

Abstract
Objectives
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is a cerebral small vessel disease caused by pathogenic variants in the NOTCH3 gene. In Finland, the majority of CADASIL patients carry the pathogenic founder variant c.397C>T, (p.Arg133Cys), but the spectrum of other NOTCH3 variants has not been investigated previously. The aim of the study was to investigate the spectrum and prevalence of NOTCH3 variants Finnish CADASIL patients and to examine the clinical features associated with them.
Materials and Methods
The spectrum of NOTCH3 variants and the clinical features associated with them were retrospectively examined in 294 Finnish CADASIL patients tested during January 1996 to October 2021 in the Medical Genetics laboratory of Department of Genomics of Turku University Hospital, where practically all samples of patients with suspected CADASIL in Finland are investigated.
Results
The most common NOTCH3 variants in the study cohort were c.397C>T, (p.Arg133Cys) (68%) and c.3206A>G p.(Tyr1069Cys) (18%), but other less common NOTCH3 variants were detected in as many as 14% of the patients. Eight of the detected NOTCH3 variants were novel: c.520T>A,p.(Cys174Ser), c.836A>G,p.(Gln279Arg), c.1369T>G,p.(Cys457Gly), c.1338C>G,p.(Cys446Trp), c.1564T>G,p.(Cys522Gly), c.2848T>G,p.(Cys950Gly), c.6102dup,p.(Gly2035Argfs*60), and c.2410+6C>G. Other NOTCH3 variants than p.Arg133Cys and p.Tyr1069Cys were more often associated with more severe clinical features.
Conclusion
This study revealed the genetic and clinical spectrum of CADASIL in the Finnish population. Sequencing of the whole NOTCH3 gene performing a gene‐panel or exome sequencing is recommended when suspecting CADASIL.
Keywords: CADASIL, Finnish, genotype, mutation, NOTCH3, phenotype
1. INTRODUCTION
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is an inherited cerebral small vessel disease caused by pathogenic variants in the NOTCH3 gene located in the chromosome region 19p13. 1 , 2 CADASIL typically presents in young or mid‐adulthood and the characteristic clinical features include migraine with aura, recurrent cerebral ischemic events, cognitive impairment and dementia, and mood disturbances. 3 Clinical presentation of CADASIL is highly variable even between patients with the same pathogenic variant and within families. 4 , 5 , 6
The NOTCH3 gene consists of 33 exons and encodes a transmembrane receptor Notch3. The Notch3 protein is comprised of an extracellular domain with 34 epidermal growth factor repeats (EGFR), three Notch/Lin12 repeats (LNR), a transmembrane domain and an intracellular domain. 2 , 7 The majority of disease‐causing variants in NOTCH3 occur in exons 2–24. Typically, the disease‐causing variants are missense variants causing an uneven number of cysteine residues within one of the 34 EGFRs of the Notch3 extracellular domain. 1 However, other types of variants, such as deletions, duplications, and splice‐site variants, including cysteine‐sparing variants have also been described in CADASIL families. 8 , 9 In Finland, most of the CADASIL patients carry the variant c.397C>T,(p.Arg133Cys), due to the founder effect. 10 Until now, the spectrum and prevalence of other NOTCH3 variants in Finnish CADASIL patients has been unknown.
The aim of this study was to study the spectrum of disease‐causing NOTCH3 variants in the Finnish population and to assess genotype–phenotype correlations of the variants.
2. MATERIALS AND METHODS
The Ethical Committee of the Hospital District of Southwest Finland and Helsinki University Hospital Ethics Committee of Medicine approved the study. Approvals for the access to medical records were obtained from the Hospital District of Southwest Finland and Helsinki University Hospital.
2.1. Patients
This retrospective study included all genetically confirmed Finnish CADASIL patients examined in the Medical Genetics laboratory of Department of Genomics of Turku University Hospital from January 1996 to October 2021 (n = 294). In all, 2324 patient samples were sent for NOTCH3 gene testing to the Medical Genetics laboratory of Department of Genomics of Turku University Hospital from January 1996 to October 2021. Of these, 1861 samples were studied at least for the Finnish founder variant c.397C>T,(p.Arg133Cys), and in part of the cases also for variants c.622C>T,p.(Arg182Cys) and c.3206A>G,p.(Tyr1069Cys). Of the 2324 samples, 463 samples were studied for other NOTCH3 variants using Sanger sequencing of specific NOTCH3 exons or sequencing the whole NOTCH3 gene using next‐generation sequencing (NGS). All patients with reported NOTCH3 variants were included in the study, including predictive cases. In Finland, practically all samples of patients with suspected CADASIL are sent for gene testing to the Medical Genetics laboratory of Department of Genomics of Turku University Hospital.
2.2. NOTCH3 gene testing
In Finland, genetic testing for NOTCH3 began in 1996 by testing for the Finnish founder mutation c.397C>T,(p.Arg133Cys) and the c.622C>T,p.(Arg182Cys) variant, and since 2008 also for the c.3206A>G,p.(Tyr1069Cys) variant, using the restriction fragment length polymorphism (RFLP) technique. Sanger sequencing of NOTCH3 exons was gradually introduced and since 2019, next‐generation sequencing (NGS) has been used for screening all NOTCH3 exons in the Laboratory of Medical Genetics of Turku University Hospital.
Hence, the patients in this study were studied for at least the variant c.397C>T,(p.Arg133Cys) and in most cases also for variant c.622C>T,p.Arg182Cys (n = 131), using RFLP or Sanger sequencing. Some of the patients were also studied for the variant c.3206A>G p.(Tyr1069Cys), using RFLP or Sanger sequencing (n = 116). Some of the samples were Sanger sequenced for variants in exon 4 (n = 2), exons 3 and 4 (n = 1), exon 10 (n = 2), exon 19 (n = 1), or for variants in exons 3–8, 11, and 18–20 (n = 24), or for variants in exons 3–8, 11–12, and 18–20 (n = 8). A minority (n = 7) of the samples were studied across all NOTCH3 exons using NGS. DNA samples of two patients with variant c.1660G>T,p.(Gly528Cys) were first analyzed in the Laboratory of Medical Genetics of Turku University Hospital, but the variant was detected in an additional analysis in France. Some uncommon variants were detected in Sanger sequencing of variants c.397C>T,(p.Arg133Cys) and c.622C>T,p.Arg182Cys (exon 4), or c.3206A>G,p.(Tyr1069Cys) (exon 20). In predictive cases, only the variant in the family was analyzed. Clinical laboratory geneticists analyzed and interpreted the gene test results, in some cases together with a clinical geneticist. In this study, variants were re‐interpreted based on the American College of Medical Genetics and Genomics (ACMG) criteria. 11 NOTCH3 variants were considered pathogenic when they resulted in cysteine‐alteration in an EGFR and they were previously reported in a CADASIL patient.
2.3. Collection of clinical data
Clinical and demographic data were collected retrospectively from medical records or genetic testing referrals. Age of onset for migraines and age of onset for ischemic events were recorded separately, as migraine usually occurs earlier than other symptoms. Risk factors included hypertension, smoking, diabetes, and hyperlipidemia. Family history was considered positive if a patient had family members with CADASIL, migraine, stroke, or dementia.
2.4. Statistical analyses
Statistical analyses were conducted using spss version 27.0 (SPSS Inc.). Age at onset and clinical features among patients carrying the variants p.Arg133Cys, p.Tyr1069Cys, and other variants were compared using Fisher's exact test for categorical variables and unpaired t‐test for continuous variables. Values of p < .05 were considered statistically significant.
3. RESULTS
3.1. Variant spectrum in Finnish CADASIL patients
This study identified 30 different NOTCH3 variants in a cohort of 294 Finnish patients with suspected CADASIL, including 24 missense variants, two splice‐site variants, one frameshift variant, and one in‐frame insertion (Table 1). The most common pathogenic NOTCH3 variants in the study cohort were c.397C>T,(p.Arg133Cys) (n = 200, 68%) in exon 4 and c.3206A>G,p.(Tyr1069Cys) (n = 52, 18%) in exon 20. Another 28 variants were identified in 42 patients (14%) and were classified either pathogenic, likely pathogenic or as of unknown significance, using the ACMG criteria. Of these, eight were novel variants that have not been reported earlier in the literature, to the best of our knowledge.
TABLE 1.
NOTCH3 variants detected in CADASIL patients in Finland. Reference sequence: NM_000435.2
| Number of patients (total n = 294) | Variant | Variant type | Note | dbSNP | Reference (PMID) | ClinVar/LOVD | Exon | EGFR | ACMG |
|---|---|---|---|---|---|---|---|---|---|
| 1 | c.200G>C,p.(Cys67Ser) | Missense | 19174371 (different nucleotide position) | Not reported | 3 | 2 | 5 | ||
| 1 | c.259T>C,p.(Cys87Arg) | Missense | rs1568362232 | 15364702 | VCV000585601.1 | 3 | 2 | 5 | |
| 1 | c.323G>A,p.(Cys108Tyr) | Missense | rs1555729584 | 15364702 33268848 | VCV000872948.2 | 3 | 2 | 5 | |
| 2 | c.328C>T,p.(Arg110Cys) | Missense | rs775836288 | 9388399 | VCV000447831.25 | 3 | 2 | 5 | |
| 1 | c.341‐2A>G | Splice site | Causes in‐frame deletion including cysteine residue | rs2046935672 | 10802807 25982499 | VCV000995318.1 | intr 3 | 2 | 5 |
| 1 | c.421C>T,p.(Arg141Cys) | Missense | rs1174625611 | 9388399 | VCV000447846.13 | 4 | 3 | 5 | |
| 1 | c.391G>T,p.(Gly131Cys) | Missense | 19006080 | #0000081682 | 4 | 3 | 5 | ||
| 200 | c.397C>T,p.(Arg133Cys) | Missense | rs137852642 | 9388399 15378071 | VCV000009225.18 | 4 | 3 | 5 | |
| 1 | c.464G>A,p.(Cys155Tyr) | Missense | 22664156 | #0000081573 | 4 | 3 | 5 | ||
| 1 a | c.520T>A,p.(Cys174Ser) | Missense | rs1599394806 | Novel | VCV000872534.9 | 4 | 4 | 4 | |
| 1 | c.622C>T,p.(Arg182Cys) | Missense | rs28933697 | 8878478 | VCV000009220.22 | 4 | 4 | 5 | |
| 5 | c.580T>C,p.(Cys194Arg) | Missense | rs1568361818 | 12146805 | VCV000585612.3 | 4 | 4 | 5 | |
| 1 | c.752G>A,p.(Cys251Tyr) | Missense | rs1555729405 | 19174371 19372454 | VCV000447872.1 | 5 | 6 | 5 | |
| 1 | c.836A>G,p.(Gln279Arg) | Missense | Cysteine‐sparing | Novel | Not reported | 6 | 7 | 3 | |
| 1 | c.931T>G,p.(Cys311Gly) | Missense | rs781158121 | 27844030 | #0000832811 | 6 | 7 | 4 | |
| 1 | c.1012T>A,p.(Cys338Ser) | Missense | 29188607 (different nucleotide position) | Not reported | 6 | 8 | 5 | ||
| 1 | c.1261C>T,p.(Arg421Cys) | Missense | rs1555729068 | 15364702 16009764 | VCV000447779.4 | 8 | 10 | 5 | |
| 1 | c.1300_1308dupGAGTGTCTG,p.(Glu434_Leu436dup) | In‐frame insertion | Causes insertion of three additional amino acids, including cysteine | 19174371 | Not reported | 8 | 11 | 5 | |
| 1 | c.1369T>G,p.(Cys457Gly) | Missense | Novel | Not reported | 8 | 11 | 4 | ||
| 1 | c.1338C>G,p.(Cys446Trp) | Missense | Novel | Not reported | 8 | 11 | 4 | ||
| 2 | c.1564T>G,p.(Cys522Gly) | Missense | Novel | #0000832896 | 10 | 13 | 4 | ||
| 3 | c.1660G>T,p.(Gly528Cys) | Missense | 19174371 | Not reported | 10 | 13 | 5 | ||
| 1 | c.2410+6C>G | Splice site | Effect unknown | rs1479891295 | Novel | Not reported | intr 15 | n/a | 3 |
| 1 | c.2848T>G,p.(Cys950Gly) | Missense | rs1378535955 | Novel | Not reported | 18 | 24 | 4 | |
| 2 | c.3043T>C,p.(Cys1015Arg) | Missense | rs1599382214 | 10371548 | VCV000803539.6 | 19 | 26 | 5 | |
| 52 | c.3206A>G p.(Tyr1069Cys) | Missense | 19174371 | VCV001510760.1 | 20 | 27 | 5 | ||
| 6 | c.3226C>T,p.(Arg1076Cys) | Missense | rs1438626607 | 11571335 12861102 | VCV000447830.4 | 20 | 27 | 5 | |
| 1 | c.3298C>T,p.(Arg1100Cys) | Missense | 34222332 | Not reported | 20 | 28 | 5 | ||
| 1 | c.5510G>A,p.(Arg1837His) | Missense | Cysteine‐sparing | rs138265894 | 32573853 | Not reported | 30 | intracellular domain | 3 |
| 1 | c.6102dup,p.(Gly2035Argfs*60) | Frameshift | Causes premature stop codon | rs771517374 | Novel | #0000660245 | 33 | intracellular domain | 3 |
Abbreviations: EGFR, epidermal growth factor repeat; LOVD, Leiden Open Variation Database; n/a, information not available.
Note: ACMG variant classification (5 = pathogenic, 4 = likely pathogenic, 3 = variant of unknown significance, 2 = likely benign, and 1 = benign).
African ethnicity.
The majority of all variants were found in exon 4 (6/29, 21%), followed by exon 3 (4/29, 14%) and 8 (4/29, 14%) (Figure 1). Variants c.5510G>A,p.(Arg1837His) and c.6102dup,p.(Gly2035Argfs*60) affected the intracellular part of Notch3, while the remaining variants affected the extracellular EGFR region. Of the 28 variants affecting the extracellular domain with EGFRs, 26 were cysteine‐altering variants. Atypical variants affecting the extracellular EGFR region included a cysteine‐sparing variant c.836A>G,p.(Gln279Arg) in exon 6, and a splice‐site variant c.2410+6C>G whose effect on gene transcription and translation is unknown.
FIGURE 1.
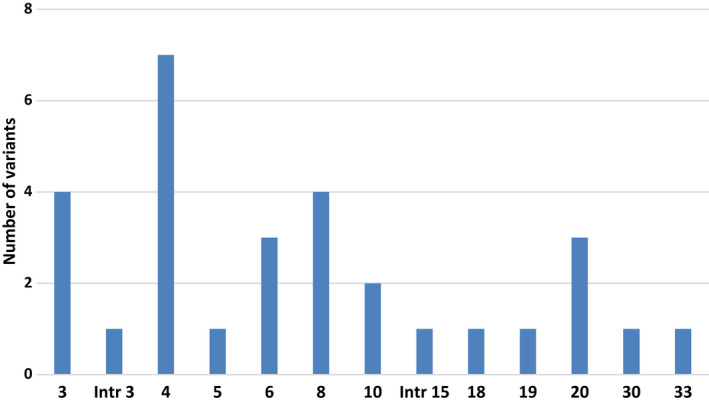
Exonic distribution of NOTCH3 variants identified in the Finnish CADASIL cohort. Total number of different variants was 30.
3.2. Clinical features of the CADASIL patients
Clinical data from medical records were available for 38% (112/294) of the patients (Figure 2). For the remaining 62% (182/294) of patients, only information from the genetic testing referral was available. In 77 cases of the 182 with only genetic testing referral available, 26 cases were predictive gene tests and in 51 cases there was no clinical information in the referral. Hence, the study cohort for analyzing clinical characteristics included 217 patients (Figure 2).
FIGURE 2.
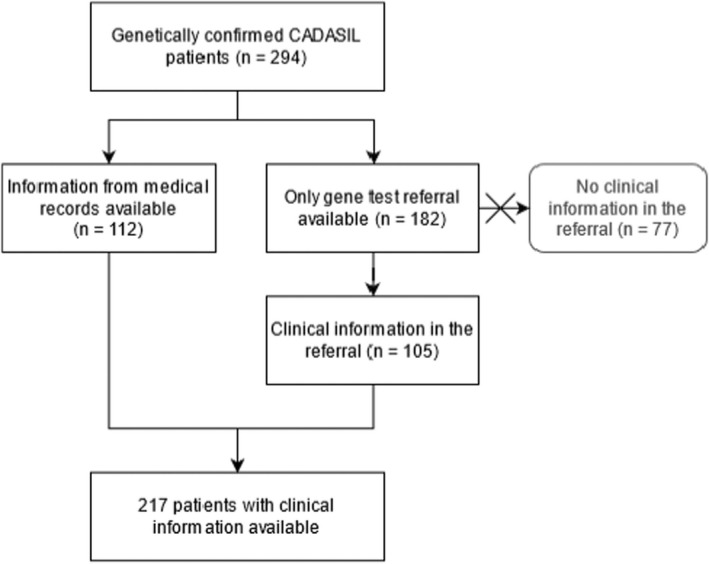
Schematic representation describing the study cohort and available clinical data
Of the patients, 53% were women (155/294). Mean age at the time of gene testing was 50 (SD ± 13.7) years old, ranging from 17 to 87. The age at onset information was available in 103 cases: mean age at onset was 47 (SD ± 11.5) years old, ranging from 17 to 77. Family history was reported positive in the referral or medical records in 59% of patients (173/294). A skin biopsy result was available for 19 patients (19/294, 6%), and GOM was detected in all the cases. All patients were Finnish, but the patient with variant c.520T>A,p.(Cys174Ser) had African ethnicity. The demographic and clinical features of the patients with variants p.Arg133Cys, p.Tyr1069Cys, and other NOTCH3 variants are summarized in Table 2. Clinical features of the 42 patients with less common NOTCH3 variants in Finland are presented in more detail in Table S1. Of these 42 patients, eight had a skin biopsy and/or autopsy report available (Table 3).
TABLE 2.
Comparison of clinical features between Finnish CADASIL patients carrying the NOTCH3 variants p.Arg133Cys and p.Tyr1069Cys and other NOTCH3 variants
| p.Arg133Cys (n = 200) | p.Tyr1069Cys (n = 52) | Other (n = 42) | p‐value (p.Arg133Cys vs. other) | p‐value (p.Tyr1069Cys vs. other) | |
|---|---|---|---|---|---|
| Sex (F/M) | 104/96 | 28/24 | 23/19 | ||
| Family history | 118 (59%) | 29 (56%) | 26 (62%) | ||
| Predictive cases | 16 (8%) | 7 (13%) | 3 (7%) | ||
| Age at the time of predictive testing, mean ± SD | 33 ± 10.8 | 43 ± 10.8 | 41 ± 18.0 | ||
| Clinical features | |||||
| Clinical information available | 142 (71%) | 35 (67%) | 40 (95%) | ||
| Age at onset, mean ± SD | 46 ± 12.5 (n = 68) | 53 ± 5.7 (n = 8) | 49 ± 9.2 (n = 27) | .208 | .235 |
| Risk factors a | 32/142 (23%) | 6/35 (17%) | 13/40 (33%) | .216 | .184 |
| Migraine/headache | 48/142 (34%) | 8/35 (23%) | 21/40 (52%) | .042 | .010 |
| Ischemic stroke/TIA | 59/142 (42%) | 8/35 (23%) | 23/40 (58%) | .105 | .004 |
| ICH | 1/142 (1%) | 2/35 (6%) | 5/40 (13%) | .002 | .438 |
| Epilepsy | 6/142 (4%) | 1/35 (3%) | 5/40 (13%) | .066 | .206 |
| Psychiatric symptom | 15/142 (11%) | 3/35 (9%) | 7/40 (18%) | .272 | .321 |
| Cognitive impairment | 44/142 (31%) | 11/35 (31%) | 15/40 (38%) | .449 | .633 |
| GOM detected in skin biopsy b | 11/142 (8%) | 1/35 (3%) | 7/40 (18%) | .078 | .061 |
Abbreviations: F, female; GOM, granular osmiophilic material; ICH, intracerebral hemorrhage; M, male; SD, standard deviation; TIA, transient ischemic attack.
Risk factors include hypertension, smoking, diabetes, and hyperlipidemia.
GOM was detected in all patients with skin biopsy result available.
TABLE 3.
Clinical features of the patients with rare NOTCH3 variants and deposition of GOM in skin biopsy, and/or neuropathological autopsy data available
| c.341‐2A>G | c.391G>T,p.(Gly131Cys) | c.1012T>A,p.(Cys338Ser) | c.1300_1308dupGAGTGTCTG,p.(Glu434_Leu436dup) | c.1660G>T,p.(Gly528Cys) | c.1660G>T,p.(Gly528Cys) | c.3226C>T,p.(Arg1076Cys) | c.6102dup,p.(Gly2035Argfs*60) | |
|---|---|---|---|---|---|---|---|---|
| Patient | C6 | C7 | C19 | C21 | C28 | C29 | C38 | C42 |
| Sex | F | F | M | M | F | F | F | F |
| Age at onset | 52 | 35 | 40 | 49 | 59 | 50? | 45 | 52 |
| Age at death | 62 | 44 | 68 | 87 | ||||
| Source of clinical information | Medical records | Referral | Medical records | Medical records | Medical records | Medical records | Referral and skin biopsy report | Medical records |
| WM changes in MRI | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Migraine/headache | Yes | n/a | Yes | No | n/a | Yes | n/a | No |
| Ischemic stroke/TIA | Yes | n/a | No | Yes | n/a | n/a | Yes | n/a |
| ICH | No | n/a | No | No | Yes | No | n/a | No |
| Epilepsy | No | n/a | Yes | No | No | Yes | n/a | No |
| Psychiatric symptom | No | n/a | n/a | n/a | Yes | Yes | n/a | Yes |
| Cognitive impairment | Yes | n/a | No | No | Yes | Yes | n/a | Yes |
| Hypertension | Yes | n/a | Yes | Yes | n/a | n/a | No | n/a |
| Hyperlipidemia | Yes | n/a | No | Yes | n/a | n/a | No | n/a |
| Smoking | No | n/a | No | Yes | n/a | n/a | No | n/a |
| Diabetes | No | n/a | No | No | n/a | n/a | No | n/a |
| Family history | Yes | n/a | n/a | Yes | Yes | n/a | n/a | Yes |
| Skin biopsy | GOM+ | GOM+ | Not performed | GOM+ | GOM+ | GOM+ | GOM+ | GOM+ |
| Neuropathology | Lacunar infarcts, WM changes, thickened arterioles; vascular changes consistent with CADASIL. Purkinje cell loss in cerebellum, gliotic foci in hippocampus | Lacunar infarcts, WM changes; most prominent lesions in basal ganglia. Thickened vessels, especially arterioles, consistent with CADASIL. Findings milder than usually found in CADASIL patients | WM changes, multiple infarcts in basal ganglia, infarcts in nucleus caudatus and putamen. Thickened vessels, especially arterioles, consistent with CADASIL. Ischemic lesion in cerebellum, AD changes in temporal lobe, β‐amyloid positivity, Braak II. |
Abbreviations: AD, Alzheimer's disease; F, female; GOM, granular osmiophilic material; ICH, intracerebral hemorrhage; M, male; n/a, information not available or not applicable; TIA, transient ischemic attack; WM, white matter.
4. DISCUSSION
The spectrum of NOTCH3 variants in CADASIL patients and clinical features associated with them have been studied only in a few studies focusing mainly on Asian 5 , 12 , 13 , 14 and some South‐European 4 , 15 , 16 populations. The aim of this study was to retrospectively investigate the prevalence of different NOTCH3 variants in Finnish CADASIL patients and examine whether there are phenotypic differences between carriers of different variants. In Finland, practically all samples of patients with suspected CADASIL are sent for gene testing to the Medical Genetics laboratory of Department of Genomics of Turku University Hospital, and therefore, this study material therefore represents the great majority of Finnish CADASIL patients. The Finnish population is genetically homogeneous due to isolation and population bottlenecks. Thus, in Finland, until the implementation of more developed genetic testing methods, patients with clinically suspicious CADASIL were often screened only for the most common NOTCH3 variants (p.Arg133Cys and p.Tyr1069Cys) or variants in specific exons encoding EGFRs and sequencing of the whole NOTCH3 gene has become routine only in recent years. The estimated prevalence of CADASIL in Finland is 4/100000, 17 which is similar to other populations. 4 , 18 , 19 , 20 In the present study, 28 NOTCH3 variants other than the most common NOTCH3 variants, p.Arg133Cys and p.Tyr1069Cys, were identified in as much as 14% of patients, despite the fact that only the minority of the patients were studied for the whole NOTCH3 gene. These results indicate that CADASIL may be somewhat underdiagnosed in Finland, as less common NOTCH3 variants may have been missed in constricted DNA analyses. 21 , 22 , 23
Overall, this study identified 30 different NOTCH3 variants in Finnish CADASIL patients. All detected variants were distributed throughout the 33 exons of the gene, including two intronic variants between exons. Of the variants, 26 were typical disease‐causing variants resulting in an uneven number of cysteine residues within one of the EGFRs. This study detected four atypical NOTCH3 variants: a cysteine‐sparing variant c.836A>G,p.(Gln279Arg) in exon 6, a cysteine‐sparing variant c.5510G>A,p.(Arg1837His) in exon 30 affecting the intracellular part of the protein, a frameshift variant c.6102dup,p.(Gly2035Argfs*60) causing premature stop codon in exon 33, and a splice‐site variant c.2410+6C>G with an unknown effect in intron 15. The clinical significance of atypical NOTCH3 variants is uncertain. 24 Unfortunately, we had very limited clinical information for patients with cysteine‐sparing missense variants; therefore, comparing them with patients carrying cysteine‐altering variants was not possible. NOTCH3 variants causing premature stop codons have been reported only in a few patients with suspected CADASIL and interpreted controversially. 25 , 26 , 27 Furthermore, truncating variants located in the last exon of NOTCH3, as in our case, have been linked to lateral meningocele syndrome, which is a rare neurological disorder affecting connective tissues, distinct from CADASIL. 28 Interestingly, the skin biopsy of the patient with c.6102dup,p.(Gly2035Argfs*60) in this study showed GOM deposits typical for CADASIL supporting the pathogenic role of the variant, unlike other cases with truncating NOTCH3 variants (nonsense variant in exon 3 and intragenic deletion of exons 3–16) reported in the literature that did not show GOM. 25 , 26 Furthermore, segregation analysis revealed that the patient's unaffected mother and sibling did not carry the variant c.6102dup,p.(Gly2035Argfs*60).
There were no remarkable differences in the clinical manifestations between patients with different rare NOTCH3 variants, between patients with variants in specific exons or between patients with variants in EGFR domains 1–6 versus patients with variants in EGFR domains 7–34. However, some observations could be made from the available clinical data. Age at onset of patients with a variant affecting EGFR 1–6 was 46 ± 11.4, which was somewhat lower than patients with an EGFR 7–34 variant, whose age at onset was 50 ± 7.8. This supports the study of Rutten et al., 23 which suggested that EGFR 1–6 pathogenic variants were associated with an earlier age at onset of stroke than variants affecting EGFRs 7–34. Some of the detected variants were identified in more than one patient, which revealed a few possible genotype–phenotype relationships. Four out of five patients (80%) with the variant c.580T>C,p.(Cys194Arg) suffered from migraine or headache. Both patients with the variant c.1660G>T,p.(Gly528Cys) had severe psychiatric symptoms including depression or anxiety and changes in behavior, and one also suffered from hallucinations and paranoia. In addition, both patients with c.1660G>T,p.(Gly528Cys) developed dementia. Of the five patients carrying the variant c.3226C>T,p.(Arg1076Cys) with clinical information available, all had ischemic strokes or TIAs, two patients also had ICH, suggesting that cerebrovascular events are common in CADASIL patients with the variant c.3226C>T,p.(Arg1076Cys).
The frequency of ischemic strokes and/or TIA was higher in the patients carrying the rare NOTCH3 variants (58%) than in patients with p.Arg133Cys (42%, p = .105) or p.Tyr1069Cys (23% p = .004), though the difference was statistically significant only when comparing to the patients carrying p.Tyr1069Cys. Intracerebral hemorrhage (ICH) was distinguished from microbleeds and was reported in 13% of the patients with rare variants, whereas in the patients carrying p.Arg133Cys and p.Tyr1069Cys the frequencies were 1% (p = .002) and 6%, respectively. Most ICH cases had cysteine‐altering missense variants, and the ICHs were located in capsula interna and thalamus (n = 1), in thalamus (n = 1), in nucleus caudatus (n = 1), and in basal ganglia (n = 1). Furthermore, recurrent ICH was detected in a patient with the cysteine‐sparing variant c.836A>G,p.(Gln279Arg) according to the gene test referral, but detailed information was unfortunately not available. Taken together, the results indicate that ICH is an important manifestation of CADASIL. Similarly, epilepsy was also reported more frequently in the patients carrying rare variants than in patients carrying p.Arg133Cys or p.Tyr1069Cys, although the differences were not statistically significant. All patients with epilepsy had cysteine‐altering missense variants, indicating that seizures might be more common in patients with typical NOTCH3 variants. Furthermore, migraine or headache were also reported more frequently in patients with rare variants (50%) compared to patient groups with p.Arg133Cys (34%, p = .042) or p.Tyr1069Cys (23%, p = .01). Psychiatric manifestations were also more common in patients with rare variants (18%) than in patients carrying p.Arg133Cys (11%) or p.Tyr1069Cys (9%), although the differences were not statistically significant.
Because we were unable to acquire clinical information from all 294 patients in the study cohort, the analysis of clinical features was performed on only 217 patients. In half of cases, the clinical information was available only from the gene test referral. Thus, due to the small sample size with limited statistical power, results of phenotype comparison between variant groups should be interpreted with caution. Although the amount of patient clinical information for the study cohort was limited, the results of the study suggest that the less common NOTCH3 variants are more often associated with more severe clinical features than the variants p.Arg133Cys and p.Tyr1069Cys. However, percentages of cognitive impairment did not differ between variant groups. This may be due to the limited clinical information available for the study. Cognitive decline may be present at the later stages of disease and therefore may not have been mentioned in a gene test referral or in medical records if only some patient notes were available.
In conclusion, this is the first study revealing the NOTCH3 variant spectrum among CADASIL patients in Finland. Although the majority of Finnish CADASIL patients carry the founder mutation p.Arg133Cys, less common variants were detected in a significant portion (14%) of patients. Sequencing of the whole NOTCH3 gene, or performing a gene panel or exome sequencing is recommended when suspecting CADASIL. More patients should be identified, and functional studies are needed to clarify the role of the novel NOTCH3 variants.
AUTHOR CONTRIBUTIONS
SM involved in design of the study, data collection, interpretation of the data, and preparation of the first draft of the study, and revised the manuscript. LK involved in design of the study and interpretation of the data, and drafted and revised the manuscript. JS involved in data collection, and drafted and revised the manuscript. LM involved in design, supervision and funding of the study and interpretation of the data, and drafted and revised the manuscript. MP involved in design and supervision of the study, data collection and interpretation of the data, and drafted and revised the manuscript.
FUNDING INFORMATION
This study was supported by Academy of Finland (341007), Liv och Hälsa Foundation and State‐funding for university‐level health research (TYH2020231).
CONFLICT OF INTEREST
None of the authors has any conflicts of interest to disclose.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/ane.13703.
Supporting information
Table S1
ACKNOWLEDGMENTS
None.
Mönkäre, S. , Kuuluvainen, L. , Schleutker, J. , Myllykangas, L. & Pöyhönen, M. (2022). Clinical features and spectrum of NOTCH3 variants in Finnish patients with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Acta Neurologica Scandinavica, 146, 643–651. 10.1111/ane.13703
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Joutel A, Vahedi K, Corpechot C, et al. Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet. 1997;350(9090):1511‐1515. doi: 10.1016/S0140-6736(97)08083-5 [DOI] [PubMed] [Google Scholar]
- 2. Joutel A, Corpechot C, Ducros A, et al. Notch3 mutations in CADASIL, a hereditary adult‐onset condition causing stroke and dementia. Nature. 1996;383(6602):707‐710. doi: 10.1038/383707a0 [DOI] [PubMed] [Google Scholar]
- 3. Chabriat H, Joutel A, Dichgans M, Tournier‐Lasserve E, Bousser MG. CADASIL. Lancet Neurol. 2009;8(7):643‐653. doi: 10.1016/S1474-4422(09)70127-9 [DOI] [PubMed] [Google Scholar]
- 4. Bianchi S, Zicari E, Carluccio A, et al. CADASIL in central Italy: a retrospective clinical and genetic study in 229 patients. J Neurol. 2015;262(1):134‐141. doi: 10.1007/s00415-014-7533-2 [DOI] [PubMed] [Google Scholar]
- 5. Chen S, Ni W, Yin XZ, et al. Clinical features and mutation spectrum in Chinese patients with CADASIL: a multicenter retrospective study. CNS Neurosci Ther. 2017;23(9):707‐716. doi: 10.1111/cns.12719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mykkänen K, Junna M, Amberla K, et al. Different clinical phenotypes in monozygotic CADASIL twins with a novel NOTCH3 mutation. Stroke. 2009;40(6):2215‐2218. doi: 10.1161/STROKEAHA.108.528661 [DOI] [PubMed] [Google Scholar]
- 7. Joutel A, Andreux F, Gaulis S, et al. The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J Clin Invest. 2000;105(5):597‐605. doi: 10.1172/JCI8047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Muiño E, Gallego‐Fabrega C, Cullell N, et al. Systematic review of cysteine‐sparing NOTCH3 missense mutations in patients with clinical suspicion of CADASIL. Int J Mol Sci. 2017;18(9):1964. doi: 10.3390/ijms18091964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rutten JW, Haan J, Terwindt GM, van Duinen SG, Boon EMJ, Lesnik Oberstein SAJ. Interpretation of NOTCH3 mutations in the diagnosis of CADASIL. Expert Rev Mol Diagn. 2014;14(5):593‐603. doi: 10.1586/14737159.2014.922880 [DOI] [PubMed] [Google Scholar]
- 10. Mykkänen K, Savontaus ML, Juvonen V, et al. Detection of the founder effect in Finnish CADASIL families. Eur J Hum Genet. 2004;12(10):813‐819. doi: 10.1038/sj.ejhg.5201221 [DOI] [PubMed] [Google Scholar]
- 11. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and The Association for Molecular Pathology. Genet Med. 2015;17(5):405‐424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hu Y, Sun Q, Zhou Y, et al. NOTCH3 variants and genotype‐phenotype features in Chinese CADASIL patients. Front Genet. 2021;12:705284. doi: 10.3389/fgene.2021.705284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim YE, Yoon CW, Seo SW, et al. Spectrum of NOTCH3 mutations in Korean patients with clinically suspicious cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Neurobiol Aging. 2014;35(3):726.e1‐726.e6. doi: 10.1016/j.neurobiolaging.2013.09.004 [DOI] [PubMed] [Google Scholar]
- 14. Ueda A, Ueda M, Nagatoshi A, et al. Genotypic and phenotypic spectrum of CADASIL in Japan: the experience at a referral center in Kumamoto University from 1997 to 2014. J Neurol. 2015;262(8):1828‐1836. doi: 10.1007/s00415-015-7782-8 [DOI] [PubMed] [Google Scholar]
- 15. Almeida MR, Elias I, Fernandes C, Machado R, Galego O, Santo G. NOTCH3 mutations in a cohort of Portuguese patients within CADASIL spectrum phenotype. Neurogenetics. 2022;23(1):1‐9. doi: 10.1007/s10048-021-00679-w [DOI] [PubMed] [Google Scholar]
- 16. Paraskevas GP, Stefanou MI, Constantinides VC, et al. CADASIL in Greece: mutational spectrum and clinical characteristics based on a systematic review and pooled analysis of published cases. Eur J Neurol. 2022;29(3):810‐819. doi: 10.1111/ene.15180 [DOI] [PubMed] [Google Scholar]
- 17. Kalimo H, Ruchoux MM, Viitanen M, Kalaria RN. CADASIL: a common form of hereditary arteriopathy causing brain infarcts and dementia. Brain Pathol. 2002;12(3):371‐384. doi: 10.1111/j.1750-3639.2002.tb00451.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moreton FC, Razvi SSM, Davidson R, Muir KW. Changing clinical patterns and increasing prevalence in CADASIL. Acta Neurol Scand. 2014;130(3):197‐203. doi: 10.1111/ane.12266 [DOI] [PubMed] [Google Scholar]
- 19. Narayan SK, Gorman G, Kalaria RN, Ford GA, Chinnery PF. The minimum prevalence of CADASIL in northeast England. Neurology. 2012;78(13):1025. doi: 10.1212/WNL.0b013e31824d586c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Razvi SSM, Davidson R, Bone I, Muir KW. The prevalence of cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL) in the west of Scotland. J Neurol Neurosurg Psychiatry. 2005;76(5):739. doi: 10.1136/jnnp.2004.051847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rutten JW, Dauwerse HG, Gravesteijn G, et al. Archetypal NOTCH3 mutations frequent in public exome: implications for CADASIL. Ann Clin Transl Neurol. 2016;3(11):844‐853. doi: 10.1002/acn3.344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rutten JW, Hack RJ, Duering M, et al. Broad phenotype of cysteine‐altering NOTCH3 variants in UK Biobank. Neurology. 2020;95(13):e1835. doi: 10.1212/WNL.0000000000010525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rutten JW, van Eijsden BJ, Duering M, et al. The effect of NOTCH3 pathogenic variant position on CADASIL disease severity: NOTCH3 EGFr 1–6 pathogenic variant are associated with a more severe phenotype and lower survival compared with EGFr 7–34 pathogenic variant. Genet Med. 2019;21(3):676‐682. doi: 10.1038/s41436-018-0088-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Coupland K, Lendahl U, Karlström H. Role of NOTCH3 mutations in the cerebral small vessel disease cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke. 2018;49(11):2793‐2800. doi: 10.1161/STROKEAHA.118.021560 [DOI] [PubMed] [Google Scholar]
- 25. Moccia M, Mosca L, Erro R, et al. Hypomorphic NOTCH3 mutation in an Italian family with CADASIL features. Neurobiol Aging. 2015;36(1):547.e5‐547.e11. doi: 10.1016/j.neurobiolaging.2014.08.021 [DOI] [PubMed] [Google Scholar]
- 26. Rutten JW, Boon EMJ, Liem MK, et al. Hypomorphic NOTCH3 alleles do not cause CADASIL in humans. Hum Mutat. 2013;34(11):1486‐1489. doi: 10.1002/humu.22432 [DOI] [PubMed] [Google Scholar]
- 27. Schubert V, Bender B, Kinzel M, Peters N, Freilinger T. A novel frameshift variant in the CADASIL gene NOTCH3: pathogenic or not? J Neurol. 2018;265(6):1338‐1342. doi: 10.1007/s00415-018-8844-5 [DOI] [PubMed] [Google Scholar]
- 28. Gripp KW, Robbins KM, Sobreira NL, et al. Truncating mutations in the last exon of NOTCH3 cause lateral meningocele syndrome. Am J Med Genet A. 2015;167(2):271‐281. doi: 10.1002/ajmg.a.36863 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.