of UTRs, their role in the maintenance of cellular homeostasis and in the onset and progression of human disorders, including cancer (30). Finally, UTRdb, providing useful information for the functional assessment of UTRs, may represent a valuable resource for the development of mRNA vaccine technology, whose recent impressive breakthrough greatly supported the fight against the COVID-19 pandemic (31).
DATA COLLECTION AND PROCESSING
Data collection
Gene annotations in GTF format and the corresponding FASTA files for 615 organisms were downloaded from Ensembl, including: 313 organisms from Ensembl rel 107 (http://ftp.ensembl.org/pub/release-107/), 125 from Ensembl plants rel 54 (http://ftp.ensemblgenomes.org/pub/plants/release-54/) and 177 from Ensembl metazoans (http://ftp.ensemblgenomes.org/pub/metazoa/release-54/) rel 54 (17). All GTF files were preliminarily evaluated and organisms lacking UTR annotations were discarded. In particular, a total of 42 species (19 from Ensembl rel 107, 13 from Ensembl plants and 10 from Ensembl metazoans) were not included in UTRdb for this reason, thus reducing the total number of organisms incorporated in our database to 573. Lists of orthologous genes, annotations of repeated elements, low complexity DNA regions and small genetic variants were downloaded from Ensembl through their Representational State Transfer (REST) API (32) (https://rest.ensembl.org/) by means of custom Python scripts. Collections of human genetic variants significantly associated with genotypic traits or disorders according to a compendium of GWAS were downloaded from the GWAS Catalog (33) (https://www.ebi.ac.uk/gwas/).
Evolutionary conserved elements identified by PhastCons were obtained from the UCSC Genome Browser database (https://hgdownload.soe.ucsc.edu/downloads.html) (34).
When multiple PhastCons annotations for an organism, were available, the one incorporating the largest number of alignments/species was selected (e.g. ‘phastConsElements135way.txt.gz’ instead of ‘phastConsElements26way.txt.gz’ in Caenorhabditis elegans entries).
Annotations of miRNA targets for the Homo sapiens GRCh38 and Mus musculus GRCm39 reference genome assemblies were downloaded from the Ensembl genome browser, through their public mysql databases, using the following commands: ‘mysql -h ensembldb.ensembl.org -u anonymous -P 5306 homo_sapiens_funcgen_107_38 -e ‘select * from mirna_target_feature’ and ‘mysql -h ensembldb.ensembl.org -u anonymous -P 5306 mus_musculus_funcgen_107_39 -e ‘select * from mirna_target_feature’, respectively. A custom script was applied to retain only target sites supported by experimental evidence according to Tarbase v8 (https://dianalab.e-ce.uth.gr/html/diana/web/index.php?r=tarbasev8) (27).
Alternative polyadenylation sites (in BED format) were obtained from APADB (http://tools.genxpro.net:9000/apadb/download/) version v2 (29) and the UCSC Liftover tool (35) (available at https://genome-store.ucsc.edu/) was used to convert genomic coordinates to different reference genome assemblies when required (e.g. hg19 to hg38 for the human genome). Additionally, miRNA target sites predicted by TargetScan (32) and miRanda (36) and included in APADB were incorporated in UTRdb as well.
Annotations of IRES (Internal Ribosome Entry Site) motifs were collected from IRESite (http://iresite.org/IRESite_web.php?page=browse_cellular_transcripts) (25), a database of experimentally validated IRES structures.
Epitranscriptome modifications, including RNA editing events due to the deamination of the adenosine (A) in inosine (I) and m6A sites were obtained from the specialized databases REDIportal (http://srv00.recas.ba.infn.it/atlas/download.html) and RMVar (https://rmvar.renlab.org/download.html) (22,23), respectively.
Known RNA functional secondary structure elements and other sequence motifs associated with UTRs were annotated according to Rfam (20) (https://rfam.xfam.org/), while mass spectrometry-based proteomic evidences of potential upstream ORFs translation were obtained from PRIDE (24) (https://www.ebi.ac.uk/pride/). Cap Analysis of Gene Expression (CAGE) tags were downloaded from fantom5 (28) (https://fantom.gsc.riken.jp/5/) in BED format for the following species: Homo sapiens (GRCh38), Mus musculus (GRCm39), Canis lupus (Dog10K_Boxer_Tasha), Gallus gallus (gca000002315v5.GRCg6a), Macaca mulatta (Mmul_10) and Rattus norvegicus (mRatBN7.2).
Construction of UTRdb relational database tables
A compendium of highly curated and accurate resources for functional annotation of UTRs were collected (see Data Collection and Processing section) and automatically processed by custom software in order to generate ad hoc tables containing UTRdb entries and attributes (Figure 1). The sequence, and corresponding annotation of gene models, in the form of a GTF file, were retrieved for every eukaryotic genome reference assembly available from the Ensembl genome browser.
Figure 1.

UTRdb data processing and database organization. A total of 26 098 657 UTR sequences were extracted from Ensembl and Ensembl Genomes included in UTRdb. Functional annotations were retrieved from their respective repositories (e.g. D1, D2, D3, .., Dn) and integrated by means of custom Python scripts. All data was organized in MySQL tables. The web-interface was developed using Bootstrap and HTML5.
Ad-hoc Python scripts were applied to retrieve genomic coordinates of UTRs and extract (by means of the pysam (37) Python module) the corresponding sequences.
Functional annotations, including polyA signals, RNA editing sites, SNPs, miRNA binding sites, PhastCons evolutionarily conserved elements and many others (see see Data Collection and Processing section), were retrieved from their respective repositories (as detailed above), formatted according to the requirements of UTRdb and integrated in the database, again by applying custom Python scripts (Figure 1).
Annotations of uORFs were obtained by performing in silico translation of 5′ UTR sequences, ORFs formed by an AUGs without an in-frame downstream stop codon (38) were also included.
DATABASE CONTENT AND WEB INTERFACE
UTRdb content and structure
The knowledge base incorporated in UTRdb 2.0 represents a more than 25-fold increase in terms of the number of UTRs and 7-fold increase in terms of the number of species represented in the database, compared to previous release (15). UTRdb 2.0 includes functional annotation for a total of 26 098 657 UTRs—12 908 478 5′UTRs (with a mean length of 300 nt) and 13 190 179 3′UTRs (with a mean length of 753 nt) (Table 1)—from 6 688 161 genes in 573 organisms. Approximately, 0.7% of the UTRs in the database are from H. sapiens and collectively 8.8% are associated with model organisms. Importantly in UTRdb 2.0 the plant repertoire of UTRs derived from plants is greatly expanded compared to previous versions; a total of 8 452 197 UTRs from 112 plant species is included in our database, while only six species were available in previous releases. An average of 22 527 5′UTRs and 23 019 3′UTRs are available for every species incorporated in the database, however the number and size of UTRs associated with different species vary greatly; probably this is due to a series of factors including: evolutionary constraints/history, the quality of the available genome annotations, the total number of genes in the genome (Table 1).
Table 1.
Distribution of UTR annotations collected in UTRdb (full values are reported for the five most represented organisms in each Ensembl group)
| Species | 5′UTR | 3′UTR | ||
|---|---|---|---|---|
| N | Average L | N | Average L | |
| Vertebrates | ||||
| Homo_sapiens.GRCh38.107 | 93 457 | 231 | 87 932 | 1241 |
| Mus_musculus.GRCm39.107 | 56 214 | 222 | 52 526 | 1135 |
| Mus_spretus.SPRET_EiJ_v1.107 | 46 627 | 194 | 44 162 | 975 |
| Danio_rerio.GRCz11.107 | 33 326 | 226 | 30 928 | 953 |
| Sus_scrofa.Sscrofa11.1.107 | 36 098 | 338 | 36 757 | 1434 |
| Metazoans | ||||
| Crassostrea_gigas.GCA902806645v1.54 | 72 508 | 211 | 72 800 | 613 |
| Ascaris_suum.ASM18702v3.54 | 52 374 | 688 | 55 488 | 1618 |
| Pomacea_canaliculata_gca003073045v1.GCA003073045v1.54 | 39 028 | 425 | 39 051 | 1686 |
| Limulus_polyphemus_gca000517525v1.Limulus_polyphemus_2.1.2.54 | 34 796 | 240 | 33 600 | 1281 |
| Drosophila_melanogaster.BDGP6.32.54 | 30 091 | 316 | 30 211 | 600 |
| Plants | ||||
| Triticum_dicoccoides.WEWSeq_v.1.0.54 | 214 610 | 350 | 240 626 | 476 |
| Aegilops_tauschii.Aet_v4.0.54 | 201 304 | 407 | 222 362 | 553 |
| Hordeum_vulgare.IBSC_v2.42 | 184 618 | 365 | 203 233 | 549 |
| Zea_mays.B73_RefGen_v4.42 | 112 659 | 534 | 113 636 | 729 |
| Triticum_aestivum.IWGSC.54 | 88 479 | 192 | 92 320 | 345 |
UTRdb provides a rich set of functional annotations for UTRs. These include, but are not limited to: 6 306 354 evolutionarily conserved elements, 359 503 A-to-I RNA editing events, 49 040 m6A modifications and 58 553 426 nucleotide variants. Additionally, 10 755 925 candidate uORFs, 341 IRESs and 80 558 CAGEs are annotated in 5′UTR entries, while 180 195 alternative polyAdenylation sites are associated with 3′UTR entries (Table 2). UTRdb 2.0 has been structured as a relational database, built on the MySQL framework and easily accessible through an ad hoc web-interface developed in Bootstrap and HTML5.
Table 2.
Distribution of UTR functional annotations available in UTRdb 2.0
| Annotation | 5′UTR | 3′UTR | ||
|---|---|---|---|---|
| Human | Others | Human | Others | |
| uORFs | 78 276 | 10 677 649 | - | - |
| IRES | 239 | 102 | - | - |
| CAGEs | 2885 | 77 673 | - | - |
| Alternative PolyA sites | - | - | 124 015 | 56 180 |
| miRNAs targets | 51 428 | 7972 | 741 407 | 205 379 |
| Conserved blocks | 431 218 | 1 048 459 | 2 095 576 | 2 731 101 |
| RNA editing | 15 293 | 253 | 336 959 | 6998 |
| m6A modifications | 8050 | 6517 | 28 | 34 445 |
| Variants | 5 890 887 | 8 789 358 | 22 821 251 | 21 051 930 |
| Rfam motifs | 159 | 24 922 | 528 | 54 478 |
Web interface
Server-side operations including MySQL querying and retrieval are handled by Python scripts with the support of dedicated modules (e.g. pyMySQL, python CGI). UTR sequences and their associated annotations are displayed through dynamic and sortable tables generated by means of the DataTables jQuery module.
The overall architecture of the database has been completely redesigned with respect to previous releases. Indeed, annotation categories (i.e. miRNA targets, Rfam motifs, RNA editing sites and so on) are now organized in multiple databases, each including a set of organism-specific tables (generally linked by the transcript id as the unique primary key). This organization increases the scalability of the database allowing straightforward updates at organism level (i.e. when new genomic assemblies or annotations are released) without the need to shut down the entire service or reindex the full database.
Queries to UTRdb 2.0 can be performed using either a basic or an advanced interface. The former has been designed for quick queries, and supports limited metadata fields/filters: the type of UTR (5′, 3′, or both), the organism and a gene identifier, which can be specified in the form of a gene symbol, Ensembl gene or transcript IDs. To facilitate the user experience, an autocomplete function for gene symbols and gene and transcript ids has been implemented. The advanced interface, instead, has been designed to allow more complex queries enabling different filtering schemes through several parameters, which can be used individually or in combination. For example, users can filter entries according to UTRs size and/or the number of exons they span, the presence of Rfam motifs, miRNA target sites or epitranscriptomics modifications (A-to-I editing or m6A). In case of 5′UTRs, users could additionally filter the results for the presence of upstream ORFs (uORFs), Internal Ribosome Entry sites (IREs) or CAGE tags. For 3′UTR queries, instead, users can extract entries containing annotations of alternative polyadenylation sites. Since each UTRdb entry is identified by a unique identifier in the format {UTR_type_genome_assembly_transcript_id}, the database can also be queried by entering the unique id of the entry. Annotations of low quality for example UTRs of <10 nt in size, are flagged by a warning at the beginning of the entry. An overview of the search interfaces, as well as a sample entry, are displayed in Figure 2. Additionally, UTRdb 2.0 features explicative, interactive graphical representations of the annotations. The various functional annotations associated with any UTR can be downloaded in the form of standard JSON tables, while all UTR and related functional annotations for an organism can be retrieved in bulk through a dedicated download web page.
Figure 2.
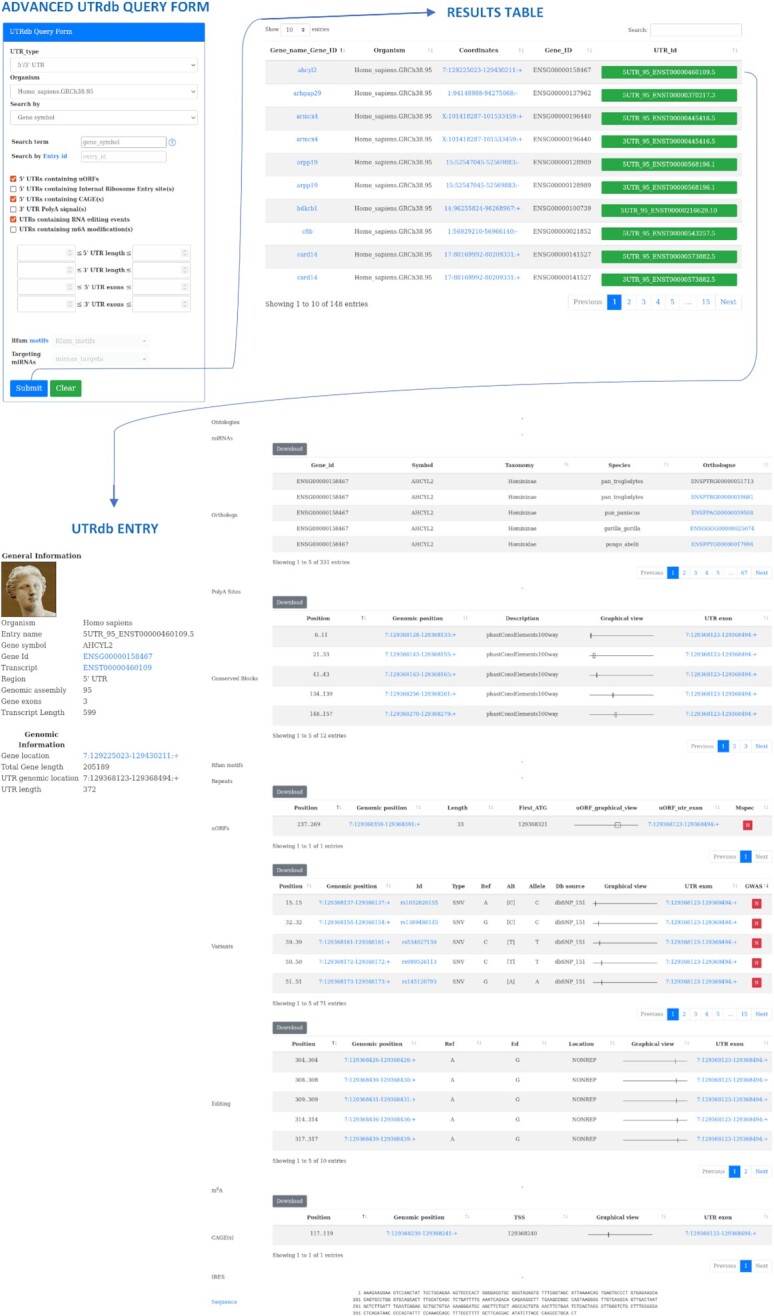
The advanced search front-end of UTRdb and an example of the tabular output as well as a sample entry.
UTRdb 2.0 UPDATE AND FUTURE PLANS
One of the key improvements of UTRdb 2.0 has been the re-design and re-implementation of the service's backend that, while less visible for users than the complete frontend overhaul, will provide solid ground for the future of UTRdb in terms of easier maintenance, more straightforward integration of new or updated annotations as they become available, and development of novel functionalities.
Considering the biological relevance of UTRs, we plan to regularly update UTRdb, in correspondence with the new releases of Ensembl, providing researchers an accurate, sustainable and accessible resource through the inclusion of annotations from novel organisms, the interoperability with other resources through crosslinks and a dedicated API. Additionally, we will work to enhance the quality of UTR annotations as new data (e.g. from long reads technologies such as Oxford Nanopore and PacBio) will become available and allow the referencing of data in both a location-independent and resource-dependent manner through resolvable identifiers (URLs) (by means of the identifiers.org registry) and constantly improving the compliance with FAIR principles (Findability, Accessibility, Interoperability and Reusability).
DATA AVAILABILITY
All the Python scripts to extract UTR sequences from Ensembl Fasta files and retrieve their associated annotations from multiple databases are available on our dedicated github repository at https://github.com/BioinfoUNIBA/UTRdb.
ACKNOWLEDGEMENTS
We kindly thank the ELIXIR-IT node through the ReCaS computing center at the University of Bari for hosting the database and providing the needed computational resources. We acknowledge the support of infrastructural facilities provided by PON CNR.Biomics (PIR01_00017) and ELIXIRxNextGenIT (IR0000010) projects. We also thank Laura Marra e Annarita Armenise for technical assistance.
Contributor Information
Claudio Lo Giudice, Department of Biosciences, Biotechnology and Environment, University of Bari A. Moro, 70126 Bari, Italy.
Federico Zambelli, Department of Biosciences, University of Milan, 20133 Milan, Italy; Institute of Biomembranes, Bioenergetics and Molecular Biotechnology, Consiglio Nazionale delle Ricerche, 70126 Bari, Italy.
Matteo Chiara, Department of Biosciences, University of Milan, 20133 Milan, Italy; Institute of Biomembranes, Bioenergetics and Molecular Biotechnology, Consiglio Nazionale delle Ricerche, 70126 Bari, Italy.
Giulio Pavesi, Department of Biosciences, University of Milan, 20133 Milan, Italy; Institute of Biomembranes, Bioenergetics and Molecular Biotechnology, Consiglio Nazionale delle Ricerche, 70126 Bari, Italy.
Marco Antonio Tangaro, Institute of Biomembranes, Bioenergetics and Molecular Biotechnology, Consiglio Nazionale delle Ricerche, 70126 Bari, Italy.
Ernesto Picardi, Department of Biosciences, Biotechnology and Environment, University of Bari A. Moro, 70126 Bari, Italy; Institute of Biomembranes, Bioenergetics and Molecular Biotechnology, Consiglio Nazionale delle Ricerche, 70126 Bari, Italy.
Graziano Pesole, Department of Biosciences, Biotechnology and Environment, University of Bari A. Moro, 70126 Bari, Italy; Institute of Biomembranes, Bioenergetics and Molecular Biotechnology, Consiglio Nazionale delle Ricerche, 70126 Bari, Italy.
FUNDING
European Life-science Infrastructure for Biological Information Italy; ELIXIR-CONVERGE [H2020-INFRADEV-2019-2]: connect and align ELIXIR Nodes to deliver sustainable FAIR life-science data management services [GA 871075]; ELIXIR-IT FOE by Ministero dell’Università e Ricerca; Italian Program PON [AIM1893457 to C.L.]: Attraction and International Mobility – Activity number 1 – Line 1 CUP H95G19000120006ATT1 funded by the Italian Ministry of Education, University and Research. Funding for open access charge: ELIXIR Italy.
Conflict of interest statement. None declared.
REFERENCES
- 1. Lonsdale J., Thomas J., Salvatore M., Phillips R., Lo E., Shad S., Hasz R., Walters G., Garcia F., Young N.et al.. The genotype-tissue expression (GTEx) project. Nat. Genet. 2013; 45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fortelny N., Overall C.M., Pavlidis P., Freue G.V.C.. Can we predict protein from mRNA levels. Nature. 2017; 547:E19–E20. [DOI] [PubMed] [Google Scholar]
- 3. Buccitelli C., Selbach M.. mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet. 2020; 21:630–644. [DOI] [PubMed] [Google Scholar]
- 4. Ghandi M., Huang F.W., Jané-Valbuena J., Kryukov G.V., Lo C.C., McDonald E.R., Barretina J., Gelfand E.T., Bielski C.M., Li H.et al.. Next-generation characterization of the cancer cell line encyclopedia. Nature. 2019; 569:503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Das S., Vera M., Gandin V., Singer R.H., Tutucci E.. Intracellular mRNA transport and localized translation. Nat. Rev. Mol. Cell Biol. 2021; 22:483–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mugridge J.S., Coller J., Gross J.D.. Structural and molecular mechanisms for the control of eukaryotic 5′–3′ mRNA decay. Nat. Struct. Mol. Biol. 2018; 25:1077–1085. [DOI] [PubMed] [Google Scholar]
- 7. Genuth N.R., Barna M.. Heterogeneity and specialized functions of translation machinery: from genes to organisms. Nat. Rev. Genet. 2018; 19:431–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pereira-Castro I., Moreira A.. On the function and relevance of alternative 3′-UTRs in gene expression regulation. WIREs RNA. 2021; 12:e1653. [DOI] [PubMed] [Google Scholar]
- 9. Leppek K., Das R., Barna M.. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2018; 19:158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sun L., Fazal F.M., Li P., Broughton J.P., Lee B., Tang L., Huang W., Kool E.T., Chang H.Y., Zhang Q.C.. RNA structure maps across mammalian cellular compartments. Nat. Struct. Mol. Biol. 2019; 26:322–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hinnebusch A.G., Ivanov I.P., Sonenberg N.. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science. 2016; 352:1413–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McGillivray P., Ault R., Pawashe M., Kitchen R., Balasubramanian S., Gerstein M.. A comprehensive catalog of predicted functional upstream open reading frames in humans. Nucleic Acids Res. 2018; 46:3326–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fabian M.R., Sonenberg N., Filipowicz W.. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010; 79:351–379. [DOI] [PubMed] [Google Scholar]
- 14. Mitschka S., Mayr C.. Context-specific regulation and function of mRNA alternative polyadenylation. Nat. Rev. Mol. Cell Biol. 2022; 10.1038/s41580-022-00507-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Steri M., Idda M.L., Whalen M.B., Orrù V.. Genetic variants in mRNA untranslated regions. Wiley Interdiscip. Rev. RNA. 2018; 9:e1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W.et al.. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018; 46:D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cunningham F., Allen J.E., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Austine-Orimoloye O., Azov A.G., Barnes I., Bennett R.et al.. Ensembl 2022. Nucleic Acids Res. 2022; 50:D988–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grillo G., Turi A., Licciulli F., Mignone F., Liuni S., Banfi S., Gennarino V.A., Horner D.S., Pavesi G., Picardi E.et al.. UTRdb and UTRsite (RELEASE 2010): a collection of sequences and regulatory motifs of the untranslated regions of eukaryotic mRNAs. Nucleic Acids Res. 2010; 38:D75–D80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yates A.D., Allen J., Amode R.M., Azov A.G., Barba M., Becerra A., Bhai J., Campbell L.I., Martinez Carbajo, Chakiachvili M.et al.. Ensembl genomes 2022: an expanding genome resource for non-vertebrates. Nucleic Acids Res. 2022; 50:D996–D1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kalvari I., Nawrocki E.P., Ontiveros-Palacios N., Argasinska J., Lamkiewicz K., Marz M., Griffiths-Jones S., Toffano-Nioche C., Gautheret D., Weinberg Z.et al.. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021; 49:D192–D200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S.et al.. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005; 15:1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mansi L., Tangaro M.A., Lo Giudice C., Flati T., Kopel E., Schaffer A.A., Castrignanò T., Chillemi G., Pesole G., Picardi E.. REDIportal: millions of novel A-to-I RNA editing events from thousands of RNAseq experiments. Nucleic Acids Res. 2021; 49:D1012–D1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Luo X., Li H., Liang J., Zhao Q., Xie Y., Ren J., Zuo Z.. RMVar: an updated database of functional variants involved in RNA modifications. Nucleic Acids Res. 2021; 49:D1405–D1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Perez-Riverol Y., Bai J., Bandla C., García-Seisdedos D., Hewapathirana S., Kamatchinathan S., Kundu D.J., Prakash A., Frericks-Zipper A., Eisenacher M.et al.. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022; 50:D543–D552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mokrejš M., Mašek T., Vopálenský V., Hlubuček P., Delbos P., Pospíšek M.. IRESite—a tool for the examination of viral and cellular internal ribosome entry sites. Nucleic Acids Res. 2010; 38:D131–D136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kozomara A., Birgaoanu M., Griffiths-Jones S.. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019; 47:D155–D162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Karagkouni D., Paraskevopoulou M.D., Chatzopoulos S., Vlachos I.S., Tastsoglou S., Kanellos I., Papadimitriou D., Kavakiotis I., Maniou S., Skoufos G.et al.. DIANA-TarBase v8: a decade-long collection of experimentally supported miRNA–gene interactions. Nucleic Acids Res. 2018; 46:D239–D245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lizio M., Harshbarger J., Shimoji H., Severin J., Kasukawa T., Sahin S., Abugessaisa I., Fukuda S., Hori F., Ishikawa-Kato S.et al.. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 2015; 16:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Müller S., Rycak L., Afonso-Grunz F., Winter P., Zawada A.M., Damrath E., Scheider J., Schmäh J., Koch I., Kahl G.et al.. APADB: a database for alternative polyadenylation and microRNA regulation events. Database J. Biol. Databases Curation. 2014; 2014:bau076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Griesemer D., Xue J.R., Reilly S.K., Ulirsch J.C., Kukreja K., Davis J.R., Kanai M., Yang D.K., Butts J.C., Guney M.H.et al.. Genome-wide functional screen of 3′UTR variants uncovers causal variants for human disease and evolution. Cell. 2021; 184:5247–5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fang E., Liu X., Li M., Zhang Z., Song L., Zhu B., Wu X., Liu J., Zhao D., Li Y.. Advances in COVID-19 mRNA vaccine development. Signal Transduct. Target. Ther. 2022; 7:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yates A., Beal K., Keenan S., McLaren W., Pignatelli M., Ritchie G.R.S., Ruffier M., Taylor K., Vullo A., Flicek P.. The ensembl REST API: ensembl data for any language. Bioinformatics. 2015; 31:143–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buniello A., MacArthur J.A.L., Cerezo M., Harris L.W., Hayhurst J., Malangone C., McMahon A., Morales J., Mountjoy E., Sollis E.et al.. The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019; 47:D1005–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee B.T., Barber G.P., Benet-Pagès A., Casper J., Clawson H., Diekhans M., Fischer C., Gonzalez J.N., Hinrichs A.S., Lee C.M.et al.. The UCSC genome browser database: 2022 update. Nucleic Acids Res. 2022; 50:D1115–D1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hinrichs A.S., Karolchik D., Baertsch R., Barber G.P., Bejerano G., Clawson H., Diekhans M., Furey T.S., Harte R.A., Hsu F.et al.. The UCSC genome browser database: update 2006. Nucleic Acids Res. 2006; 34:D590–D598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Enright A.J., John B., Gaul U., Tuschl T., Sander C., Marks D.S.. MicroRNA targets in Drosophila. Genome Biol. 2003; 5:R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gilman P., Janzou S., Guittet D., Freeman J., DiOrio N., Blair N., Boyd M., Neises T., Wagner M.. PySAM (Python Wrapper for system advisor model ‘SAM’). 2019; 10.11578/dc.20190903.1. [DOI]
- 38. Iacono M., Mignone F., Pesole G.. uAUG and uORFs in human and rodent 5′untranslated mRNAs. Gene. 2005; 349:97–105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the Python scripts to extract UTR sequences from Ensembl Fasta files and retrieve their associated annotations from multiple databases are available on our dedicated github repository at https://github.com/BioinfoUNIBA/UTRdb.