

Abstract
Objective
T cells are critical in the pathogenesis of systemic lupus erythematosus (SLE) in that they secrete inflammatory cytokines, help autoantibody production, and form autoreactive memory T cells. Although the contribution of T cells to several forms of organ‐mediated damage in SLE has been previously demonstrated, the role of T cells in neuropsychiatric SLE (NPSLE), which involves diffuse central nervous system manifestations and is observed in 20–40% of SLE patients, is not known. Therefore, we conducted this study to evaluate how behavioral deficits are altered after depletion or transfer of T cells, to directly assess the role of T cells in NPSLE.
Methods
MRL/lpr mice, an NPSLE mouse model, were either systemically depleted of CD4+ T cells or intracerebroventricularly injected with choroid plexus (CP)–infiltrating T cells and subsequently evaluated for alterations in neuropsychiatric manifestations. Our study end points included evaluation of systemic disease and assessment of central nervous system changes.
Results
Systemic depletion of CD4+ T cells ameliorated systemic disease and cognitive deficits. Intracerebroventricular injection of CP–infiltrating T cells exacerbated depressive‐like behavior and worsened cognition in recipient mice compared with mice who received injection of splenic lupus T cells or phosphate buffered saline. Moreover, we observed enhanced activation in CP–infiltrating T cells when cocultured with brain lysate–pulsed dendritic cells in comparison to the activation levels observed in cocultures with splenic T cells.
Conclusion
T cells, and more specifically CP–infiltrating antigen‐specific T cells, contributed to the pathogenesis of NPSLE in mice, indicating that, in the development of more targeted treatments for NPSLE, modulation of T cells may represent a potential therapeutic strategy.
INTRODUCTION
T cells are integral in the pathogenesis of systemic lupus erythematosus (SLE), a complex, systemic autoimmune disease that primarily affects women (1, 2). As a result of aberrant activation and defects in peripheral tolerance, SLE T cells promote chronic inflammation by secreting inflammatory cytokines, helping autoantibody production, and forming autoreactive memory T cells. Furthermore, the infiltration of SLE T cells into nonlymphoid target organs, such as the skin, kidneys, and the brain, perpetuates localized damage (2).
The pathogenicity of SLE T cells has been extensively established in studies that used monoclonal antibodies (3, 4, 5), fusion proteins (6, 7), and gene‐targeted deletion (8, 9) involving both specific T cell subsets and costimulatory pathways. In addition to their role in the increased expression of the major histocompatibility complex (10) and the oligoclonality present in lupus nephritis (11), kidney‐specific SLE CD4+ T cell clones have been shown to accelerate kidney damage without increasing anti–double‐stranded DNA (anti‐dsDNA) antibodies (12). This suggests that organ‐infiltrating T cells are autospecific for tissue‐dependent antigens.
Neuropsychiatric systemic lupus erythematosus (NPSLE) affects ~20–40% of patients with SLE and encompasses a wide spectrum of clinical symptoms, including those related to mood and cognitive disorders (13). The issue of appropriate attribution of symptoms to disease or therapeutic side effects further complicates the clinical care of patients with NPSLE. Despite their established contribution to systemic disease and organ‐mediated damage, T cells have not been shown to play a role in the underlying mechanisms responsible for the central nervous system (CNS) manifestations in SLE patients (14, 15).
In some NPSLE patients and NPSLE mouse models, a leukocytic infiltrate has been noted in the choroid plexus (CP) (16, 17, 18, 19). The CP forms the blood–cerebrospinal fluid barrier and is increasingly considered to be a neuroimmune interface (20). Furthermore, resolution of the CP infiltrate was associated with improvement in behavior (21, 22). In particular, the CP in the MRL/lpr strain, a widely used NPSLE mouse model that develops cognitive and affective behavioral deficits similar to neuropsychiatric manifestations in human lupus, becomes heavily infiltrated with T cells while the brain parenchyma is mostly unaffected (16, 19, 23). O'Sullivan et al have previously described amelioration of the CP infiltrate with CD4+ T cell depletion but did not detail whether behavioral deficits were similarly improved (24). Furthermore, our recent findings of unique T cell receptor (TCR) clonality in the CP compared with other affected tissue sites in MRL/lpr mice suggest that brain‐infiltrating T cells may be locally contributing to NPSLE and may have specific reactivity to brain‐specific antigens (25).
Using a mouse model of lupus, we investigated the potential contribution of T cells to the pathogenesis of NPSLE by performing systemic depletion of CD4+ T cells or by locally injecting CP‐infiltrating T cells intracerebroventricularly (ICV) in mice to evaluate whether neuropsychiatric disease was attenuated or promoted, respectively. We conducted further evaluations to understand the mechanisms by which T cells may be contributing to NPSLE. As T cell therapies are currently being evaluated in the treatment of lupus, our study examines brain‐infiltrating T cells as specific contributors to NPSLE.
METHODS
Mice
Female MRL/MpJ‐Faslpr/lpr (MRL/lpr) mice were either purchased from The Jackson Laboratory at ~5–6 weeks of age or bred at the Albert Einstein College of Medicine animal facilities. We used only female MRL/lpr mice for experiments, since, similar to human lupus, the disease phenotype is more penetrant and severe in females. We used baseline serum anti‐dsDNA indices to normalize and assign mice into treatment groups. Mice were housed at 21–23°C on a 12‐hour light/dark cycle. All animal protocols were approved by the Albert Einstein College of Medicine Animal Care and Use Committee.
Stimulation and expansion of T cells
We generated pooled (nonsorted) single‐cell suspensions from phosphate buffered saline (PBS)–perfused spleen or CP tissue excised from donor 16‐ to 18‐week‐old female MRL/lpr mice (7–10 mice), as previously described (17). Cells were initially stimulated and expanded on plates precoated with 0.5 μg/ml of anti‐CD3 antibodies (BD Pharmingen) and with 0.5 μg/ml of anti‐CD28 antibodies (BD Pharmingen) added to RPMI 1640 medium supplemented with 10% fetal bovine serum (17). We then added 10 units of murine interleukin‐2 (IL‐2) (R&D Systems) every other day to further stimulate T cell proliferation for 7 days, after which cells were prepared for phenotyping and/or injection.
Systemic depletion of CD4+ T cells
Female MRL/lpr mice were systemically depleted of CD4+ T cells as previously described (24). In brief, the 2 cohorts of 6‐week‐old female mice (~7 mice per group) were intraperitoneally (IP) injected with a bolus of 2 mg of either anti‐CD4 (Clone GK1.5; Bioxcell) or anti–keyhole limpet hemocyanin IgG2b isotype (Clone LTF‐2; Bioxcell) as control, with the initial injection subsequently followed by weekly 1‐mg IP injections. At 14 weeks of age, mice were assessed for cognitive and affective deficits. For postmortem assessments of mice at end of study, 1 mouse cohort was used for histologic analyses and the other for transcriptome analyses. Only one isotype‐treated mouse was excluded from behavioral results because of insufficient exploration.
ICV adoptive transfer
We administered a single ICV injection of CP T cells, splenic T cells, or PBS into 6‐week‐old MRL/lpr female mice (4 cohorts of 4–8 mice per group because of technical considerations related to the surgery) (26). In brief, we implanted a single‐guide syringe (Hamilton) into the right lateral ventricle at the following coordinates: 0.34 mm anteroposterior, 1.00 mm mediolateral, and 2 mm dorsal ventricular. The T cells, which underwent 3 washes in PBS, were injected at a rate of 0.35 μl/minute for a total count of 200,000 cells and a volume of 2 μl; PBS alone was used as a control. We performed the surgeries during daytime hours and provided postsurgical care to the mice in accordance with the approved animal protocol. After 4 weeks, injected mice underwent behavioral testing. After testing, we conducted postmortem histology, transcriptome, and flow cytometry assessments.
Carboxyfluorescein succinimidyl ester (CFSE) labeling and in vivo tracking
After stimulation and expansion of cells for 7 days, we prepared the CP and splenic T cells into pellets and discarded the supernatant. We resuspended the T cells in CFSE staining solution (Invitrogen) and incubated the cells at 37°C for 20 minutes. Stained cells were quenched with the addition of medium and further incubated at 37°C for 5 additional minutes. Cells were robustly washed and resuspended in sterile PBS. We then intravenously injected the recipient 6‐ to 7‐week‐old MRL/lpr female mice with 500,000 cells; mice were killed 2 or 4 days after injection to evaluate CFSE‐labeled T cell infiltration in various organs by flow cytometry.
Isolation and coculture of pulsed dendritic cells with T cells
Single‐cell suspensions of MRL/lpr splenocytes were generated, and CD11c+ dendritic cells were subsequently isolated from the splenocytes using CD11c+ microbeads (Militenyi Biotec) (27). MRL/lpr splenocytes or CD11c+ dendritic cells were pulsed for 24 hours with lysates containing either 200 μg/ml brain tissue from MRL/lpr or MRL/MpJ mice or 200 μg/ml liver tissue from MRL/lpr mice. After 1–2 hours of incubation, we added 100 ng/ml of lipopolysaccharide to activate and mature the dendritic cells. We added CFSE‐labeled splenic or CP T cells retrieved from the 16‐ to 18‐week‐old MRL/lpr mice at a ratio of ~1:1 and cocultured the cells with the pulsed dendritic cells for 28 hours. After we added cell activation cocktail with brefeldin A (Biolegend) for the final 6 hours of incubation, we evaluated T cell proliferation and activation markers by flow cytometry.
Behavioral tests
Object placement (OP) and object recognition (OR) tests
We used OP and OR tests to evaluate spatial memory and recognition memory, respectively. The mice were subjected to 2 trials; these “training periods” were meant to initially expose the mice to 2 identical objects in fixed positions. After a retention interval, we placed the mice back into the same field but with one of the objects moved (OP test) or replaced (OR test). Interactions of mice with either the “novel” object or the “old” object were manually recorded. Experimenters were blinded to the experimental assignment of tested mice. Due to a mouse's innate preference to explore novel objects in their environment, the OP and OR tests were evaluated categorically as a “pass” (>55% novel object preference) or a “fail” (<55% novel object preference); this robust cutoff was previously determined for this behavioral test in the MRL/lpr strain (28). We calculated the OP or OR preference score as follows: exploration time of novel object/total interaction time with either object × 100 (expressed as a percentage). We excluded mice if they insufficiently explored the field and objects (<3 seconds in either trial).
Porsolt swim test
Mice underwent the Porsolt swim test to assess depressive‐like behavior, with assessments based on a mouse's immobility when swimming. In brief, we placed each mouse into a transparent cylindrical tank with water at 27°C. Animals were allowed to initially adjust to the environment for 1 minute, after which we scored the mice over three 3‐minute intervals. The immobility percentage was calculated as the amount of time a mouse was immobile/total time evaluated. Mice that failed to meet prespecified inclusion criteria were excluded from the behavioral test analyses.
Flow cytometry
Cells were stained for flow cytometry as previously described (17). In brief, we generated single‐cell suspensions after the final PBS perfusion using a 0.25% trypsin‐EDTA digestion buffer (Gibco) for the CP or a mechanical dissociation method for the spleen. After lysis of red blood cells (Thermo Fisher Scientific), cells were washed and then Fc blocked with anti‐CD16 (BD Pharmingen) in 3% fetal bovine serum–PBS for 15–30 minutes on ice. Multiple panels were used to stain cells, including phenotyping of activated/proliferating T cells and phenotyping of the CP infiltrate for lymphoid and myeloid cells.
Enzyme‐linked immunosorbent assays (ELISAs)
We measured IgG anti‐dsDNA antibodies, IgG anti‐chromatin antibodies, and total immunoglobulins by ELISA in serum samples obtained at baseline and/or at study end as previously described (29, 30, 31). We conducted blood urea nitrogen ELISAs according to the manufacturer's instructions (BioAssay Systems).
Histology and immunofluorescence staining
Staff at the Albert Einstein Histopathology and Comparative Pathology Core conducted hematoxylin and eosin (H&E) staining. For immunofluorescence staining, brain and kidney sections were deparaffinized and rehydrated, followed by antigen retrieval in citrate buffer (pH 6) at 90–95°C for 10 minutes. Slides were subsequently washed in PBS and blocked in 20% horse serum–2% Triton in PBS for 1 hour at room temperature. Slides were incubated with antibodies against either CD3 (1:200; Invitrogen), B220 (1:200; Becton Dickinson, now known simply as BD), CD4 (1:200; eBioscience), CD8a (1:200; eBioscience), ionized calcium–binding adapter molecule 1 (IBA‐1) (1:200; Fujifilm Wako), glial fibrillary acidic protein (GFAP) (1:200; Invitrogen), or C3 (1:200; MP Biomedicals) for 2 days, with the first day at room temperature and the second day at 4°C. Slides were washed in PBS and incubated with either donkey anti‐rat or donkey anti‐rabbit secondary antibodies (1:200; JacksonImmuno Research). For final steps, slides were washed, stained with DAPI, and mounted using Fluoromount‐G (Southern Biotech). Sections were imaged using the EVOS Fl Auto 2 and quantified with either ImageJ or Volocity software.
RNA isolation, complementary DNA (cDNA) synthesis, and polymerase chain reaction (PCR) arrays
Retrieved tissue was flash frozen in liquid nitrogen. RNA was homogenized and isolated from tissue using TRIzol and either the Zymo Mini kit or the Micro‐prep kit, according to the manufacturer's instructions. We synthesized cDNA from 400 ng of RNA using the Qiagen RT2 First‐Strand kit. We used the Qiagen RT2 Profiler PCR array according to the manufacturer's instructions, with arrays run on the real‐time quantitative PCR ViiA 7 system (Thermo Fisher Scientific). We normalized and analyzed data using the manufacturer's provided online tool. We identified the top differentially expressed genes based on either >2‐fold change in expression level or significant difference in expression based on a threshold of P < 0.05 (determined by Student's t‐test).
Statistical analysis
We used GraphPad Prism 9 software for all data analyses. For each experiment, we included multiple replications or mouse cohorts. When comparing groups, we first tested data for normality and then performed either a Student's t‐test or one‐way analysis of variance. For the OP and OR behavioral tests, we performed chi‐square tests. We used Tukey's or Dunn's tests for multiple comparisons based on normality results. Throughout the results, P values less than 0.05 were considered significant.
RESULTS
Control of systemic disease in MRL/lpr mice by systemic depletion of CD4+ T cells
To confirm the successful depletion of CD4+ T cells, we performed flow cytometry on blood samples from mice after they had undergone several weeks of IP injection of anti‐CD4 antibodies or isotype control. Blood samples from mice that received anti‐CD4 antibody treatment were significantly depleted of CD4+ T cells compared with the isotype control group, whereas the percentage of CD8+ T cells was not significantly different between the anti‐CD4–treated and control groups (Figure 1A). Postmortem assessments of the treated mice indicated that splenomegaly and anti‐dsDNA autoantibodies were significantly decreased in the CD4+ T cell–depleted mice versus the isotype control group (Figures 1B and C, respectively). Furthermore, kidney disease was similarly significantly attenuated (Figure 1D). These results confirm that depletion of CD4+ T cells ameliorated systemic and kidney disease in MRL/lpr mice.
Figure 1.
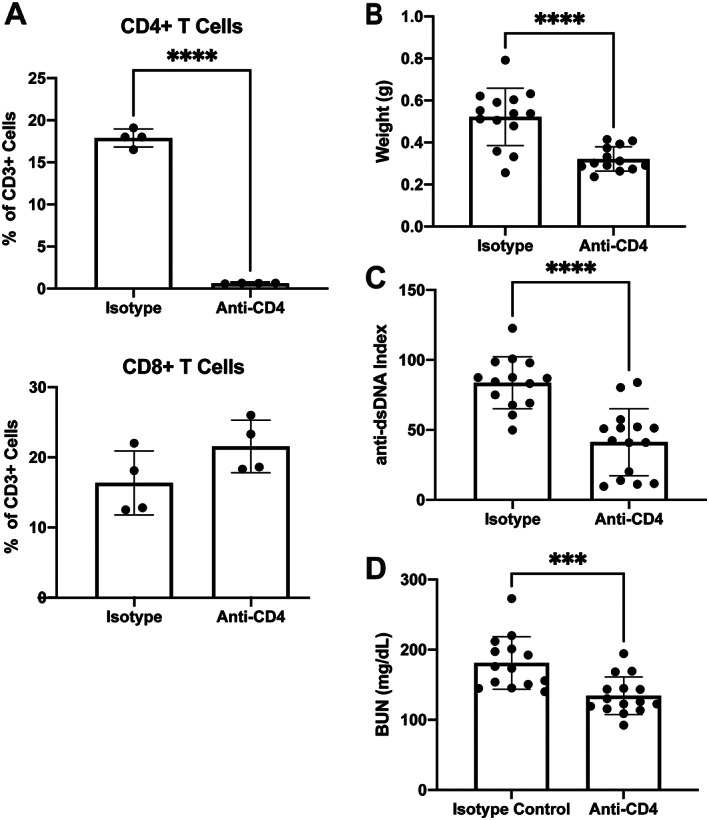
Attenuation of systemic disease in MRL/lpr mice after depletion of CD4+ T cells. A, Percentage of CD4+ and CD8+ T cells among peripheral blood mononuclear cells in blood samples obtained from mice 4 weeks after treatment with anti‐CD4 or isotype control. B, Weight of spleens obtained postmortem from mice to assess presence of splenomegaly. C, Assessment of postmortem titer levels of serum anti–double‐stranded DNA (anti‐dsDNA) autoantibodies in mice. D, Measurement of blood urea nitrogen (BUN) in postmortem serum samples from mice to assess lupus nephritis. In A–D, solid symbols show individual mice; bars show the mean ± SD. The treatment groups were compared using Student's t‐test. *** = P < 0.001; **** = P < 0.0001.
Amelioration of neuropsychiatric manifestations by systemic depletion of CD4+ T cells
Our evaluation of cognitive deficits and affective behavior in MRL/lpr mice systemically depleted of CD4+ T cells showed that, compared with mice in the isotype control group without CD4+ T cell depletion, the CD4+ T cell–depleted mice demonstrated significantly improved spatial and recognition memory (Figures 2A and B). Furthermore, we observed a modest improvement in depressive‐like behavior according to the Porsolt swim test in CD4+ T cell–depleted mice (Supplementary Figure 1, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42252). Overall, these results suggested that CD4+ T cells do contribute, whether directly or indirectly, to the neuropsychiatric manifestations in MRL/lpr mice.
Figure 2.
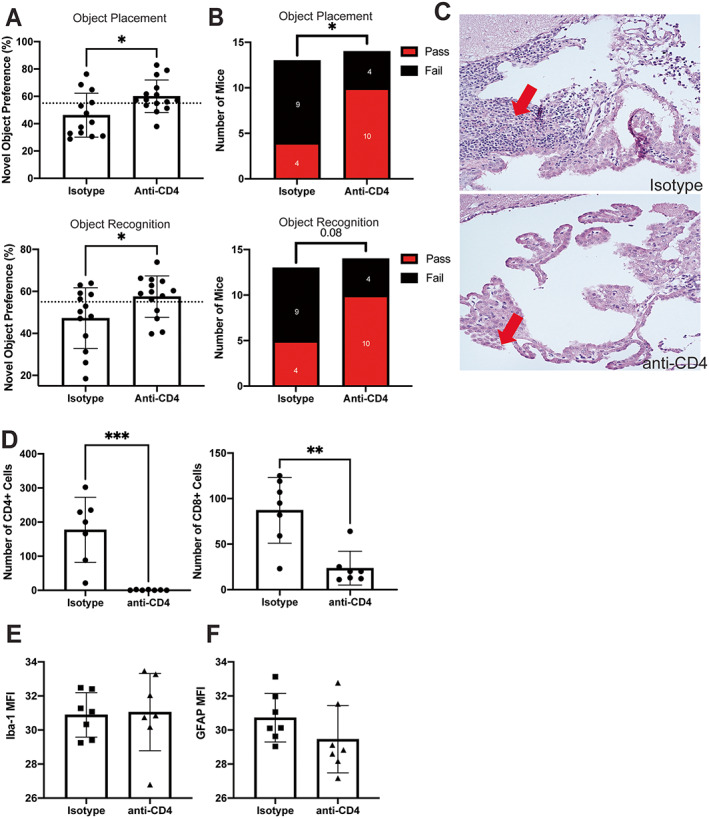
Improved cognition in MRL/lpr mice after systemic depletion of CD4+ T cells. A, Percentage of mice exhibiting novel object preference in assessments of spatial memory (object placement test) and recognition memory (object recognition test). Dashed line indicates a cutoff level of 55% for pass/fail, where those above the line had a passing score (i.e., showed novel object preference). B, Number of mice with a passing or failing score, based on 55% cutoff, during novel object preference tests for either spatial memory (n = 13 mice) (top) or recognition memory (n = 14 mice) (bottom). C, Representative images of hematoxylin and eosin–stained brain tissue from mice treated with isotype control or anti‐CD4, with arrows indicating the choroid plexus (CP) infiltrate. D–F, Immunofluorescence staining (expressed as mean fluorescence intensity [MFI]) of CD4+ and CD8+ in T cells in the CP (D), of ionized calcium–binding adapter molecule 1 (IBA‐1) in the hippocampus to assess microglial activation (n = 7 mice) (E), and of glial fibrillary acidic protein (GFAP) in the hippocampus to assess astrocyte activation (F). In A and D–F, solid symbols show individual mice; bars show the mean ± SD. The treatment groups were compared using Student's t‐test or chi‐square test. * = P < 0.05; ** = P < 0.01; *** = P < 0.001. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42252/abstract.
Systemic CD4+ T cell depletion significantly attenuated the CP infiltrate present in MRL/lpr mice. H&E staining of brain tissue from CD4+ T cell–depleted mice showed that very few lymphocytes were present in the CP (Figure 2C). Concordantly, immunofluorescence staining indicated that CD4+ T cells were absent and the number of CD8+ T cells was significantly decreased in the CP (Figure 2D). Hippocampal tissue was stained for IBA‐1 and GFAP as markers of microglial and astrocyte activation, respectively. We found no significant differences in the mean fluorescence intensity results for IBA‐1 or GFAP (Figures 2E and F, respectively) between the groups. Thus, CD4+ T cell–depleted MRL/lpr mice had attenuated systemic and neuropsychiatric disease with significant resolution of the CP infiltrate.
Accelerated neuropsychiatric disease by local adoptive transfer of CP‐infiltrating T cells
To demonstrate the local and specific effects of brain‐infiltrating T cells on neuropsychiatric disease, we cultured CP T cells or splenic T cells from older MRL/lpr mice (14 weeks of age) and then injected the cells by ICV into prediseased, young MRL/lpr mice (6 weeks of age). At 4 weeks after adoptive transfer surgery, young MRL/lpr mice underwent behavioral assessments to evaluate whether neuropsychiatric disease was accelerated. The cultured T cells injected into these mice were phenotyped, with the results indicating that, compared with the phenotype of expanded splenic T cells, expanded CP T cells were mostly double negative (Figure 3A). Mice that received CP T cells had significantly worse spatial memory than mice injected with PBS and significantly worse recognition memory than both the PBS‐injected mice and splenic T cell recipient mice (Figures 3B and C). In addition, mice that received CP T cells had worse depressive‐like behavior than the other 2 mouse groups (Figure 3D).
Figure 3.
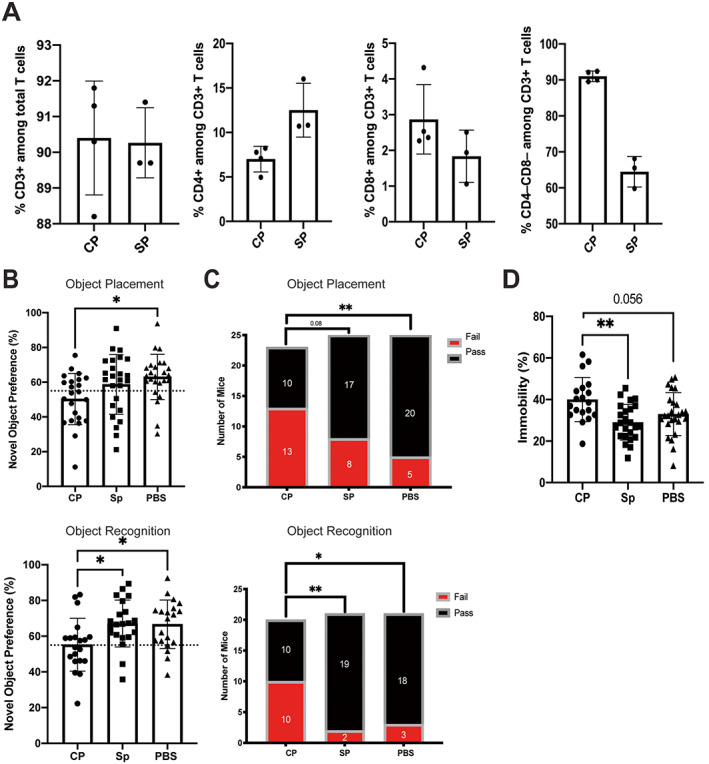
Accelerated neuropsychiatric disease in MRL/lpr mice that received local adoptive transfer of cultured choroid plexus (CP)–infiltrating T cells or splenic (SP) T cells obtained from older MRL/lpr mice or that received phosphate buffered saline (PBS) as control. A, Phenotypes of the cultured CP or splenic T cells injected into prediseased, younger MRL/lpr mice. B and C, Results of assessments of spatial memory (object placement test) and recognition memory (objection recognition test) showing percentages of mice exhibiting novel object preference based on the 55% pass/fail cutoff (dashed line) (B) and numbers of mice with a passing or failing score based on the 55% cutoff (C). D, Assessments of depressive‐like behavior with the Porsolt swim test, showing percentage of time that a mouse spent immobile. In A, B, and D, solid symbols show individual mice; bars show the mean ± SD. Groups were compared using one‐way analysis of variance or chi‐square test. * = P < 0.05; ** = P < 0.01. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42252/abstract.
Saccharin preference was also decreased in the CP T cell recipient mouse group compared with mice that received splenic T cells (Supplementary Figure 2A, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42252). This latter finding points to anhedonia, a feature of murine depressive‐like behavior. Importantly, no significant behavioral differences were observed between the PBS‐injected group and the mice that did not undergo surgical manipulation (Supplementary Figures 2B and C). Thus, we found that local adoptive transfer of CP‐infiltrating T cells but not splenic T cells exacerbated the cognitive deficits and worsened the affective behavior of MRL/lpr mice injected with these cells, a finding that is consistent with the neuropsychiatric phenotype in this mouse model.
No effect from ICV T cell delivery on systemic disease markers
A potential advantage to the experimental ICV delivery of T cells is the ability to directly affect the CNS without significantly altering systemic disease. Across the treatment groups, there was no difference in splenomegaly, a hallmark of the mouse model (Supplementary Figure 3A, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42252). Furthermore, spleens from mice that received either CP T cells or splenic T cells showed no differences in the composition of either innate or adaptive immune cells compared with spleens from mice that received PBS (Supplementary Figure 3B). Interestingly, there was a significant decrease in titers of anti‐dsDNA antibodies in mice that received CP T cells compared with the other 2 treatment groups (Supplementary Figure 3C). However, this finding does not align with titer results for other antinuclear antibodies as there were no changes in titer levels of antichromatin antibodies in postmortem samples (Supplementary Figure 3D) and total IgG antibodies (Supplementary Figure 3E). Finally, the severity of nephritis was not significantly different among the treatment groups (Supplementary Figure 3F). Therefore, although neuropsychiatric disease was accelerated in the CP T cell recipient mice, systemic disease was generally unaffected.
Exacerbation of select markers of neuroinflammation in ICV CP T cell recipient mice
Because we found robust differences in behavior among the 3 ICV recipient groups, which was consistent with and complementary to the results shown in the CD4+ T cell depletion studies, we next evaluated markers of neuroinflammation in response to different treatments, as there may be a correlation between modulation of these markers and T cell–induced neurobehavioral deficits. IBA‐1 and GFAP staining of the hippocampus and the cortex revealed no significant differences in astrocyte and microglial activation levels between the groups (Figures 4A–C). Further evaluation of the hippocampus and the cortex revealed several differentially expressed genes across the ICV recipient groups. In hippocampal tissue, we observed up‐regulation of NFKB1, TGFB2, and JAK1 genes in mice that received ICV injection of T cells compared with mice that received ICV injection of PBS (Figure 4D). In the cortex, the top up‐regulated genes of CP T cell recipient mice were ADM and CD9, which are involved in vasorelaxation and cell adhesion, respectively (Figure 4E). After hierarchical clustering of the genes to identify potentially significant general pathways, we found that the T cell recipient groups were more closely aligned in their gene expression profiles compared with the PBS and nonsurgical control groups (Supplementary Figure 4, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42252).
Figure 4.
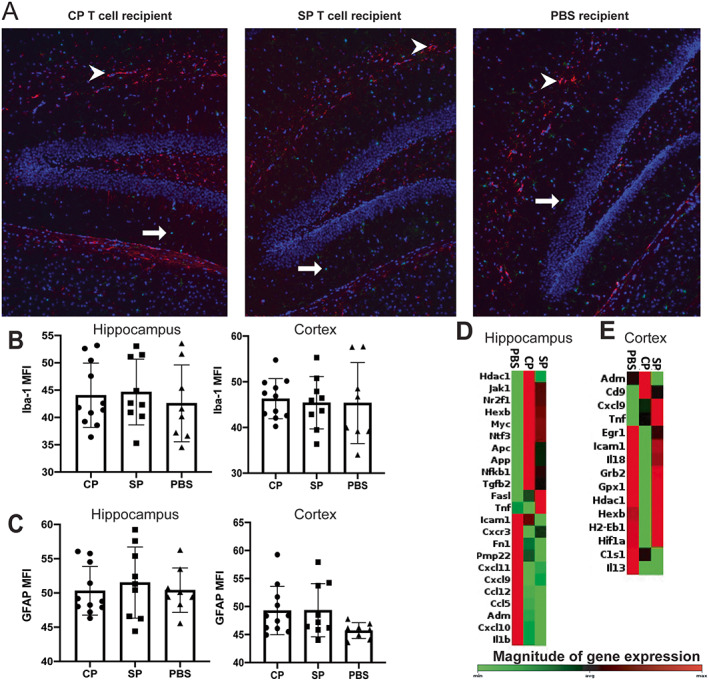
Identification of differential gene expression in MRL/lpr mice that received intracerebroventricular (ICV) injection of choroid plexus–infiltrating T cells (CP), splenic T cells (SP), or phosphate buffered saline (PBS). A, Staining of the hippocampus for IBA‐1 and GFAP in a representative CP T cell recipient mouse, SP T cell recipient mouse, and PBS recipient mouse. Microglia are shown in green (IBA‐1+) (arrows), astrocytes in red (GFAP+) (arrowheads), and nuclear areas in blue (DAPI+). B and C, Immunofluorescence staining of the hippocampus and cortex for IBA‐1 (B) and for GFAP (C) in recipient mice. D and E, Heatmaps of genes up‐regulated in the hippocampus (D) and in the cortex (E) of mice after ICV treatment (CP, SP, or PBS). In B and C, solid symbols show individual mice; bars show the mean ± SD. See Figure 2 for other definitions. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42252/abstract.
No difference in composition of CP across ICV‐injected mouse groups
We next evaluated the effects of local injection of T cells on the CP of recipient mice. Initial histologic scoring of the H&E‐stained brain tissue revealed no apparent differences between the 3 treatment groups (Figure 5A). Further quantification of T and B cells with CD3 and B220 immunofluorescence staining revealed no differences in the percentage of CD3+ T cells and B220+ B cells in the 2 T cell recipient groups (Figures 5B and C). We performed flow cytometry in one recipient mouse cohort to further characterize both the lymphoid and myeloid populations present. Similarly, no significant differences were observed in percentages of these populations, including CD4, CD8, or double‐negative T cells, between the 3 experimental groups (Supplementary Figure 5, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42252).
Figure 5.
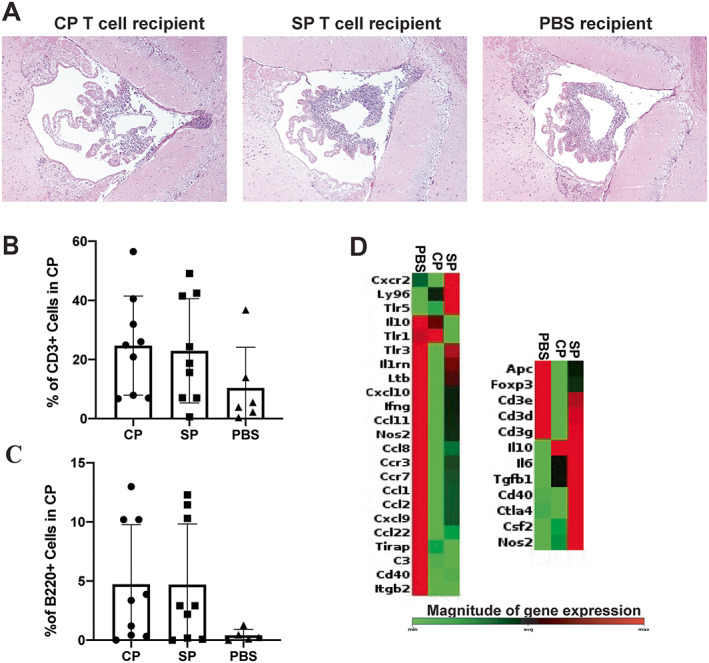
Similarity in the characteristics of the choroid plexus (CP) infiltrate across the MRL/lpr mouse treatment groups that received intracerebroventricular (ICV) injection of CP‐infiltrating T cells, splenic (SP) T cells, or phosphate buffered saline (PBS). A, Representative images of hematoxylin and eosin–stained brain tissue samples showing composition of CP in the 3 treatment groups. B and C, Percentage of CD3+ cells (B) and B220+ cells (C) among total cells by immunofluorescence staining in the CP of mice by treatment group. D, Heatmap of chemokine and receptor gene expression down‐regulated in the CP of mice after ICV treatment (CP, SP, PBS). In B and C, solid symbols show individual mice; bars show the mean ± SD. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42252/abstract.
To evaluate gene expression, we conducted PCR array analyses on CP from ICV‐injected mice. Interestingly, when we compared the spleens of mice that received T cells versus mice that received PBS, we found down‐regulation of many chemokines, cytokines, and their respective receptors in the CP T cell recipient group (Figure 5D). With regard to genes related to T and B cell activation, Foxp3 expression was significantly decreased in the CP T cell recipient mice, suggesting potential regulatory T cell dysfunction; however IL10 expression levels were significantly elevated, indicating their potential production by other immune cells (i.e., macrophages).
Enhanced CP T cell activation upon major histocompatibility complex presentation of brain lysate, supporting brain‐specific antigenicity
We previously demonstrated that the CP in MRL/lpr mice has enhanced clonality of the TCRß third complementary‐determining region, skewed V gene usage, and increased sequence homology compared with other tissue sites in MRL/lpr mice, suggesting that the CP T cells may be preferentially infiltrating and/or proliferating in CP tissue (25). We hypothesized that T cells were selectively infiltrating the CP and proliferating in mice with neuropsychiatric lupus because of specific autoreactivity to brain‐specific antigens. In our evaluation of the presence of CFSE‐labeled T cells in CP or spleen tissue of recipient mice that were intravenously injected with either CP‐ or spleen‐derived T cells 2 days earlier, we observed no differences in the percentage of CFSE‐labeled T cells in the CP of mice that received either CP T cells or splenic T cells (Figures 6A and B). These results suggest that there may not be a difference between CP and splenic T cells in migration into the CP.
Figure 6.
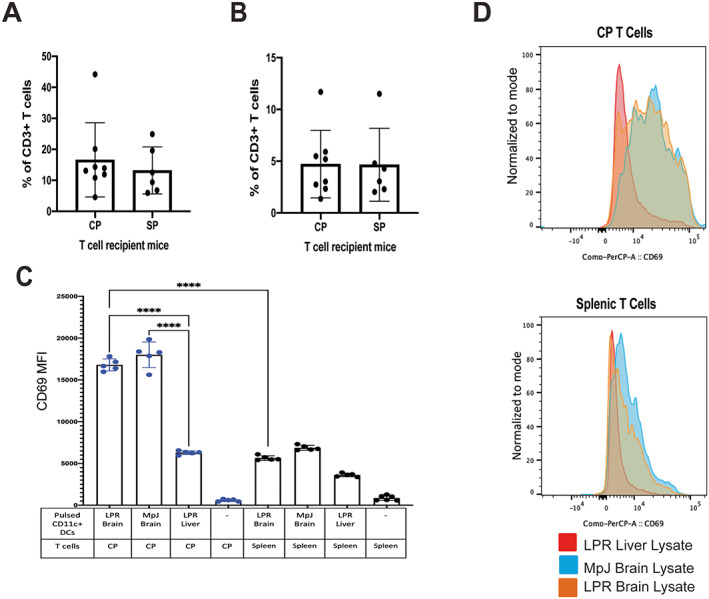
Enhanced activation of choroid plexus (CP)–infiltrating T cells in brain tissue from MRL/lpr mice that received intracerebroventricular injection of CP‐infiltrating T cells (CP) or splenic T cells (SP). A, Percentage of carboxyfluorescein succinimidyl ester (CFSE)–labeled CP or SP T cells in the CP (A) and in spleens (B) of recipient mice. No differences in the infiltration of T cells are shown. C, Mean fluorescence intensity (MFI) of CD69 expression in T cells cocultured with pulsed dendritic cells. CP T cells are labeled in blue and splenic T cells in black. D, Overlays showing expression of CD69 by CP T cells or SP T cells incubated with CD11c+ dendritic cells pulsed with either LPR mouse liver lysates, MpJ mouse brain lysates, or LPR mouse brain lysates. Solid symbols show individual mice (A, B) or pooled samples from 2 or 3 mice each (C); bars show the mean ± SD. Groups were compared using one‐way analysis of variance or Student's t‐test. **** = P < 0.0001. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42252/abstract.
To evaluate the antigen reactivity of CP‐infiltrating T cells, pooled CFSE‐labeled CP T cells were cocultured with brain lysate–pulsed or liver lysate–pulsed CD11c+ dendritic cells, enriched from the splenocytes of MRL/lpr mice. Compared with expression levels at baseline, expression levels of CD69, an early T cell activation marker, had significantly increased in cocultured CP T cells in the presence of brain lysate–loaded dendritic cells (Figures 6C and D). Specifically, CP T cells cocultured with either MRL/lpr or MRL/MpJ mouse brain lysate demonstrated significantly enhanced expression of CD69 compared with CP T cells cocultured with liver lysate. Furthermore, the CP T cell group had increased CD69 expression compared with the respective T cell splenocyte group in the presence of dendritic cells primed with brain lysates (Figures 6C and D). Similar findings were observed when T cells were cocultured with lysate‐primed splenocytes (Supplementary Figure 6, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42252). These findings suggest that the potential mechanism for contribution of CP T cells to NPSLE is through their autoreactivity to brain‐specific antigens.
DISCUSSION
T cells are integral to SLE but have not been previously implicated in playing a role in the pathogenesis of NPSLE. Our results showed that systemic depletion of CD4+ T cells significantly normalized both systemic disease and neurobehavioral deficits in a mouse model. Given this effect on systemic disease, the adoptive transfer method was then chosen to specifically address the contribution of brain‐infiltrating T cells to NPSLE. Mice that underwent ICV injection of brain‐infiltrating T cells demonstrated exacerbated behavior deficits compared with mice that received ICV injection of other autoreactive T cells, suggesting that CP T cells uniquely contribute to neuropsychiatric disease. However, although NPSLE manifestations were affected, we observed no changes in systemic disease in mice that received T cell ICV injection versus PBS. No differences between the groups were seen in the histologic characteristics or composition of the infiltrate. Nevertheless, although the results did not demonstrate a greater infiltration of the CP with CFSE‐labeled CP T cells when compared with splenic T cells, we did observe enhanced activation of CP T cells when cocultured with dendritic cells primed with whole brain lysate, suggesting brain‐specific reactivity.
Our results showed that cognitive deficits were attenuated or worsened upon depletion or transfer of T cells, respectively. Remarkably, although the lack of a significant improvement in depressive‐like behavior with systemic anti‐CD4 antibody treatment might lead to a conclusion that T cells are not relevant in affective aspects of neuropsychiatric lupus, depressive‐like behavior in mice was worse upon adoptive transfer of CP T cells. These seemingly discordant results in studying the effects of T cells on depressive‐like behavior may simply be reflective of the different methods used for systemic depletion versus direct ICV injection of T cells. Alternatively, different mechanisms may underlie distinct neuropsychiatric manifestations (32), and thus additional studies are necessary to further elucidate the contribution of T cells to depressive‐like behavior.
Interestingly, despite some variability in the representation of T cell subsets in injected cell pools depending on their original source (CP versus spleen), no significant differences were observed regarding the composition of infiltrating cells present in the CP or in the migration capacity of CP‐ versus spleen‐derived T cells. Therefore, we surmise that the enhanced pathogenicity of CP T cells, compared with splenic T cells, was attributable to the differential autoreactivity that we had demonstrated with brain antigens.
The identification of the antigens involved, specifically in target organs, continues to interest SLE researchers. Recent studies have reported vimentin‐reactive T cells in the urine of patients with lupus nephritis and demonstrated that kidney‐infiltrating T cells can initiate lupus nephritis after their adoptive transfer to mice (12, 33). Previously, our group described increased clonality and sequence homology present in the CP of NPSLE mice, suggesting that the TCR repertoire is being specifically shaped (25). Results of our experiments with brain lysate–pulsed dendritic cells suggested that CP T cells respond with enhanced activation to whole brain lysate, from both healthy and inflamed brains. Therefore, in combination with the behavioral results, NPSLE appears to be driven by antigen‐specific T cells in the CP. Accumulation of CP T cells does not therefore reflect nonspecific lymphoproliferation or infiltration.
In human NPSLE, the role of T cells has not been elucidated or robustly evaluated. In many clinical trials that used T cell therapies, primary outcomes were related to general disease activity (e.g., the SLE Disease Activity Index) or lupus nephritis specifically and not to NPSLE (34). This omission may be because of the challenges with including appropriate attribution of neuropsychiatric manifestations, variability of symptoms and their onset, and the lack of standardized diagnostic or prognostic tools for NPSLE (35). Nevertheless, despite limited studies, there is some evidence to support the use of T cell–modulating therapies such as azathioprine and cyclosporine, alone or in combination, in NPSLE (28, 36, 37, 38). These studies further support the possibility that targeting T cells might be a more specific, yet insufficiently explored, approach to treatment of NPSLE.
Our analysis into the specific contribution and autoreactivity of brain‐infiltrating T cells in NPSLE has several limitations. Although the MRL/lpr mouse model is the mostly widely accepted and utilized mouse model to evaluate SLE and NPSLE pathogenesis, it does not recapitulate the entire diverse neuropsychiatric presentations of NPSLE or focal disease caused by antiphospholipid syndrome (39, 40, 41). We considered locally depleting T cells with osmotic pumps; however, in this study, we implemented the adoptive transfer method in mice by a single ICV injection to demonstrate the specific contribution of CP‐infiltrating T cells. Because of the limited number of cells that could be retrieved from a single donor mouse's CP, the pooling and stimulating expansion of CP T cells and splenic T cells were necessary to yield enough cells to inject into recipient mice. A regimen of anti‐CD3/anti‐CD28 antibody stimulation and IL‐2 was utilized to selectively stimulate the expansion of T cells alone. Despite differences in the phenotype of stimulated cells versus the composition of the T cell infiltrate in situ in the CP of MRL/lpr mice (17, 25), the differential expansion of CD4+, CD8+, and double‐negative T cells upon stimulation as well as loss of marker expression (i.e., CD4 or CD8) upon activation can explain the increased percentage of double‐negative T cells in culture (42, 43).
Our initial studies suggested that it is CD4+ T cells that drive neuropsychiatric disease, but the subsequent ICV experiments would also be consistent with the double‐negative (CD4−CD8−) T cell subset as a prominent contributor to NPSLE. We had previously described a population of double‐negative T cells in the CP of MRL/lpr mice (17, 25), and this subset was prominent in the ICV‐injected T cells in our present study. In the periphery, double‐negative T cells in SLE have been shown to be clonal and to secrete IL‐17 and interferon‐γ (44). Furthermore, double‐negative T cells can promote immunoglobulin and anti‐dsDNA antibody production (45). In the CNS, double‐negative T cells are increased in postischemic mouse brains and shown to enhance neuroinflammation responses (46). Whether these cells contribute to neuropsychiatric disease and how they might do so are so far unknown. Another interesting point is that, after systemic anti‐CD4 depletion, CD8+ and CD4+ T cells were decreased in the CP. We believe that the depletion of CD4 T cells reasonably decreased not only their number but also their inflammatory contribution to the local brain environment, thus indirectly affecting the infiltration of CD8+ T cells. Therefore, future studies (to include, for example, having subsets of cultured T cells before ICV injection) are needed to continue to parse the specific subset contribution of CP‐infiltrating CD4+, CD8+, and particularly double‐negative T cells in NPSLE.
In conclusion, we have demonstrated that T cells, specifically CP‐infiltrating T cells, contribute and drive NPSLE in the MRL/lpr mouse model. Several additional studies are warranted to further elucidate the specific mechanisms by which these cells affect the CNS parenchyma and subsequent clinical symptoms, as well as to evaluate how T cell modulation affects NPSLE in human disease.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Putterman had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Moore, Huang, Macian, Putterman.
Acquisition of data
Moore, Huang, Reynolds.
Analysis and interpretation of data
Moore, Huang, Reynolds, Macian, Putterman.
Supporting information
Disclosure Form
Supplemental Figure 1 Systemic depletion of CD4+ T cells modestly improved depressive‐like behavior. Percentage of time that a mouse spent immobile to assess depressive‐like behavior (Porsolt Swim test, n = 14 each). Each dot represents an individual mouse, bars represent mean with standard deviation.
Supplemental Figure 2. Behavioral assessments on ICV‐injected and nonsurgical mice. A) Percentage of saccharin water preference indicating anhedonia behavior (n = 17/18/20). B) Percentage of novel object preference to assess spatial memory (Object Placement Test, left, n = 25/16) and recognition memory (Object Recognition Test, right, n = 25/16), with the dashed line indicating 55% cutoff. C) Percentage of time that a mouse spent immobile to assess depressive‐like behavior (Porsolt Swim test, n = 25/16). Each dot indicates a single mouse. CP, CP T cell recipient mice; SP, spleen T cell recipient mice; PBS, PBS recipient mice; Ctrl, nonsurgical MRL/lpr mice.
Supplemental Figure 3. Systemic disease markers are not affected by ICV delivery. A) Terminal spleen weight to assess splenomegaly (n = 25 each). B) Percentage of immune cells present in the spleen of recipient mice (n = 6/7/8). C) Terminal serum anti‐double stranded DNA index to assess autoantibody production (n = 25 each). D) Terminal serum anti‐chromatin index (n = 25 each). E) Terminal serum total IgG titers (n = 17/13/15). F) Terminal serum BUN levels to evaluate kidney disease in recipient mice (n = 25 each). Each dot indicates a single mouse, bars represent mean with standard deviation. Multiple comparisons made between groups using one‐way ANOVA. ** p < 0.01. CP, CP T cell recipient mice; SP, spleen T cell recipient mice; PBS, PBS recipient mice.
Supplemental Figure 4. Similar hierarchical clustering of ICV T cell groups by gene expression profiles. A) Clustering of neuroinflammatory and CNS‐related genes by heatmap in the hippocampus of ICV‐injected and nonsurgical groups. B) Clustering of neuroinflammatory and CNS‐related genes by heatmap in the cortex of ICV‐injected and nonsurgical groups. CP, CP T cell recipient mice; SP, spleen T cell recipient mice; PBS, PBS recipient mice; Ctrl = nonsurgical MRL/lpr mice.
Supplemental Figure 5. Composition of CP immune cells in ICV recipient mice. Percentage of immune cells in the CP of ICV recipient mice (n = 6/7/8). Each dot indicates a single mouse. CP, CP T cell recipient mice; SP, spleen T cell recipient mice; PBS, PBS recipient mice; DCs, dendritic cells; DN T cells, double negative T cells.
Supplemental Figure 6. Enhanced activation of CP T cells by brain tissue‐pulsed splenocytes. A) Mean fluorescent intensity (MFI) of CD69 expression in T cells co‐cultured with pulsed splenocytes, CP T cells are labeled in blue, splenic T cells labeled in black (n = 4 each). B) Overlay expression of CD69 in CP T cells incubated with LPR splenocytes pulsed with either LPR liver lysate (red), MpJ brain lysate (blue), or LPR brain (orange). Each dot indicates pooled samples of 2‐3 mice each, bars represent mean with standard deviation. Multiple comparisons between groups using one‐way ANOVA and Student's T‐test. **** p < 0.0001. CP, choroid plexus; SP, spleen.
ACKNOWLEDGMENTS
We acknowledge Noelie Cayla and Dr. Kamran Khodakhah for their expertise and use of their stereotaxic instruments to perform the ICV injections.
Dr. Moore's work was supported by the Gina M. Finzi Memorial Student Summer Fellowship Program and the Medical Scientist Training Program (award T32‐GM007288).
Author disclosures are available at https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fart.42252&file=art42252‐sup‐0001‐Disclosureform.pdf.
REFERENCES
- 1. Tsokos GC. Systemic lupus erythematosus. N Engl J Med 2011;365:2110–21. [DOI] [PubMed] [Google Scholar]
- 2. Comte D, Karampetsou MP, Tsokos GC. T cells as a therapeutic target in SLE. Lupus 2015;24:351–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Merino R, Fossati L, Iwamoto M, et al. Effect of long‐term anti‐CD4 or anti‐CD8 treatment on the development of lpr CD4‐ CD8‐ double negative T cells and of the autoimmune syndrome in MRL‐lpr/lpr mice. J Autoimmun 1995;8:33–45. [DOI] [PubMed] [Google Scholar]
- 4. Jabs DA, Burek CL, Hu Q, et al. Anti‐CD4 monoclonal antibody therapy suppresses autoimmune disease in MRL/Mp‐lpr/lpr mice. Cell Immunol 1992;141:496–507. [DOI] [PubMed] [Google Scholar]
- 5. Early GS, Zhao W, Burns CM. Anti‐CD40 ligand antibody treatment prevents the development of lupus‐like nephritis in a subset of New Zealand black x New Zealand white mice. Response correlates with the absence of an anti‐antibody response. J Immunol 1996;157:3159–64. [PubMed] [Google Scholar]
- 6. Furie R, Nicholls K, Cheng TT, et al. Efficacy and safety of abatacept in lupus nephritis: a twelve‐month, randomized, double‐blind study. Arthritis Rheumatol 2014;66:379–89. [DOI] [PubMed] [Google Scholar]
- 7. Merrill JT, Burgos‐Vargas R, Westhovens R, et al. The efficacy and safety of abatacept in patients with non–life‐threatening manifestations of systemic lupus erythematosus: results of a twelve‐month, multicenter, exploratory, phase IIb, randomized, double‐blind, placebo‐controlled trial. Arthritis Rheum 2010;62:3077–87. [DOI] [PubMed] [Google Scholar]
- 8. Bubier JA, Sproule TJ, Foreman O, et al. A critical role for IL‐21 receptor signaling in the pathogenesis of systemic lupus erythematosus in BXSB‐Yaa mice. Proc Natl Acad Sci U S A 2009;106:1518–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kyttaris VC, Zhang Z, Kuchroo VK, et al. Cutting edge: IL‐23 receptor deficiency prevents the development of lupus nephritis in C57BL/6‐lpr/lpr mice. J Immunol 2010;184:4605–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheah PL, Looi LM, Chua CT, et al. Enhanced major histocompatibility complex (MHC) class II antigen expression in lupus nephritis. Malays J Pathol 1997;19:115–20. [PubMed] [Google Scholar]
- 11. Massengill SF, Goodenow MM, Sleasman JW. SLE nephritis is associated with an oligoclonal expansion of intrarenal T cells. Am J Kidney Dis 1998;31:418–26. [DOI] [PubMed] [Google Scholar]
- 12. Okamoto A, Fujio K, Tsuno NH, et al. Kidney‐infiltrating CD4+ T‐cell clones promote nephritis in lupus‐prone mice. Kidney Int 2012;82:969–79. [DOI] [PubMed] [Google Scholar]
- 13. Schwartz N, Stock AD, Putterman C. Neuropsychiatric lupus: new mechanistic insights and future treatment directions. Nat Rev Rheumatol 2019;15:137–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Govoni M, Hanly JG. The management of neuropsychiatric lupus in the 21st century: still so many unmet needs? Rheumatology (Oxford) 2020;59 Suppl:v52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bendorius M, Po C, Muller S, et al. From systemic inflammation to neuroinflammation: the case of neurolupus. Int J Mol Sci 2018;19:3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stock AD, Der E, Gelb S, et al. Tertiary lymphoid structures in the choroid plexus in neuropsychiatric lupus. JCI Insight 2019;4:e124203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jain S, Stock A, Macian F, et al. A distinct T follicular helper cell subset infiltrates the brain in murine neuropsychiatric lupus. Front Immunol 2018;9:487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. James WG, Hutchinson P, Bullard DC, et al. Cerebral leucocyte infiltration in lupus‐prone MRL/MpJ‐fas lpr mice—roles of intercellular adhesion molecule‐1 and P‐selectin. Clin Exp Immunol 2006;144:299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ma X, Foster J, Sakic B. Distribution and prevalence of leukocyte phenotypes in brains of lupus‐prone mice. J Neuroimmunol 2006;179:26–36. [DOI] [PubMed] [Google Scholar]
- 20. Meeker RB, Williams K, Killebrew DA, et al. Cell trafficking through the choroid plexus. Cell Adh Migr 2012;6:390–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Duprez T, Nzeusseu A, Peeters A, et al. Selective involvement of the choroid plexus on cerebral magnetic resonance images: a new radiological sign in patients with systemic lupus erythematosus with neurological symptoms. J Rheumatol 2001;28:387–91. [PubMed] [Google Scholar]
- 22. Atkins CJ, Kondon J, Quismorio F, et al. The choroid plexus in systemic lupus erythematosus. Ann Rheum Dis 1971;30:333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ariel DS, Sivan G, Ofer P, et al. The blood brain barrier and neuropsychiatric lupus. Autoimmun Rev 2017;16:612–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. O'Sullivan FX, Vogelweid CM, Besch‐Williford CL, et al. Differential effects of CD4+T cell depletion on inflammatory central nervous system disease, arthritis and sialadenitis in MRL/lpr mice. J Autoimmun 1995;8:163–75. [DOI] [PubMed] [Google Scholar]
- 25. Moore E, Huang MW, Jain S, et al. The T cell receptor repertoire in neuropsychiatric systemic lupus erythematosus. Front Immunol 2020;11:1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wen J, Chen CH, Stock A, et al. Intracerebroventricular administration of TNF‐like weak inducer of apoptosis induces depression‐like behavior and cognitive dysfunction in non‐autoimmune mice. Brain Behav Immun 2016;54:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mukherjee G, Geliebter A, Babad J, et al. DEC‐205‐mediated antigen targeting to steady‐state dendritic cells induces deletion of diabetogenic CD8+ T cells independently of PD‐1 and PD‐L1. Int Immunol 2013;25:651–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Papachristos DA, Oon S, Hanly JG, et al. Management of inflammatory neurologic and psychiatric manifestations of systemic lupus erythematosus: a systematic review. Semin Arthritis Rheum 2021;51:49–71. [DOI] [PubMed] [Google Scholar]
- 29. Chalmers SA, Wen J, Shum J, et al. CSF‐1R inhibition attenuates renal and neuropsychiatric disease in murine lupus. Clin Immunol 2017;185:100–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mike EV, Makinde HM, Gulinello M, et al. Lipocalin‐2 is a pathogenic determinant and biomarker of neuropsychiatric lupus. J Autoimmun 2019;96:59–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Putterman C, Diamond B. Immunization with a peptide surrogate for double‐stranded DNA (dsDNA) induces autoantibody production and renal immunoglobulin deposition. J Exp Med 1998;188:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gao HX, Campbell SR, Cui MH, et al. Depression is an early disease manifestation in lupus‐prone MRL/lpr mice. J Neuroimmunol 2009;207:45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tesch S, Abdirama D, Grießbach AS, et al. Identification and characterization of antigen‐specific CD4+ T cells targeting renally expressed antigens in human lupus nephritis with two independent methods. Sci Rep 2020;10:21312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nandkumar P, Furie R. T‐cell‐directed therapies in systemic lupus erythematosus. Lupus 2016;25:1080–5. [DOI] [PubMed] [Google Scholar]
- 35. Moore E, Huang MW, Putterman C. Advances in the diagnosis, pathogenesis and treatment of neuropsychiatric systemic lupus erythematosus. Curr Opin Rheumatol 2020;32:152–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Germano V, Diamanti AP, Ferlito C, et al. Cyclosporine A in the long‐term management of systemic lupus erythematosus. J Biol Regul Homeost Agents 2011;25:397–403. [PubMed] [Google Scholar]
- 37. Fargetti S, Ugolini‐Lopes MR, Pasoto SG, et al. Short‐ and long‐term outcome of systemic lupus erythematosus peripheral neuropathy: bimodal pattern of onset and treatment response. J Clin Rheumatol 2021;27:S212. [DOI] [PubMed] [Google Scholar]
- 38. Magro‐Checa C, Zirkzee EJ, Huizinga TW, et al. Management of neuropsychiatric systemic lupus erythematosus: current approaches and future perspectives. Drugs 2016;76:459–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gulinello M, Putterman C. The MRL/lpr mouse strain as a model for neuropsychiatric systemic lupus erythematosus. J Biomed Biotech 2011;2011:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jeltsch‐David H, Muller S. Neuropsychiatric systemic lupus erythematosus and cognitive dysfunction: the MRL‐lpr mouse strain as a model. Autoimmun Rev 2014;13:963–73. [DOI] [PubMed] [Google Scholar]
- 41. Moore E, Putterman C. Are lupus animal models useful for understanding and developing new therapies for human SLE? J Autoimmun 2020;112:102490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bristeau‐Leprince A, Mateo V, Lim A, et al. Human TCR α/β+ CD4‐CD8‐ double‐negative T cells in patients with autoimmune lymphoproliferative syndrome express restricted Vβ TCR diversity and are clonally related to CD8+ T cells. J Immunol 2008;181:440–8. [DOI] [PubMed] [Google Scholar]
- 43. Ford MS, Zhang ZX, Chen W, Zhang L. Double‐negative T regulatory cells can develop outside the thymus and do not mature from CD8+ T cell precursors. J Immunol 2006;177:2803–9. [DOI] [PubMed] [Google Scholar]
- 44. Crispín JC, Oukka M, Bayliss G, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL‐17 and infiltrate the kidneys. J Immunol 2008;181:8761–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shivakumar S, Tsokos GC, Datta SK. T cell receptor α/β expressing double‐negative (CD4‐/CD8‐) and CD4+ T helper cells in humans augment the production of pathogenic anti‐DNA autoantibodies associated with lupus nephritis. J Immunol 1989;143:103–12. [PubMed] [Google Scholar]
- 46. Meng H, Zhao H, Cao X, et al. Double‐negative T cells remarkably promote neuroinflammation after ischemic stroke. Proc Natl Acad Sci U S A 2019;116:5558–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclosure Form
Supplemental Figure 1 Systemic depletion of CD4+ T cells modestly improved depressive‐like behavior. Percentage of time that a mouse spent immobile to assess depressive‐like behavior (Porsolt Swim test, n = 14 each). Each dot represents an individual mouse, bars represent mean with standard deviation.
Supplemental Figure 2. Behavioral assessments on ICV‐injected and nonsurgical mice. A) Percentage of saccharin water preference indicating anhedonia behavior (n = 17/18/20). B) Percentage of novel object preference to assess spatial memory (Object Placement Test, left, n = 25/16) and recognition memory (Object Recognition Test, right, n = 25/16), with the dashed line indicating 55% cutoff. C) Percentage of time that a mouse spent immobile to assess depressive‐like behavior (Porsolt Swim test, n = 25/16). Each dot indicates a single mouse. CP, CP T cell recipient mice; SP, spleen T cell recipient mice; PBS, PBS recipient mice; Ctrl, nonsurgical MRL/lpr mice.
Supplemental Figure 3. Systemic disease markers are not affected by ICV delivery. A) Terminal spleen weight to assess splenomegaly (n = 25 each). B) Percentage of immune cells present in the spleen of recipient mice (n = 6/7/8). C) Terminal serum anti‐double stranded DNA index to assess autoantibody production (n = 25 each). D) Terminal serum anti‐chromatin index (n = 25 each). E) Terminal serum total IgG titers (n = 17/13/15). F) Terminal serum BUN levels to evaluate kidney disease in recipient mice (n = 25 each). Each dot indicates a single mouse, bars represent mean with standard deviation. Multiple comparisons made between groups using one‐way ANOVA. ** p < 0.01. CP, CP T cell recipient mice; SP, spleen T cell recipient mice; PBS, PBS recipient mice.
Supplemental Figure 4. Similar hierarchical clustering of ICV T cell groups by gene expression profiles. A) Clustering of neuroinflammatory and CNS‐related genes by heatmap in the hippocampus of ICV‐injected and nonsurgical groups. B) Clustering of neuroinflammatory and CNS‐related genes by heatmap in the cortex of ICV‐injected and nonsurgical groups. CP, CP T cell recipient mice; SP, spleen T cell recipient mice; PBS, PBS recipient mice; Ctrl = nonsurgical MRL/lpr mice.
Supplemental Figure 5. Composition of CP immune cells in ICV recipient mice. Percentage of immune cells in the CP of ICV recipient mice (n = 6/7/8). Each dot indicates a single mouse. CP, CP T cell recipient mice; SP, spleen T cell recipient mice; PBS, PBS recipient mice; DCs, dendritic cells; DN T cells, double negative T cells.
Supplemental Figure 6. Enhanced activation of CP T cells by brain tissue‐pulsed splenocytes. A) Mean fluorescent intensity (MFI) of CD69 expression in T cells co‐cultured with pulsed splenocytes, CP T cells are labeled in blue, splenic T cells labeled in black (n = 4 each). B) Overlay expression of CD69 in CP T cells incubated with LPR splenocytes pulsed with either LPR liver lysate (red), MpJ brain lysate (blue), or LPR brain (orange). Each dot indicates pooled samples of 2‐3 mice each, bars represent mean with standard deviation. Multiple comparisons between groups using one‐way ANOVA and Student's T‐test. **** p < 0.0001. CP, choroid plexus; SP, spleen.