


Abstract
CRAMdb (a database for composition and roles of animal microbiome) is a comprehensive resource of curated and consistently annotated metagenomes for non-human animals. It focuses on the composition and roles of the microbiome in various animal species. The main goal of the CRAMdb is to facilitate the reuse of animal metagenomic data, and enable cross-host and cross-phenotype comparisons. To this end, we consistently annotated microbiomes (including 16S, 18S, ITS and metagenomics sequencing data) of 516 animals from 475 projects spanning 43 phenotype pairs to construct the database that is equipped with 9430 bacteria, 278 archaea, 2216 fungi and 458 viruses. CRAMdb provides two main contents: microbiome composition data, illustrating the landscape of the microbiota (bacteria, archaea, fungi, and viruses) in various animal species, and microbiome association data, revealing the relationships between the microbiota and various phenotypes across different animal species. More importantly, users can quickly compare the composition of the microbiota of interest cross-host or body site and the associated taxa that differ between phenotype pairs cross-host or cross-phenotype. CRAMdb is freely available at (http://www.ehbio.com/CRAMdb).
INTRODUCTION
Animal microbiomes comprise bacteria, viruses, fungi, archaea, and protozoa, which colonize almost all body parts of animals and play important roles in the health, diseases (1–5), growth (6), development (7) and traits (8–12) of their animal hosts. In recent years, the number of animal metagenomic datasets (including both amplicon and metagenomic sequencing datasets) has increased rapidly. Most of the related raw sequencing data have been deposited in public databases, such as the EBI European Nucleotide Archive (ENA) (13) and NCBI Sequence Read Archive (SRA) (14). These public resources greatly facilitate the reuse of the rapidly increasing amounts of data among different authors. The exploration of animal microbiomes offers the potential to promote animal health, production and environmental adaptation through the directed regulation of the microbiota. For example, diarrhea is treated by regulating gut microbes in wild or domestic animals (15,16), and milk production can be increased by regulating gut microbes in dairy cows (17).
There are also several databases that contain metagenomic data from various animal hosts, including Qiita (18), Metagenomic Rapid Annotations using Subsystems Technology (MG-RAST) (19), Animal Microbiome Database (AMDB) (20), A Database for the Domestic Animal Gut Microbiome Atlas (ADDAGMA) (21). However, Qiita and MG-RAST contain processed data collected from various habitats, making them difficult to identify the relationships between the gut microbiota and animal hosts. AMDB provides data linking host phylogeny and diet to the gut microbiota, but lacked microbiome-phenotype associations data. ADDAGMA from our group provides microbe-phenotype association data, but only focuses on domestic animal species. Table 1 summarized their main features. However, a comprehensive database for microbiome composition including data on bacteria, archaea, fungi and viruses, and microbiome roles in health, diseases and production in various animal species has not been established.
Table 1.
Main features of CRAMdb and comparison with similar databases
| Database | Main features | No. microbes | No. phenotypes | No. animals | No. datasets | Reference |
|---|---|---|---|---|---|---|
| CRAMdb | 1. Provide the integrated data of cross-host, cross-body site and cross-phenotype comparisons. 2. Illustrate the landscape of the microbiota (bacteria, archaea, fungi, and viruses) in various animal species. 3. Reveale the relationships between the microbiota and various phenotypes across different animal species. |
12382 (9430 bacteria, 278 archaea, 2216 fungi and 458 viruses) | 48 | 516 | 475 | This study |
| Qiita | 1. Easily share and reuse existing data-sets in the form of studies. 2. Easily interface with the EBI repository for automated deposition. Query and interact with Qiita data programmatically. |
/ | / | / | 660 | (18) |
| MG-RAST | 1. Store large amounts of metagenomic data. 2. Support the analysis and download of metagenomic data. |
/ | / | / | 150000 | (19) |
| AMDB | 1. Provides a taxonomic overview of the gut microbiota of various animal species. 2. Gene profiles of bacterial 16S rRNAs covering animal species to assess the relationship between gut microbiota and animal hosts. |
10478 (only bacteria) | / | 467 | 34 | (20) |
| ADDAGMA | 1. Focus on changes in the gut microbiome of domestic animals. 2. Provides links between gut microbes and various phenotypes in domestic animals. |
3215 (only bacteria) | 48 | 4 | 356 | (21) |
To address these limitations in existing databases, we created CRAMdb, which is the first comprehensive resource of particularly focusing on the microbiome composition of animals and associations between the microbiota and various phenotypes of animals with manually curated metadata. Overall, we collected metagenomic data (including 16S, 18S, ITS, and metagenomic sequencing data) of 516 animal species from 475 projects spanning 43 phenotype pairs to construct the database that is equipped with 9430 bacteria, 278 archaea, 2216 fungi and 458 viruses. CRAMdb enables researchers to quickly acquire data on microbiota of interest and provide an important reference for exploring the composition and roles of the microbiome in animals.
DATABASE CONSTRUCTION
Data extraction and curation
Supplementary Figure S1 shows the pipeline for the construction of CRAMdb. We manually selected raw sequencing reads and metadata from the ENA and SRA databases based on the following criteria. For microbiome composition data: (i) the samples were from healthy animal hosts and collected from any body parts (including feces, intestine, skin, rumen samples and so on); (ii) amplicons and metagenomic data had to be sequenced using Illumina or 454 pyrosequencing instruments; and (iii) raw data had to be linked to at least one published article. For microbiome association data, (i) the samples had to include at least one phenotype pair, such as health versus diarrhea (Health/Diarrhea) and (ii) other criteria were the same as those for the microbiome composition data.
Processing of raw sequencing reads
For amplicon sequences, as shown in Supplementary Figure S1A, VSEARCH (version 2.15.1) (22) was used to merge paired-end sequencing reads and perform the quality control, control of the sequencing error rate ≤0.01 with the specified parameeter ‘–fastq_maxee_rate 0.01’. USEARCH (version 10.0.240) (23) was used to denoise the clean data to generate ASVs (Amplicon Sequence Variants). ASVs were compared with the RDP (version 16) (24) or UNITE (version 8.2) (25) database by using VSEARCH for taxonomy annotation. The relative abundance of all ASVs were quantified using VSEARCH and in-house scripts as in our previous report (26).
For virus metagenomic sequences, as shown in Supplementary Figure S1B, the clean reads were quality controlled using Fastp (version 0.20.1) (27) and host reads were dopped out using Bowtie2 (version 2.2.4) (28). Spades (29), Virsorter/Virfinder (30,31), and Drep (32) were used to assemble, forecast and de-redundancy virus contigs, respectively. Finally, the processed data were aligned against the NR database by using Diamond (version 0.9.9) (33) to obtain the corresponding annotations.
For taxonomic analysis and statistical analysis, 475 projects and 25356 samples included in CRAMdb were annotated using two methods, respectively. For amplicon sequences, each ASV was 97% matched when quantified, and reads were counted for these ASVs using VSEARCH. And the confidence interval of the taxonomic annotation is 0.6. For viral sequences, diamond annotation was used, with 40% identity for clustering, and the constraints are E-value ≤0.00001. For screening associated taxa, abundance matrix in raw read counts were processed using the edgeR package for normalization and differential analysis with computed P value <0.05 as the threshold.
Supplementary Video is a tutorial video for using CRAMdb. And the curated metadata for projects and analysis codes are publicly shown at https://github.com/Tong-Chen/CRAMdb.
Statistical methods
edgeR was used to fit a negative binomial generalized log-linear model to the species read-counts matrix. The likelihood ratio tests was performed in the linear model to get the differential ananlysis P-values. Multiplicity correction is performed by applying the Benjamini-Hochberg method (BH) on the P-values to control the false discovery rate (FDR). E-value represents the number of expected hits of similar quality (score) for query sequences that could be found just by chance. 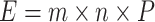 where m = number of residues in the query sequence, n = total number of residues in a database and P = probability of an alignment result from random chance.
where m = number of residues in the query sequence, n = total number of residues in a database and P = probability of an alignment result from random chance.
Web implementation
CRAMdb is implemented as a web application using Javascript and HTML for frontend development. The used core JavaScript libraries include Vue.js (https://vuejs.org/) as the main frontend framework, plotly.js (https://plotly.com/), D3.js (https://d3js.org/) and Echarts.js (https://echarts.apache.org/zh/index.html) for interactive visualizations and data explorations. High-level web framework Django (https://www.djangoproject.com/) is used for backend data preprocess and data analysis. The global search function is based on the Elasticsearch module (https://www.elastic.co/elasticsearch/). Open-source data management system Mysql (https://www.mysql.com/) is used for curated metadata saving and accessing. Abundance profiles of taxons were stored in hdf5 format for fast access and calculation.
DATA CONTENT
Objectives and functional features of CRAMdb
CRAMdb provides two main types of content: ‘Microbiome composition’ and ‘Microbiome associations’ (Figure 1). The objectives of providing the ‘Microbiome composition’ data are to allow the host microbial composition to be analyzed and the distribution of microbiota members in different hosts or at different body sites to be explored. The data on ‘Microbiome associations’ were assembled with the aim of revealing the roles of the microbiota among different phenotypes (health, disease, and production traits). To achieve these aims, CRAMdb provides an easy-to-use platform for users to browse, search, and analyse the composition of the microbial communities (e.g. bacteria, archaea, fungi and viruses) harboured by 25356 samples from 516 animals in 475 projects but also provides the 419 associated taxa from 43 phenotype pairs (e.g. healthy versus disease) in animals. These projects were performed in 233 cities in 48 countries. The samples were from multiple body sites (including feces, intestine, skin, and rumen samples) or from the whole body in some species (e.g. Mosquito and Drosophila).
Figure 1.

The objectives and functional features of CRAMdb.
Microbiome composition data cover bacteria, archaea, fungi and viruses
The ‘Microbiome composition’ content consists of data on 12382 taxa of bacteria, archaea, fungi and viruses. The most abundant of these groups is ‘bacteria’, representing 76.16% (9430) of the total taxa, followed by ‘archaea’, ‘fungi’ and ‘viruses’, which represent 2.25% (278), 17.89% (2216) and 3.7% (458) of the taxa, respectively. Supplementary Figure S2 shows the summary of the composition of taxa included in CRAMdb. The homepage shows the total numbers of bacteria, archaea, fungi, and viruses included in the database. By clicking on one of these four groups, users can obtain a detailed list of the members of the group of interest. For example, by clicking on ‘Bacteria’, users can browse the bacterial taxa in the database, including the name and classification of a given taxon and the number of related hosts, projects or samples (Supplementary Figure S3).
Microbiome composition data allow for cross-host or cross-body site comparisons
To allow users to check if a microorganism is unique to a specific host (or body site) or is shared by multiple hosts (body sites) and if it differs in abundance between hosts (or body sites), the database supports cross-host or cross-body site comparisons of the microbiome composition. Taking Oxalobacter formigenes as an example, users can see that this bacterium has been identified in 20 of the hosts included in the database and can view trends in its relative abundance across different hosts (Figure 2A). In addition, a view is provided that allows users to not only visualize the mean abundance of O. formigenes in different hosts by the same body sites (Figure 2B), but also compare the mean abundance of O. formigenes in the same host by different body sites (Supplementary Figure S4). For example, it can be seen that the relative abundance of O. formigenes in Sus scrofa (Pig) is higher in the feces and lower in the intestine, suggesting that it may be easier to monitor or isolate O. formigenes from feces samples of Sus scrofa.
Figure 2.

Microbiome composition cross-host comparisons. (A) Relative abundance of O. formigenes in 20 animals. (B) The mean abundance of O. formigenes in different hosts by the same body sites.
Microbial prevalence cross-host comparisons
The database also supports analyses of microbial prevalence cross-host comparisons. As an example, we have calculated the prevalence of O. formigenes in various hosts at different taxonomic levels. At the class level of host, Figure 3A and B shows that O. formigenes is the most common in ‘Mammalia’ (53.2%), followed by ‘Insecta’ (47.9%), and is least prevalent in ‘Aves’ (24.8%). At the species level of host, Figure 3C and D shows that O. formigenes is the most common in ‘Diceros bicornis’ and ‘Nycticebus pygmaeus’ (100%) and is least prevalent in ‘Sceloporus undulatus’ (2.0%). Moreover, users can visualize the prevalence of O. formigenes at genus, family, or order levels of animal hosts.
Figure 3.

Microbial prevalence cross-host comparisons. (A, B) The prevalence of O. formigenes in hosts at the class level. (C, D) The prevalence of O. formigenes in hosts at the species level. User also view the prevalence of O. formigenes in hosts at the genus, family or order level (see also http://www.ehbio.com/CRAMdb/Detail?detailParameter=Oxalobacter%20formigenes&taxType=Species&module=com&detailInterface=com_tax_detail&type=Bacteria).
Associated taxa identified between two phenotypes and cross-phenotype comparisons
To allow a more in-depth exploration of the relationships between the microbiota and animal health, diseases or production traits, we identified the associated taxa between two phenotypes (e.g. health versus diarrhea) for selected projects. So far, In CRAMdb, ‘associated taxa’ included ‘associated bacteria’ and ‘associated fungi’, which refer to taxon that show significant differences in relative abundances between phenotypes (P < 0.05). Since the identification of the associated taxa was on per-project basis, users can visualize the associated taxa between pairs of the phenotypes recorded in each project. Figure 4A shows the associated taxa (species and genus) identified between individuals of the S. scrofa with a healthy status or diarrhea in project PRJNA492210. Researchers can choose to show the associated taxa at either the species or genus level or at both levels together.
Figure 4.

Associated taxa identified between two phenotypes and cross-phenotype comparisons. (A) Associated taxa were identified between two phenotypes. This data from project PRJNA492210 was used as an example. The associated taxa were enriched in health group (in red) or diarrhea group (in green). (B) Cross-phenotype comparisons of associated taxa. Comparison of the relative abundance of Ruminococcus in six phenotype pairs from five different hosts. Fold change (log2FC) of abundance was highlighted in the red box (see also http://www.ehbio.com/CRAMdb/Detail?type=Bacteria&detailInterface=fun_tax_detail&taxType=Genus&module=fun&detailParameter=Ruminococcus). ‘Associated taxa’ refer to taxon (at species and genus levels) that show significant differences in relative abundances between phenotypes (P < 0.05).
We performed in-depth analysis to identify the associated taxa cross phenotype pair. Take Ruminococcus as an example, it has been identified as an associated taxon in six phenotype pairs from different host animals (Figure 4B). Ruminococcus was significantly reduced in multiple diseases or traits, including weaned, diarrhea, Diabetes mellitus type 2, infection of salmonella enteritidis, Colitis and radiation exposure, when compared with the healthy phenotypes. Interestingly, Ruminococcus are consistently depleted in human with Crohn or Colitis (34–36). Thus, these results suggest that Ruminococcus may be important in maintaining intestinal homeostasis and has potential probiotic effect on animals and human health. In addition, CRAMdb also provides users with the associated taxa of the host under different phenotypes. Users can compare associated taxa under the same phenotype to identify key microbes that may affect host health.
Global search widget
The global search widget allows users to enter keywords of interest and shows the search results in a dropdown list, and allows users to click and go to the corresponding page in our database. Users can use it to find any of the following entries in our database including microbes, hosts, projects, and phenotypes. For example, users enter ‘Colitis’ and then obtain all of the following entries about ‘Colitis’ in our database, (i) colitis in Microbiome Composition Projects or species; (ii) colitis in Microbiome Associations Phenotype pairs or projects (Supplementary Figure S5). In addition, if one might be interested in Megamonas funiformis, users can obtain the results including the taxon, associated hosts, sample types, and associated phenotypes of M. funiformis by using the global search of CRAMdb. CRAMdb could provide a basis for users to determine the best host and sample types to study M. funiformis. It can also help users judge the effects of M. funiformis on the health of the host.
Interactive map
‘Interactive map’ is a dot distribution map showing the distribution of animal metagenomic researches across the world. In the CRAMdb, the majority of studies were conducted in China, and the United States is the second most common place for the researches. Users could click or drag out a rectangle or polygon to select one or more dots on the map to obtain a list of related projects with the information of hosts, phenotype pairs, project accession, body sites, and so on in the corresponding area. Within the table, users could click projects or hosts to explore detail informations.
FUTURE DEVELOPMENT
New metagenomic data from various animals will continue to be added to CRAMdb in the future. In addition, we plan to add new content to CRAMdb, especially metagenomic data on the microbiome composition of protozoa and metagenomic data on the microbiome associations of viruses, fungi and protozoa. We also plan to add functionalities to the platform that will allow users to perform cross-projects, cross-host, or cross-body site comparisons. These changes will provide an integrated landscape of the composition and roles of animal microbiomes, which will further facilitate the reuse of metagenomic data by providing an up-to-date central repository of animal microbiome data and advance the understanding of microbiome-animal host relationships.
CONCLUSION
CRAMdb is a database for exploring the composition and roles of the microbiome in various animal species, and it provides an option to search for the composition or roles of a microbe. With 516 animals from 475 projects (including 16S, 18S, ITS and metagenomic sequencing data), CRAMdb is the first comprehensive database of data on bacteria, archaea, fungi, and viruses associated with animals. One of the main features of CRAMdb is that users can quickly obtain the average composition of microbiota from a particular context, such as a specific sample type, across different hosts or projects. In addition, we introduced the associated taxa for each phenotype pair or cross-phenotype. Currently, CRAMdb includes 419 associated taxa from 43 phenotype pairs. We believe that CRAMdb has the potential to become a very useful and important database for biologists and bioinformaticians seeking to study animal microbiomes. In the future, we will continuously update CRAMdb to provide the latest content and include more features.
DATA AVAI LABILITY
CRAMdb is freely available at http://www.ehbio.com/CRAMdb/. The curated metadata for projects and the analysis codes are publicly shown at https://github.com/Tong-Chen/CRAMdb.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Pu Xue and Yu Liu in EHBIO Gene Technology (Beijing) Co., Ltd for their help on the construction of the database CRAMdb.
Contributor Information
Bingbing Lei, College of Life Sciences, Shihezi University, Shihezi, Xinjiang 832003, China.
Yueren Xu, College of Life Sciences, Shihezi University, Shihezi, Xinjiang 832003, China.
Yunjiao Lei, College of Life Sciences, Shihezi University, Shihezi, Xinjiang 832003, China.
Cunyuan Li, College of Life Sciences, Shihezi University, Shihezi, Xinjiang 832003, China; Key Laboratory of Ecological Corps for Oasis City and Mountain Basin System, College of Science, Shihezi University, Shihezi, Xinjiang 832000, China.
Ping Zhou, State Key Laboratory of Sheep Genetic Improvement and Healthy Production, Xinjiang Academy of Agricultural and Reclamation Science, Xinjiang 832003, China.
Limin Wang, State Key Laboratory of Sheep Genetic Improvement and Healthy Production, Xinjiang Academy of Agricultural and Reclamation Science, Xinjiang 832003, China.
Qing Yang, Key Laboratory of Ecological Impacts of Hydraulic-Projects and Restoration of Aquatic Ecosystem of Ministry of Water Resources, Institute of Hydroecology Ministry of Water Resources and Chinese Academy of Sciences, Wuhan 430079, China.
Xiaoyue Li, College of Life Sciences, Shihezi University, Shihezi, Xinjiang 832003, China.
Fulin Li, College of Life Sciences, Shihezi University, Shihezi, Xinjiang 832003, China.
Chuyang Liu, College of Life Sciences, Shihezi University, Shihezi, Xinjiang 832003, China.
Chaowen Cui, College of Life Sciences, Shihezi University, Shihezi, Xinjiang 832003, China.
Tong Chen, State Key Laboratory Breeding Base of Dao-di Herbs, National Resource Center for Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing, China.
Wei Ni, College of Life Sciences, Shihezi University, Shihezi, Xinjiang 832003, China.
Shengwei Hu, College of Life Sciences, Shihezi University, Shihezi, Xinjiang 832003, China; State Key Laboratory of Sheep Genetic Improvement and Healthy Production, Xinjiang Academy of Agricultural and Reclamation Science, Xinjiang 832003, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Third Xinjiang Scientific Expedition Program [2021xjkk0605 to S.W.H., 2021xjkk0504 to W.N.]; Foundation of State Key Laboratory for Sheep Genetic Improvement and Healthy Production [2021ZD08 to S.W.H.]; Bingtuan Science and Technology Project [2021CB033 to W.N.]; Scientific and Technology Innovation Project of the China Academy of Chinese Medical Sciences [C12021A04115 to T.C.]; Fundamental Research Funds for the Central Public Welfare Research Institutes [ZZ13-YQ-095 to T.C.]. Funding for open access charge: Third Xinjiang Scientific Expedition Program [2021xjkk0605 to S.W.H., 2021xjkk0504 to W.N.]; Foundation of State Key Laboratory for Sheep Genetic Improvement and Healthy Production [2021ZD08 to S.W.H.]; Bingtuan Science and Technology Project [2021CB033 to W.N.]; Scientific and Technology Innovation Project of the China Academy of Chinese Medical Sciences [C12021A04115 to T.C.]; Fundamental Research Funds for the Central Public Welfare Research Institutes [ZZ13-YQ-095 to T.C.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Fan Y., Pedersen O.. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021; 19:55–71. [DOI] [PubMed] [Google Scholar]
- 2. Roager H.M., Licht T.R.. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018; 9:3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Van Treuren W., d Dodd D.. Microbial contribution to the human metabolome: implications for health and disease. Annu. Rev. Pathol. Mech. Dis. 2020; 15:345–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xiao J., Fiscella K.A., Gill S.R.. Oral microbiome: possible harbinger for children's health. Int. J. Oral Sci. 2020; 12:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Altmäe S., Franasiak J.M., Mändar R.. The seminal microbiome in health and disease. Nat. Rev. Urol. 2019; 16:703–721. [DOI] [PubMed] [Google Scholar]
- 6. Wang X., Tsai T., Deng F., Wei X., Chai J., Knapp J., Apple J., Maxwell C.V, Lee J.A., Li Y.et al.. Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome. 2019; 7:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sampson T.R., Mazmanian S.K.. Control of brain development, function, and behavior by the microbiome. Cell Host Microbe. 2015; 17:565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grant A., Gay C.G., Lillehoj H.S.. Bacillus spp. as direct-fed microbial antibiotic alternatives to enhance growth, immunity, and gut health in poultry. Avian Pathol. 2018; 47:339–351. [DOI] [PubMed] [Google Scholar]
- 9. Calik A., Omara I.I., White M.B., Li W., Dalloul R.A.. Effects of dietary direct fed microbial supplementation on performance, intestinal morphology and immune response of broiler chickens challenged with coccidiosis. Front. Vet. Sci. 2019; 6:463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Garcia-Mazcorro J.F., Ishaq S.L., Rodriguez-Herrera M.V, Garcia-Hernandez C.A., Kawas J.R., Nagaraja T.G.. Review: are there indigenous in the digestive tract of livestock animal species? Implications for health, nutrition and productivity traits. Animal. 2020; 14:22–30. [DOI] [PubMed] [Google Scholar]
- 11. Ogunade I.M., McCoun M., Idowu M.D., Peters S.O.. Comparative effects of two multispecies direct-fed microbial products on energy status, nutrient digestibility, and ruminal fermentation, bacterial community, and metabolome of beef steers. J. Anim. Sci. 2020; 98:skaa201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Buntyn J.O., Schmidt T.B., Nisbet D.J., Callaway T.R.. The role of direct-fed microbials in conventional livestock production. Annu. Rev. Anim. Biosci. 2016; 4:335–355. [DOI] [PubMed] [Google Scholar]
- 13. Toribio A.L., Alako B., Amid C., Cerdeño-Tarrága A., Clarke L., Cleland I., Fairley S., Gibson R., Goodgame N., ten Hoopen P.et al.. European nucleotide archive in 2016. Nucleic Acids Res. 2017; 45:D32–D36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Collaboration, on behalf of the I.N.S.D. Leinonen R., Sugawara H., Shumway M.. The sequence read archive. Nucleic Acids Res. 2011; 39:D19–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu J., Ma L., Nie Y., Chen J., Zheng W., Wang X., Xie C., Zheng Z., Wang Z., Yang T.et al.. A microbiota-derived bacteriocin targets the host to confer diarrhea resistance in early-weaned piglets. Cell Host Microbe. 2018; 24:817–832. [DOI] [PubMed] [Google Scholar]
- 16. Kim H.S., Whon T.W., Sung H., Jeong Y.-S., Jung E.S., Shin N.-R., Hyun D.-W., Kim P.S., Lee J.-Y., Lee C.H.et al.. Longitudinal evaluation of fecal microbiota transplantation for ameliorating calf diarrhea and improving growth performance. Nat. Commun. 2021; 12:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bainbridge M.L., Cersosimo L.M., Wright A.-D.G., Kraft J.. Rumen bacterial communities shift across a lactation in holstein, jersey and holstein × jersey dairy cows and correlate to rumen function, bacterial fatty acid composition and production parameters. FEMS Microbiol. Ecol. 2016; 92:fiw059. [DOI] [PubMed] [Google Scholar]
- 18. Gonzalez A., Navas-Molina J.A., Kosciolek T., McDonald D., Vázquez-Baeza Y., Ackermann G., DeReus J., Janssen S., Swafford A.D., Orchanian S.B.. Qiita: rapid, web-enabled microbiome meta-analysis. Nat. Methods. 2018; 15:796–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Keegan K.P., Glass E.M., Meyer F.. MG-RAST, a metagenomics service for analysis of microbial community structure and function. Microbial Environmental Genomics (MEG). 2016; Springer; 207–233. [DOI] [PubMed] [Google Scholar]
- 20. Yang J., Park J., Jung Y., Chun J.. AMDB: a database of animal gut microbial communities with manually curated metadata. Nucleic Acids Res. 2022; 50:D729–D735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu Y., Lei B., Zhang Q., Lei Y., Li C., Li X., Yao R., Hu R., Liu K., Wang Y.. ADDAGMA: a database for domestic animal gut microbiome atlas. Comput. Struct. Biotechnol. J. 2022; 20:891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rognes T., Flouri T., Nichols B., Quince C., Mahé F.. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016; 4:e2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Edgar R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010; 26:2460–2461. [DOI] [PubMed] [Google Scholar]
- 24. Cole J.R., Wang Q., Fish J.A., Chai B., McGarrell D.M., Sun Y., Brown C.T., Porras-Alfaro A., Kuske C.R., Tiedje J.M.. Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014; 42:D633–D642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nilsson R.H., Larsson K.-H., Taylor A.F.S., Bengtsson-Palme J., Jeppesen T.S., Schigel D., Kennedy P., Picard K., Glöckner F.O., Tedersoo L.. The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019; 47:D259–D264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu Y.-X., Qin Y., Chen T., Lu M., Qian X., Guo X., Bai Y.. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell. 2021; 12:315–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen S., Zhou Y., Chen Y., Gu J.. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018; 34:i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Langmead B., Salzberg S.L.. Fast gapped-read alignment with bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D.. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012; 19:455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roux S., Enault F., Hurwitz B.L., Sullivan M.B.. VirSorter: mining viral signal from microbial genomic data. PeerJ. 2015; 3:e985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ren J., Ahlgren N.A., Lu Y.Y., Fuhrman J.A., Sun F.. VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome. 2017; 5:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Olm M.R., Brown C.T., Brooks B., Banfield J.F.. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017; 11:2864–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buchfink B., Xie C., Huson D.H.. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2015; 12:59–60. [DOI] [PubMed] [Google Scholar]
- 34. Lewis J.D., Chen E.Z., Baldassano R.N., Otley A.R., Griffiths A.M., Lee D., Bittinger K., Bailey A., Friedman E.S., Hoffmann C.. Inflammation, antibiotics, and diet as environmental stressors of the gut microbiome in pediatric Crohn's disease. Cell Host Microbe. 2015; 18:489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Franzosa E.A., Sirota-Madi A., Avila-Pacheco J., Fornelos N., Haiser H.J., Reinker S., Vatanen T., Hall A.B., Mallick H., McIver L.J.. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019; 4:293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dai D., Zhu J., Sun C., Li M., Liu J., Wu S., Ning K., He L., Zhao X.-M., Chen W.-H.. GMrepo v2: a curated human gut microbiome database with special focus on disease markers and cross-dataset comparison. Nucleic Acids Res. 2022; 50:D777–D784. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.