

Abstract
Motivation
Functional interpretation of high-throughput metabolomic and transcriptomic results is a crucial step in generating insight from experimental data. However, pathway and functional information for genes and metabolites are distributed among many siloed resources, limiting the scope of analyses that rely on a single knowledge source.
Results
RaMP-DB 2.0 is a web interface, relational database, API and R package designed for straightforward and comprehensive functional interpretation of metabolomic and multi-omic data. RaMP-DB 2.0 has been upgraded with an expanded breadth and depth of functional and chemical annotations (ClassyFire, LIPID MAPS, SMILES, InChIs, etc.), with new data types related to metabolites and lipids incorporated. To streamline entity resolution across multiple source databases, we have implemented a new semi-automated process, thereby lessening the burden of harmonization and supporting more frequent updates. The associated RaMP-DB 2.0 R package now supports queries on pathways, common reactions (e.g. metabolite-enzyme relationship), chemical functional ontologies, chemical classes and chemical structures, as well as enrichment analyses on pathways (multi-omic) and chemical classes. Lastly, the RaMP-DB web interface has been completely redesigned using the Angular framework.
Availability and implementation
The code used to build all components of RaMP-DB 2.0 are freely available on GitHub at https://github.com/ncats/ramp-db, https://github.com/ncats/RaMP-Client/ and https://github.com/ncats/RaMP-Backend. The RaMP-DB web application can be accessed at https://rampdb.nih.gov/.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The impact of metabolomics and multi-omics on biomedical and translational research continues to grow. Multi-omic studies that combine metabolomics data with genomic, transcriptomic or proteomic data provide additional perspectives that capture the many complex interactions occurring among genes, proteins and metabolites (Barberis et al., 2020; Chen et al., 2020; Eicher et al., 2020; Zhao et al., 2020). However, the interpretation of multi-omic data raises many hurdles for researchers. Challenges associated with multi-omic integration include the large variety of identifier types for metabolites, genes and proteins, the scarcity of up-to-date comprehensive and integrated gene/protein and metabolite annotation sources, and the tools to work across these omics types. With these issues in mind, we created RaMP-DB, the relational database of metabolic pathways (Zhang et al., 2018). The original RaMP-DB release in 2018 integrated functional and other biologically relevant annotations for metabolites, genes and proteins, where the latter were aggregated across the multiple sources HMDB (Wishart et al., 2007, 2009, 2013, 2018, 2022), Reactome (Fabregat et al., 2018; Jassal et al., 2020), WikiPathways (Kutmon et al., 2016; Slenter et al., 2018) and KEGG (Kanehisa and Goto, 2000; Kanehisa et al., 2016, 2017) (through HMDB). Our intent for building RaMP-DB was to provide up-to-date and comprehensive annotations that could be readily used to interpret metabolomic and multi-omic data. Interpretation is facilitated through the associated R package and web interface, or by integrating the publicly available relational database (e.g. available as a mySQL dump) directly into one’s own tools.
Among tools that harmonize multiple sources and support multi-omic analyses, RaMP-DB is notable for its inclusion of multi-omic pathways from multiple sources, its ability to accept mixed ID types for genes, proteins and metabolites, and its ability to compute enrichment statistics on multi-omic pathways (Supplementary Table S1). It is worth noting that public knowledge sources and associated tools that draw information from multiple databases are oftentimes built for a specific purpose. For example, ConsensusPathDB (Kamburov et al., 2009) focuses on physical interactions and supports interaction network exploration and single-omic enrichment analyses. IMPaLA (Kamburov et al., 2011) focuses on multi-omic pathway analysis of transcripts/proteins and metabolites but lacks a special focus on metabolite functional enrichment in the form of lipid and chemical structure annotations. RaMP-DB 2.0 was specifically built to address these gaps and supports multi-omic biological pathway enrichment and metabolite chemical class enrichment by integrating information from multiple sources into a publicly available, user-friendly resource.
We present here on the recent enhancements to our RaMP-DB 2.0 ecosystem (Table 1). RaMP-DB 2.0's contents have been updated to reflect expansions of its constituent pathway databases. For example, the most recent ontologies from HMDB 5.0 (Wishart et al., 2022), such as relevant portions of the new chemical functional ontology, are now included. Further, RaMP-DB 2.0 now utilizes a new semi-automated entity resolution method that verifies compound structural elements when mapping metabolite entries across different databases. This entity resolution method is augmented by manual curation to verify metabolite identifier cross-references when aggregating or merging metabolite records from different data sources. The semi-automated process allows for more frequent updates to be performed, while still facilitating quality checking of cross-database links listed by RaMP-DB’s constituent sources.
Table 1.
Comparison of features in RaMP-DB 1.0 versus RaMP-DB 2.0
| Feature | Available in RaMP-DB 1.0 | Available in RaMP-DB 2.0 |
|---|---|---|
| Pathway annotations from KEGG, HMDB, WikiPathways and Reactome | X | X |
| Reactions and metabolite ontology from HMDB | X | X |
| Redundancy clustering of pathways | X | X |
| RShiny online interface | X | |
| Angular online interface | X | |
| API | X | |
| Chemical structure annotations from HMDB, ChEBI and LIPID MAPS | X | |
| Chemical class enrichment analysis | X | |
| Semi-automated entity resolution pipeline | X |
RaMP-DB 2.0 now also includes chemical structure and class annotations for metabolites with an increased focus on lipids, which can be used for exploring the breadth of chemical classes and space covered by a collection of metabolites/lipids under study. As an additional new feature, the over-representation of chemical classes in a metabolite set of interest relative to a larger collection of quantified metabolites can now be calculated, providing complementary information to biological pathway enrichment, which typically informs on a much smaller fraction of metabolites (Barupal and Fiehn, 2017). Pathway and chemical enrichment analyses support the inclusion of a custom background (e.g. metabolites evaluated in a study). Lastly, an updated user-friendly web interface (https://rampdb.nih.gov/) interacts with RaMP-DB 2.0 through a public application programming interface (API) and supports queries and enrichment analyses. All components of RaMP-DB 2.0 are thoroughly documented, and its underlying processes are fully transparent within GitHub.
2 Materials and methods
2.1 Parsing data sources and RaMP-DB 2.0 schema
Each primary data source incorporated into RaMP-DB 2.0 has a dedicated python class that fetches on-line data files to a local data store, then reads and parses the data source-specific input data and writes the key data to a collection of intermediate files of a standard format. The primary sources incorporated into RaMP-DB 2.0 include HMDB v5.0 (Wishart et al., 2007, 2009, 2013, 2018, 2022), Reactome v81 (Fabregat et al., 2018; Jassal et al., 2020), WikiPathways v20220710 (Kutmon et al., 2016; Slenter et al., 2018), ChEBI (release August 3, 2022) (Degtyarenko et al., 2008; Hastings et al., 2013, 2016) and LIPID MAPS (release July 13, 2022) (Fahy et al., 2009; Sud et al., 2007). A processing class reads the collection of all intermediate files for all data sources and populates a collection of entity classes that are organized into natural relationships among genes, metabolites, pathways and related information. This data structure allows a well-controlled process of entity resolution, with error checking at redundancy checks. This entity resolution step uses a configuration file that controls the database refresh and a python class for database loading to write a final set of files that are formatted to ease database upload (see Section 2.2).
Supplementary Figure S1A diagrams the RaMP-DB 2.0 database schema. Each table in the RaMP-DB 2.0 database described below is a mySQL table, linked together by internal RaMP IDs (see Section 2.3) that represent entities harmonized from across sources. The analyte table contains a list of internal and unique RaMP analyte IDs that correspond to genes, proteins and metabolites. Meta-information associated with these analytes are featured in the source table and the analytesynonym table. The source table includes all IDs and common names that map to each RaMP analyte entity. Analytesynonyms provides 1 006 301 common name synonyms for the metabolites and genes in RaMP. Two mapping tables (analytehaspathway and analytehasontology) connect our analyte entity IDs with tables that contain information on pathways and metabolite annotations held in the ontology table. Separate tables contain metabolite information on chemical classes from HMDB (v5.0) (Wishart et al., 2007, 2009, 2013, 2018, 2022) and LIPID MAPS (Fahy et al., 2009; Sud et al., 2007) (release July 13, 2022), and chemical properties from HMDB (Wishart et al., 2007, 2009, 2013, 2018, 2022) (v5.0), ChEBI (Degtyarenko et al., 2008; Hastings et al., 2013, 2016) (release August 3, 2022) and LIPID MAPS (Fahy et al., 2009; Sud et al., 2007) (release July 13, 2022). Three tables contain meta-information that characterizes the current database build: the db_version table holds the build version and build timestamp of the entire RaMP 2.0 database, the version_information table holds information on each data source including the data sources version and release date, and the entity_status_info table contains a tally of current RaMP 2.0 entities within the build. Entities include counts for unique metabolites, genes/proteins, pathways, chemical property records for metabolites and mappings between analytes and pathways in RaMP. The catalyzed table contains 1 541 996 associations between metabolites and genes that participate in metabolic reactions together.
All scripts to build the MySQL database are available through a public GitHub repository at https://github.com/ncats/RaMP-BackEnd. The link to the most recently available MySQL database dump is available through the public RaMP R package (https://github.com/ncats/RaMP-DB/).
2.2 Semi-automated entity resolution
To faithfully represent entities across various data sources, we have implemented a data model that accurately encapsulates gene, protein, metabolite entities and their associated meta-data (e.g. identifiers, synonyms, chemical properties, data source tags, etc.). The data intake process starts with reading configuration files that instruct back-end processes to fetch data from external data sources. The source data files are then parsed with data-source-specific parsers into intermediate consistently formatted files prior to combining and resolving data from the various data sources. As a special case, metabolite entity resolution collapses metabolite records from different data sources if the metabolite records from each source share at least one external id reference. A previously generated and manually curated list of incorrect metabolite id mappings is referenced to eliminate improper collapsing of metabolite records. A special class for this task named ‘EntityBuilder’ resolves the data by compiling all source data into a data model that deduplicates metabolites and holds associations between metabolites and their chemical properties, genes, pathways and associated information. The resolved data is written into final files prior to bulk loading into the relational database schema (Supplementary Fig. S1B).
Most data sources link metabolite entities to a collection of additional ID types, such as PubChem CID, ChemSpider, HMDB ID, ChEBI ID and LIPID MAPS ID. ID cross-references of mappings help to suggest metabolites that are in common across different data sources. Two metabolites drawn from different sources that share a common metabolite ID are mapped to a common RaMP-DB 2.0 metabolite entity and assigned an internal RaMP ID (see Section 2.3 for more details). Thus, a metabolite entity contains all molecules cross-referenced in the constituent resources comprising RaMP-DB 2.0. Following the construction of these entities and relationships, the linked metabolites in the data model are verified by comparing molecular weights taken from the data source that publishes the identifier (HMDB, ChEBI, PubChem, Lipid Maps or KEGG). We found that a molecular weight difference of 10% or more from the lower molecular weight metabolite was sufficient to capture many mismappings while minimizing the number of ‘false positive’ mismapping flags (candidate mapping errors that are sent for manual inspection). Thus, any two resolution candidates with greater than 10% difference in molecular weight are flagged as a bad annotation and subsequently manually verified. During this automated assessment of all ID-based compound associations, associations between metabolites suspected as being faulty (e.g. high molecular weight variance) are flagged and exported to a list for manual curation. If it is deemed through subsequent manual curation that two IDs refer to different metabolites, then the metabolite ID pair goes into a list of associations to skip. After manual curation of all such issues, the data are built, skipping associations within the exclusion list. This results in both entities being represented but skipping any merge suggested by the bad cross reference. This mismapping exclusion list is referenced on every subsequent database build. Reported discrepancies in the association between two metabolites IDs are added to the exclusion list as they are reported so that later database builds will use the latest curation patches (https://github.com/ncats/RaMP-BackEnd/blob/master/config/curation_mapping_issues_list.txt).
2.3 RaMP IDs
During entity resolution and database loading, internal RaMP IDs are generated. These RaMP IDs are not intended to be used by the general user, so as to streamline the data accession process (however, the interested user can easily access the identifiers using the mySQL database associated with the package). Instead, the RaMP IDs represent database-internal values for compound, gene, ontology, and pathway entities that act as keys that relate entities to one another, which are sourced from multiple sources, in the database. The values are used to reference RaMP entities within the database. While consistent within a database update, these IDs are not conserved across database versions. Entity IDs from external data sources are maintained as primary authoritative IDs to be used in result tables and in work derived from RaMP-DB 2.0 analyses. The RaMP-DB 2.0 primary analyte source information table and pathway table maintain a field that tracks the associated primary data source so that all RaMP-DB 2.0 entities maintain a record of data provenance.
2.4 API
The API serves as an interface between the R package and external applications that use the package’s capabilities. The API was created using the Plumber API generator (v 1.1.0) (Schloerke and Allen, 2022), which creates the necessary urls, as well as documentation. The API receives HTTPS requests, makes the necessary calls to RaMP functions and returns the resulting data back to the application that made the request. All API requests support the JSON format to return data, but in addition, many queries have optional parameters to allow the return of data in TSV format. The calls supported by the API are publicly provided on the RaMP-DB web interface at https://ramp-api-alpha.ncats.io/__docs__/.
2.5 Web interface
The web interface makes HTTPS requests to the API based on the user interactions and renders the data returned by the API. It was developed using the Angular framework (currently version 13) and Material Design methodology created by Google. Users are given examples and descriptions for each supported RaMP query but may also substitute their own identifiers. The interface is a single-page application, allowing the bulk of the application to be loaded when the user first visits the website. To minimize loading times, any subsequent interactions load the minimum amount of data needed to fulfill its functions. The code repository along with instructions on how to run both the application and the API can be found at https://github.com/ncats/RaMP-Client. The web interface can be accessed at https://rampdb.nih.gov.
2.6 R Package
RaMP-DB 2.0 functions are annotated with roxygen v7.1.2 blocks to generate Rd help files, with working examples that can be opened in R. The package includes an extensive Vignette tutorial (https://ncats.github.io/RaMP-DB/RaMP_Vignette.html) that features the primary functions available within the package. Instructions on the GitHub page describe setting up the MySQL database locally and installing the RaMP-DB 2.0 R package. Importantly, the RaMP-DB 2.0 MySQL full database dump is included in the RaMP-DB 2.0 Package GitHub site and can also be explored independently of the R package. Like all parts of RaMP-DB 2.0, the RaMP-DB 2.0 R Package is open-source and available at https://github.com/ncats/RaMP-DB. A summary of the key queries that can be performed by RaMP, as well as the front-end and API methods performing these queries, is supplied in Supplementary Table S2.
2.7 Pathway and chemical enrichment analysis
Enrichments are calculated using a Fisher’s exact test, based on a 2 × 2 contingency table. These contingency tables are used to test the null hypothesis that the number of altered metabolites belonging to that pathway is less than or equal to the number expected by random chance. Where n is the total number of metabolites in the reference background, a is the count of differentially expressed analytes in the pathway being tested, b is the count of background analytes in the pathway being tested c is the count of differentially expressed analytes outside of the pathway being tested, and d is the count of background analytes outside the pathway being tested, the probability of observing a particular input contingency table is calculated using Equation 1.
| (1) |
By default, RaMP-DB 2.0 uses the one-tailed version of the Fisher’s exact test P-value calculation. However, the two-tailed hypothesis is also supported by the package. Multiple test correction for pathway analysis results is performed using either the Holm (Holm, 1979) or Benjamini-Hochberg (Benjamini and Hochberg, 1995) method. Pathway analysis results can be clustered using the Heuristic Multiple Linkage Clustering algorithm developed for redundancy clustering of gene ontology terms employed by DAVID (Huang et al., 2007). In this method, pathways are grouped into provisional clusters based on a user-defined threshold of overlap of the pathways’ constituent analytes. Users can also define the minimum number of overlapping pathways required to form a provisional cluster. Provisional clusters are then merged based on a user-defined threshold of overlap of the constituent pathways in each cluster. This process is repeated until there are no remaining cluster overlap values that are higher than the cluster merge threshold. This clustering algorithm is used to generate groups for our pathways in enrichment results and plots, which displays adjusted Fisher’s P values, the number of altered analytes per pathway, database of origin for each pathway and cluster membership for each pathway.
3 Results
3.1 RaMP-DB 2.0 ecosystem
RaMP-DB 2.0 has three main components: (i) Python code for constructing the back-end MySQL database that draws from multiple sources, (ii) an R package that supports queries and analyses using RaMP-DB 2.0 and (iii) an online interface and associated API for programmatic access. The RaMP R package supports four different query types (biological pathways, ontologies, chemical classes/properties and reactions) and two different enrichment analyses (biological pathway and chemical) (Fig. 1B). Queries are available for lists of genes, proteins and metabolites, returning pathways, biochemical reactions and/or ontologies that contain those analytes (Supplementary Table S2). Users may also query a list of pathways and return analytes associated with those pathways. Lastly, for lists of metabolites only, users may query ontologies from HMDB, chemical structure or chemical class information. Mappings returned from the pathway and chemical class queries can be leveraged for functional enrichment analysis using the Fisher’s exact test.
Fig 1.
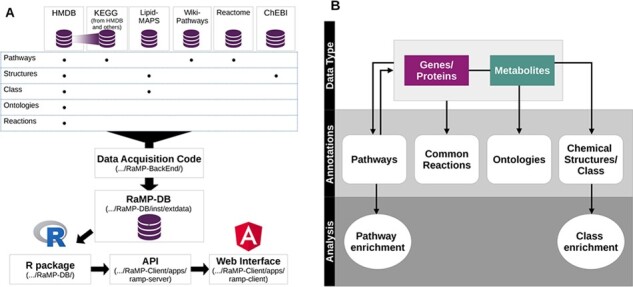
Overview of RaMP-DB. (A) Relationships between metabolites/gene data, annotation types and enrichment options in RaMP. Certain annotations, such as chemical structure, are only available for metabolites. (B) RaMP data sources, and relationships between different components of RaMP, including links to publicly accessible code
3.2 RaMP-DB 2.0 structure and contents
The data for RaMP-DB 2.0 are drawn from six distinct sources (Fig. 1A) and parsed using python scripts available at https://github/ncats/RaMP-BackEnd. These data include annotations associated with pathways, common reactions, functional ontologies, chemical structures and chemical classes on human metabolites and genes/proteins. The parsed data are organized into an analyte-centric relational database containing 13 tables (Supplementary Fig. S1A). As in previous iterations, the relational structure offered by MySQL is designed for efficient retrieval of annotations related to a list of analytes of interest input by the user.
RaMP-DB 2.0 incorporates updated pathway annotations from four popular public metabolite pathway databases: HMDB (v5.0) (Wishart et al., 2007, 2009, 2013, 2018, 2022), Reactome (v81) (Fabregat et al., 2018; Jassal et al., 2020), WikiPathways (v20220710) (Kutmon et al., 2016; Slenter et al., 2018) and KEGG (Kanehisa and Goto, 2000; Kanehisa et al., 2016, 2017) (through HMDB) (Table 2). RaMP-DB 2.0 contains pathway associations for both genes/proteins and metabolites. New additions to RaMP-DB 2.0 include ontologies derived from HMDB (v5.0) (Wishart et al., 2007, 2009, 2013, 2018, 2022), chemical structure and class information. RaMP-DB 2.0 now contains 256 952 chemical structures associated with its collection of metabolites. Many metabolite entities in RaMP-DB 2.0 have multiple structures associated with them. For example, ‘RAMP_C_000000004’ (2-Hydroxybutyric acid) is associated with one structure from HMDB, as well as both enantiomer forms apiece from LIPID MAPS and ChEBI, for a total of five structures. Most notably, the inclusion of 44 430 lipids from LIPID MAPS has greatly expanded the number of lipids for which information is available in RaMP-DB 2.0. These structures, obtained from HMDB (v5.0) (Wishart et al., 2007, 2009, 2013, 2018, 2022), ChEBI (release August 3, 2022) (Degtyarenko et al., 2008; Hastings et al., 2013, 2016) and LIPID MAPS (release July 13, 2022) (Fahy et al., 2009; Sud et al., 2007), provide a rich source of information for chemical compounds of interest and can be used as a basis for cheminformatics analysis.
Table 2.
Number of analytes and pathways (A) and chemical properties (B) available through RaMP-DB 2.0
| A | |||||
|---|---|---|---|---|---|
| Totala | HMDB v5.0 | KEGG (from HMDB 5.0) | Reactome v81 | WikiPathways v20220710 | |
| # Distinct metabolites | 256 086 (+142 361) | 216 683 | 5898 | 2355 | 3695 |
| # Distinct genes/enzymes | 15 827 (+410) | 7111 | – | 11 227 | 13 393 |
| # Distinct pathways | 53 831 (+2035) | 49 613 | 363 | 2583 | 1272 |
| #Metabolite-pathway mappings | 412 775 (+343 120) | 367 609 | 1714 | 30 804 | 12 648 |
| # Gene-pathway mappings | 401 303 (−695 287) | 208 211 | 8479 | 125 171 | 59 442 |
|
| |||||
| B | |||||
|
| |||||
| Total distinct compoundsb | HMDB v5.0 | ChEBI release 212 | LIPID MAPS release July 13, 2022 | ||
|
| |||||
| Chemical propertiesc | 256 592 | 217 776 | 13 066 | 44 981 | |
The number in parentheses represents the difference in numbers compared to the previous RaMP version (1.1.0).
Distinct InChIKeys.
Chemical properties are only captured for compounds referenced within RaMP.
RaMP-DB 2.0 also contains chemical class, superclass and subclass as dictated by the ClassyFire (Djoumbou Feunang et al., 2016) taxonomy and the LIPID MAPS database, which are used for chemical class enrichment analysis. Lastly, RaMP-DB 2.0 contains a collection of 1 541 996 metabolic enzyme/metabolite mutual reaction relationships and 699 succinct functional ontologies from HMDB (Wishart et al., 2007, 2009, 2013, 2018, 2022) (v 5.0). Functional ontologies comprise a subset of the Chemical Functional Ontology within HMDB 5.0 (Wishart et al., 2022) (outlined in Supplementary Table S3). The Chemical Functional Ontology, similar to the Gene Ontology (The Gene Ontology Consortium, 2019) resource for transcriptomics and proteomics, was developed to provide an additional collection of annotations for metabolite functions and origins.
3.3 Resolving entity mismappings
Integrating data from multiple resources expands the number and type of annotations for analytes. This integration requires accurate mapping of gene, protein, metabolite entities across the different resources. To this end, we initially group together individual entities that represent the same molecule, based on a shared ID as prescribed by the source data (Fig. 2). However, this reliance on accurate mappings from source databases (e.g. ‘Glucose’ mapping to hmdb: HMDB0304632, hmdb: HMDB0000122, kegg: C00031, chemspider: 58238, and others) can lead to errors.
Fig 2.

Approach to entity resolution of names across different database sources. The figure depicts the mappings of IDs and metabolites from two different source databases (e.g. denoted as Source 1 and Source 2 Dictionary). Source 2 Dictionary has two instances of ID C, mapping to two different metabolites (1 and 3). In this case, Metabolites 1 and 3 will be merged and considered the same metabolite, which may or may not be accurate
As an example, we identified an instance where a data source provides a metabolite record for a diglyceride as well as a corresponding valid PubChem ID (Supplementary Fig. S2). Another metabolite record from the same source represents 11-Oxo-androsterone glucuronide but has an external ID reference to the PubChem diglyceride record. Naively following ID associations would collapse these two metabolites into one entity due to the common PubChem CID. In this case, the steroid-based compound has a molecular weight of 480.55 Da, while the diglyceride has a molecular weight of 681.12 Da, resulting in the error in linking being identified automatically through molecular weight comparison.
Notably, these mis-mappings would propagate to errors in pathways or other annotation mappings and introduce false positives in enrichment analyses. To address this issue, we have developed a heuristic based on molecular weight (MW) to automatically flag potential mis-mappings that could occur between database sources (described in Section 2.2). MW was chosen as a coarse measure of matching to ensure that gross errors in mapping would be flagged at a high rate while smaller discrepancies would be permitted. This is a result of the fact that many of the entities being merged are represented as generic structures in their source databases, meaning that small differences in molecular weight are often present between members of the same entity. Other heuristics based on structure (i.e. SMILES string and InChi Key prefix) were found to either not permit any uncertainty, or to be too coarse so as to miss some mismappings. Flagged mis-mappings are manually investigated and potentially discarded as appropriate. Mis-mappings are recorded and used to automatically correct future updates of RaMP-DB 2.0, thereby expediting subsequent database rebuilds.
RaMP-DB 2.0 curation revealed a total of 955 distinct metabolites involved in incorrect associations linking disparate molecules. Within the 955 metabolites, 351 metabolites had incorrect ID-based cross-reference associations to 604 distinct metabolites. Curation eliminated the propagation of these errors and the resulting mis-associations that could arise between these metabolites and pathways.
3.4 Overlap of entities across data sources
Following entity resolution, RaMP-DB 2.0 contains a total of 256 086 metabolites, 15 827 genes and 53 831 pathways. This entity resolution allows for an analysis of overlapping content in each of the constituent databases, highlighting the value of a united database for these annotations. Out of the 256 086 unique metabolites found in RaMP-DB 2.0, 109 754 metabolites (43%) are only found in one of the source databases (99% of these unique metabolites are derived from HMDB, Fig. 3). Of the 57 302 metabolites that have at least one pathway association in RaMP-DB 2.0, 12 966 are only found with pathway mappings in one source database (23%). Conversely, out of 14 301 genes/proteins in RaMP-DB 2.0, 686 genes/proteins (5%) are unique to their source database. Notably, entity resolution allows users to input mixed ID types when performing batch queries, which is particularly relevant for metabolomic data that seldom report a single ID type. The different ID types supported for each analyte type are shown in Table 3.
Fig 3.
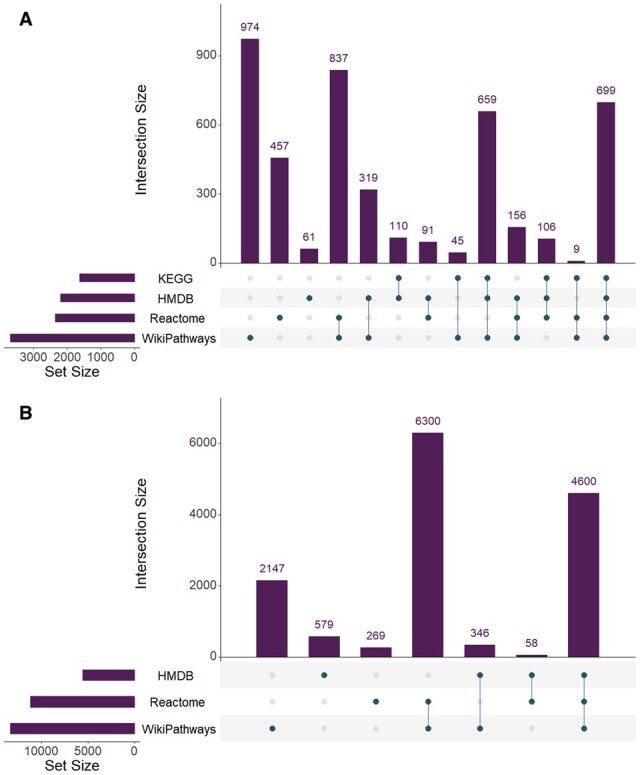
Overlap in content among source databases. Only analytes mapping to pathways are considered, as HMDB contains a large number of metabolites associated only with ontologies, which are not relevant to Reactome and Wikipathways as pathway-centric databases. (A) Overlap in metabolites associated with at least one pathway between source databases in RaMP. (B) Overlap of genes associated with at least one pathway. The filled circle(s) underneath each bar in the plots demonstrate the source databases that the analyte counts are drawn from
Table 3.
Supported ID types for analytes in RaMP
| Analyte type | Supported ID types |
|---|---|
| Metabolites | PubChem, HMDB, ChEBI, WikiData, ChemSpider, CAS, SwissLipids, LIPID MAPS, KEGG, LipidBank, PlantFA |
| Genes/proteins | HMDB, UniProt, Entrez, Gene Symbol, Ensembl, NCBIprotein, EN, WikiData, ChEBI |
Note: The ID types (in lower case) need to be prepended to the ID for queries and analyses (e.g. hmdb: HMDB0000064).
3.5 Upgrades to the user interface
The new RaMP-DB interface was designed with modularity in mind, and an emphasis on matching functionality in the RaMP package. Separate pages are available for Biological Pathways (pathways from analytes, analytes from pathways), Chemical Classes (chemical class queries or general properties such as InChiKEY and SMILES string), Ontology Queries (ontologies from metabolites or metabolites from ontologies), Reactions (reactions shared by analytes) and Enrichment Analysis (chemical class or pathway). Results of mySQL queries to the underlying database as well as enrichment analyses are downloadable in the TSV format. To improve reproducibility, the functions used by the R package for each query/analysis are displayed. This allows users to recreate analyses locally if desired. Figure 4A showcases the design and features of the updated user interface. The API that interacts with the user interface provides another option for programmatic access to the information stored in RaMP-DB 2.0.
Fig 4.

Screenshots of the updated RaMP-DB web interface. (A) The web interface implements all the new features of the RaMP-DB 2.0 package. A new, more streamlined splash screen organizes the functionalities of the interface into queries for pathways, chemical properties, reactions and ontologies, as well as enrichment analyses. (B) Example pathway enrichment ‘lollipop’ plot generated in the web interface (cropped), using an example query of 20 metabolites and 4 genes. The new plotting function simplifies the process of identifying functional redundancies in pathway analysis output
3.6 Enrichment analyses
We have implemented functions for testing the enrichment of pathways and chemical classes in a list of metabolites and/or genes and proteins. Chemical class enrichment analysis is a noteworthy addition to RaMP-DB 2.0’s capabilities, as chemical class annotation coverage in RaMP for metabolites (100%) far outstrips that of pathway annotation for metabolites (22.3%). Increasing the coverage of metabolites considered by functional enrichment analysis broadens the scope of output, potentially providing better insight.
RaMP-DB 2.0 also now offers several options for defining the analytical background used for the Fisher’s exact test in pathway enrichment analysis. First, it can be defined as the number of metabolites contained in the pathway database that the pathway being evaluated is found in (e.g in this case, the entire RaMP-DB 2.0). Second, the reference background can be described as the full set of metabolites that were identified in the study. Last, it can be defined as the collection of metabolites that are known to be present in the biospecimen of interest (e.g. urine or blood metabolites only).
We have implemented a ‘lollipop plot’ (Fig. 4B) function for plotting pathway analysis results. This function uses the pathway redundancy clustering algorithm developed for RaMP-DB 1.0 (see Section 2) to group together highly overlapping pathways, highlighting potential redundancies in enrichment results. Specifically, the plotting function displays pathway cluster membership, database of origin for pathways, the number of altered analytes mapping to each pathway and Fisher’s P value with multiple test correction of choice (including the Holm (1979) and Benjamini and Hochberg (1995) methods).
3.7 Comparison against other functional enrichment analysis resources
RaMP-DB 2.0 is a new addition to an already active ecosystem of open-source software devoted to multi-omics functional enrichment analysis. Popular existing resources include MetaboAnalyst (Chong et al., 2018; Xia et al., 2015), a preeminent multi-omics pathway analysis tool, as well as ChemRich (Barupal and Fiehn, 2017), a tool for chemical class enrichment analysis of metabolomics data. RaMP-DB 2.0 offers several features and insights that are not available through these resources. A major advantage of the RaMP-DB platform is the flexibility in input formatting that RaMP-DB accepts. MetaboAnalyst cannot accept mixed input types (i.e. all input analytes must be present in the same source database), and only recognizes 3 identifier types for genes as well as 2 for metabolites, compared to the 9 identifiers for genes and 11 for metabolites that RaMP-DB can recognize. Similarly, ChemRich requires SMILES strings for each input metabolite, whereas RaMP-DB can perform chemical class enrichment analysis on all 11 metabolite identifier types.
We found that even when RaMP-DB’s superior capabilities for mapping input analytes were ignored, RaMP-DB returns a larger pool of relevant pathways than MetaboAnalyst based on the same input. To test this, we supplied the same query used to generate Figure 4B, formatted to ensure that all input analytes would map to MetaboAnalyst’s underlying databases. While we identified 27 significant pathways (False Discovery Rate < 0.05) using RaMP-DB, MetaboAnalyst found only 6 significant pathways. RaMP-DB found many pathways not identified by MetaboAnalyst, including Glyoxylate metabolism pathways, Glycolysis/gluconeogenesis pathways and Urea cycle-related pathways. It is important to note that many of RaMP-DB’s pathways are highly overlapping between or even within sources, which inflates RaMP-DB’s returned pathway total. This makes the redundancy clustering provided by RaMP-DB useful for interpreting results and an important feature that is unique to our tool.
4 Discussion
To the best of our knowledge, RaMP-DB 2.0 is the only knowledge source and associated tool that supports batch queries of analyte annotations, multi-omic pathway and chemical class enrichment analysis with ability to input mixed ID types, and batch queries of pathway and chemical annotations using mixed identifier schemes for both genes and metabolites. Incorporating multiple sources into enrichment analyses greatly expands the mappability of analytes to pathways, thereby enhancing the user’s ability to functionally interpret complex data. RaMP-DB 2.0 verifies the accuracy of mapping analyte entities across its various source databases using a semi-automated process followed by manual curation. The updated RaMP-DB 2.0 now includes 256 086 metabolites, 15 827 genes, 53 828 pathways, 412 775 mappings between metabolites and pathways, and 401 303 mappings between genes and pathways. Improving the accuracy of mappings between analyte identifiers will increase the accuracy of downstream insights gleaned from data.
Other recent efforts in metabolomic software development have noted that chemical class and substructure enrichment analysis can provide functional insight where pathway annotations are unavailable, as chemical structure annotations for metabolites typically offer better coverage (Barupal and Fiehn, 2017; Wanichthanarak et al., 2017). The primary benefit of class enrichment is the superior coverage of chemical class annotations available for metabolites, thanks to the ClassyFire taxonomy (Djoumbou Feunang et al., 2016). Increased annotation coverage for metabolites allows for the incorporation of more experimental information into test results. Class enrichment also allows for the testing of different hypotheses than pathway analysis. For example, in studies where the objective is to identify putative therapeutic targets, the discovery of altered classes can suggest enzymes acting upon generic species of that class as potential inhibition targets (Gao et al., 2019). Integrating this functionality into RaMP-DB 2.0 gives users another option for gaining functional insight into their data. This gain in functionality is particularly pertinent for lipidomics research, where resources for functional annotations are scarce (Molenaar et al., 2019).
We also note the importance of using an appropriate background/reference list of analytes for pathway enrichment analysis (Huang et al., 2007; Mubeen et al., 2019). Typically, ‘background’ metabolites used for the Fisher’s exact test are defined as all metabolites in the original database the pathway being tested was derived from. However, an alternative definition is the list of all metabolites identified in a study. Many Fisher’s pathway analysis tools such as DAVID (Huang et al., 2007) and MetaboAnalyst (Xia et al., 2015; Chong et al., 2018) enable users to select their choice of background. Accordingly, we have implemented the option for either background selection in RaMP 2.0. We have also implemented a third option for backgrounds, comprising all metabolites known to occur in a given biospecimen (as determined by HMDB ontology). Example biospecimen types include ‘Adipose Tissue’, ‘Blood’ and ‘Heart’. We note that using a broad background could include metabolites that should be excluded from the analyses because they are absent in the biospecimen under study, or were not detected for some reason (e.g. failure to ionize or exclusion due to the extraction protocol used). As such, a more appropriate hypothesis to test is to use a custom background of only those metabolites detected in the study, or those appropriate for the biospecimen of interest. A recent study of metabolomics pathway analysis strategies confirms that the choice of background in the Fisher’s exact test exerts large effects on the list of significant pathways returned by the Fisher’s exact test (Wieder et al. 2021) . Enabling users to choose their background based on the information available could thus lead to more reliable outputs for enrichment analysis.
Despite our recent enhancements, RaMP-DB 2.0 has some limitations, particularly regarding its coverage and entity resolution. Currently, RaMP-DB 2.0 is limited to human pathways, although metabolite coverage does include microbial, food, and other exogenous metabolites. Furthermore, the resolution of metabolites across the different resources is not foolproof. Metabolite identification in large-scale metabolomic experiments is still an unresolved issue, and experiments often yield a mix of different levels of certainty and structural resolution in identified metabolites. These factors are not taken into account during mapping of metabolites across sources. Users should thus carefully assess their input list of metabolites, particularly for enrichment analysis and double check that mapping of metabolites is correct. This process is facilitated by the modular design of running enrichment analyses, as described above, which requires users to review database mappings from their input IDs as well as pathway mappings before seeing enrichment results. In all cases, we recommend that users input IDs, rather than names for analyses (this is the default implementation by design).
Further enhancements to RaMP-DB being explored include the incorporation of pathway network-level information, such as complete chemical reactions (to complement current gene/protein and metabolite pairs from enzymatic reactions) linked to specific pathways, to allow for the use of topological pathway analysis algorithms. Other methods for performing enrichment analysis will also be explored, noting the drawback of Fisher’s exact test as it treats all metabolites in a pathway as equivalent parts of a set. This is a misrepresentation, as pathways are a collection of metabolites undergoing reactions that result in some signal or biochemical product. We also anticipate the inclusion of more data types for genes and proteins such as gene ontology annotations or additional reactions to expand the coverage of expert-curated reactions in RaMP-DB. As with other annotations in RaMP-DB 2.0, functions for batch querying and enrichment analysis of new annotations will be implemented. Further, the R package currently relies on a local instance of RaMP-DB 2.0 rather than calling upon the API, which forces users to install and store their own copy of the database. In the future, we intend to remove this limitation. Lastly, we emphasize the important role of users in improving RaMP-DB and its functionalities, and thus encourage users to reach out with any comments, suggestions, and/or issues.
5 Conclusions
RaMP-DB 2.0 is a multi-sourced relational database comprising pathway and chemical annotations for metabolites, genes and proteins. An improved resolution of metabolite and gene/protein mappings across the databases has been implemented and is supplemented with manual curation. Associated and improved R package and web user-friendly interface have been constructed to query the database and perform chemical and pathways enrichment analyses. All steps of RaMP-DB 2.0 are reproducible with all the code used to build or use the database publicly available in GitHub.
Supplementary Material
Acknowledgements
We would like to thank the Individual Graduate Partnership Program of the National Institutes of Health for training and mentoring provided to T.E. and K.S.
Funding
This work was supported in part by the Intramural research program of the National Center for Advancing Translational Sciences [ZIC TR000410-03], National Institutes of Health.
Conflict of Interest: none declared.
Contributor Information
John Braisted, Division of Preclinical Innovation, National Center for Advancing Translational Sciences, Rockville, MD 20850, USA.
Andrew Patt, Division of Preclinical Innovation, National Center for Advancing Translational Sciences, Rockville, MD 20850, USA.
Cole Tindall, Division of Preclinical Innovation, National Center for Advancing Translational Sciences, Rockville, MD 20850, USA.
Timothy Sheils, Division of Preclinical Innovation, National Center for Advancing Translational Sciences, Rockville, MD 20850, USA.
Jorge Neyra, Somatus, McLean, VA 22102, USA.
Kyle Spencer, Division of Preclinical Innovation, National Center for Advancing Translational Sciences, Rockville, MD 20850, USA; Department of Biomedical Informatics, The Ohio State University, Columbus, OH 43210, USA.
Tara Eicher, Division of Preclinical Innovation, National Center for Advancing Translational Sciences, Rockville, MD 20850, USA; Department of Computer Science and Engineering, The Ohio State University, Columbus OH 43210, USA.
Ewy A Mathé, Division of Preclinical Innovation, National Center for Advancing Translational Sciences, Rockville, MD 20850, USA.
Data availability
RaMP-DB 2.0 is an open source project and its code is available through the following GitHub repositories: 1) Code for the R package: https://github.com/ncats/ramp-db, 2) Python code for constructing the RaMP-DB 2.0 mySQL database: https://github.com/ncats/RaMP-BackEnd, and 3) code for building the front-end web application: https://github.com/ncats/RaMP-Client/. The link to the current RaMP-DB 2.0 dump is available at https://figshare.com/ndownloader/files/36760461, and the latest version link is made available through the R package code ReadMe file.
References
- Barberis E. et al. (2020) Large-scale plasma analysis revealed new mechanisms and molecules associated with the host response to SARS-CoV-2. IJMS, 21, 8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B, 57, 289–300. [Google Scholar]
- Chen Y.-M. et al. (2020) Blood molecular markers associated with COVID-19 immunopathology and multi-organ damage. EMBO J., 39, e105896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J. et al. (2018) MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res., 46, W486–W494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degtyarenko K. et al. (2008) ChEBI: a database and ontology for chemical entities of biological interest. Nucleic Acids Res., 36, D344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djoumbou Feunang Y. et al. (2016) ClassyFire: automated chemical classification with a comprehensive, computable taxonomy. J. Cheminform., 8, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barupal D.K., Fiehn O. (2017) Chemical similarity enrichment analysis (ChemRICH) as alternative to biochemical pathway mapping for metabolomic datasets. Sci. Rep., 7. 10.1038/s41598-017-15231-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eicher T. et al. (2020) Metabolomics and multi-omics integration: a survey of computational methods and resources. Metabolites, 10, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A. et al. (2018) The reactome pathway knowledgebase. Nucleic Acids Res., 46, D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahy E. et al. (2009) Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res., 50Suppl (April), 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B. et al. (2019) Multi-omics analyses detail metabolic reprogramming in lipids, carnitines, and use of glycolytic intermediates between prostate small cell neuroendocrine carcinoma and prostate adenocarcinoma. Metabolites, 9, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium. (2019) The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res., 47, D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings J. et al. (2016) ChEBI in 2016: improved services and an expanding collection of metabolites. Nucleic Acids Res., 44, D1214–D1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings J. et al. (2013) The ChEBI reference database and ontology for biologically relevant chemistry: enhancements for 2013. Nucleic Acids Res., 41, D456–D463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm S. (1979) A simple sequentially rejective multiple test procedure. Scand. J. Stat., 6, 65–70. [Google Scholar]
- Huang D.W. et al. (2007) The DAVID gene functional classification tool: a novel biological Module-Centric algorithm to functionally analyze large gene lists. Genome Biol., 8, R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jassal B. et al. (2020) The reactome pathway knowledgebase. Nucleic Acids Res., 48, D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamburov A. et al. (2011) Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics (Oxford, Engl.), 27, 2917–18. [DOI] [PubMed] [Google Scholar]
- Kamburov A. et al. (2009) ConsensusPathDB–a database for integrating human functional interaction networks. Nucleic Acids Res., 37, D623–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M. et al. (2017) KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res., 45, D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res., 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M. et al. (2016) KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res., 44, D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutmon M. et al. (2016) WikiPathways: capturing the full diversity of pathway knowledge. Nucleic Acids Res., 44, D488–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar M.R. et al. (2019) LION/web: a web-based ontology enrichment tool for lipidomic data analysis. GigaScience, 8. 10.1093/gigascience/giz061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mubeen S. et al. (2019) The impact of pathway database choice on statistical enrichment analysis and predictive modeling. Front. Genet., 10, 1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloerke B., Allen J. (2022) Plumber: An API Generator for R.
- Slenter D.N. et al. (2018) WikiPathways: a multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res., 46, D661–D667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sud M. et al. (2007) LMSD: LIPID MAPS structure database. Nucleic Acids Res., 35, D527–D532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanichthanarak K. et al. (2017) Metabox: a toolbox for metabolomic data analysis, interpretation and integrative exploration. PLoS One, 12, e0171046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieder,C. et al. (2021) Pathway analysis in metabolomics: Recommendations for the use of over-representation analysis. Plos Computational Biology, 17(9):e1009105, 10.1371/journal.pcbi.1009105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D.S. et al. (2018) HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res, 46, D608–D617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D.S. et al. (2013) HMDB 3.0–the human metabolome database in 2013. Nucleic Acids Res., 41, D801–D807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D.S. et al. (2009) HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res., 37, D603–D610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D.S. et al. (2007) HMDB: the human metabolome database. Nucleic Acids Res., 35, D521–D526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D.S. et al. (2022) HMDB 5.0: the human metabolome database for 2022. Nucleic Acids Res., 50, D622–D631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J. et al. (2015) MetaboAnalyst 3.0 – making metabolomics more meaningful. Nucleic Acids Res., 43, W251–W257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B. et al. (2018) RaMP: a comprehensive relational database of metabolomics pathways for pathway enrichment analysis of genes and metabolites. Metabolites, 8, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao , Yin Y.. et al. (2020) Omics study reveals abnormal alterations of breastmilk proteins and metabolites in puerperant women with COVID-19. Sig. Transduct. Target. Ther., 5, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RaMP-DB 2.0 is an open source project and its code is available through the following GitHub repositories: 1) Code for the R package: https://github.com/ncats/ramp-db, 2) Python code for constructing the RaMP-DB 2.0 mySQL database: https://github.com/ncats/RaMP-BackEnd, and 3) code for building the front-end web application: https://github.com/ncats/RaMP-Client/. The link to the current RaMP-DB 2.0 dump is available at https://figshare.com/ndownloader/files/36760461, and the latest version link is made available through the R package code ReadMe file.