

Abstract
In this study, we describe a synthetic method for incorporating arenes into closed tubes that we name capsularenes. First, we prepared vase‐shaped molecular baskets 4–7. The baskets comprise a benzene base fused to three bicycle[2.2.1]heptane rings that extend into phthalimide (4), naphthalimide (6), and anthraceneimide sides (7), each carrying a dimethoxyethane acetal group. In the presence of catalytic trifluoroacetic acid (TFA), the acetals at top of 4, 6 and 7 change into aliphatic aldehydes followed by their intramolecular cyclization into 1,3,5‐trioxane (1H NMR spectroscopy). Such ring closure is nearly a quantitative process that furnishes differently sized capsularenes 1 (0.7×0.9 nm), 8 (0.7×1.1 nm;) and 9 (0.7×1.4 nm;) characterized by X‐Ray crystallography, microcrystal electron diffraction, UV/Vis, fluorescence, cyclic voltammetry, and thermogravimetry. With exceptional rigidity, unique topology, great thermal stability, and perhaps tuneable optoelectronic characteristics, capsularenes hold promise for the construction of novel organic electronic devices.
Keywords: Acenes, Aromatic Compounds, Molecular Electronics, Supramolecular Catalysis, Trioxanes
Arenes can now be incorporated into closed tubes—capsularenes. These rigid, electron deficient, thermally stable, and tubular molecules with three arenes or acenes at a close distance can lend themselves to the construction of organic electronic devices.
Introduction
Over last few decades, the preparation and study of tube‐shaped organics have drawn a significant interest across chemical disciplines. [1] For instance, carbon nanotubes [2] (CNTs) with a cylindrical structure and opened or closed ends [3] possess excellent mechanical, [4] thermal and electronic [5] characteristics leading to their use in the construction of novel microelectronic devices, sensors or composite materials. With a goal of creating mimics of CNTs, organic chemists have developed synthetic methods for accessing cyclic p‐cyclophenylenes, [6] nanobelts,[ 1a , 1b , 7 ] and phenine nanotubes. [8] In this vein, Sanchez and co‐workers have recently extended resorcin[4]arenes into tubularenes [9] with π‐conjugated aromatics lining the top segment of a deep‐cavity cavitand. While tube‐shaped organics possess intriguing optoelectronic [10] and supramolecular [11] characteristics their multi‐step synthesis in addition to low yields limit the corresponding application studies. With a facile access to gram quantities of molecular baskets (basket 2 with phthalimide sides and acetals at the rim is shown in Figure 1A), [12] we wondered if their acetals could, in the presence of an acidic catalyst, be linked via the formation of 1,3,5‐trioxane ring. [13] In this way three aromatic sides (i.e., acenes, Figure 1B) become fixed and at a close distance to comprise the walls of a closed tube that we name capsularenes. So far, acenes [14] have been used as charge carriers for constructing organic field‐effect transistors (OFETs) and organic light‐emitting diodes (OLEDs) in addition to organic photovoltaic devices. [15] Tuning the electronic characteristics of acenes [16] (i.e. the band gap) and engineering their assembly in the solid state is essential for obtaining materials with desired properties (conductivity, emission characteristics, stability, etc.). Fascinatingly, macrocyclic dimers [17] and trimers [18] (Figure 1C) [19] of acenes have shown emerging electronic characteristics [20] with smaller band gaps, [17a] RGB luminescence [21] and the unique ability to undergo singlet fission [19] —a process in which an excited singlet state breaks into two triplets. [22] It follows that capsularenes having acene chromophores within a rigid tube‐shaped scaffold (Figure 1B), could give rise to robust organic electronic materials with excellent chemical and thermal stability as well as long‐range order in the solid state. [15] Moreover, their inner space might host a guest [23] for tuning the electronic interaction of the acenes. [17a] To obtain capsularenes, we hypothesized that basket 2 [24] (Figure 1A) or its deeper congeners could, in the presence of a catalyst, [25] swing the aromatic arms inward [26] to facilitate intramolecular cyclotrimerization of the carbonyls into 1,3,5‐trioxane. As a precedent, the formation of [16]‐starand (Figure 1D) [27] from the corresponding hexakis‐ketone ([16]‐ketonand) was reported to take place in the presence of an acid [28] with the release of strain driving the reaction. [29] By taking advantage of remarkable dynamic characteristics of trioxanes [30] and the flexible bicyclic framework of molecular baskets, [12] we herein and for the first time describe a synthetic methodology for incorporating acenes into closed tubes—capsularenes—and characterize their optoelectronic characteristics.
Figure 1.

A) Acid promoted conversion of basket 2 into capsularene 1. B) Van der Waals and stick representations of energy minimized capsularene (PM3) with anthraceneimide sides. C) A cyclotrimer of pentacene was used for studying singlet fission. D) Chemical structure of [1 6]‐starand.
Results and Discussion
Kinetics and Thermodynamics of 1,3,5‐Trioxane Ring Formation
Cyclotrimerization of aliphatic aldehydes into 1,3,5‐trioxanes is an exothermic reaction (ΔH°<0, Figure 2) [31] for which the equilibrium favors reactants at higher temperatures [32] while cyclotrimers at lower temperatures. [33] Since the condensation encompasses three particles combining into one (ΔS°<0), highly concentrated solutions of aldehydes drive the equilibrium forward thereby increasing the yield of 1,3,5‐trioxanes.[ 25 , 34 ] In terms of kinetics, both aldehydes and their cyclotrimers are persistent at ambient conditions with Lewis [35] and Bronsted [36] acids acting as catalysts and facilitating the interconversion. For instance, Denmark and co‐workers found [30] that acetaldehyde would in the presence of BF 3 ⋅Et2O in CDCl3/CD2Cl2 cyclize into paraldehyde at ‐80 °C and then fully convert back into monomer at 20 °C. Indeed, our computational results suggested that the conversion of acetaldehyde into paraldehyde is an exothermic reaction with ΔH°=−17 kcal mol−1 (DFT/B3LYP : 6‐31+G*; Figure 2). In addition, the cyclotrimerization of our model phthalimidoacetaldehyde into 1,3,5‐trioxane 3 was also found to be exothermic with ΔH°=−8.8 kcal mol−1 (DFT/B3LYP : 6‐31+G*; Figure 2). The computed entropic cost was highly unfavorable (ΔS°=−67.7 e.u.); with no consideration of solvation, the overall loss of two translational degrees of freedom (c.a., 60 e.u.) accounts for the result. Further, the intramolecular conversion of tris‐aldehyde basket 2 into capsularene 1 (Figure 2) was found to be heat consuming with ΔH°=4.7 kcal mol−1, at the same level of theory. The unfavorable enthalpic change is unsurprising since the formation of 1 requires bending of the bicyclic platform which brings the carbonyls to a short “striking” distance [12] partaking in the capsularene's 13.6 kcal mol−1 of the strain energy (DFT/B3LYP : 6‐31+G*, Figure S46 and Table S16). With no consideration of solvation, the calculation also showed the formation of 1 being accompanied with an adverse entropic change (ΔS°=−57.4 e.u., Figure 2). To sum up, the theory suggests that the conversion of basket 2 into capsularene 1 is an unfavorable process with ΔG°=21.8 kcal mol−1 at 298 K.
Figure 2.
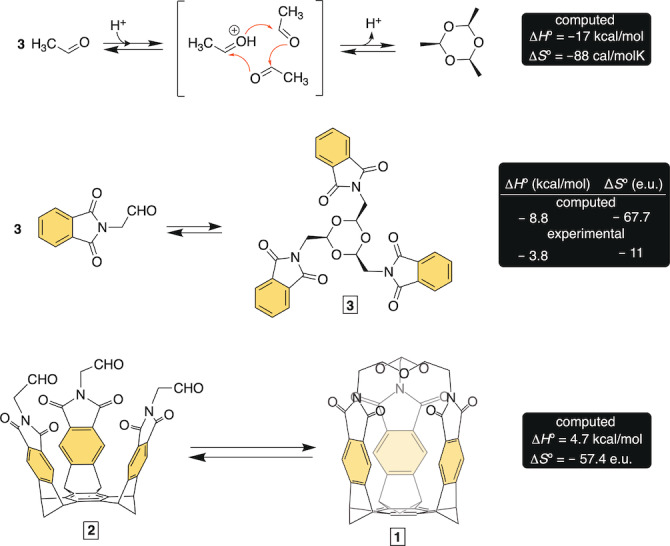
Computed (DFT/B3LYP:6‐31+G*) and experimental (NMR) data pertaining the conversion of acetaldehyde (top), phthalimidoacetaldehyde (middle) and basket 2 (bottom) into corresponding 1,3,5‐trioxanes.
Kinetics and Thermodynamics of Cyclotrimerization of Model Phthalimidoacetaldehyde
After phthalimidoacetaldehyde was placed in dichloromethane containing excess of trifluoroacetic acid (TFA), 1H NMR spectrum of the mixture revealed an immediate formation of 1,3,5‐trioxane product 3 (Figure 2, Figure S20). The interconversion was taking place on the millisecond time scale with 1H‐1H EXSY spectrum showing corresponding exchange cross signals (Figure S21). The proportion of cyclotrimer 3 increased after cooling the solution (from 298 to 254 K; Figure S22), with the data fitting well to the van't Hoff equation (Figure S22). From this, the intermolecular formation of the 1,3,5‐trioxane was found to be driven by enthalpy (ΔH°=−3.8 kcal mol−1) and thus in agreement with theory (Figure 2). A release of solvent molecules ought to, in part, account for the difference in computed and experimental entropy (ΔΔS°=57 e.u., Figure 2).
Preparation, Characterization and Thermodynamics in the Formation of Capsularene 1
A condensation of tris‐anhydride and dimethyl acetal of aminoacetaldehyde gave tris‐acetal basket 4 in 60 % yield (Figure 3A). Upon dissolving this compound in dichloromethane and exposing the solution to hexane vapors, single crystals grew over time. X‐ray diffraction analysis of the sample revealed two C 3 symmetric cavitands occupying the unit cell with their hexasubstituted benzene rings facing each other (d=4.063 Å, Figure 3B). [37] The cavity of each host held a disordered molecule of dichloromethane while the acetals were geared into left‐ and right‐handed propellers. For deprotecting tris‐acetal basket 4 and obtaining 2, we used an excess of TFA. After ca. 12 h, 1H NMR spectrum showed no signal(s) from acetal/aldehyde protons with resonances corresponding to the sole formation of C 3v symmetric 1 (Figure 3C). Small vicinal coupling of H A and H B (3 J H(A/B)=1.2 Hz) was in line with their gauche arrangement (ϕ=60°, Figure 3A). Phthalimidoacetaldehyde trioxane 3 (Figure 2) with unrestricted rotation about single C1–C2 bonds, and a greater proportion of H A‐C1‐C2‐H B antiperiplanar conformers (ϕ=180°), showed 3 J H(A/B)=5.2 Hz (Figure S20). Interestingly, a nucleophilic acyl substitution of tris‐anhydride with tris‐amine trioxane would also give capsularene 1 (Figure 3A). However, when tris‐imide basket 5 was used as a nucleophile in the reaction with tris‐chloro trioxane (Figure 3B) there was no desired product observed.
Figure 3.

A) Synthesis of tris‐acetal 4 and its TFA catalyzed conversion into capsularene 1, with basket 2 forming as an intermediate. The condensation of tris‐anhydride and tris(methylamine)‐trioxane gave capsule 1 in 50 % yield. B) The coupling of tris‐imide 5 with tris(chloromethyl)‐trioxane gave no desired products. ORTEP plots (50 % probability) of X‐ray structures of tris‐imide basket 5 (top) and tris‐acetal basket 4 (bottom). C) 1H NMR spectra (850 MHz, 298 K) of tris‐acetal basket 4 (CDCl3) and capsularene 1 (CD2Cl2).
A slow evaporation of 1 in dichloromethane gave single crystals. X‐ray diffraction analysis revealed the capsularene shaped as a hexagonal prism with three phthalimides standing almost perpendicular with respect to the benzene “floor” (Figure 4A). Capsularene 1 has phthalimide sides at 5 Å centroid‐to‐centroid distance with practically no space between their edges (C=O⋅⋅⋅O=C distance is 3 Å, Figure 4A/B). Along the crystallographic a axis (Figure 4B), capsularenes reside on top of one another forming 1D arrays [1]n held by C−H⋅⋅⋅π contacts (C−H⋅⋅⋅πcentroid=4.073<C−H⋅⋅⋅πcentroid=163°; Figure 4B). [38] Within the crystal, [1]n assemblies reside at π‐π stacking distance (3.5 Å, Figure 4B) along crystallographic b and c axes, with groups of four surrounding a channel filled with disordered molecules of CH2Cl2.
Figure 4.
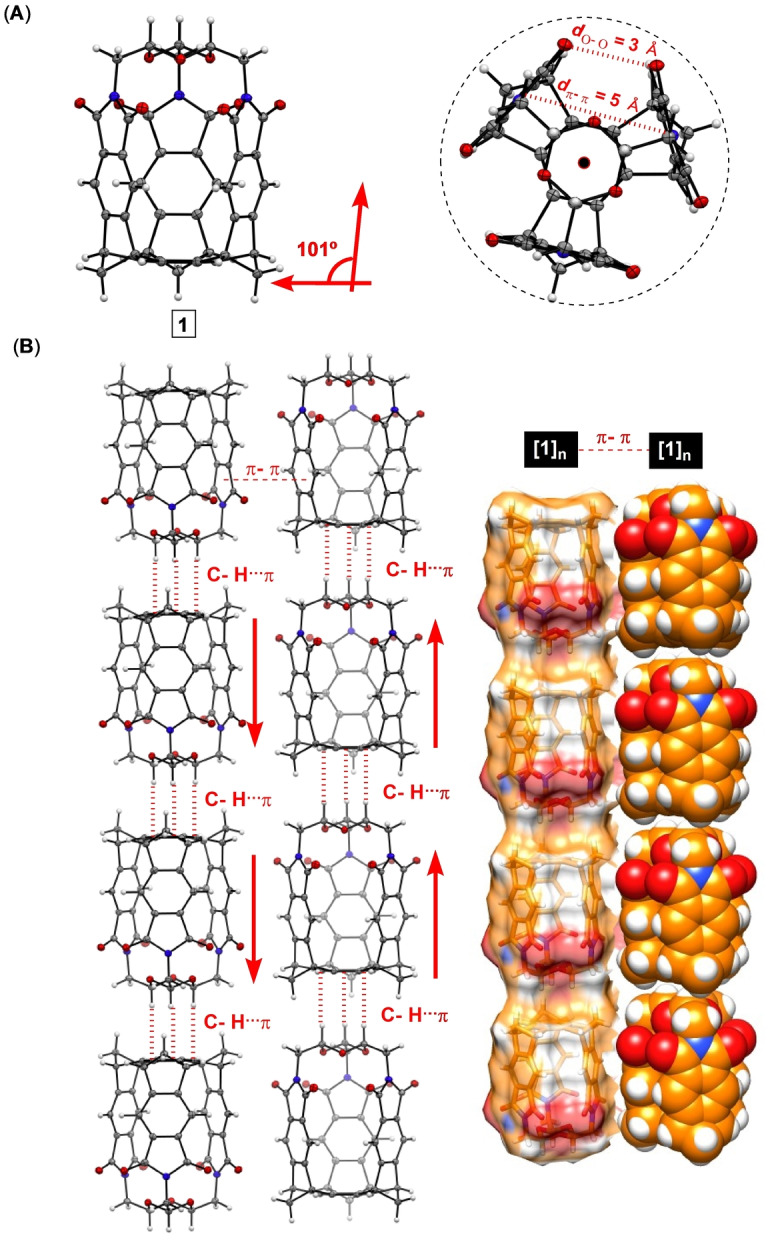
A) ORTEP (50 % probability) plots of capsularene 1 with B) these molecules packing into 1D arrays along the crystallographic a axis.
On the basis of theory (Figure 2), the conversion of basket 2 into capsularene 1 is an endergonic process (ΔG°=21.8 kcal mol−1 at 298 K) in which ΔH°=4.7 kcal mol−1 and TΔS°=−17.1 kcal mol−1. This disagrees with the experiment in which the conversion of 2 to 1 is a quantitative process (ΔG°<0). To account for the observation, we note that the results from X‐ray diffraction analyses show the cavity of basket 4 (similar in size and shape to 2) being occupied by a molecule of CH2Cl2 (Figure 3B) while no electron density can be found within capsularene 1 (Figure 4A). It follows that desolvation of the inner space of 2 takes place in its conversion into 1. We suggest that this entropically favored process (ΔS°>0), unaccounted by theory, is sufficient to overcome the vibrational/translational losses in the formation of capsularenes making the conversion exergonic.
Supramolecular Catalysis in the Formation of 1
To monitor the conversion of tris‐acetal basket 4 into capsularene 1, we used 1H NMR spectroscopy (Figure 5A; Figures S23). The occurrence of two consecutive reactions was evident, with a rapid acetal hydrolysis preceding a slower conversion of tris‐aldehyde basket 2 into 1. By varying the amount of TFA (1 to 6.5 M) and keeping the starting concentration of 2 constant (2.0 mM), we used variable time normalization analysis [39] to find that the reaction's order in acid is 0.64 (Figure S24). In a similar manner, [40] the reaction's order in 2 was found to be 0.8 (Figure S26). From the experimental rate law (rate=k obs [TFA]0.6[2]0.8, Figure 5B), the fractional order in acid could be related to both TFA and 2 participating in equilibria outside the catalytic cycle and in a nonlinear manner. Indeed, TFA is known to undergo a dimerization in nonpolar media with K d=0.7 M in 1,2‐dichloroethane. [41] About the partial reaction order in tris‐aldehyde 2, we noted that the reaction's rate became increasingly slower for reactions having the same starting concentrations of reactants 2 and TFA but greater amount of product 1 in the system (Figure S29). Moreover, a steady magnetic perturbation of proton nuclei from 2 in the presence of greater quantities of 1 (1H NMR spectroscopy, Figure S31) suggested the inclusion of capsularene 1 into basket 2 (Figure 5B). [42] So, it follows that the product inhibition contributes to the observed fractional order in the substrate. In accord with the Michealis‐Menten scenario (Figure 5C), [43] tris‐aldehyde 2 could in first step of the catalytic cycle form a hydrogen‐bonding complex with TFA. The formation of such [TFA⊂2] intermediate is in line with the notion that we had to use an excess of acid to make the reaction proceed at a reasonable rate (i.e. increase the population of [TFA⊂2]). A protonation of aliphatic aldehyde 2 (pK a<−4) [44] with TFA (pK a=0.52) could, under the experimental conditions ([TFA]≫[2]), also take place giving rise to an ion pair. In the rate‐limiting step k 2, three carbonyl groups in [TFA⊂2] undergo cyclotrimerization to give the product. We used differential calculus to derive the expected change in the concentration of 2 over time (see Supporting Information). Despite that fitting the experimental data to a simplified kinetic model (Figure 5C) was satisfactory, a rather poor fit to extended models (i.e., more variables) of the mechanism in Figure 5B suggested that the proposed scenario may necessitate further adjustments.
Figure 5.

A) The conversion of 1.92 mM tris‐acetal basket 4 in CD2Cl2, containing 3.27 M TFA, into 1 was monitored with 1H NMR spectroscopy (850 MHz, 298 K). A segment of recorded spectra for a period of 338 min is shown. B) The proposed catalytic cycle for the formation of capsularene 1 from tris‐aldehyde basket 2 with the corresponding kinetic parameters and energy minimized structures (OPLS3) of the participating molecules. C) Changes in the concentration of 0.25 mM basket 2, measured for varying initial concentrations of basket 1, were fitted as a function of time to a model of product inhibition to determine the rate coefficient k app and the dissociation constant K d2.
The intramolecular attack of the carbonyl oxygen to the adjacent and activated carbonyl in 2 (Figure 5B) requires for two groups to meet at the bonding distance. Indeed, the inward motion of the phthalimide sides would enable the transformation but only after squeezing out the encapsulated solvent from the inner space of 2. If we assume that [TFA⊂2] comprises TFA occupying the cavity of basket 2 (Figure 5B), then a partial in‐to‐out pivoting of this guest could allow a closer approach of the phthalimide arms without solvent molecules entering the concave interior. TFA is in this way acting as encapsulation catalyst to facilitate the formation of the macrocyclic product. In line with these claims, the conversion of 2 to 1 was found to be much slower in CDCl3 then CD2Cl2 (Figure S32). That is to say, with larger chloroform known to have greater binding affinity for occupying the inner space of baskets, [45] we reason that the capacity of TFA to act as catalyst decreased due to a smaller degree of encapsulation. All the same, preliminary data suggests that differently sized trichloroacetic acid (pK a=0.77), trifluoromethanesulfonic acid (pK a=−15) and HCl (pK a=−6.3) produce only trace quantities of 1 as, we posit, their inability to properly occupy the cavity of 2.
As opposed to intermolecular formation of 1,3,5‐trioxanes, the acid‐catalyzed formation of capsularene 1 appeared to be a highly favored transformation. Two weeks after monitoring the macrocyclization with 1H NMR spectroscopy, there was a complete disappearance of resonances from tris‐aldehyde 2. When capsularene 1 was subjected to refluxing methanol having catalytic HCl we could not observe the appearance of tris‐acetal basket 4 (1H NMR spectroscopy). Allegedly, capsularene 1 would under the experimental conditions produce an insufficient quantity of tris‐aldehyde 2 to become trapped by the alcoholic solvent.
Longer Capsularenes
To obtain longer capsularenes holding naphthalimide 8 and anthraceneimide 9 sides (Figure 6A), we prepared tris‐acetal baskets 6 and 7 using already established procedures. [46] After 6 or 7 were each dissolved in CH2Cl2 and subjected to an excess of TFA, there followed the exclusive formation of 8 and 9, respectively (Figure S16/S19). Importantly, a set of 1H NMR resonances from tris‐acetal baskets 6 or 7 undergoes similar magnetic perturbations upon the formation of 8 and 9 (Figure 6A). The observation is in line with the acene walls approaching each other in the conversion to bring their nuclei close enough to became magnetically perturbed by the adjacent aromatics. [47]
Figure 6.

A) Capsularenes 8 and 9 were obtained from tris‐acetal baskets 6 and 7, respectively. 1H NMR spectra (850 MHz, 298 K) of 6–9 in CDCl3 are shown at the bottom; note that CDCl3 solution of capsularenes 9 contains TFA (for improving its solubility). B) ORTEP plot (50 % probability) and CPK representation of capsularene 8 obtained from MicroED analysis. (Right) Capsularenes 8 form 1D arrays along crystallographic b axis.
Using a variety of experimental conditions, we failed to grow monocrystals of 8 and 9. However, when a solid sample of 8 was subjected to microcrystal electron diffraction (MicroEd) analysis [48] the data revealed its structure and packing in solid state (Figure 6B). As in the case of capsularene 1, longer capsularene 8 has naphthalimide sides at 5 Å centroid‐to‐centroid distance, C=O⋅⋅⋅O=C separation at 3 Å, and the inner space (78 Å3) without a guest occupant (Figure 6B). The packing of 8 is similar to that observed for 1 (Figure 4) with capsularenes assembling into 1D supramolecular polymer along crystallographic b axis. Along this same axis, capsularenes are offset forming C−H⋅⋅⋅π contacts (C−H⋅⋅⋅πcentroid=3.764 Å <C−H⋅⋅⋅πcentroid=158°; Figure 6B). [38] In perpendicular dimension, they are held by π‐π stacking contacts (3.5 Å, Figure 6B). In contrast to 1, microcrystals of 8 are without channels holding solvent molecules.
Trapping a Guest within Capsularenes
The absence of guest molecule(s) populating the cylindrical interior of larger capsularenes (78 and 120 Å3, Figure 7A; see ESI‐MS in Supporting Information) agrees with desolvation (ΔS°>0) acting as the driving force in their formation. Preliminary measurements (NMR and MS) showed no incorporation of differently sized He (7 Å3), NO+ (23 Å3) or CH4 (33 Å3) inside capsularene 1 (49 Å3) when the preparation was run in the presence of these guests (at atmospheric pressure and room temperature). If we consider that removal of solvent molecule(s) from the inner space of basket 2 drives the formation of 1 as well as TFA being the encapsulation catalyst, the absence of templation by probed guests makes sense.
Figure 7.

A) Energy‐minimized structures (DFT:B3LYP/6‐31+G*) of capsularenes 1, 8 and 9 with their inner volumes. B) UV/Vis spectra of model compounds tris‐acetal baskets 4,6 and 7 and capsularenes 1, 8 and 9 in CH2Cl2 at 298 K. C) Emission spectra (λ exc=280 nm) of model compounds, tris‐acetal baskets 4, 6 and 7 and capsularenes 1, 8 and 9 in CH2Cl2. D) Cyclic voltammograms of tris‐acetal basket 4, tris‐acetal basket 6, capsularene 1 and capsularene 8 in DMF containing TBAPF6.
Optical and Redox Characteristics of Capsularenes
Rigid and hollow (49–120 Å3, Figure 7A) capsularenes 1, 8 and 9 are short and closed tubes, [49] 0.7 nm in width and 0.9–1.4 nm in length. In contrast to CNTs, they encompass axial but not radial conjugation of π electrons. [9a] Concurrently, acene chromophores are fixed at 5 Å centroid‐to‐centroid distance (Figure 4A) and might perturb each other's electron clouds. [17a] Indeed, UV/Vis spectra of capsularenes are somewhat red shifted with respect to the corresponding model compounds in Figure 7B. [50] The computed UV/Vis spectra (TD‐DFT: CAM‐B3LYP/def2svpd; Figure S47–S49) were in good agreement with experimental data thereby revealing for the lowest observable energy transitions to be of π‐π* type. [51] Using these π‐π* absorptions, [52] we estimated for band gaps to be 4.0, 3.4 eV and 3.0 eV for monomeric models while lower (3.5 eV, 3.3 and 2.9 eV) for capsularenes 1, 8 and 9 (Figure 7B). [14] Emission spectra showed an almost uniform red shift in the series: models→tris‐acetal baskets→capsularenes (Figure 7C). Anthraceneimides within 9 radiated bright blue light (Figure 7C), making these molecules good candidates for engineering OLEDs. [21] In this regard, the Stokes shifts are predominantly increasing in the series from model compounds (140, 23 and 33 nm) to tris‐acetal baskets 4/6/7 (169, 23 and 34 nm) and capsularenes 1/8/9 (162, 34 and 42 nm).
The electrochemical characteristics of acenes within 1 and 8 were also probed with cyclic voltammetry (CV) and bulk electrolysis; note that 9 is poorly soluble in organic solvents used for electrochemical measurements. The standard reduction potentials (E 1/2, Figure 7D) of baskets 4 and 6 are between 1.93–1.98 V, indicating that the formation of their carbonyl radical anions [53] is practically unperturbed by the nature of the conjugated aromatic. The results of bulk electrolysis of 4 suggest that three electrons transferred in the process (Figure S33). [54] In contrast to baskets 4 and 6, reduction of capsularenes 1 and 8 (Figure S35) was more energy demanding thereby requiring more negative (190 mV) potentials (−2.12 to −2.18 V, Figure 7D). We reason that the proximity of six carbonyls within capsularenes along with their structural rigidity must have contributed to the observation. That is to say, the formation of 4 3−⋅ and 6 3−⋅ could be offset by the aromatic arms flexing out and moving away from one another. Capsularenes 1 3−⋅ and 8 3−⋅ are fixed in space and small bonding changes are all that these molecules have at their disposal for balancing the electrostatic repulsion.
Thermal Stability of Capsularenes
Thermogravimetric analysis (TGA) of capsularenes 1, 8 and 9 revealed two distinguishable phase transitions that began at 273, 339 and 338 °C, respectively (Figures S42–S44). On the contrary, trioxane model compounds 3, 10 and 11 (Figures S39–S41) had the onset of their mass loss at lower temperatures (141, 268 and 245 °C, respectively). Evidently, the thermal stability of acenes is improved by their incorporation into rigid capsularenes.
Conclusion
In summary, acenes [14] can be incorporated into closed tubes “capsularenes” to, as side walls, reside within 5 Å from one another. The formation of such rigid and tubular nanostructures [16] from flexible molecular baskets is a quantitative process driven by desolvation in which TFA acts as encapsulation catalyst. [55] While optical and redox characteristics of acenes are, within capsularenes, somewhat perturbed, [19] their thermal stability is significantly improved. [15] Moreover, the unique mode of packing of capsularenes in solid‐state, dominated by C−H‐π and π‐π intermolecular contacts, arises from their tubular shape and rigidity. Since accessibility, thermal stability, the mode of packing and conductivity play key roles in building OLEDs, [17a] FETs [15] and photovoltaic cells,[ 7a , 19 ] we reason that capsularenes may, down the road, help with addressing efficiency, lifetime, and mechanical characteristics of organic electronic materials. In this regard, supramolecular chemistry of capsularenes (i.e., doping with guests, complexation with macrocycles into pseudorotaxanes, etc.) [11] is expected to provide additional avenues for property optimization. That is to say, noncovalent modifications shall allow tuning solubility, self‐assembly, packing in solid state and optoelectronic characteristics of here described or even longer analogues of capsularenes. [10]
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgements
This work was supported with funds from NSF under CHE‐2002781. This study made use of the Campus Chemical Instrument Center NMR facility at the Ohio State University. The work was also supported by The Foundation for a Better World. Electron microscopy was performed at the Center for Electron Microscopy and Analysis (CEMAS).
Dedicated to Professor Fraser Stoddart on the occasion of his 80th birthday
R. Z. Pavlović, L. Zhiquan, T. J. Finnegan, C. A. Waudby, X. Wang, V. W. L. Gunawardana, X. Zhu, C. M. Wong, T. Hamby, C. E. Moore, N. Hoefer, D. W. McComb, C. S. Sevov, J. D. Badjić, Angew. Chem. Int. Ed. 2022, 61, e202211304; Angew. Chem. 2022, 134, e202211304.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.
- 1a. Guo Q.-H., Qiu Y., Wang M.-X., Stoddart J. F., Nat. Chem. 2021, 13, 402; [DOI] [PubMed] [Google Scholar]
- 1b. Li Y., Kono H., Maekawa T., Segawa Y., Yagi A., Itami K., Acc. Mater. Res. 2021, 2, 681; [Google Scholar]
- 1c. Ball M., Zhong Y., Wu Y., Schenck C., Ng F., Steigerwald M., Xiao S., Nuckolls C., Acc. Chem. Res. 2015, 48, 267; [DOI] [PubMed] [Google Scholar]
- 1d. Zhang B., Hernandez Sanchez R., Zhong Y., Ball M., Terban M. W., Paley D., Billinge S. J. L., Ng F., Steigerwald M. L., Nuckolls C., Nat. Commun. 2018, 9, 1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Dai H., Acc. Chem. Res. 2002, 35, 1035; [DOI] [PubMed] [Google Scholar]
- 2b. Dai W., Wang D., J. Phys. Chem. C 2021, 125, 9593; [Google Scholar]
- 2c. Yao J., Yang M., Duan Y., Chem. Rev. 2014, 114, 6130. [DOI] [PubMed] [Google Scholar]
- 3. Kawasaki S., Komatsu K., Okino F., Touhara H., Kataura H., Phys. Chem. Chem. Phys. 2004, 6, 1769. [Google Scholar]
- 4. De Volder M. F. L., Tawfick S. H., Baughman R. H., Hart A. J., Science 2013, 339, 535. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Deng J., Li M., Wang Y., Green Chem. 2016, 18, 4824; [Google Scholar]
- 5b. Tuček J., Kemp K. C., Kim K. S., Zbořil R., ACS Nano 2014, 8, 7571; [DOI] [PubMed] [Google Scholar]
- 5c. Yang Z., Ren J., Zhang Z., Chen X., Guan G., Qiu L., Zhang Y., Peng H., Chem. Rev. 2015, 115, 5159; [DOI] [PubMed] [Google Scholar]
- 5d. Zhao W., Jiang M., Wang W., Liu S., Huang W., Zhao Q., Adv. Funct. Mater. 2021, 31, 2009136. [Google Scholar]
- 6.
- 6a. Omachi H., Segawa Y., Itami K., Acc. Chem. Res. 2012, 45, 1378; [DOI] [PubMed] [Google Scholar]
- 6b. Xia J., Jasti R., Angew. Chem. Int. Ed. 2012, 51, 2474; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 2524; [Google Scholar]
- 6c. Jasti R., Bhattacharjee J., Neaton J. B., Bertozzi C. R., J. Am. Chem. Soc. 2008, 130, 17646; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. Darzi E. R., Jasti R., Chem. Soc. Rev. 2015, 44, 6401; [DOI] [PubMed] [Google Scholar]
- 6e. Zhang X., Shi H., Zhuang G., Wang S., Wang J., Yang S., Shao X., Du P., Angew. Chem. Int. Ed. 2021, 60, 17368; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 17508. [Google Scholar]
- 7.
- 7a. Bergman H. M., Kiel G. R., Handford R. C., Liu Y., Tilley T. D., J. Am. Chem. Soc. 2021, 143, 8619; [DOI] [PubMed] [Google Scholar]
- 7b. Zhang Y., Tong S., Wang M.-X., Angew. Chem. Int. Ed. 2020, 59, 18151; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 18308. [Google Scholar]
- 8. Sun Z., Ikemoto K., Fukunaga T. M., Koretsune T., Arita R., Sato S., Isobe H., Science 2019, 363, 151. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Mirzaei S., Castro E., Sanchez R. H., Chem. Sci. 2020, 11, 8089; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Castro E., Mirzaei S., Hernandez Sanchez R., Org. Lett. 2021, 23, 87. [DOI] [PubMed] [Google Scholar]
- 10. Hermann M., Wassy D., Esser B., Angew. Chem. Int. Ed. 2021, 60, 15743; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 15877. [Google Scholar]
- 11. Xu Y., von Delius M., Angew. Chem. Int. Ed. 2020, 59, 559; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 567. [Google Scholar]
- 12. Yan Z., McCracken T., Xia S., Maslak V., Gallucci J., Hadad C. M., Badjic J. D., J. Org. Chem. 2008, 73, 355. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Shi G., Gadhe C. G., Park S.-W., Kim K. S., Kang J., Seema H., Singh N. J., Cho S. J., Org. Lett. 2014, 16, 334; [DOI] [PubMed] [Google Scholar]
- 13b. Tikhonova I. A., Gribanyov D. A., Tugashov K. I., Dolgushin F. M., Smol′yakov A. F., Peregudov A. S., Klemenkova Z. S., Shur V. B., Organometallics 2009, 28, 6567. [Google Scholar]
- 14. Ye Q., Chi C., Chem. Mater. 2014, 26, 4046. [Google Scholar]
- 15. Anthony J. E., Chem. Rev. 2006, 106, 5028. [DOI] [PubMed] [Google Scholar]
- 16. Ball M., Zhang B., Zhong Y., Fowler B., Xiao S., Ng F., Steigerwald M., Nuckolls C., Acc. Chem. Res. 2019, 52, 1068. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Bula R., Fingerle M., Ruff A., Speiser B., Maichle-Moessmer C., Bettinger H. F., Angew. Chem. Int. Ed. 2013, 52, 11647; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 11861; [Google Scholar]
- 17b. Schaack C., Evans A. M., Ng F., Steigerwald M. L., Nuckolls C., J. Am. Chem. Soc. 2022, 144, 42; [DOI] [PubMed] [Google Scholar]
- 17c. Zhang B., Trinh M. T., Fowler B., Ball M., Xu Q., Ng F., Steigerwald M. L., Zhu X. Y., Nuckolls C., Zhong Y., J. Am. Chem. Soc. 2016, 138, 16426. [DOI] [PubMed] [Google Scholar]
- 18. Bergman H. M., Kiel G. R., Witzke R. J., Nenon D. P., Schwartzberg A. M., Liu Y., Tilley T. D., J. Am. Chem. Soc. 2020, 142, 19850. [DOI] [PubMed] [Google Scholar]
- 19. Kuroda K., Yazaki K., Tanaka Y., Akita M., Sakai H., Hasobe T., Tkachenko N. V., Yoshizawa M., Angew. Chem. Int. Ed. 2019, 58, 1115; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1127. [Google Scholar]
- 20. Hele T. J. H., Fuemmeler E. G., Sanders S. N., Kumarasamy E., Sfeir M. Y., Campos L. M., Ananth N., J. Phys. Chem. A 2019, 123, 2527. [DOI] [PubMed] [Google Scholar]
- 21. Yuan C., Saito S., Camacho C., Irle S., Hisaki I., Yamaguchi S., J. Am. Chem. Soc. 2013, 135, 8842. [DOI] [PubMed] [Google Scholar]
- 22. Smith M. B., Michl J., Chem. Rev. 2010, 110, 6891. [DOI] [PubMed] [Google Scholar]
- 23. Frydrych R., Lis T., Bury W., Cybinska J., Stepien M., J. Am. Chem. Soc. 2020, 142, 15604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lei Z., Gunther M. J., Liyana Gunawardana V. W., Pavlovic R. Z., Xie H., Zhu X., Keenan M., Riggs A., Badjic J. D., Chem. Commun. 2020, 56, 10243. [DOI] [PubMed] [Google Scholar]
- 25. Zhu Z., Espenson J. H., Synthesis 1998, 417. [Google Scholar]
- 26. Zhiquan L., Xie H., Border S. E., Gallucci J., Pavlovic R. Z., Badjic J. D., J. Am. Chem. Soc. 2018, 140, 11091. [DOI] [PubMed] [Google Scholar]
- 27. Lee W. Y., Park C. H., J. Org. Chem. 1993, 58, 7149. [Google Scholar]
- 28. Lee W. Y., Park C. H., Kim S., J. Am. Chem. Soc. 1993, 115, 1184. [Google Scholar]
- 29.
- 29a. Lee W. Y., Synlett 1994, 765; [Google Scholar]
- 29b. Cho S. J., Hwang H. S., Park J. M., Oh K. S., Kim K. S., J. Am. Chem. Soc. 1996, 118, 485. [Google Scholar]
- 30. Denmark S. E., Wilson T., Willson T. M., J. Am. Chem. Soc. 1988, 110, 984. [Google Scholar]
- 31. Wiberg K. B., Wang Y., Arkivoc 2011, 45. [Google Scholar]
- 32.
- 32a. Bew S. P., Cheesman M. R., Sharma S. V., Chem. Commun. 2008, 5731; [DOI] [PubMed] [Google Scholar]
- 32b. Corrochano P., Garcia-Rio L., Poblete F. J., Rodriguez-Dafonte P., Tetrahedron Lett. 2010, 51, 1761. [Google Scholar]
- 33. Wakasugi T., Miyakawa T., Suzuki F., Itsuno S., Ito K., Synth. Commun. 1993, 23, 1289. [Google Scholar]
- 34. Augé J., Gil R., Tetrahedron Lett. 2002, 43, 7919. [Google Scholar]
- 35. Arias Ugarte R., Devarajan D., Mushinski R. M., Hudnall T. W., Dalton Trans. 2016, 45, 11150. [DOI] [PubMed] [Google Scholar]
- 36. Hon Y. S., Lee C. F., Tetrahedron 2001, 57, 6181. [Google Scholar]
- 37. Maslak V., Yan Z., Xia S., Gallucci J., Hadad C. M., Badjic J. D., J. Am. Chem. Soc. 2006, 128, 5887. [DOI] [PubMed] [Google Scholar]
- 38.
- 38a. Nishio M., Umezawa Y., Fantini J., Weiss M. S., Chakrabarti P., Phys. Chem. Chem. Phys. 2014, 16, 12648; [DOI] [PubMed] [Google Scholar]
- 38b. Xie H., Zhiquan L., Pavlovic R. Z., Gallucci J., Badjic J. D., Chem. Commun. 2019, 55, 5479. [DOI] [PubMed] [Google Scholar]
- 39. Burés J., Angew. Chem. Int. Ed. 2016, 55, 2028; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2068. [Google Scholar]
- 40. Burés J., Angew. Chem. Int. Ed. 2016, 55, 16084; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16318. [Google Scholar]
- 41. Christian S. D., Stevens T. L., J. Phys. Chem. 1972, 76, 2039. [Google Scholar]
- 42. Zhiquan L., Polen S., Hadad C. M., RajanBabu T. V., Badjic J. D., J. Am. Chem. Soc. 2016, 138, 8253. [DOI] [PubMed] [Google Scholar]
- 43. Van Hooidonk C., Breebaart-Hansen J. C. A. E., Recl. Trav. Chim. Pays-Bas 1970, 89, 289. [Google Scholar]
- 44.
- 44a. Kelly C. P., Cramer C. J., Truhlar D. G., J. Phys. Chem. B 2006, 110, 16066; [DOI] [PubMed] [Google Scholar]
- 44b. Stewart R., Yates K., J. Am. Chem. Soc. 1958, 80, 6355; [Google Scholar]
- 44c. Yates K., Stewart R., Can. J. Chem. 1959, 37, 664. [Google Scholar]
- 45. Ruan Y., Wang B.-Y., Erb J. M., Chen S., Hadad C. M., Badjic J. D., Org. Biomol. Chem. 2013, 11, 7667. [DOI] [PubMed] [Google Scholar]
- 46. Gunther M. J., Pavlovic R. Z., Finnegan T. J., Wang X., Badjic J. D., Angew. Chem. Int. Ed. 2021, 60, 25075; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 25279. [Google Scholar]
- 47. Pavlović R. Z., Border S. E., Finnegan T. J., Zhiquan L., Gunther M. J., Mūnoz E., Moore C. E., Hadad C. M., Badjić J. D., J. Am. Chem. Soc. 2019, 141, 16600. [DOI] [PubMed] [Google Scholar]
- 48. Jones C. G., Martynowycz M. W., Hattne J., Fulton T. J., Stoltz B. M., Rodriguez J. A., Nelson H. M., Gonen T., ACS Cent. Sci. 2018, 4, 1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Scott L. T., Jackson E. A., Zhang Q., Steinberg B. D., Bancu M., Li B., J. Am. Chem. Soc. 2012, 134, 107. [DOI] [PubMed] [Google Scholar]
- 50. Hermann K., Sardini S., Ruan Y., Yoder R. J., Chakraborty M., Vyas S., Hadad C. M., Badjic J. D., J. Org. Chem. 2013, 78, 2984. [DOI] [PubMed] [Google Scholar]
- 51.
- 51a. Stojanović S., Turner D. A., Hadad C. M., Badjić J. D., Chem. Sci. 2011, 2, 752; [Google Scholar]
- 51b. Gawronski J., Kazmierczak F., Gawronska K., Rychlewska U., Norden B., Holmen A., J. Am. Chem. Soc. 1998, 120, 12083. [Google Scholar]
- 52. Mohebbi A. R., Munoz C., Wudl F., Org. Lett. 2011, 13, 2560. [DOI] [PubMed] [Google Scholar]
- 53. Stojanović S., Turner D. A., Share A. I., Flood A. H., Hadad C. M., Badjić J. D., Chem. Commun. 2012, 48, 4429. [DOI] [PubMed] [Google Scholar]
- 54. Becker B., Bohnen A., Ehrenfreund M., Wohlfarth W., Sakata Y., Huber W., Muellen K., J. Am. Chem. Soc. 1991, 113, 1121. [Google Scholar]
- 55.
- 55a. Catti L., Zhang Q., Tiefenbacher K., Chem. Eur. J. 2016, 22, 9060; [DOI] [PubMed] [Google Scholar]
- 55b. Brown C. J., Toste F. D., Bergman R. G., Raymond K. N., Chem. Rev. 2015, 115, 3012. [DOI] [PubMed] [Google Scholar]
- 56.Deposition Numbers 2157559 (for 1), 2157560 (for 4), 2157558 (for 5), and 2193439 (for MicrED of capsularene 8) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.