

Abstract
Morphological features including infiltrative growth, tumour budding (TB), and poorly differentiated clusters (PDCs) have a firmly established negative predictive value in colorectal cancer (CRC). Despite extensive research, the mechanisms underlying different tumour growth patterns remain poorly understood. The aim of this study was to investigate the involvement of epithelial–mesenchymal transition (EMT) in TB and PDCs in CRC. Using laser‐capture microdissection, we obtained distinct parts of the primary CRC including TB, PDCs, expansive tumour front, and the central part of the tumour, and analysed the expression of EMT‐related markers, i.e. the miR‐200 family, ZEB1/2, RND3, and CDH1. In TB, the miR‐200 family and CDH1 were significantly downregulated, while ZEB2 was significantly upregulated. In PDCs, miR‐141, miR‐200c, and CDH1 were significantly downregulated. No significant differences were observed in the expression of any EMT‐related markers between the expansive tumour front and the central part of the tumour. Our results suggest that both TB and PDCs are related to partial EMT. Discrete differences in morphology and expression of EMT‐related markers between TB and PDCs indicate that they represent different manifestations of partial EMT. TB seems to be closer to complete EMT than PDCs. © 2022 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: colon cancer, colorectal cancer, epithelial–mesenchymal transition, miRNA, partial epithelial–mesenchymal transition, poorly differentiated clusters, tumour budding
Introduction
In recent decades, our knowledge about the pathogenesis of colorectal carcinoma (CRC) has greatly expanded. It is now clear that the invasive tumour front represents the gateway for CRC progression and metastasis, and the negative prognostic value of infiltrative tumour growth exhibiting cancer cell dissociation is now firmly established [1, 2, 3]. Despite extensive research, the mechanisms underlying different tumour growth patterns, and the association between infiltrative growth and unfavourable clinical outcomes, remain poorly understood [4, 5, 6].
Tumour growth at the invasive front is traditionally divided into an expansive and an infiltrative pattern. The former is characterized by well‐circumscribed tumour margins and a clear boundary between the tumour and the host tissue, while the latter features widespread dissociation and dissemination of cancer cells into the neighbouring normal tissue [5, 7, 8]. An infiltrative pattern has long been associated with an unfavourable prognosis [9, 10]. Further studies in CRC specified two morphological variants of cancer cell dissociation, i.e. tumour budding (TB) and poorly differentiated clusters (PDCs), which serve as independent prognostic markers and upon which novel grading systems for CRC have been proposed [11, 12, 13].
TB is defined as the presence of cancer cells detached from the main tumour either as single cells or as clusters of fewer than five cells [14]. Studies suggest that TB is a strong predictor of nodal metastasis, disease recurrence, and cancer‐related death in patients with CRC [12, 15]. PDCs are defined as clusters of more than five cancer cells in tumour stroma lacking gland formation [11]. They were found to correlate significantly with lymphatic invasion, occult nodal metastasis, cancer‐specific survival, and recurrence‐free survival [16, 17, 18].
The true nature of and the pathogenetic mechanisms associated with TB and PDCs are not completely understood. On the one hand, it has been argued that TB and PDCs share many morphological and biological features and may be distinguished only by an arbitrary numerical cut‐off [17]. On the other hand, it has been hypothesized that PDCs reflect the least differentiated component of CRC [19], while TB represents a histological process unrelated to the degree of differentiation [20, 21]. Additionally, it has been proposed that TB and PDCs might be related to complete or partial epithelial–mesenchymal transition (EMT) [19, 22, 23].
Complete (full) EMT implies a switch from an epithelial to a spindle cell morphology, accompanied by a switch in intermediate filament expression, an alteration in the composition of intercellular junctions, and upregulation of EMT transcription factors [24, 25, 26]. EMT‐related markers thus include intermediate filaments (loss of epithelial markers and expression of mesenchymal markers), classical and desmosomal cadherins (loss of E‐cadherin and expression of N‐cadherin, loss of desmosomal cadherins), transcription factors [ZEB1/2, TWIST1/2, SNAI1 (SNAIL) and SNAI2 (SLUG)], microRNAs (miRNAs), etc. It is now clear that full EMT rarely occurs in human tumours, whereas partial EMT is more common at various stages of tumour development and progression. Partial EMT generates a spectrum of intermediate cellular phenotypes with varied morphology, simultaneously expressing both epithelial and mesenchymal markers and various transcription factors [24, 25, 26]. In our experience, detection of EMT should be based on morphology and a panel of various EMT‐related markers, including EMT transcription factors and their regulatory molecules [27, 28]. Among them, miRNAs have been demonstrated to be sensitive markers of EMT [29].
miRNAs are small non‐coding RNAs that regulate gene expression at the post‐transcriptional level. Among the miRNAs studied, the miR‐200 family was identified as one of the major contributors of EMT activation. The miR‐200 family includes two clusters of miRNAs located on different chromosomes: miR‐200b, miR‐200a, and miR‐429 on chromosome 1; and cluster miR‐200c and miR‐141 on chromosome 12 [30]. All these five members of the miR‐200 family regulate translation of both ZEB1 and ZEB2, which are important EMT transcription factors [26, 30].
The aim of this study was to investigate the involvement of EMT in TB and PDCs in CRC. We hypothesized that EMT may be activated in TB and PDCs but not in the central parts of the tumour or at the invasive tumour front with an expansive growth pattern. We used laser‐capture microdissection to obtain distinct parts of the primary tumour, including TB and PDCs, for further analysis. Based on previous studies including ours, the following EMT‐related markers were chosen: the miR‐200 family, ZEB1/2, RND3, and CDH1 [29, 30]. Their expression was compared in TB versus PDCs, as well as with the central part of the tumour and expansive growth pattern at the invasive front.
Materials and methods
Patients and tissue samples
This study was approved by the National Medical Ethics Committee, Republic of Slovenia, Ministry of Health (reference number 0120‐88/2020/3) and carried out following the rules of the Declaration of Helsinki. Archival histopathological slides and tissue blocks from a cohort of patients treated surgically for CRC at the University Medical Centre Maribor, Slovenia during the period from 1 January to 31 December 2019 were included. After surgery, resection specimens were fixed in formalin for 24 h and processed according to standard routine practice: at least one tumour block per 1 cm of tumour diameter was taken, including sections from the central part of the tumour, tumour–stroma interface, transition zone between the tumour and the adjacent mucosa, and the deepest point of invasion, including serosa. Resection margins and lymph nodes were also taken. Tissue samples were embedded in paraffin wax, cut into 3‐ to 4‐μm‐thick sections and stained with haematoxylin and eosin. CRC cases were classified according to the pTNM classification of malignant tumours [31]. Clinical information including age, gender, tumour location, metastasis, history of radio‐/chemo‐therapy, and co‐morbidity was available for all patients as well as an average follow‐up of 35 months.
Only patients with CRC staged ≥T2 were included. A total of 70 patients were available. Patients with a history of radio‐/chemo‐therapy for rectal cancer (n = 3) or other prior/concurrent malignancies (n = 2) were excluded. After re‐examining the histopathological slides, patients with carcinomas exhibiting a monotonic invasive tumour front, which was either purely infiltrative with no expansive pattern (n = 26) or vice versa (n = 6); mucinous carcinomas (n = 10); and microsatellite unstable cancers (n = 2) were excluded. A case of an early CRC, with minimal invasion of the muscularis propria, was also excluded (n = 1). A total of 20 cases were finally included in the study.
Histological grading of TB and PDCs
Histopathological slides were re‐examined and analysed by two pathologists (AP and NZ). Tumour budding (TB) was defined as single tumour cells or clusters of fewer than five cells at the invasive tumour front, and counted and graded as Bd1–Bd3 (Bd1, 0–4 buds/0.785 mm2; Bd2, 5–9 buds/0.785 mm2; Bd3, ≥10 buds/0.785 mm2) according to recommendations [12]. Poorly differentiated clusters (PDCs) were defined as clusters of more than five tumour cells at the invasive front or within the tumour lacking gland formation; they were counted in the hotspot area identified at scanning magnification and graded as G1–G3 (G1, <5; G2, 5–9; G3, ≥10 PDCs in a microscopic field of objective lens ×20) as previously reported [11, 17]. Areas with significant electrocoagulation‐induced cytological artefacts and areas with prominent necrosis and/or inflammation were not considered in the grading assessment.
From each tumour, representative slides of the central part, expansive tumour front (Figure 1A), and areas with the highest numbers of PDCs (Figure 1B) and TB (Figure 1C,D) were selected, and corresponding paraffin blocks retrieved from the archive. Samples of the central part of the tumour were searched for in the area with the greatest tumour thickness and taken from the same distance towards the surface and the deepest point of invasion. According to inclusion criteria, only tumours containing the three patterns crucial for our investigation, i.e. expansive growth pattern, TB, and PDCs, were selected for further analysis.
Figure 1.
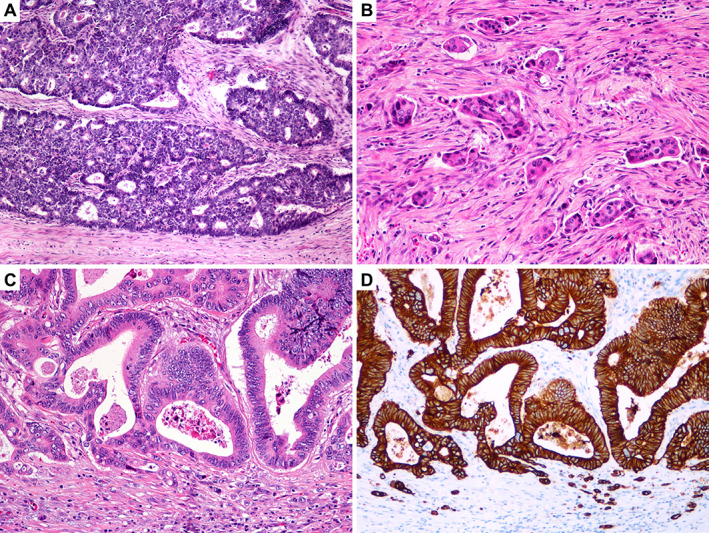
Growth patterns at the invasive tumour front. (A) Expansive front: well‐circumscribed tumour margin, with a clear boundary between the tumour and host tissue. (B) Poorly differentiated clusters: clusters of more than five tumour cells lacking gland formation, sharply demarcated from the stroma, and surrounded by clefting artefacts. (C) Tumour budding: single tumour cells and clusters of fewer than five cells. (D) Tumour budding highlighted by immunohistochemistry for cytokeratin AE1/AE3.
Laser‐capture microdissection
Using laser‐capture microdissection (LMD6; Leica Microsystems, Wetzlar, Germany), tissue was obtained separately from TB, PDCs, the expansive tumour front, and the central part of the tumour. For the purpose of laser‐capture microdissection, 7‐μm‐thick slides were cut from formalin‐fixed, paraffin‐embedded (FFPE) tissue blocks and mounted on PEN membranes (2.0 μm, nucleases and human nucleic acid‐free, cat. No. 11505189, Leica Microsystems). The slides were dried overnight at 56 °C and then stained using Mayer's haematoxylin according to the recommended protocol [32]. After staining, the slides were air‐dried and either immediately prepared for laser‐capture microdissection or first stored at −80 °C until use for laser‐capture microdissection.
RNA isolation
RNA isolation was performed using an miRNeasy FFPE Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The deparaffinization solution used was hexadecane and overnight digestion was performed in proteinase K. Elution was performed using 17 μl of RNase‐free water. The successful isolation procedure was confirmed by adding 1 μl of a mixture of spike‐ins (UniSp2, UniSp4, and UniSp6) in proteinase K digestion buffer, followed by quantification of these spike‐ins. The concentration and quality of isolated RNA were measured spectrophotometrically using a NanoDrop One Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) at A 260, A 230, and A 280.
miRNA expression analysis
The miRNAs miR‐141, miR‐200a, miR‐200b, miR‐200c, and miR‐429 were analysed using the miRCURY LNA miRNA PCR system (Qiagen) based on SYBR Green detection. As reference genes (RGs), miR‐27a‐3p, miR‐193a‐5p, and let‐7g‐5p were used according to published literature [33]. All reagents were from Qiagen unless otherwise indicated. For reverse transcription, an miRCURY LNA RT Kit was used according to the manufacturer's instructions in a 10 μl final volume containing the reaction master mix and 10 ng of total RNA including the UniSp6 spike‐in.
The resulting cDNA was diluted ten‐fold and 3 μl was used in a 10 μl final qPCR reaction volume according to the manufacturer's instructions. qPCR was carried out using the QuantStudio 7 Pro Real‐Time PCR System (Thermo Fisher Scientific). All qPCR reactions were run in duplicates. To test the qPCR efficiency, cDNA samples were first pooled and then qPCR was performed as described above. qPCR reaction efficiency was tested in triplicates for each analysed miRNA using three‐fold dilutions. The signal was collected after each cycle. Following amplification, melting curve analysis of PCR products was performed to verify the specificity and identity. Melting curves were acquired on the SYBR channel using a ramping rate of 0.7 °C/60 s for 60–95 °C.
mRNA expression analysis
CDH1, RND3, ZEB1, and ZEB2 mRNAs were assessed using TaqMan probes and abundance was expressed relative to B2M mRNA, which was found to be suitable for CRC expression analyses in previous studies including ours [34, 35, 36]. Samples were reverse‐transcribed using M‐MLV Reverse Transcriptase (Thermo Fisher Scientific) as reported previously [37]. After mixing 6 μg of random hexamers and 4 or 5 μl of RNA isolate, reverse transcription (RT) was started by incubation at 65 °C for 10 min. After this random priming step, the RT Master mix, including 250 U of M‐MLV RT, 1× First Strand Buffer, 1 mm dNTP, 10 mm DTT, 4 U of RNase inhibitor, and 4.5 mm MgCl2, was added to achieve a final 20 μl volume. The RT reaction conditions were 25 °C for 10 min, 37 °C for 60 min, and 70 °C for 15 min.
Due to the small amounts of RNA isolated from micro‐dissected samples, we were unable to obtain enough RNA for successful amplification of mRNAs. Therefore, following RT, we performed pre‐amplification using the TaqMan PreAmp Master Mix (Applied Biosystems, Foster City, CA, USA) in a 12 μl reaction according to the manufacturer's instructions. The resulting PreAmp reactions were diluted five‐fold. In the qPCR reaction, 4.5 μl of diluted sample was used with 5.0 μl of 2× FastStart Essential DNA Probe Master Mix (Roche, Basel, Switzerland) and 0.5 μl of TaqMan 20× probe in a 10 μl total reaction volume. Thermal conditions were as follows: 50 °C for 2 min, initial denaturation at 95 °C for 10 min, and 45 cycles of denaturation at 95 °C for 15 s and annealing/extension at 60 °C for 1 min. All qPCR analyses were performed on a QuantStudio Pro7 in duplicates. The signal was collected at the endpoint of each cycle. To test the qPCR efficiency, cDNA samples were first pooled and then qPCR was performed as described above. qPCR reaction efficiency was tested in triplicates for each analysed mRNA using five‐fold dilutions.
Immunohistochemistry
We used the commercially available antibodies anti‐RhoE/Rnd3 (Clone 4, dilution 1:350; Merck Millipore, MA, USA) and anti‐CKAE1/AE3 (Clone NCL‐L‐AE1/AE3, dilution 1:50; Novocastra Leica Biosystems, Newcastle, UK). Sections of 4 μm thickness were cut from paraffin blocks and processed for automated antigen retrieval and staining using an automatic immunostainer (BenchMark GX; Ventana Medical Systems, Tuscon, AZ, USA). Deparaffinization was performed using the EZ Prep reagent, followed by antigen retrieval with Cell Conditioning Buffer 1 (Ventana Medical Systems) at 100 °C for 56 min. Slides were cooled to 36 °C and incubated with primary antibody against the antigen (dilution 1:350). Immunoreactivity was visualized using the OptiView DAB Detection Kit (Ventana Medical Systems) according to the manufacturer's instructions. Sections were counterstained with haematoxylin II (Ventana Medical Systems) for 8 min and Ventana Bluing Reagent for 4 min. Negative controls omitting the primary antibodies were also included.
The percentage of staining in the tumour was scored as 0 (no staining), 1 (between 1% and 25%), 2 (between 26% and 50%), and 3 (more than 50% of the tumour). The staining intensity was evaluated separately in the central part of the tumour, TB, and PDCs, and scored as 0 (no staining), 1 (weak staining), 2 (moderate staining) or 3 (strong staining). In cases with a variable staining intensity, the strongest intensity was noted. The intensity scores were compared between the central part of the tumour, TB, and PDCs.
Statistical analyses
Results were presented as the relative gene expression. Expression of the gene of interest (GOI, CqGOI) was presented as ΔCq, which was calculated relative to the geometric mean of RGs (CqRG). In CRC samples, mRNA and miRNA expression differences were compared between the central part and the expansive tumour front, PDCs, and TB, respectively. We further compared the expression of mRNA and miRNA in PDCs and TB with the expression in the expansive tumour front as well as between PDCs and TB. For all statistical comparisons, ΔCq and the Wilcoxon rank test were used. For all investigated correlations/associations, Pearson's correlation coefficient was used. Statistical analysis of data was performed using SPSS version 27 (SPSS Inc, Chicago, IL, USA). Differences were considered significant at p ≤ 0.05. The results are shown as relative expression of mRNAs and miRNAs at the expansive tumour front, PDCs, and TB relative to the central part of the tumour.
Results
Patients and tissue samples
In total, we analysed tissue samples from 20 patients (13 males and 7 females) with CRC, aged 54–87 years (mean 72.8 years). Demographic and clinicopathological features are presented in Table 1.
Table 1.
Demographic and clinicopathological features of patients
| Case No. | Age (years) | Gender | Location | pTNM | TB | PDCs | NED (months) | Follow‐up (months) |
|---|---|---|---|---|---|---|---|---|
| 1 | 75 | M | Transverse colon | T2N0M0 | Bd1 | G3 | 34 | 34 |
| 2 | 74 | F | Caecum | T2N0M0 | Bd1 | G1 | 27 | 27 |
| 3 | 79 | F | Descending colon | T2N1bM0 | Bd3 | G1 | 31 | 31 |
| 4 | 81 | F | Descending colon | T3N0M0 | Bd1 | G1 | 32 | 32 |
| 5 | 70 | M | Ascending colon | T3N0M0 | Bd2 | G1 | 30 | 30 |
| 6 | 71 | M | Ascending colon | T3N0M0 | Bd2 | G1 | 3 | 4; DUC |
| 7 | 71 | M | Transverse colon | T3N0M0 | Bd1 | G2 | 38 | 38 |
| 8 | 81 | F | Caecum | T3N0M0 | Bd1 | G1 | 37 | 37 |
| 9 | 69 | M | Caecum | T3bN0M0 | Bd3 | G1 | 36 | 36 |
| 10 | 70 | M | Transverse colon | T3N1M0 | Bd2 | G1 | 33 | 33 |
| 11 | 81 | M | Transverse colon | T3N1bM0 | Bd2 | G2 | 0 | 0; DPC |
| 12 | 73 | F | Transverse colon | T3N1bM0 | Bd2 | G2 | 27 | 27 |
| 13 | 63 | M | Descending colon | T3N1bM0 | Bd2 | G3 | 34 | 34 |
| 14 | 57 | M | Transverse colon | T3N1bM0 | Bd2 | G3 | 30 | 35; LR + DM |
| 15 | 58 | M | Ascending colon | T3N1bM0 | Bd2 | G3 | 37 | 37 |
| 16 | 54 | F | Transverse colon | T3cN1bM0 | Bd1 | G1 | 35 | 35 |
| 17 | 75 | M | Transverse colon | T3N2bM1b | Bd2 | G3 | 6 | 11; DM + DD |
| 18 | 87 | M | Ascending colon | T4aN0M0 | Bd2 | G3 | 28 | 28 |
| 19 | 83 | M | Sigmoid colon | T4N1bM0 | Bd3 | G3 | 38 | 38 |
| 20 | 84 | F | Transverse colon | T4aN2aM0 | Bd1 | G1 | 0 | 0; DPC |
DD, died of disease; DM, distant metastasis; DPC, died of perioperative complications, DUC, died of unrelated causes; LR, local disease recurrence; NED, no evidence of disease; TB, tumour budding; PDCs, poorly differentiated clusters.
Expression of the miR‐200 family
When the central part of CRC and the expansive tumour front were compared, no significant differences were observed in the expression of analysed miRNAs from the miR‐200 family. However, we observed significant downregulation of all analysed miRNAs in TB compared with the central part of CRC: p < 0.001 for miR‐141, miR‐200b, and miR‐200c; p = 0.004 for miR‐200a and p = 0.001 for miR‐429. Additionally, all miRNAs were downregulated in TB compared with the expansive tumour front.
Interestingly, in PDCs, compared with the central part of CRC, the only significant downregulation was observed for miR‐141 and miR‐200c (p = 0.001 and p = 0.017, respectively). No significant differences were observed in the expression patterns of the other three miRNAs. Furthermore, all analysed miRNAs were significantly downregulated in TB compared with PDCs. Results are summarized in Figure 2 and supplementary material, Table S1.
Figure 2.
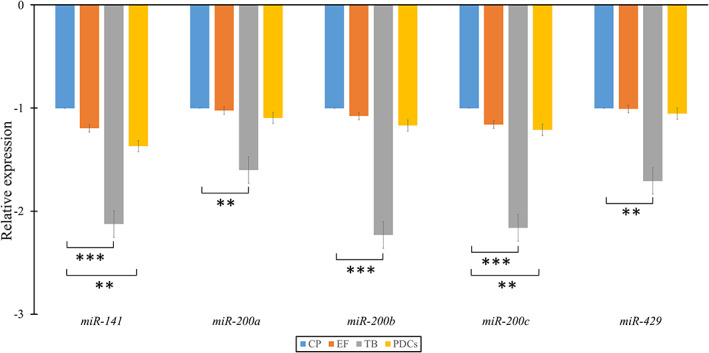
Fold‐change of the miR‐200 family at the expansive tumour front, tumour budding, and poorly differentiated clusters compared with the central part of the tumour. CP, central part; EF, expansive tumour front; PDCs, poorly differentiated clusters; TB, tumour budding. *p < 0.05; **p < 0.01; ***p < 0.001.
Expression of mRNAs
When the central part of CRC was compared with the expansive tumour front, none of the other mRNAs, namely CDH1, RND3, and ZEB2, showed any statistically significant change in expression. ZEB1 was below the detection limit, irrespective of the sampling area. However, we observed significant downregulation of CDH1 (p = 0.001) and upregulation of ZEB2 (p = 0.021) in TB compared with the central part of CRC. Although we observed upregulation of RND3, it was not significant. The same expression pattern was observed when TB was compared with the expansive tumour front.
When PDCs were compared with the central part of CRC, the only change observed was significant downregulation of CDH1 (p = 0.012). Results are summarized in Figure 3.
Figure 3.
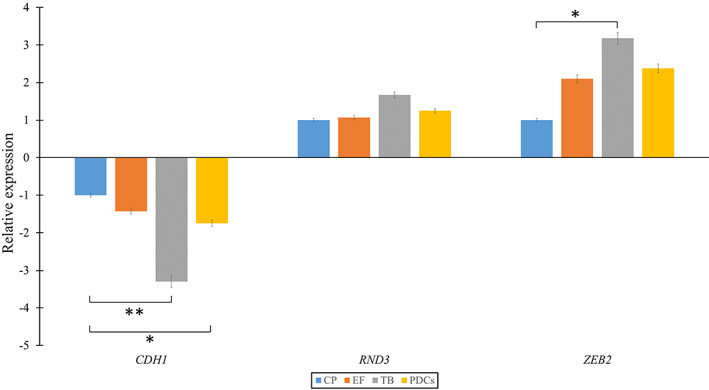
Fold‐change of the CDH1, RND3, and ZEB2 family at the expansive tumour front, tumour budding, and poorly differentiated clusters in comparison to the central part of the tumour. CP, central part; EF, expansive tumour front; PDCs, poorly differentiated clusters; TB, tumour budding. *p < 0.05; **p < 0.01; ***p < 0.001.
Correlation between miRNAs in mRNAs
We investigated the correlation between the expression of the miR‐200 family and their target genes RND3 and ZEB2 as well as an EMT‐related marker, CDH1. No significant correlation was detected between miR‐200b and its target RND3. However, we observed a significant positive correlation between all of the analysed miRNAs and the expression of CDH1 (r s = 0.262, p = 0.021 for miR‐141; r s = 0.356, p = 0.001 for miR‐200a; r s = 0.230, p = 0.0043 for miR‐200b; r s = 0.376, p = 0.001 for miR‐200c; r s = 0.305, p = 0.007 for miR‐429). Moreover, we observed a significant negative correlation between the expression of four out of five members of the miR‐200 family and its target ZEB2 (r s = −0.422, p = 0.007 for miR‐141; r s = −0.338, p = 0.033 for miR‐200a; r s = −0.409, p = 0.009 for miR‐200c; r s = −0.325, p = 0.041 for miR‐429). Results are summarized in supplementary material, Figures S1 and S2.
Immunohistochemistry
Immunohistochemistry for CKAE1/AE3 was used to highlight TB. Immunohistochemistry for RND3 showed moderate staining in inflammatory cells observed in all cases. The percentage of tumour staining was evaluated as follows: score 0 in two cases, score 1 in six cases, score 2 in eight cases, and score 3 in four cases. In tumour cells, the cytoplasmic dot staining was of variable intensity. The staining intensity was scored separately in the central part of the tumour, TB, and PDCs. In 15/20 cases, a similar staining intensity was observed between the central part of the tumour, TB, and PDCs (score 0 in two cases, score 1 in five cases, score 2 in seven cases, and score 3 in one case). In 5/20 cases, staining intensity at least one score higher was observed in TB and/or PDCs compared with the central part of the tumour: score 1 in TB and/or PDCs and score 0 in the central part of the tumour in one case; score 2 in TB and/or PDCs and score 1 in the central part of the tumour in two cases; score 3 in TB and/or PDCs and score 1 in the central part of the tumour in one case; and score 3 in TB and/or PDCs and score 2 in the central part of the tumour in one case (Figure 4).
Figure 4.
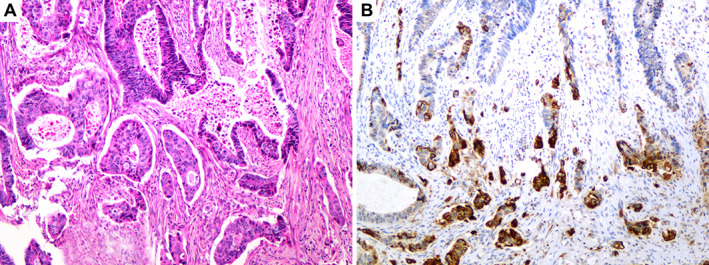
(A) Invasive tumour front with tumour budding and poorly differentiated clusters. (B) Immunohistochemistry for RND3: the intensity of staining in tumour budding and poorly differentiated clusters is stronger compared with the main tumour.
Discussion
The results of our study suggest the involvement of partial EMT in TB and PDCs in CRC, based on significant downregulation of the miR‐200 family and CDH1 as well as upregulation of ZEB2 in TB, and on significant downregulation of miR‐141, miR‐200c, and CDH1 in PDCs. The lack of spindle cell morphology in both TB and PDCs is consistent with partial EMT. Conversely, there was no evidence of EMT at the expansive tumour front, which exhibited no significant differences in the expression of any EMT‐related markers compared with the central part of the tumour. To our knowledge, this is the first study to characterize and compare the expression of the miR‐200 family and other EMT‐related markers between TB and PDCs in CRC.
Importantly, we found that TB and PDCs exhibit different expression patterns of EMT‐related markers, indicating that TB and PDCs might represent different manifestations of partial EMT. While miR‐200c, miR‐141, and CDH1 were significantly downregulated in both TB and PDCs, TB additionally exhibited significant downregulation of miR‐200a/b as well as miR‐429 and significant upregulation of ZEB2. Recent studies showed that partial EMT generates a broad spectrum of cellular states exhibiting intermediate/hybrid morphological, transcriptional, and epigenetic features between epithelial and mesenchymal cells [38]. Increasing evidence suggests that cancer cells with different degrees of EMT exhibit different functional roles during tumourigenesis, invasion, and metastasis [39]. As dysregulation of EMT‐related markers was more extensive in TB than in PDCs, we speculate that TB is closer to complete EMT than PDCs. However, it is still unclear whether TB and PDCs are cellular states undergoing a gradual transition from an epithelial to a mesenchymal phenotype or only distinct cell populations expressing different EMT‐related markers. It is also possible that pathogenetic mechanisms other than EMT contribute to TB and PDCs.
The speculation that TB and PDCs represent different manifestations of partial EMT is further supported by the morphological differences between them [11]. Although they are both composed of dedifferentiated cells, there are several features typical of either TB or PDCs [3, 40]. Firstly, TB is commonly present at the infiltrative tumour front, particularly in the areas of CRC exhibiting finger‐like projections into the neighbouring stroma [41]. In contrast, PDCs often appear both within the tumour and at the invasive tumour front [11]. Secondly, TB cells appear to be actively detaching from the larger neoplastic glands [41, 42], whereas PDCs appear to be unrelated to the main tumour, are sharply demarcated from the stroma, and are often surrounded by clefting artefacts. The morphology of PDCs overlaps substantially with that of micropapillary carcinoma [17, 43]. Finally, TB cells are round, elongated, or atypical [42, 44]. They have poorly developed or absent junctional complexes and absent microvilli [45]. Most TB cells lack mitotic and apoptotic activity [23]. Conversely, PDCs consist of cells with abundant eosinophilic cytoplasm, retaining an epithelioid phenotype.
In this study, we analysed the expression of several EMT‐related markers, i.e. the miR‐200 family, ZEB1/2, RND3, and CDH1. The different EMT‐related marker expression profiles between TB and PDCs found in this study emphasize the importance of using multiple EMT‐related markers to analyse EMT. Previous research has shown that the miR‐200 family is among the most sensitive EMT‐related markers and can be used for studying partial and complete EMT [29, 46, 47]. As the main EMT regulator, the miR‐200 family exerts much of its effect through targeting ZEB1/2 [48]. ZEB1 and ZEB2 act as E‐cadherin repressors and their expression promotes cell migration and invasion [30]. RND3 is a modulator of cell migration and proliferation [49]. Studies describing the expression of RND3 in CRC were based on immunohistochemistry and produced conflicting results. Notably, Luo et al reported that RND3 acts as a tumour suppressor [50], whereas Zhou et al described RND3 as an independent prognostic marker in CRC, correlating with the depth of invasion, lymph node metastasis, and distant metastasis [51]. ZEB2 and RND3 are targets of miR‐200b [52, 53]. Since we observed significant downregulation of miR‐200b in TB, we also performed immunohistochemistry for RND3 to assess whether the staining reflects the underlying molecular changes. Only a few cases showed a stronger positive reaction in TB compared with the main tumour. The evaluation in most other cases was inconclusive and difficult to interpret due to patchy or variable staining.
The involvement of EMT in TB in CRC has already been suggested by several authors [24, 54, 55, 56, 57, 58]. Those investigating the expression of miR‐200 as EMT‐related markers provided the most conclusive results. Martinez‐Ciarpaglini et al reported overexpression of ZEB1/2 and reduction of miR‐200a/b/c in areas with TB [54]. Additionally, Paterson et al observed downregulation of miR‐200b/c in areas with TB [57]. Knudsen et al demonstrated downregulation of miR‐200b and loss of membranous E‐cadherin in TB [58]. In contrast, studies analysing the expression of proteins (E‐cadherin, beta‐catenin, TGF‐beta) or EMT‐related transcription factors (ZEB1/2, SNAI1/2, TWIST1/2) sometimes resulted in inconclusive or contradictory observations [59]. For example, Yamada et al observed that EMT‐related transcription factors were not expressed in the regions of TB and concluded that TB in CRC might not represent EMT [60]. Likewise, Bronsert et al showed that only a few TB cells expressed ZEB1 and almost no TB cells had a complete absence of E‐cadherin expression. They interpreted these findings as consistent with partial EMT [61].
A few studies have investigated the involvement of EMT in PDCs. Hong et al concluded that similarities in the expression of E‐cadherin and beta‐catenin in TB and PDCs could indicate the activation of EMT in both processes [62]. Similarly, Barresi et al proposed the involvement of EMT in PDCs based on the loss of cytoplasmic E‐cadherin [63]. By analysing the expression of E‐cadherin and beta‐catenin, Bertoni et al suggested possible partial EMT in PDCs in the tumour centre and complete EMT in PDCs at the tumour periphery [19]. Although these studies indicate the involvement of EMT in PDCs, they were all based on the expression of proteins as EMT biomarkers, which is less reliable in defining partial EMT. It has already been shown that aberrant expression of E‐cadherin is not restricted to dissociated cells but is also present in other parts of CRC, which makes the interpretation of its expression problematic [63]. In our study, we found significant downregulation of CDH1 in PDCs.
The true nature of TB and PDCs is not completely understood and the relationship between them remains speculative [64, 65]. Attempts to analyse cell dissociation were first challenged by 3D models showing that most cells and clusters that appear isolated in pathological practice represent 2D artefacts, as they are in fact connected to adjacent glands [61, 66]. Likewise, TB and PDCs could only appear as separate entities on 2D sections [17]. This was reinforced by studies that showed a significant overlap in the protein expression and molecular profiles between TB and PDCs [17]. Haddad et al also considered that TB may arise either from PDCs or from the main tumour [67]. On the contrary, acknowledging the morphological differences between TB and PDCs, Reggiani Bonetti et al proposed that TB and PDCs could represent sequential steps in tumour growth [68]. Additionally, the mitotic activity in PDCs is lower compared with the main tumour but higher compared with TB, which led to speculation that PDCs are an intermediate grade between the neoplastic glands and TB [62].
Previous studies including ours have reported activation of EMT at the invasive tumour front of CRC [27, 28, 58, 69, 70]. In this study, we demonstrated that the involvement of EMT along the invasive tumour front is irregular and, importantly, correlates with tumour morphology. In contrast to TB and PDCs, the expansive growth pattern is probably not associated with EMT as we found no significant differences in the expression of EMT markers between the expansive tumour front and the central part of the tumour. This is in accordance with Paterson et al, who showed uniform expression of miR‐200b from the tumour core to the tumour–host interface in CRCs with intact basement membranes [57]. These observations together emphasize the presence of intratumour heterogeneity at the invasive tumour front.
There are several advantages and limitations of our study. The main advantage is the use of laser‐capture microdissection, which enabled us to obtain tissue from specific locations in CRC, including small clusters and even single cells, constituents of TB, and PDCs. Using this technique, stromal and inflammatory cells surrounding TB and PDCs that could influence the results were not captured. Furthermore, the inclusion criteria were designed to enable comparison of the expression of EMT‐related markers between distinct locations and growth patterns within the same tumour. The comparison of miRNA expression between different tumours or patients would be inappropriate, since miRNAs might be tissue‐specific, and their expression age‐ and gender‐dependent [71]. Finally, the use of multiple EMT‐related markers enabled us to recognize and characterize different states of partial EMT. The main study limitation is the relatively small number of patients, which was the result of the stringent inclusion criteria. Additionally, patient selection was biased because only those with tumours displaying all the required morphological patterns were included. We also excluded patients with T1 carcinomas, because of the limited amount of cancer tissue. Finally, although areas with intratumour TB and PDCs were avoided when sampling the central part of the tumour, some degree of heterogeneity is a possibility.
In conclusion, our results suggest that both TB and PDCs are related to partial EMT. Discrete differences in morphology and EMT‐related marker expression between TB and PDCs indicate that they represent different manifestations of partial EMT. TB seems to be closer to complete EMT than PDCs (Figure 5).
Figure 5.
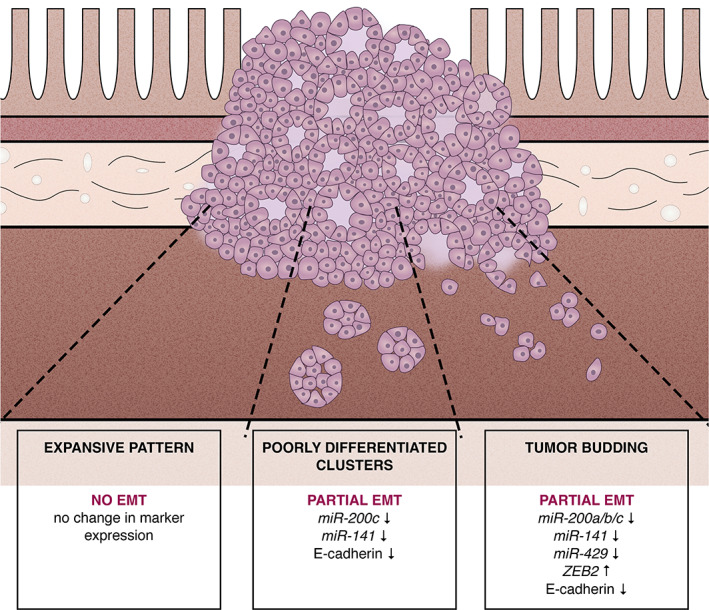
Involvement of the epithelial–mesenchymal transition in different tumour growth patterns in colorectal cancer.
Author contributions statement
AP, EB and NZ conceived the study. RK and BI provided tissue samples and performed data collection and literature search. AP, EB, SB and FZ carried out experiments, data analysis, and data interpretation. AP, EB and NZ wrote the manuscript. All the authors reviewed and approved the submitted and published versions.
Supporting information
Figure S1. Correlation between ΔCq values of the miR‐200 family and ΔCq value of CDH1
Figure S2. Correlation between ΔCq values of the miR‐200 family and ΔCq values of ZEB2
Table S1. Heat map of the relative ratio of the miR‐200 family in different parts of the tumour compared with the central part of the tumour in each of the analysed samples of colorectal carcinoma
Acknowledgements
We wish to thank Cristina Bottin for LCMD assistance and Eva Pavlič for the creation of Figure 5. This work was supported by the Slovenian Research Agency (ARRS) under research core funding numbers P3‐0054 and J3‐1754.
No conflicts of interest were declared.
Data availability statement
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
References
- 1. Karamitopoulou E, Zlobec I, Koelzer VH, et al. Tumour border configuration in colorectal cancer: proposal for an alternative scoring system based on the percentage of infiltrating margin. Histopathology 2015; 67: 464–473. [DOI] [PubMed] [Google Scholar]
- 2. Carr I, Levy M, Watson P. The invasive edge: invasion in colorectal cancer. Clin Exp Metastasis 1986; 4: 129–139. [DOI] [PubMed] [Google Scholar]
- 3. Gabbert H, Wagner R, Moll R, et al. Tumor dedifferentiation: an important step in tumor invasion. Clin Exp Metastasis 1985; 3: 257–279. [DOI] [PubMed] [Google Scholar]
- 4. Park KJ, Choi HJ, Roh MS, et al. Intensity of tumor budding and its prognostic implications in invasive colon carcinoma. Dis Colon Rectum 2005; 48: 1597–1602. [DOI] [PubMed] [Google Scholar]
- 5. Zlobec I, Lugli A. Invasive front of colorectal cancer: dynamic interface of pro‐/anti‐tumor factors. World J Gastroenterol 2009; 15: 5898–5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Morikawa T, Kuchiba A, Qian ZR, et al. Prognostic significance and molecular associations of tumor growth pattern in colorectal cancer. Ann Surg Oncol 2012; 19: 1944–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jass JR, Love SB, Northover JM. A new prognostic classification of rectal cancer. Lancet 1987; 1: 1303–1306. [DOI] [PubMed] [Google Scholar]
- 8. Bryne M. Is the invasive front of an oral carcinoma the most important area for prognostication? Oral Dis 1998; 4: 70–77. [DOI] [PubMed] [Google Scholar]
- 9. Halvorsen TB, Seim E. Association between invasiveness, inflammatory reaction, desmoplasia and survival in colorectal cancer. J Clin Pathol 1989; 42: 162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gabbert HE, Meier S, Gerharz CD, et al. Tumor‐cell dissociation at the invasion front: a new prognostic parameter in gastric cancer patients. Int J Cancer 1992; 50: 202–207. [DOI] [PubMed] [Google Scholar]
- 11. Ueno H, Kajiwara Y, Shimazaki H, et al. New criteria for histologic grading of colorectal cancer. Am J Surg Pathol 2012; 36: 193–201. [DOI] [PubMed] [Google Scholar]
- 12. Lugli A, Kirsch R, Ajioka Y, et al. Recommendations for reporting tumor budding in colorectal cancer based on the International Tumor Budding Consensus Conference (ITBCC) 2016. Mod Pathol 2017; 30: 1299–1311. [DOI] [PubMed] [Google Scholar]
- 13. Barresi V, Bonetti LR, Ieni A, et al. Histologic grading based on counting poorly differentiated clusters in preoperative biopsy predicts nodal involvement and pTNM stage in colorectal cancer patients. Hum Pathol 2014; 45: 268–275. [DOI] [PubMed] [Google Scholar]
- 14. Ueno H, Murphy J, Jass JR, et al. Tumour ‘budding’ as an index to estimate the potential of aggressiveness in rectal cancer. Histopathology 2002; 40: 127–132. [DOI] [PubMed] [Google Scholar]
- 15. Rogers AC, Winter DC, Heeney A, et al. Systematic review and meta‐analysis of the impact of tumour budding in colorectal cancer. Br J Cancer 2016; 115: 831–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barresi V, Reggiani Bonetti L, Ieni A, et al. Poorly differentiated clusters: clinical impact in colorectal cancer. Clin Colorectal Cancer 2017; 16: 9–15. [DOI] [PubMed] [Google Scholar]
- 17. Shivji S, Conner JR, Barresi V, et al. Poorly differentiated clusters in colorectal cancer: a current review and implications for future practice. Histopathology 2020; 77: 351–368. [DOI] [PubMed] [Google Scholar]
- 18. Konishi T, Shimada Y, Lee LH, et al. Poorly differentiated clusters predict colon cancer recurrence: an in‐depth comparative analysis of invasive‐front prognostic markers. Am J Surg Pathol 2018; 42: 705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bertoni L, Barresi V, Bonetti LR, et al. Poorly differentiated clusters (PDC) in colorectal cancer: does their localization in tumor matter? Ann Diagn Pathol 2019; 41: 106–111. [DOI] [PubMed] [Google Scholar]
- 20. Zlobec I, Dawson HE, Blank A, et al. Are tumour grade and tumour budding equivalent in colorectal cancer? A retrospective analysis of 771 patients. Eur J Cancer 2020; 130: 139–145. [DOI] [PubMed] [Google Scholar]
- 21. Jass JR, O'Brien J, Riddell RH, et al. Recommendations for the reporting of surgically resected specimens of colorectal carcinoma: Association of Directors of Anatomic and Surgical Pathology. Am J Clin Pathol 2008; 129: 13–23. [DOI] [PubMed] [Google Scholar]
- 22. Zlobec I, Lugli A. Tumour budding in colorectal cancer: molecular rationale for clinical translation. Nat Rev Cancer 2018; 18: 203–204. [DOI] [PubMed] [Google Scholar]
- 23. Martinez‐Ciarpaglini C, Fernandez‐Sellers C, Tarazona N, et al. Improving tumour budding evaluation in colon cancer by extending the assessment area in colectomy specimens. Histopathology 2019; 75: 517–525. [DOI] [PubMed] [Google Scholar]
- 24. Grigore AD, Jolly MK, Jia D, et al. Tumor budding: the name is EMT. Partial EMT. J Clin Med 2016; 5: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pastushenko I, Brisebarre A, Sifrim A, et al. Identification of the tumour transition states occurring during EMT. Nature 2018; 556: 463–468. [DOI] [PubMed] [Google Scholar]
- 26. Prieto‐García E, Díaz‐García CV, García‐Ruiz I, et al. Epithelial‐to‐mesenchymal transition in tumor progression. Med Oncol 2017; 34: 122. [DOI] [PubMed] [Google Scholar]
- 27. Pavlič A, Urh K, Štajer K, et al. Epithelial–mesenchymal transition in colorectal carcinoma: comparison between primary tumor, lymph node and liver metastases. Front Oncol 2021; 11: 662806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ranković B, Zidar N, Žlajpah M, et al. Epithelial–mesenchymal transition‐related microRNAs and their target genes in colorectal cancerogenesis. J Clin Med 2019; 8: 1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zidar N, Boštjančič E, Malgaj M, et al. The role of epithelial–mesenchymal transition in squamous cell carcinoma of the oral cavity. Virchows Arch 2018; 472: 237–245. [DOI] [PubMed] [Google Scholar]
- 30. Humphries B, Yang C. The microRNA‐200 family: small molecules with novel roles in cancer development, progression and therapy. Oncotarget 2015; 6: 6472–6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brierley J, Gospodarowicz MK, Wittekind C. TNM Classification of Malignant Tumours (8th edn). John Wiley & Sons, Inc: Chichester and Hoboken, 2017. [Google Scholar]
- 32. Espina V, Milia J, Wu G, et al. Laser capture microdissection. Methods Mol Biol 2006; 319: 213–229. [DOI] [PubMed] [Google Scholar]
- 33. Eriksen AH, Andersen RF, Pallisgaard N, et al. MicroRNA expression profiling to identify and validate reference genes for the relative quantification of microRNA in rectal cancer. PLoS One 2016; 11: e0150593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sørby LA, Andersen SN, Bukholm IR, et al. Evaluation of suitable reference genes for normalization of real‐time reverse transcription PCR analysis in colon cancer. J Exp Clin Cancer Res 2010; 29: 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kheirelseid EA, Chang KH, Newell J, et al. Identification of endogenous control genes for normalisation of real‐time quantitative PCR data in colorectal cancer. BMC Mol Biol 2010; 11: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lu X, van der Straaten T, Tiller M, et al. Evidence for qualified quantitative mRNA analysis in formalin‐fixed and paraffin‐embedded colorectal carcinoma cells and tissue. J Clin Lab Anal 2011; 25: 166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nardon E, Donada M, Bonin S, et al. Higher random oligo concentration improves reverse transcription yield of cDNA from bioptic tissues and quantitative RT‐PCR reliability. Exp Mol Pathol 2009; 87: 146–151. [DOI] [PubMed] [Google Scholar]
- 38. Teeuwssen M, Fodde R. Cell heterogeneity and phenotypic plasticity in metastasis formation: the case of colon cancer. Cancers (Basel) 2019; 11: 1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pastushenko I, Blanpain C. EMT transition states during tumor progression and metastasis. Trends Cell Biol 2019; 29: 212–226. [DOI] [PubMed] [Google Scholar]
- 40. Ammendola S, Turri G, Marconi I, et al. The presence of poorly differentiated clusters predicts survival in stage II colorectal cancer. Virchows Arch 2021; 478: 241–248. [DOI] [PubMed] [Google Scholar]
- 41. Morodomi T, Isomoto H, Shirouzu K, et al. An index for estimating the probability of lymph node metastasis in rectal cancers. Lymph node metastasis and the histopathology of actively invasive regions of cancer. Cancer 1989; 63: 539–543. [DOI] [PubMed] [Google Scholar]
- 42. Dawson H, Lugli A. Molecular and pathogenetic aspects of tumor budding in colorectal cancer. Front Med (Lausanne) 2015; 2: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Barresi V, Branca G, Ieni A, et al. Poorly differentiated clusters (PDCs) as a novel histological predictor of nodal metastases in pT1 colorectal cancer. Virchows Arch 2014; 464: 655–662. [DOI] [PubMed] [Google Scholar]
- 44. Gurzu S, Silveanu C, Fetyko A, et al. Systematic review of the old and new concepts in the epithelial–mesenchymal transition of colorectal cancer. World J Gastroenterol 2016; 22: 6764–6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mitrovic B, Schaeffer DF, Riddell RH, et al. Tumor budding in colorectal carcinoma: time to take notice. Mod Pathol 2012; 25: 1315–1325. [DOI] [PubMed] [Google Scholar]
- 46. O'Brien SJ, Carter JV, Burton JF, et al. The role of the miR‐200 family in epithelial–mesenchymal transition in colorectal cancer: a systematic review. Int J Cancer 2018; 142: 2501–2511. [DOI] [PubMed] [Google Scholar]
- 47. Vu T, Datta PK. Regulation of EMT in colorectal cancer: a culprit in metastasis. Cancers (Basel) 2017; 9: 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Perdigão‐Henriques R, Petrocca F, Altschuler G, et al. miR‐200 promotes the mesenchymal to epithelial transition by suppressing multiple members of the Zeb2 and Snail1 transcriptional repressor complexes. Oncogene 2016; 35: 158–172. [DOI] [PubMed] [Google Scholar]
- 49. Paysan L, Piquet L, Saltel F, et al. Rnd3 in cancer: a review of the evidence for tumor promoter or suppressor. Mol Cancer Res 2016; 14: 1033–1044. [DOI] [PubMed] [Google Scholar]
- 50. Luo H, Zou J, Dong Z, et al. Up‐regulated miR‐17 promotes cell proliferation, tumour growth and cell cycle progression by targeting the RND3 tumour suppressor gene in colorectal carcinoma. Biochem J 2012; 442: 311–321. [DOI] [PubMed] [Google Scholar]
- 51. Zhou J, Yang J, Li K, et al. RhoE is associated with relapse and prognosis of patients with colorectal cancer. Ann Surg Oncol 2013; 20: 175–182. [DOI] [PubMed] [Google Scholar]
- 52. Peng Q, Cheng M, Li T, et al. Integrated characterization and validation of the prognostic significance of microRNA‐200s in colorectal cancer. Cancer Cell Int 2020; 20: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xia W, Li J, Chen L, et al. MicroRNA‐200b regulates cyclin D1 expression and promotes S‐phase entry by targeting RND3 in HeLa cells. Mol Cell Biochem 2010; 344: 261–266. [DOI] [PubMed] [Google Scholar]
- 54. Martinez‐Ciarpaglini C, Oltra S, Roselló S, et al. Low miR200c expression in tumor budding of invasive front predicts worse survival in patients with localized colon cancer and is related to PD‐L1 overexpression. Mod Pathol 2019; 32: 306–313. [DOI] [PubMed] [Google Scholar]
- 55. Zlobec I, Lugli A. Epithelial mesenchymal transition and tumor budding in aggressive colorectal cancer: tumor budding as oncotarget. Oncotarget 2010; 1: 651–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. De Smedt L, Palmans S, Andel D, et al. Expression profiling of budding cells in colorectal cancer reveals an EMT‐like phenotype and molecular subtype switching. Br J Cancer 2017; 116: 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Paterson EL, Kazenwadel J, Bert AG, et al. Down‐regulation of the miRNA‐200 family at the invasive front of colorectal cancers with degraded basement membrane indicates EMT is involved in cancer progression. Neoplasia 2013; 15: 180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Knudsen KN, Lindebjerg J, Nielsen BS, et al. MicroRNA‐200b is downregulated in colon cancer budding cells. PLoS One 2017; 12: e0178564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lugli A, Zlobec I, Berger MD, et al. Tumour budding in solid cancers. Nat Rev Clin Oncol 2021; 18: 101–115. [DOI] [PubMed] [Google Scholar]
- 60. Yamada N, Sugai T, Eizuka M, et al. Tumor budding at the invasive front of colorectal cancer may not be associated with the epithelial–mesenchymal transition. Hum Pathol 2017; 60: 151–159. [DOI] [PubMed] [Google Scholar]
- 61. Bronsert P, Enderle‐Ammour K, Bader M, et al. Cancer cell invasion and EMT marker expression: a three‐dimensional study of the human cancer–host interface. J Pathol 2014; 234: 410–422. [DOI] [PubMed] [Google Scholar]
- 62. Hong M, Kim JW, Shin MK, et al. Poorly differentiated clusters in colorectal adenocarcinomas share biological similarities with micropapillary patterns as well as tumor buds. J Korean Med Sci 2017; 32: 1595–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Barresi V, Branca G, Vitarelli E, et al. Micropapillary pattern and poorly differentiated clusters represent the same biological phenomenon in colorectal cancer: a proposal for a change in terminology. Am J Clin Pathol 2014; 142: 375–383. [DOI] [PubMed] [Google Scholar]
- 64. De Smedt L, Palmans S, Sagaert X. Tumour budding in colorectal cancer: what do we know and what can we do? Virchows Arch 2016; 468: 397–408. [DOI] [PubMed] [Google Scholar]
- 65. Zlobec I, Berger MD, Lugli A. Tumour budding and its clinical implications in gastrointestinal cancers. Br J Cancer 2020; 123: 700–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Prall F, Ostwald C, Linnebacher M. Tubular invasion and the morphogenesis of tumor budding in colorectal carcinoma. Hum Pathol 2009; 40: 1510–1512. [DOI] [PubMed] [Google Scholar]
- 67. Haddad TS, Lugli A, Aherne S, et al. Improving tumor budding reporting in colorectal cancer: a Delphi consensus study. Virchows Arch 2021; 479: 459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Reggiani Bonetti L, Barresi V, Bettelli S, et al. Poorly differentiated clusters (PDC) in colorectal cancer: what is and ought to be known. Diagn Pathol 2016; 11: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hur K, Toiyama Y, Takahashi M, et al. MicroRNA‐200c modulates epithelial‐to‐mesenchymal transition (EMT) in human colorectal cancer metastasis. Gut 2013; 62: 1315–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Muto Y, Suzuki K, Kato T, et al. Heterogeneous expression of zinc‐finger E‐box‐binding homeobox 1 plays a pivotal role in metastasis via regulation of miR‐200c in epithelial–mesenchymal transition. Int J Oncol 2016; 49: 1057–1067. [DOI] [PubMed] [Google Scholar]
- 71. Sheinerman K, Tsivinsky V, Mathur A, et al. Age‐ and sex‐dependent changes in levels of circulating brain‐enriched microRNAs during normal aging. Aging (Albany NY) 2018; 10: 3017–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Correlation between ΔCq values of the miR‐200 family and ΔCq value of CDH1
Figure S2. Correlation between ΔCq values of the miR‐200 family and ΔCq values of ZEB2
Table S1. Heat map of the relative ratio of the miR‐200 family in different parts of the tumour compared with the central part of the tumour in each of the analysed samples of colorectal carcinoma
Data Availability Statement
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.