


Abstract
The National Institute of Allergy and Infectious Diseases (NIAID) established the Bioinformatics Resource Center (BRC) program to assist researchers with analyzing the growing body of genome sequence and other omics-related data. In this report, we describe the merger of the PAThosystems Resource Integration Center (PATRIC), the Influenza Research Database (IRD) and the Virus Pathogen Database and Analysis Resource (ViPR) BRCs to form the Bacterial and Viral Bioinformatics Resource Center (BV-BRC) https://www.bv-brc.org/. The combined BV-BRC leverages the functionality of the bacterial and viral resources to provide a unified data model, enhanced web-based visualization and analysis tools, bioinformatics services, and a powerful suite of command line tools that benefit the bacterial and viral research communities.
INTRODUCTION
In 2004, the National Institute of Allergy and Infectious Diseases (NIAID) established the Bioinformatic Resource Center (BRC) program to facilitate research on pathogens by integrating genomic and other biological data (1). The goal was to provide the necessary data environment, bioinformatics tools, and workflows to enhance ongoing basic and applied research. The centers have evolved over the years, such that there are currently two BRCs, one that supports research on eukaryotic pathogens and invertebrate vectors (VEuPathDB) (2) and one that supports research on bacterial and viral pathogens (BV-BRC).
The Bacterial and Viral Bioinformatics Research Center (BV-BRC) was formed in 2019 through a merger of three BRC resources: the PAThosystems Resource Integration Center (PATRIC) (3), the Influenza Research Database (IRD) (4) and the Virus Pathogen Database and Analysis Resource (ViPR) (5). PATRIC was one of the original BRCs and was designed to support bioinformatics for bacterial pathogens (6). In 2012, the National Microbial Pathogen Database Resource (NMPDR) (7) was merged into the PATRIC BRC, bringing in the well-known SEED and RAST annotation resources (8). IRD, which was originally part of the BioHealthBase BRC (9), and the ViPR BRC (5,10) were designed to support analyses on influenza and a variety of other important human viral pathogens, respectively. The current, combined BV-BRC resource is supported by teams of researchers from the University of Chicago, the J. Craig Venter Institute, the University of Virginia and the Fellowship for Interpretation of Genomes, as well as many close collaborators at other institutions.
The PATRIC and IRD/ViPR resources differed in their approaches to aiding the research community. For many years, the PATRIC resource has been providing a unified browsing environment covering bacterial pathogens, along with publicly available non-pathogenic bacterial and archaeal genomes, plasmids, and phages for comparison. The underlying protein annotation system, RAST (11), enabled the comparison of orthologous genes across large phylogenetic distances and powered many of the website visualizations. Following on the success of RAST, the PATRIC resource developed a robust set of over 15 analysis services enabling genome assembly, RNA-Seq analysis, whole genome alignment, phylogenetic tree construction, and many other complex bioinformatic workflows. IRD and ViPR were more narrowly focused on developing user visual analytics and tools that were tailored to the major category A-C viral priority pathogens. This enabled the development of robust and focused searching and comparative analyses website tools. IRD and ViPR were also well known for the development of their meta-CATS tool for comparing sets of sequences based on metadata (12), VIGOR tool for annotating viral genomes (13), and Archaeopteryx.js tool for visualizing phylogenetic trees (14). Thus, one of the goals in merging the resources was to retain the functionality of these bacterial and viral resources and extend their functionality to other user communities.
While the merger of these BRC resources presented significant engineering challenges, it also provided important advantages to the user communities supported. First, there is enhanced efficiency in providing a single unified website, suite of analysis tools, and back-end database and computing architecture. Second, it enables the synergistic development of tools that help researchers studying the gamut of bacterial and viral pathogens. Third, providing a centralized resource with educational and outreach materials helps lower the learning curve for those studying a variety of pathogens using various analysis methods. Finally, as exemplified by the SARS-CoV-2 pandemic, it provides a hub for tailoring data storage and retrieval, analysis tools, services, and reporting in response to infectious disease outbreaks with public health implications.
In this paper, we describe the BV-BRC resource for the first time. We highlight the new functionality that the merger provides for the bacterial and viral research communities, as well as the improvements to the underlying computing architecture. We also describe new functionality that has been developed since the merger that is unique to the BV-BRC.
DATA
Genomes
The BV-BRC hosts genomic and other related data types for bacterial and viral pathogens with an original focus on the NIAID Category A-C Priority Pathogens (https://www.niaid.nih.gov/research/emerging-infectious-diseases-pathogens). To allow researchers to compare these human pathogens to their non-pathogenic relatives and understand the virulence and pathogenicity characteristics that set them apart, the BV-BRC integrates all publicly available bacterial and archaeal genome sequences and features as well as all genome sequences and features from the virus families containing human pathogens. In addition, the BV-BRC hosts all publicly available bacteriophage genome sequences and their bacterial host metadata to support research on bacteriophages and phage therapy, as well as a select set of eukaryotic host genome sequences to support research on host-pathogen interactions and host response to bacterial and viral infections. As of August 2022, the BV-BRC hosts over 600 000 bacterial genomes, 11 000 archaeal genomes and 8.5 million viral genomes, including over 6 million SARS-CoV-2 genomes, 22 000 phage genomes, and 10 eukaryotic host genomes (Figure 1).
Figure 1.
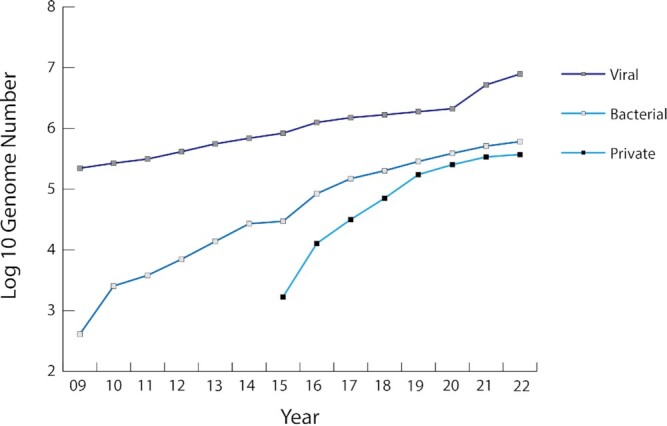
Growth of genome sequences in the BRC resources. Dates prior to 2020 include separate data for IRD, ViPR and PATRIC; dates after 2020 include data from BV-BRC. The large jump in viral genomes in 2020 coincides with the SARS-CoV-2 pandemic. Private genomes represent sequences uploaded and analyzed by individual users.
Most of these genomes are gathered from the NCBI GenBank database (15), with new genomes being integrated on a daily basis. In addition, many genomes with useful metadata have been assembled from the NCBI Sequence Read Archive (16) and then integrated into the BV-BRC (17).
Genome annotation
All of the bacterial and archaeal genomes in the BV-BRC are consistently annotated using the BV-BRC Annotation Service, which uses RASTtk (11) to provide accurate and high-quality annotations for genes, protein functions, protein families, metabolic pathways and subsystems (18). The annotation also includes the identification of genes of special interest to the infectious disease research community, such as antimicrobial resistance genes (19–21), virulence factors (22,23), essential genes, drug targets (24,25), transporters (26), and human homologs. The consistent annotation across all bacterial and archaeal genomes allows researchers to perform comparative genomic analysis in closely related as well as distant taxa using local or global protein families, metabolic pathways, and subsystems. Similarly, for select viral families, consistent annotations are provided using VIGOR4 (13), which uses manually curated reference databases to annotate genes, proteins, protein functions, and mature peptides. In the beginning of the COVID-19 pandemic, a specialized SARS-CoV-2 reference database was developed for VIGOR4, which has been used to provide accurate and consistent annotations across the SARS-CoV-2 genomes in BV-BRC. The VIGOR4 reference database collection was then expanded to support five subgenera in the genus Betacoronavirus (Embecovirus, Hibecovirus, Novecovirus, Merbecovirus, Sarbecovirus), three families in the order Bunyavirales (Arenaviridae, Phenuiviridae, Phasmaviridae), and viruses belonging to the species Monkeypox. Consistent annotations across all bacteriophage genomes are provided using Phanotate (27) in the BV-BRC Annotation Service.
Genome metadata
The BV-BRC uses a combination of automated and manual metadata collection and curation to collect and augment high value metadata attributes relating to the bacterial and viral pathogens, host, and other clinical metadata to ensure consistency and accuracy, which are crucial for supporting comparative genomics, epidemiological analysis, and development of predictive machine learning models. The genome ingestion process uses automated scripts to gather relevant metadata from the GenBank (15) and BioSample (16) records. In addition, rule-based parsers are used to derive missing metadata, such as host name, geographic location, and collection year, from strain name or comment fields. Select metadata attributes, such as isolation country, geographic group, host common name, and host group, are harmonized using authoritative data sources (e.g., bird species from the Integrated Taxonomic Information System – ITIS and eBird) and manually curated metadata mapping tables to provide for consistent and comprehensive metadata annotations. In addition, the BV-BRC currently maintains a collection of laboratory-derived antimicrobial susceptibility test (AST) results for 90 829 bacterial genomes curated from NCBI and ∼250 publications (17).
Non-genomic data
In addition to consistently annotated genomes and metadata, the BV-BRC provides several other data types which are linked to these genomes, such as domains and motifs, immune epitopes, and protein structures. These data types were available in ViPR and IRD, but not PATRIC. In the BV-BRC, these data types are now available for both bacteria and viruses and are accessible from the genome and protein-level pages. The domains and motifs are computed for all proteins from NCBI-designated reference and representative genomes by running an InterProScan search (28). The experimentally characterized epitopes are gathered from the Immune Epitope Database (IEDB) (29), mapped to corresponding proteins in BV-BRC using UniProt ID mapping (30) and exact sequence matching. The epitopes have played an important role in assessing the emerging variations in SARS-CoV-2 Spike protein and their impact on public health. The experimentally derived protein structures are collected from PDB (31) and then mapped to the corresponding bacterial and viral proteins in the BV-BRC. As of August 2022, the BV-BRC hosts over 170 million predicted domains and motifs, 300 000 experimentally characterized epitopes and 77 000 protein structures. Other non-genomic data types include protein-protein interactions, transcriptomics and host-response, and surveillance and serology data inherited from PATRIC, IRD and ViPR. These data types are hosted using a unified data model and database schema to provide equitable search and analysis capabilities across bacterial and viral datasets.
USER INTERFACE/WEBSITE
One of the key undertakings of the BV-BRC was the development of a unified BV-BRC website, which supports the needs of both the bacterial and viral research communities. The BV-BRC website was built by leveraging the existing PATRIC website framework and user interface (UI) design, and then extending it to incorporate the novel data types, analysis tools, and use cases from IRD and ViPR, thus combining the strengths of these existing resources and reusing existing components. The website maintains a unified look and feel across all organisms by using common UI elements to support searching and browsing of the data, while supporting context-driven customizations where they are needed to highlight the data or metadata attributes that are important for a given organism. For example, the BV-BRC uses tabbed navigation to provide access to various data types available for a given taxon, genome, or protein. The tabs shown for a given taxon are customized for bacteria and viruses based on the applicability and availability of certain data types. Similarly, the tables and table filters also use context-driven customizations to show default attributes that are most relevant for bacteria or viruses.
New home page
The new BV-BRC home page (https://www.bv-brc.org) is designed to provide easy access to all available data and analysis tools and to help existing PATRIC and IRD/ViPR users successfully transition to the new BV-BRC website (Figure 2). The home page highlights key aspects of the BV-BRC website, including popular searches, analysis tools and services, the private user workspace function, batch access via the data API, Command-line Interface (CLI), the FTP site, and extensive help documentation. The home page also provides access to special instructional pages that provide mapping of key website functionality and tools between the new BV-BRC website and the legacy PATRIC, IRD and ViPR websites and highlight the new features for bacterial and viral researchers.
Figure 2.

The BV-BRC home page. Various ways for accessing bacterial and viral data are shown.
New advanced searches
The legacy PATRIC website offered data browsing by taxon with data-type-specific tabs and global searching as the primary way of finding data of interest. The ViPR and IRD websites allowed users to first select the viral family of interest and then used data-type-specific advanced searches as primary entry point for searching or browsing the available data for a viral family. The new BV-BRC website provides a global search as well as data type specific advanced searches to support both of these user workflows. The global search allows users to quickly find data of interest by using simple keywords or identifiers as search terms, select the data matching those terms, and then further refine the results as needed using progressive filters available from the result table. The advanced searches allow users to restrict the scope of their search based on the organism or taxon of interest and define one or more search criteria based on attributes relevant for a given data type. Together, these search mechanisms combine the best of the search capabilities provided by PATRIC, IRD and ViPR.
VISUAL ANALYTICS TOOLS
The BV-BRC resource provides a variety of interactive visual analytics tools to allow researchers to visually explore the complex data and gain further insights from the analysis results. Most of these tools existed in the legacy PATRIC, IRD and ViPR websites and were designed primarily to support either bacterial or viral data. The BV-BRC has now combined the tools from these resources and expands their scope to support both bacterial and viral analyses. Below is a brief description of the tools that have been significantly enhanced or are new to either PATRIC or IRD/ViPR (Figure 3).
Figure 3.

New and enhanced visual analytics tools at BV-BRC. Top left: the surveillance map viewer based on Google Maps, top right: the Archaeopteryx Tree Viewer for the Gene tree and Phylogenetic tree services, bottom left: the enhanced compare regions viewer, bottom right: The new protein structure viewer.
New phylogenetic tree viewer
The BV-BRC includes the Archaeopteryx Tree Viewer (14) for both the Gene Tree and Phylogenetic Tree services. Archaeopteryx.js is a software tool for the visualization and analysis of highly annotated phylogenetic trees that runs in the web browser (source code available at https://github.com/cmzmasek/archaeopteryx-js). The visualization provided by Archaeopteryx.js allows specifying and coloring the annotation field(s) to be used as the tip labels. These include taxonomic classifications, host names, geographic locations, and protein functions. Users can zoom, scroll, change label sizes, and optionally hide overlapping tip labels. Selected tree nodes can be collapsed, and selected tips can be collected into groups and saved to the workspace or can be viewed in a tabular page showing extensive data for each entity. The tool can be used to display fairly large trees (up to 3500 external nodes on a medium-range desktop or notebook computer).
New protein structure viewer
The BV-BRC has a new interactive protein structure viewer, implemented using Mol* (32). This viewer replaces the JSMol Viewer (33) previously used in ViPR and IRD and provides better performance and functionality. The new viewer shows the protein sequence and 3D structure simultaneously and allows users to select a single amino acid or region of interest on the protein sequence and directly zoom in to that region in the 3D-structure view or select a position in the 3D-structure view and highlight that position in the sequence view. This feature is quite helpful when assessing the impact of mutations on the structure and function of the protein. The viewer also integrates domains, motifs, epitopes, and other manually curated sequence features and allows users to highlight them in the protein structure.
New surveillance map viewer
The BV-BRC hosts surveillance data for Influenza virus, collected from human, animals, and birds as part of the NIAID CEIRS program (34). The data are accessible using either the Surveillance Search or the Surveillance Tab from the Influenza taxon-level pages and are displayed as an interactive table, which allows users to find the records of interest using metadata-based progressive filters, select one or more records and then view them in the interactive Surveillance Map Viewer. The viewer is implemented using Google Maps, which is overlaid with Bird Migration Flyways. The viewer provides a summary of the number of records by location and allows users to filter by location to see total samples collected from that location and percentage of the samples that tested positive for Influenza virus. In the future, this viewer will be generalized to visualize surveillance records for other viruses and antimicrobial resistance (AMR) records for bacterial pathogens.
Genome browser
The BV-BRC has an interactive linear genome browser, implemented using JBrowse (35), which allows users to visually explore both bacterial and viral genomes. Users can zoom out to see entire genomes and contigs or zoom in to see individual nucleotides. The viewer loads data dynamically by querying the database in real-time, eliminating the need to maintain precomputed files and images. It also allows users to load a variety of computational and experimental datasets as separate tracks and view them simultaneously, providing an integrated view of the data. The results from several services, including Variation Analysis, RNA-Seq Analysis and Tn-seq Analysis, are linked to the genome browser to enable users to visually explore the content of corresponding BAM, WIG or VCF files.
Enhanced MSA viewer
Nucleotide and protein alignments are an important part of any genomic analysis, and the three original BRCs provided ways to both generate and view alignments using extensions to the BioJS MSAViewer for the visualization (36). The ViPR and IRD resources included the advanced ability of allowing researchers to view the metadata associated with the genome or strain for each of the proteins in the alignment. The BV-BRC has incorporated this functionality and now bacterial and viral researchers can see the gene or genome ID, genome name, accession number, species, or strain, geographic or host group, isolation country, collection year, subtype, lineage or clade available for their data mapped to the alignment.
Enhanced compare region viewer
Originally part of the RAST server (37), the Compare Region Viewer was designed to show the genomic neighborhood of a protein-coding gene across a phylogenetic distance. Along with the ability to view the genes in a particular region and filter on reference, representative or all public genomes in the resource, this viewer also enables selecting the number of regions, size, and type of protein family. New improvements have been added in BV-BRC that include the ability to view a genome group or feature group of interest. The Compare Region Viewer also includes a high resolution SVG image that can be exported.
Tools for SARS-CoV-2 analysis
The SARS-CoV-2 pandemic began shortly after the creation of the merged BV-BRC resource. The outbreak elicited an unprecedented global response and a subsequent explosion in viral genome sequence data and epidemiological metadata. Along with these new data came the need for bioinformatic tools and services to understand the outbreak. The BV-BRC currently mirrors the publicly available and reusable collection of SARS-CoV-2 genomes from GenBank (38), updating the collection of genomes daily. The genomes are uniformly annotated with VIGOR4 (13) and can be browsed on the website. At the time of writing, there over 6 million public SARS-CoV-2 genomes in the BV-BRC.
In addition to simply hosting the genomes, a SARS-CoV-2 Variant Tracker component (https://www.bv-brc.org/view/VariantLineage/#view_tab=overview) was developed in the BV-BRC (Figure 4) to enable users to browse current and past lineages of concern and interest, variants, covariants, and their prevalence by isolation date, geographic location, and other metadata fields. Important genomic regions are also displayed in the genome browser with over 100 manually curated tracks, including gene and protein annotations, functional features, immune epitopes, primer and probe sites, variants of concern (VOC) and variants of interest (VOI), drug resistant mutations, experimental mutational scanning and binding affinity data, and positive and negative selection sites. The tracker also employs a heuristic that is used to compute growth rates for variants to highlight those that may cause future waves of infection (39).
Figure 4.

Overview of the SARS-CoV-2 Variant Tracker. The top left panel shows the landing page for the Delta variant of concern, the top right panel shows the prevalence of the D614G variant, the bottom left panel shows variant frequency by month in the USA, and the bottom right shows tracks that are available for viewing from the genome browser, which include open reading frames, active sites, antibody epitopes, and antibody binding escape data.
SERVICES
The BV-BRC provides access to a variety of complex bioinformatic workflow services. These services capture common bioinformatic protocols and enable them through the website user interface and the command line. Users provide input data and receive outputs from each service through their private workspace. Computing for these services and data storage in the workspace are free to the public. Access to the workspace and services requires users to register for an account; the BV-BRC currently has over 35 000 registered users (Figure 5). Access to these bioinformatic services was a major driver of PATRIC usage. Prior to the merger in September 2019, over 270 000 private user jobs had been submitted to PATRIC, with an average of 4657 jobs per month. Since then, over 650 000 jobs have been submitted by our users, averaging 19 716 jobs per month (Figure 6). In addition to increases in users and data volume, this large increase in user jobs is also due to the incorporation of several IRD/ViPR workflows into the service architecture, and the development of a fast-track service queue for shorter jobs, such as small BLAST service jobs, which were originally run outside of the service architecture in PATRIC.
Figure 5.
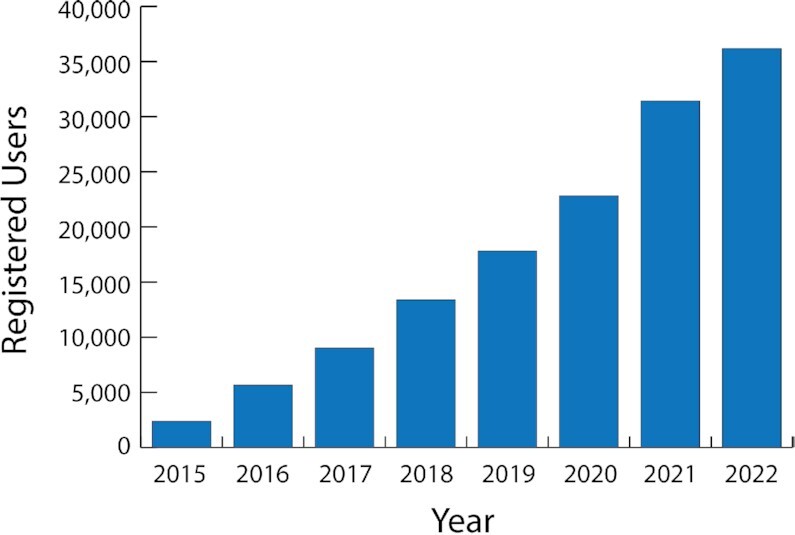
Cumulative growth in registered users for BV-BRC. For dates prior to 2020, data have been combined for PATRIC, IRD and ViPR.
Figure 6.
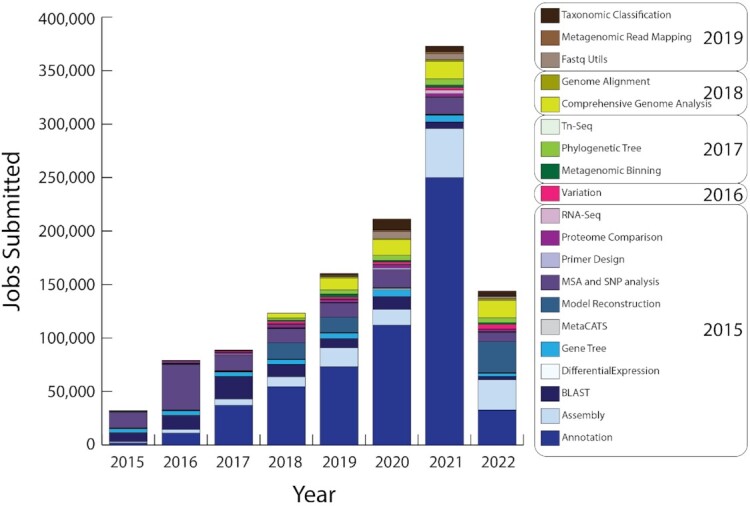
Growth in user jobs and services. The bar chart shows the growth in jobs per year starting with the PATRIC services in 2015. Boxes show the approximate year in which each service was either developed or integrated.
New services for PATRIC users
Many of the BV-BRC services are new to either the PATRIC or IRD/ViPR user communities. For PATRIC users, there are three new services that have been brought in from IRD and ViPR (Table 1). The Meta-CATS (Metadata driven Comparative Analysis Tool for Sequences) service (12) allows users to identify statistically significant SNPs between two or more feature groups of genes or proteins. The Primer Design service uses Primer3 (40) to enable the design of PCR primers for a given sequence. The Gene Tree service enables users to build phylogenetic trees from a feature group of genes or proteins of their choice using MAFFT (41) to build the alignment, and RaxML (42), PhyML (43), or FastTree (44) to infer the phylogenetic relationships. It can also align and build trees from entire viral genomes or segments as large as Monkeypox.
Table 1.
Notable improvements in BV-BRC that either did not exist in PATRIC, IRD/ViPR, or both prior to the merger
| PATRIC | IRD/ViPR | BV-BRC | |
|---|---|---|---|
| Data types | |||
| Domains and motifs | No | Yes | Yes |
| Epitopes | No | Yes | Yes |
| Protein structures | No | Yes | Yes |
| Surveillance and serology | No | Yes | Yes |
| Services | |||
| Assembly (de novo) | Yes | No | Yes |
| SARS-Cov-2 assembly and annotation | No | No | Yes |
| SARS-CoV-2 variant tracking | No | No | Yes |
| Similar genome finder | Yes | No | Yes |
| Meta-CATS | No | Yes | Yes |
| Whole genome alignment | Yes | No | Yes |
| Primer design | No | Yes | Yes |
| Gene tree (user-selected genes/proteins) | No | Yes | Yes |
| Proteome comparison | Yes | No | Yes |
| Taxonomic classification | Yes | No | Yes |
| Metagenomic binning | Yes | No | Yes |
| RNA-Seq analysis | Yes | No | Yes |
| Fastq utilities | Yes | No | Yes |
| Command line interface | Yes | No | Yes |
New services for IRD and ViPR users
For IRD and ViPR users, there are a variety of new services that are now accessible through the BV-BRC (Table 1), including the Fastq Utilities, which enables quality filtering and assessment of sequence reads; Assembly, which preforms de novo assembly of long or short reads, single cell sequencing reads, or metagenomes; RNA-Seq Analysis, which enables the comparison of expression differences between sets of reads in an RNA-Seq study; Whole Genome Alignment, which uses Mauve (45) to align whole genomes; Proteome Comparison, which enables a bidirectional BLAST (46) best hits search between two or more genomes; Similar Genome Finder, which uses Mash (47) to identify the most similar genomes in the database; Taxonomic Classification, which uses Kraken 2 to identify the taxa in a set of reads or contigs (48); and Metagenomic Binning, which attempts to bin whole genomes from metagenomic assemblies and uses CheckV to evaluate genome quality and completeness for viral metagenome assembled genomes (49).
New SARS-CoV-2 assembly and annotation service
Following the merger of PATRIC, IRD, and ViPR, the SARS-CoV-2 Assembly and Annotation service was developed (https://www.bv-brc.org/app/ComprehensiveSARS2Analysis) to enable users to perform reference-guided assemblies of SARS-CoV-2 genomes against the Wuhan-Hu-1 reference genome (GenBank ID: NC_045512.2). The assemblies are constructed by performing quality trimming with seqtk (https://github.com/lh3/seqtk.git), primer trimming with iVar (50), aligning reads against the reference with minimap2 (51), and calling the consensus sequence with iVar (50). Users can select from several popular primer schemes for the primer trimming step. The assembly output consists of the consensus sequence, list of variations, images of the read depths, and several intermediate files including the read pileup plots (52). The assembled consensus sequence is then annotated using VIGOR4 (13), which calls all the open reading frames and mature peptides. Finally, lineages are called using PANGOLIN (https://cov-lineages.org/resources/pangolin.html). All privately annotated genomes are indexed in the BV-BRC Solr database and can be compared to genomes in the public collection.
SYSTEM ARCHITECTURE AND INFRASTRUCTURE
The merger of PATRIC, IRD and ViPR to form the BV-BRC has required considerable engineering of the backend environment to maintain the data and functionality of each of the original resources and to adapt to the resulting increase in user volume. Preexisting user accounts have been seamlessly merged into a unified private BV-BRC workspace environment. Updates to the backend computing environment have provided rapid web browsing and efficient and equitable turnaround times for user-submitted jobs. Software engineering was also required to create an environment where the underlying software from all services can be run harmoniously on different operating systems, as well as being FAIR compliant (53). The overall BV-BRC system consists of several key components including the database, workspace, application framework, authentication service, and command line interface.
Database
The BV-BRC database provides a highly scalable and efficient system for the storage, management, search, and retrieval of all structured data and related metadata. The BV-BRC data model unifies the data models that were previously used by the PATRIC and IRD/ViPR systems to support a variety of bacterial and viral data types, including genomes, genomic features (genes, RNAs, proteins, mature peptides, etc.), transcriptomics, protein functions, metabolic pathways, protein families, domains, protein structures, etc. It also supports data types that are of special interest for epidemiological analysis, such as sequence typing, pathogen-specific clade and lineage assignments, AMR genotypes and phenotypes, surveillance data, immune epitopes, and serology data. The data model uses community-accepted data and metadata standards (54) and is highly extensible to support new data types and evolving metadata standards.
The database system is implemented using the state-of-the-art open-source indexing platform, Apache Solr. The database instance is hosted using a distributed SolrCloud architecture to meet high performance and scalability requirements. The database has 38 collections, and each collection has two or more replicas. The largest collection, “genome_feature”, which hosts the genes and proteins from all the genomes in BV-BRC, has more than 6.1 billion records. As of August 2022, the BV-BRC SolrCloud consists of 24 Solr instances hosted on over six machines, each with Xeon(R) Gold 6248 CPU @ 2.50GHz processors, 80 compute cores, 790GB memory, and 10TB SSD.
Workspace
The BV-BRC workspace provides bulk data upload, storage, and download services for private user data and analysis results and allow users to share data with their collaborators or make it public. The same workspace service has been successfully used in PATRIC for over 10 years and currently hosts over 225 TB of private user data. The data storage for the workspace utilizes a NetApp FAST8040 network storage appliance configured with 1.4 petabytes of storage; access to this storage is provided via a Shock service (55). The metadata storage for the workspace is provided via a replicated MongoDB database server. The workspace is accessible using an interactive web browser, programmatic APIs, or an FTP gateway. The data stored in the workspace is private to the owner of the data. The owner can grant or revoke access to the data to other users or make it public.
Application framework
The BV-BRC application framework is a modular and extensible microservice architecture for integrating high-throughput analysis services, such as genome assembly, annotation, multiple sequence alignment, phylogenetic trees, etc. The application framework runs on a model of asynchronous computation to support all non-trivial analyses, from quick BLAST (46) searches to multi-day metagenomic analyses. These computations are modeled as tasks; each task has a set of parameters encoded in a compact textual form. The parameters define the inputs to the task (which come from the BV-BRC workspace, from the BV-BRC Solr database, or directly from external sources such as the NCBI Sequence Read Archive), the location for the outputs in the BV-BRC workspace, and any required application parameters. The tasks are managed by a scheduler that maintains metadata about the tasks (ownership, execution status, runtime and memory use for completed jobs, application used, task parameter data, etc.) and schedules the jobs for execution.
By default, a local computational cluster is used as the backend for the task scheduler. The cluster is comprised of a collection of standard Linux servers ranging in processor size from 8 to 128 cores, with available memory from 16 GB to 768 GB. During typical load times, the system has ∼24 nodes and 3166 cores allocated. The cluster is managed by a SLURM scheduler (56); a module in the task scheduler submits task instances to the SLURM scheduler as job requests. The SLURM scheduler is configured to support fair-share allocation to enable users to submit many jobs to the BV-BRC without preventing job allocation to other users. It is also configured with fast-run queues to enable quick turnaround for small jobs.
The BV-BRC analysis backend is enabled by two key technologies. The first is the BV-BRC workspace. Our ability to enable transfer of input data from the workspace to the compute node and results from the compute node back to the workspace frees the compute environment from any tight binding to specific shared resources such as network filesystems. The second is our use of Singularity containers (57) to encapsulate the software environment required for the analysis. The analysis backend is comprised of code from multiple modules, each managed as an independent module in our GitHub repository (https://github.com/BV-BRC). The BV-BRC build system provides the integration logic to combine these multiple modules into a single deployable system. The BV-BRC developers can deploy this system on their own machines to do testing; for production, the build system creates Singularity containers from the modules. These containers are subject to a rigorous quality assurance process, and when they pass, they are configured in the scheduler to be used first in our alpha and/or beta test systems. When we have final approval from our test users, the container is promoted to be the new production backend. The use of containers also isolates the analysis code from the Linux distribution and version found on the compute nodes. This enables the seamless allocation of additional compute resources when job volumes are high.
Website and authentication
The BV-BRC website provides interactive access to the public and private data, user workspace, analysis services, and visual analytics tools. The website is developed using a JavaScript/Node.js stack and other modern web technologies. It utilizes OAuth 2.0 bearer tokens to provide authorized access to private data. The authentication service manages a user database and allow users to request the creation of a token given a username and password pair using a REST API.
Application programming and command-line interfaces
While many users access the BRC resources only through the BV-BRC website, there are users who prefer to use these resources via a programming interface. The BV-BRC Data API provides programmatic access to all the indexed data hosted in the BV-BRC database via easy-to-use REST API (https://www.bv-brc.org/api/doc/). The BV-BRC Command-line Interface (CLI, https://github.com/BV-BRC/BV-BRC-CLI/releases) provides command line access to all the public and private data as well as analysis services. We maintain and distribute CLI Toolkits for Mac, Windows, and Linux operating systems that expose command line programs to access all the data managed by the BV-BRC Solr database, including private user data, allow batch upload and download of the data to and from the BV-BRC workspace, and enable batch submissions for analysis services.
LEARNING TO USE THE BV-BRC
The BV-BRC team provides a variety of educational materials so that users may become proficient at using the tools that are available in the resource. Extensive help documentation (https://www.bv-brc.org/docs/) provides access to reference guides and tutorials describing how to use website tools, each service, and the command line. Tutorial links are also provided on the landing page of each service, along with information guides stepping the users through the inputs for each job. The BV-BRC team regularly conducts webinars and in-person workshops that are open to the public and are often recorded and placed under the “Webinars and Videos” link on the website and available from the BV-BRC YouTube channel (https://www.youtube.com/c/BVBRC). The webinars and workshops are usually driven by a research theme, with the intention of covering as much of the functionality of the resource as possible. We have also developed a bacterial bioinformatics Massive Online Open Course (MOOC) course that teaches the fundamentals of bacterial bioinformatics while stepping students through analyses on the resource (https://www.coursera.org/learn/informatics). At the time of writing, the bacterial MOOC has had over 7000 registrants.
For individuals seeking help with specific job submissions, there is a ‘Report Issue’ link for each job on the jobs page. This link generates a structured form reporting the necessary information for tracking problems with the job. There is also a ‘Rerun’ button that will reload the submission page with the previously submitted data and selections, allowing the user to adjust their original job parameters rather than starting from scratch. The BV-BRC also now provides a service suggestion tool for groups of data in the workspace. Clicking on this will provide a list of possible tools and services that can be deployed for the grouped data set. For individual help with more general issues, users can contact the team directly under the ‘Contact Us’ link under the ‘About’ tab, and the query will be routed to the appropriate team member with the expertise to answer the question.
FUTURE DIRECTIONS
There are several tasks remaining in the merger of PATRIC, IRD and ViPR that are required for providing complete cross functionality for bacterial and viral analyses, which will be the focus of efforts over the next year. For example, the original PATRIC Compare Regions and Protein Family Sorter tools use protein families to compare sets of orthologous genes, and the original PATRIC Phylogenetic Tree service uses protein families to build concatenated alignments. In order to make these tools work for viruses, we will be updating our protein family generation algorithm (58) to incorporate the Strict Ortholog Groups concept (59,60) and recomputing protein families to accommodate the viral genomes (58). We will also begin incorporating predicted protein structures for select bacterial and viral reference genomes using AlphaFold (61). The Genome Assembly service is another example where coverage is incomplete. Since this tool was developed out of PATRIC, the focus had been on de novo assembly of reads from whole genome shotgun sequencing. However, amplicon sequencing with reference-guided assembly is often more useful for studying viruses, so the assembly service will be updated to better accommodate common viral assembly protocols. We are also working to harmonize metadata and browsing, including curating host metadata and other metadata fields. This in turn will fuel the development of comparative analysis tools, such as those that rely on artificial intelligence techniques, to better leverage the metadata collection. Additional efforts, including the generation and curation of new protein annotations in the SEED, integration of antimicrobial resistance metadata, further development of tools and visualizations for aiding in the SARS-CoV-2 pandemic, and providing education and outreach are important tasks that we continue to carry forward.
DATA AVAILABILITY
All data and links to the FTP site and command line interface tools can be found on the BV-BRC website, https://www.bv-brc.org/. All project code can be found at our GitHub repo, https://github.com/BV-BRC.
Notes
In memory of Michael Fonstein (1959–2022).
Present address: James J. Davis, Argonne National Laboratory Computing, Environment and Life Sciences 9700 S. Cass Avenue Argonne, IL 60439, USA.
Contributor Information
Robert D Olson, Consortium for Advanced Science and Engineering, University of Chicago, Chicago, IL 60637, USA; Division of Data Science and Learning, Argonne National Laboratory, Argonne, IL 60439, USA.
Rida Assaf, Department of Computer Science, American University of Beirut, Beirut, Lebanon.
Thomas Brettin, Consortium for Advanced Science and Engineering, University of Chicago, Chicago, IL 60637, USA; Computing Environment and Life Sciences, Argonne National Laboratory, Argonne, IL 60439, USA.
Neal Conrad, Consortium for Advanced Science and Engineering, University of Chicago, Chicago, IL 60637, USA; Division of Data Science and Learning, Argonne National Laboratory, Argonne, IL 60439, USA.
Clark Cucinell, University of Virginia Biocomplexity Institute, Charlottesville, VA 22904, USA.
James J Davis, Consortium for Advanced Science and Engineering, University of Chicago, Chicago, IL 60637, USA; Division of Data Science and Learning, Argonne National Laboratory, Argonne, IL 60439, USA.
Donald M Dempsey, Department of Microbiology, University of Alabama at Birmingham School of Medicine, Birmingham, AL 35294, USA.
Allan Dickerman, University of Virginia Biocomplexity Institute, Charlottesville, VA 22904, USA.
Emily M Dietrich, Consortium for Advanced Science and Engineering, University of Chicago, Chicago, IL 60637, USA; Division of Data Science and Learning, Argonne National Laboratory, Argonne, IL 60439, USA.
Ronald W Kenyon, University of Virginia Biocomplexity Institute, Charlottesville, VA 22904, USA.
Mehmet Kuscuoglu, Department of Informatics, J. Craig Venter Institute, La Jolla, CA 92037, USA.
Elliot J Lefkowitz, Department of Microbiology, University of Alabama at Birmingham School of Medicine, Birmingham, AL 35294, USA.
Jian Lu, J. Craig Venter Institute, Rockville, MD 20850, USA.
Dustin Machi, University of Virginia Biocomplexity Institute, Charlottesville, VA 22904, USA.
Catherine Macken, Department of Statistics, University of Auckland, Auckland, New Zealand.
Chunhong Mao, University of Virginia Biocomplexity Institute, Charlottesville, VA 22904, USA.
Anna Niewiadomska, Department of Informatics, J. Craig Venter Institute, La Jolla, CA 92037, USA.
Marcus Nguyen, Consortium for Advanced Science and Engineering, University of Chicago, Chicago, IL 60637, USA; Division of Data Science and Learning, Argonne National Laboratory, Argonne, IL 60439, USA.
Gary J Olsen, Department of Microbiology, University of Illinois, Urbana, IL 61801, USA.
Jamie C Overbeek, Consortium for Advanced Science and Engineering, University of Chicago, Chicago, IL 60637, USA; Division of Data Science and Learning, Argonne National Laboratory, Argonne, IL 60439, USA.
Bruce Parrello, Consortium for Advanced Science and Engineering, University of Chicago, Chicago, IL 60637, USA; Fellowship for Interpretation of Genomes, Burr Ridge, IL 60527, USA.
Victoria Parrello, Fellowship for Interpretation of Genomes, Burr Ridge, IL 60527, USA.
Jacob S Porter, Computing Environment and Life Sciences, Argonne National Laboratory, Argonne, IL 60439, USA.
Gordon D Pusch, Fellowship for Interpretation of Genomes, Burr Ridge, IL 60527, USA.
Maulik Shukla, Consortium for Advanced Science and Engineering, University of Chicago, Chicago, IL 60637, USA; Division of Data Science and Learning, Argonne National Laboratory, Argonne, IL 60439, USA.
Indresh Singh, J. Craig Venter Institute, Rockville, MD 20850, USA.
Lucy Stewart, Department of Informatics, J. Craig Venter Institute, La Jolla, CA 92037, USA.
Gene Tan, Department of Informatics, J. Craig Venter Institute, La Jolla, CA 92037, USA.
Chris Thomas, Consortium for Advanced Science and Engineering, University of Chicago, Chicago, IL 60637, USA; Division of Data Science and Learning, Argonne National Laboratory, Argonne, IL 60439, USA.
Margo VanOeffelen, Fellowship for Interpretation of Genomes, Burr Ridge, IL 60527, USA.
Veronika Vonstein, Fellowship for Interpretation of Genomes, Burr Ridge, IL 60527, USA.
Zachary S Wallace, Department of Microbiology, University of Alabama at Birmingham School of Medicine, Birmingham, AL 35294, USA; Department of Computer Science and Engineering, University of California, San Diego, CA 92039, USA.
Andrew S Warren, University of Virginia Biocomplexity Institute, Charlottesville, VA 22904, USA.
Alice R Wattam, University of Virginia Biocomplexity Institute, Charlottesville, VA 22904, USA.
Fangfang Xia, Consortium for Advanced Science and Engineering, University of Chicago, Chicago, IL 60637, USA; Division of Data Science and Learning, Argonne National Laboratory, Argonne, IL 60439, USA.
Hyunseung Yoo, Consortium for Advanced Science and Engineering, University of Chicago, Chicago, IL 60637, USA; Division of Data Science and Learning, Argonne National Laboratory, Argonne, IL 60439, USA.
Yun Zhang, Department of Informatics, J. Craig Venter Institute, La Jolla, CA 92037, USA.
Christian M Zmasek, Department of Informatics, J. Craig Venter Institute, La Jolla, CA 92037, USA.
Richard H Scheuermann, Department of Informatics, J. Craig Venter Institute, La Jolla, CA 92037, USA; Department of Pathology, University of California, San Diego, CA 92093, USA; Division of Vaccine Discovery, La Jolla Institute for Immunology, La Jolla, CA 92037, USA; Global Virus Network, Baltimore, MD 21201, USA.
Rick L Stevens, Computing Environment and Life Sciences, Argonne National Laboratory, Argonne, IL 60439, USA; Department of Computer Science, University of Chicago, Chicago, IL 60637, USA.
FUNDING
Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services [75N93019C00076 to R.L.S. and R.H.S., U24AI162625 to E.J.L.]. Funding for open access charge: UC indirects.
Conflict of interest statement. None declared.
REFERENCES
- 1. Greene J.M., Collins F., Lefkowitz E.J., Roos D., Scheuermann R.H., Sobral B., Stevens R., White O., Di Francesco V.. National Institute of Allergy and Infectious Diseases bioinformatics resource centers: new assets for pathogen informatics. Infect. Immun. 2007; 75:3212–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amos B., Aurrecoechea C., Barba M., Barreto A., Basenko E.Y., Belnap R., Blevins A.S., Böhme U., Brestelli J., Brunk B.P.. VEuPathDB: the eukaryotic pathogen, vector and host bioinformatics resource center. Nucleic Acids Res. 2022; 50:D898–D911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Davis J.J., Wattam A.R., Aziz R.K., Brettin T., Butler R., Butler R.M., Chlenski P., Conrad N., Dickerman A., Dietrich E.M.. The PATRIC Bioinformatics Resource Center: expanding data and analysis capabilities. Nucleic Acids Res. 2020; 48:D606–D612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang Y., Aevermann B.D., Anderson T.K., Burke D.F., Dauphin G., Gu Z., He S., Kumar S., Larsen C.N., Lee A.J.. Influenza Research Database: An integrated bioinformatics resource for influenza virus research. Nucleic Acids Res. 2017; 45:D466–D474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pickett B.E., Sadat E.L., Zhang Y., Noronha J.M., Squires R.B., Hunt V., Liu M., Kumar S., Zaremba S., Gu Z.. ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012; 40:D593–D598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Snyder E., Kampanya N., Lu J., Nordberg E.K., Karur H., Shukla M., Soneja J., Tian Y., Xue T., Yoo H.. PATRIC: the VBI pathosystems resource integration center. Nucleic Acids Res. 2007; 35:D401–D406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McNeil L.K., Reich C., Aziz R.K., Bartels D., Cohoon M., Disz T., Edwards R.A., Gerdes S., Hwang K., Kubal M.. The National Microbial Pathogen Database Resource (NMPDR): a genomics platform based on subsystem annotation. Nucleic Acids Res. 2007; 35:D347–D353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Overbeek R., Olson R., Pusch G.D., Olsen G.J., Davis J.J., Disz T., Edwards R.A., Gerdes S., Parrello B., Shukla M.. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014; 42:D206–D214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Squires B., Macken C., Garcia-Sastre A., Godbole S., Noronha J., Hunt V., Chang R., Larsen C.N., Klem E., Biersack K.. BioHealthBase: informatics support in the elucidation of influenza virus host–pathogen interactions and virulence. Nucleic Acids Res. 2008; 36:D497–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pickett B.E., Greer D.S., Zhang Y., Stewart L., Zhou L., Sun G., Gu Z., Kumar S., Zaremba S., Larsen C.N.. Virus pathogen database and analysis resource (ViPR): a comprehensive bioinformatics database and analysis resource for the coronavirus research community. Viruses. 2012; 4:3209–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brettin T., Davis J.J., Disz T., Edwards R.A., Gerdes S., Olsen G.J., Olson R., Overbeek R., Parrello B., Pusch G.D.. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015; 5:8365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pickett B., Liu M., Sadat E., Squires R., Noronha J., He S., Jen W., Zaremba S., Gu Z., Zhou L.. Metadata-driven comparative analysis tool for sequences (meta-CATS): an automated process for identifying significant sequence variations that correlate with virus attributes. Virology. 2013; 447:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang S., Sundaram J.P., Stockwell T.B.. VIGOR extended to annotate genomes for additional 12 different viruses. Nucleic Acids Res. 2012; 40:W186–W192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Han M.V., Zmasek C.M.. phyloXML: XML for evolutionary biology and comparative genomics. BMC Bioinf. 2009; 10:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sayers E.W., Cavanaugh M., Clark K., Pruitt K.D., Schoch C.L., Sherry S.T., Karsch-Mizrachi I.. GenBank. Nucleic Acids Res. 2021; 49:D92–D96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sayers E.W., Beck J., Bolton E.E., Bourexis D., Brister J.R., Canese K., Comeau D.C., Funk K., Kim S., Klimke W.. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2021; 49:D10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. VanOeffelen M., Nguyen M., Aytan-Aktug D., Brettin T., Dietrich E.M., Kenyon R.W., Machi D., Mao C., Olson R., Pusch G.D.. A genomic data resource for predicting antimicrobial resistance from laboratory-derived antimicrobial susceptibility phenotypes. Briefings Bioinf. 2021; 22:bbab313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Overbeek R., Begley T., Butler R.M., Choudhuri J.V., Chuang H.-Y., Cohoon M., de Crécy-Lagard V., Diaz N., Disz T., Edwards R.. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005; 33:5691–5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alcock B.P., Raphenya A.R., Lau T.T., Tsang K.K., Bouchard M., Edalatmand A., Huynh W., Nguyen A.-L.V., Cheng A.A., Liu S.. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020; 48:D517–D525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Feldgarden M., Brover V., Gonzalez-Escalona N., Frye J.G., Haendiges J., Haft D.H., Hoffmann M., Pettengill J.B., Prasad A.B., Tillman G.E.. AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci. Rep. 2021; 11:12728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Antonopoulos D.A., Assaf R., Aziz R.K., Brettin T., Bun C., Conrad N., Davis J.J., Dietrich E.M., Disz T., Gerdes S.. PATRIC as a unique resource for studying antimicrobial resistance. Briefings Bioinf. 2019; 20:1094–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sayers S., Li L., Ong E., Deng S., Fu G., Lin Y., Yang B., Zhang S., Fa Z., Zhao B.. Victors: a web-based knowledge base of virulence factors in human and animal pathogens. Nucleic Acids Res. 2019; 47:D693–D700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu B., Zheng D., Zhou S., Chen L., Yang J.. VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 2022; 50:D912–D917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z.. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018; 46:D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhou Y., Zhang Y., Lian X., Li F., Wang C., Zhu F., Qiu Y., Chen Y.. Therapeutic target database update 2022: facilitating drug discovery with enriched comparative data of targeted agents. Nucleic Acids Res. 2022; 50:D1398–D1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saier M.H. Jr, Reddy V.S., Moreno-Hagelsieb G., Hendargo K.J., Zhang Y., Iddamsetty V., Lam K.J.K., Tian N., Russum S., Wang J.. The transporter classification database (TCDB): 2021 update. Nucleic Acids Res. 2021; 49:D461–D467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McNair K., Zhou C., Dinsdale E.A., Souza B., Edwards R.A.. PHANOTATE: a novel approach to gene identification in phage genomes. Bioinformatics. 2019; 35:4537–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jones P., Binns D., Chang H.-Y., Fraser M., Li W., McAnulla C., McWilliam H., Maslen J., Mitchell A., Nuka G.. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014; 30:1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dhanda S.K., Mahajan S., Paul S., Yan Z., Kim H., Jespersen M.C., Jurtz V., Andreatta M., Greenbaum J.A., Marcatili P.. IEDB-AR: immune epitope database—analysis resource in 2019. Nucleic Acids Res. 2019; 47:W502–W506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. UniProt Consortium UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019; 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Burley S.K., Berman H.M., Kleywegt G.J., Markley J.L., Nakamura H., Velankar S.. Protein Data Bank (PDB): the single global macromolecular structure archive. Protein Crystallogr. 2017; 1607:627–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sehnal D., Bittrich S., Deshpande M., Svobodová R., Berka K., Bazgier V., Velankar S., Burley S.K., Koča J., Rose A.S.. Mol* Viewer: modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021; 49:W431–W437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hanson R.M., Prilusky J., Renjian Z., Nakane T., Sussman J.L.. JSmol and the next-generation web-based representation of 3D molecular structure as applied to proteopedia. Isr. J. Chem. 2013; 53:207–216. [Google Scholar]
- 34. Moore K.A., Ostrowsky J.T., Mehr A.J., Osterholm M.T., Committee C.P.P., Compans R.W., García-Sastre A., Orenstein W.A., Pekosz A., Perez D.R.. Influenza response planning for the centers of excellence for influenza research and surveillance: Science preparedness for enhancing global health security. Influenza Other Respir. Viruses. 2020; 14:444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Buels R., Yao E., Diesh C.M., Hayes R.D., Munoz-Torres M., Helt G., Goodstein D.M., Elsik C.G., Lewis S.E., Stein L.. JBrowse: a dynamic web platform for genome visualization and analysis. Genome Biol. 2016; 17:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yachdav G., Wilzbach S., Rauscher B., Sheridan R., Sillitoe I., Procter J., Lewis S.E., Rost B., Goldberg T.. MSAViewer: interactive JavaScript visualization of multiple sequence alignments. Bioinformatics. 2016; 32:3501–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aziz R.K., Bartels D., Best A.A., DeJongh M., Disz T., Edwards R.A., Formsma K., Gerdes S., Glass E.M., Kubal M.. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008; 9:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sayers E.W., Cavanaugh M., Clark K., Ostell J., Pruitt K.D., Karsch-Mizrachi I.. GenBank. Nucleic Acids Res. 2019; 47:D94–D99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wallace Z.S., Davis J., Niewiadomska A.M., Olson R.D., Shukla M., Stevens R., Zhang Y., Zmasek C.M., Scheuermann R.H.. Early detection of emerging SARS-CoV-2 variants of interest for experimental evaluation. Front. Bioinform. 2022; 10.1101/2022.08.08.22278553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Untergasser A., Cutcutache I., Koressaar T., Ye J., Faircloth B.C., Remm M., Rozen S.G.. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012; 40:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Katoh K., Standley D.M.. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013; 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014; 30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guindon S., Dufayard J.-F., Lefort V., Anisimova M., Hordijk W., Gascuel O.. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010; 59:307–321. [DOI] [PubMed] [Google Scholar]
- 44. Price M.N., Dehal P.S., Arkin A.P.. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One. 2010; 5:e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Darling A.C., Mau B., Blattner F.R., Perna N.T.. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004; 14:1394–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T.L.. BLAST+: architecture and applications. BMC Bioinf. 2009; 10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ondov B.D., Treangen T.J., Melsted P., Mallonee A.B., Bergman N.H., Koren S., Phillippy A.M.. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016; 17:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wood D.E., Lu J., Langmead B.. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019; 20:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nayfach S., Camargo A.P., Schulz F., Eloe-Fadrosh E., Roux S., Kyrpides N.C.. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2021; 39:578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Grubaugh N.D., Gangavarapu K., Quick J., Matteson N.L., De Jesus J.G., Main B.J., Tan A.L., Paul L.M., Brackney D.E., Grewal S.. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019; 20:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018; 34:3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.. The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wilkinson M.D., Dumontier M., Aalbersberg I.J., Appleton G., Axton M., Baak A., Blomberg N., Boiten J.-W., da Silva Santos L.B., Bourne P.E.. The FAIR Guiding Principles for scientific data management and stewardship. Sci Data. 2016; 3:160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dugan V.G., Emrich S.J., Giraldo-Calderón G.I., Harb O.S., Newman R.M., Pickett B.E., Schriml L.M., Stockwell T.B., Stoeckert C.J. Jr, Sullivan D.E.. Standardized metadata for human pathogen/vector genomic sequences. PLoS One. 2014; 9:e99979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bischof J., Wilke A., Gerlach W., Harrison T., Paczian T., Tang W., Trimble W., Wilkening J., Desai N., Meyer F.. 2015 IEEE/ACM 2nd International Symposium on Big Data Computing (BDC). 2015; IEEE; 68–72. [Google Scholar]
- 56. Yoo A.B., Jette M.A., Grondona M.. Workshop on Job Scheduling Strategies for Parallel Processing. 2003; Springer; 44–60. [Google Scholar]
- 57. Kurtzer G.M., Sochat V., Bauer M.W.. Singularity: Scientific containers for mobility of compute. PLoS One. 2017; 12:e0177459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Davis J.J., Gerdes S., Olsen G.J., Olson R., Pusch G.D., Shukla M., Vonstein V., Wattam A.R., Yoo H.. PATtyFams: protein families for the microbial genomes in the PATRIC database. Front. Microbiol. 2016; 7:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zmasek C.M., Lefkowitz E.J., Niewiadomska A., Scheuermann R.H.. Genomic evolution of the Coronaviridae family. Virology. 2022; 570:123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zmasek C.M., Knipe D.M., Pellett P.E., Scheuermann R.H.. Classification of human Herpesviridae proteins using Domain-architecture Aware Inference of Orthologs (DAIO). Virology. 2019; 529:29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A.. Highly accurate protein structure prediction with AlphaFold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data and links to the FTP site and command line interface tools can be found on the BV-BRC website, https://www.bv-brc.org/. All project code can be found at our GitHub repo, https://github.com/BV-BRC.