

Abstract
Objective
Duchenne muscular dystrophy (DMD) exon 45–55 deletion (del45–55) has been postulated as a model that could treat up to 60% of DMD patients, but the associated clinical variability and complications require clarification. We aimed to understand the phenotypes and potential modifying factors of this dystrophinopathy subset.
Methods
This cross‐sectional, multicenter cohort study applied clinical and functional evaluation. Next generation sequencing was employed to identify intronic breakpoints and their impact on the Dp140 promotor, intronic long noncoding RNA, and regulatory splicing sequences. DMD modifiers (SPP1, LTBP4, ACTN3) and concomitant mutations were also assessed. Haplotypes were built using DMD single nucleotide polymorphisms. Dystrophin expression was evaluated via immunostaining, Western blotting, reverse transcription polymerase chain reaction (PCR), and droplet digital PCR in 9 muscle biopsies.
Results
The series comprised 57 subjects (23 index) expressing Becker phenotype (28%), isolated cardiopathy (19%), and asymptomatic features (53%). Cognitive impairment occurred in 90% of children. Patients were classified according to 10 distinct index‐case breakpoints; 4 of them were recurrent due to founder events. A specific breakpoint (D5) was associated with severity, but no significant effect was appreciated due to the changes in intronic sequences. All biopsies showed dystrophin expression of >67% and traces of alternative del45–57 transcript that were not deemed pathogenically relevant. Only the LTBP4 haplotype appeared associated the presence of cardiopathy among the explored extragenic factors.
Interpretation
We confirmed that del45–55 segregates a high proportion of benign phenotypes, severe cases, and isolated cardiac and cognitive presentations. Although some influence of the intronic breakpoint position and the LTBP4 modifier may exist, the pathomechanisms responsible for the phenotypic variability remain largely unresolved. ANN NEUROL 2022;92:793–806
Mutations in the DMD gene that cause a severe deficiency of the dystrophin protein are responsible for Duchenne muscular dystrophy (DMD), which evolves with muscle degeneration and weakness starting in early childhood, loss of ambulation by the age of 13 years, and death around the patient's 30s. Partial dystrophin deficiency gives rise to Becker muscular dystrophy (BMD), defined by the preservation of ambulation beyond 16 years and a broader spectrum of the age of onset and severity. 1 Other reported dystrophin phenotypes include isolated cardiomyopathy and isolated hyperCKemia occasionally associated with pseudometabolic manifestations. Brain involvement expressed as neurodevelopmental, cognitive, or behavioral abnormalities can also be present. 2
The clinical severity caused by a given DMD gene mutation depends, to a large extent, on its impact on the open reading frame. 3 However, the location within the gene is also relevant for maintaining transcript stability and the conformational structure of the protein necessary for its adequate assembly and interaction with other proteins. 4 Additionally, genetic factors in trans, including variants of genes related to the inflammatory response, regeneration, and fibrosis (SPP1, LTBP4, CD40, and TSHBS1) and to muscle endurance (ACTN3), can act as modulators. 5
Among the diverse DMD mutations replicable for DMD therapy, the mega‐deletion of exons 45 to 55 (del45–55) may be able to correct up to 47 to 62% of total DMD cases recorded in the mutation databases. 6 , 7 , 8 This in‐frame deletion in the dystrophin central domain has been observed in asymptomatic subjects and patients with more severe muscular involvement or presenting with significant cardiopathy. 6 , 7 , 9 , 10 , 11 , 12 , 13 Currently, there is no explanation for this clinical heterogeneity. Therefore, although rescue of functional dystrophin at the cellular and organismal levels has been achieved by mimicking this mutation by means of multiexon skipping (MES) with cocktails of antisense oligonucleotides (AOs) 14 , 15 , 16 , 17 or applying genome‐editing technology, 18 , 19 it is necessary to deepen the underlying pathogenic mechanisms to make this model a feasible alternative to current advanced therapies. 20
The breakpoint (BP) position of del45–55 in the flanking introns has been invoked as a potential determinant of phenotypic variability. On the one hand, it is known that these introns contain regulatory elements, such as the promoter of dystrophin isoform Dp140, which plays a role in brain development and whose deficiency may entail a risk of cognitive impairment, 21 and also hosts several long noncoding RNAs (lncRNAs) that regulate DMD expression. 22 On the other hand, changes in the intronic architecture at the deletion junctions may alter splicing signals that could promote aberrant splicing events. 23 To address these issues in the pre–next generation sequencing (NGS) era represented a cyclopean task because of the large size of the introns involved combined with the rarity of this mutation.
In this paper, we aimed to evaluate the different phenotypes associated with del45–55 by conducting a multicenter study and analyzing the intronic BPs changes that could account for the clinical variability. Simultaneously we explored concurrent gene trans‐modifying factors, including the SPP1, LTBP4, and ACTN3 DMD genetic modifiers.
Patients and Methods
Patients
A search for patients harboring del45–55 in the DMD gene was carried out via a cooperative study across Spanish neuromuscular centers led by the Universitary and Polytechnic La Fe Hospital in Valencia and Hospital of Sant Pau in Barcelona. In addition, to inquire about the genetic and clinical department files, the patient registries held by the Carlos III Institute of the National Spanish Health Service (Center for Biomedical Network Research on Rare Diseases registry, CIBERER) and the one supported by the Duchenne Parent Project Spain association were also screened. Index patients were contacted by their respective neurologists or pediatric neurologists and were invited to participate in the study. Data from the hospital or primary care files were obtained from the attending physician after acquiring consent from the patient or the closest relative available, and the approval of the local health service supervisor was also obtained. The protocol of the study was approved by the ethics committee of Hospital UiP La Fe (2018/0200), and informed consent was obtained according to the Helsinki Declaration and signed by the patients and their respective parents or guardians.
Clinical Aspects
A protocol including relevant clinical features (demographic data, family history, development milestones, age at onset, age at symptom presentation, and age at significant disease milestones), full neurologic and functional assessment using a modified Vignos scale (Supplementary Table 1), and cardiologic and respiratory evaluations was applied to every patient by their respective specialist at their treatment center.
For cognitive evaluation in adults, the protocol included a checklist inquiring about neurodevelopmental milestones, school and social performance, and behavioral and psychiatric disturbance. When any consistent abnormality was reported, a full assessment was recommended using the Wechsler Adult Intelligence Scale. A formal cognitive evaluation was executed systematically by the same neuropsychologist on all pediatric patients. The study comprised a battery of validated tests employed in DMD evaluation 24 covering different cognitive functions as follows: general intelligence assessed with the Raven's Colored Progressive Matrices test, working memory evaluated using the Digit Span subtest of Wechsler Intelligence Scale for Children version IV, phonological processing and verbal fluency evaluated using the respective Nepsy II battery subtests, and executive functions and problem‐solving capacity using the Tower of London test. Emotional and behavioral problems were evaluated using the Child Behavior Checklist parent's questionnaire.
A structured questionnaire inquiring about family data, main clinical features, and age at presentation of significant milestones was administered via phone interview with patients or close relatives when the patient was not able to attend a face‐to‐face visit.
Subjects harboring the deletion were classified into one of the following phenotypes: (1) isolated hyperCKemia, including plainly asymptomatic or oligo‐/paucisymptomatic when associated with minor symptoms such as pseudometabolic symptoms (myalgia and cramps or rhabdomyolysis induced by exercise or strenuous factors); (2) BMD phenotype; and (3) isolated cardiopathy. Myopathic severity impairment was classified into 4 grades according to the modified Vignos scale as follows: I, asymptomatic (grade 0–1); II, mild (2–4); III, mild–severe (5, 6); IV, severe (≥7). Cardiac involvement was defined according to international guidelines, 25 based on the presence of left ventricular ejection fraction < 55% or fractional shortening < 25% or the existence of morphological abnormalities in the ventricular walls evaluated via echocardiography or clinical diagnosis by the attending cardiologist. Respiratory dysfunction was defined as forced vital capacity (FVC) < 60% or ventilator use to treat restrictive pulmonary insufficiency.
NGS Targeted Panel Design and Sequencing
A 2.683Mbp custom panel was designed using SureDesign software (Agilent Technologies, Santa Clara, CA). Probes targeted all the DMD exons and the complete 44‐ and 55‐intron sequences. It also contained the SPP1, LTBP4, and ACTN3 DMD modifiers and genes involved in myopathy. 26 DNA libraries were prepared according to the SureSelectQXT Target Enrichment for Illumina Multiplexed Sequencing protocol and sequenced on a Miseq platform (Illumina, San Diego, CA) using the V3‐150 cycles cartridges. Files were obtained using the MiSeq Reporter 2.6.2.3 and Illumina RTA 1.18.54. Reads were mapped to the human reference genome (GRCh37/hg19). Data analysis was performed using Alissa and SureCall (Agilent Technologies).
BPs and lncRNA Identification
Primer pairs were designed to amplify 500bp of the deletion junctions according to each BP position inferred from NGS alignment in introns 44 and 55 and to detect the lncRNAs nclINT44s, nclINT44s2, nclINT55s, and nclINT55a. 22 Polymerase chain reaction (PCR) was performed using the PCR Master MIX (Thermo Fisher Scientific, Waltham, MA), and the products were resolved on 1% agarose gel. BP junction amplicons were purified using ExoSAP‐IT (Applied Biosystems, Waltham, MA) and analyzed via Sanger sequencing.
Dystrophin Expression Analysis
Available diagnostic muscle biopsies stored in liquid nitrogen were used for dystrophin expression analysis, RNA isolation, and reverse transcription PCR (RT‐PCR). RNA was extracted from muscle cryosections (RNeasy mini kit; QIAGEN, Hilden, Germany). RT was performed using 750ng of total RNA with SuperScript IV Reverse Transcriptase (Invitrogen, Waltham, MA), and nested PCR of cDNA samples was carried out using specific primer pairs (hybridizing in DMD exons 41, 43, 44–58, 59, and 60) as previously described. 27 PCR products were analyzed on 1% agarose gels, and DNA was purified (Gel Extraction Kit; Omega Bio‐Tek, Norcross, GA) and validated via Sanger sequencing.
Duplex droplet digital PCR (ddPCR) was performed with 4μl of cDNA, using the QX200 Droplet Digital PCR system (Bio‐Rad Laboratories, Hercules, CA). Two specific ddPCR custom assays were used in each reaction, with probes designed to hybridize the DMD 44–56 exon junction (FAM‐labeled) and the 44–58 junction (HEX‐labeled). Data were analyzed with QuantaSoft Analysis Pro Software (Bio‐Rad Laboratories). The concentration (copies/μl) of both transcripts was used to determine the percentage of the del45–57 transcript in each sample as follows: [(copies/ul 44–58)/(copies/ul 44–58 + copies/ul 44–56)]*100 . 28
For muscle immunostaining, 7μm cryosections were coincubated with the primary monoclonal antibodies antidystrophin (NCL‐Dys2; Novocastra Laboratories, Newcastle Upon Tyne, UK; 1:50), and rat monoclonal anti–laminin‐2 (Sigma‐Aldrich, Burlington, MA; 1:500). The appropriate secondary antibodies were used (Alexa Fluor 594 goat antimouse and Alexa Fluor 488 goat antirat; 1:200, Invitrogen). Sixteen‐bit images were acquired using an LSM800 confocal microscope (Zeiss, Oberkochen, Germany) at ×200 magnification.
Dystrophin detection via Western blotting from the skeletal muscle biopsies was performed as previously described, 29 using the following primary antibodies: antidystrophin (NCL‐Dys1, 1:40, Novocastra) and anti–α‐actinin (A7732, 1:3,000, Sigma‐Aldrich). Horseradish peroxidase–conjugated antimouse IgG (ab6808, 1:2,000, Abcam, Cambridge, UK) was used as the secondary antibody. Membranes were visualized using chemiluminescence (ECL PLUS, Thermo Fisher Scientific) and Amershand Imager 600 (GE Healthcare, Chicago, IL). Dystrophin intensity was normalized to α‐actinin using the ImageJ software.
Deletion Junction Characterization
In the intronic sequences near BPs, repetitive elements were searched using the RepeatMasker program (http://repeatmasker.org/cgi-bin/WEBRepeatMasker) and inverted repeats were obtained using the Palindrome program (http://emboss.bioinformatics.nl/cgi-bin/emboss/palindrome). The Human Splicing Finder V3 tool was used to analyze the splicing regulatory elements (SREs) and the potential pseudoexon activation at the deletion junctions, as previously described. 30 For these analyses, we searched for SREs and splice sites in patient sequences, including rare variants, rather than reference ones.
Founder Effect Haplotype Analysis
Haplotype analysis was performed for the groups of index patients who shared the same intronic BPs. Variants were called using the GATK HaplotypeCaller tool 31 following best practices from the Broad Institute. Single nucleotide polymorphism (SNP) genotypes were extracted from all positions in the DMD‐sequenced region. Variants were annotated using Genome Aggregation Database allele frequency (European non‐Finnish population). Only variants with allele frequencies of ≥0.3 and <0.5 were kept for further analysis. For haplotype construction of each group, the SNP (v) and reference (r) forms of each subject were considered. A plot representing only the predominant form (r/v) in each deletion group was plotted.
DMD Modifiers
Within the known DMD genetic modifiers, we selected to explore SPP1, LTBP4, and ACTN3 for relevance and logistic reasons. For that purpose, the NGS gene panel was used in the index cases and ad hoc Sanger sequencing in the secondary cases. The criteria applied were as follow 5 : the presence of G risky allele in SPP1 (dominant model), the LTBP4 IAAM/IAAM beneficial haplotype, and ACTN3; in Model 1, we included the TT and TC as harmful alleles, whereas in Model 2, the heterozygous allele was taken as an intermediate. Presence of concurrent mutations in the other genes within the working panel were also analyzed (Supplementary Table 2).
Statistical Analysis
Nonparametric tests were used to compare continuous variables within the different groups (p < 0.005). A Bayesian ordinal regression model was applied to assess the effect of deletion location, lncRNA preservation profile groups, and DMD‐modifying factors on the severity of skeletal muscular involvement. Bayesian logistic regression was used to evaluate the effect of dichotomous variables (cardiac involvement). Cognitive involvement was only exploratory due to the difference in diagnostic methodology between adult and pediatric populations. The criteria applied are listed in Table1. All statistical analyses were performed using R (v3.5.3), R package brms (v2.8.0), and clickR (v0.4.32).
TABLE 1.
Bayesian Regression Analyses Evaluating the Effect of Deletion Location, lncRNA Pattern, and Modifying Factors on Muscle Severity Impairment Grade and Cardiac Involvement
| Muscle Severity Impairment a | Cardiopathy b | |
|---|---|---|
| Deletion, n = 55 | ||
| D1, n = 25 | Ref. | Ref. |
| D2, n = 5 | 0.525 (0.067–3.817) | 0.396 (0.028–4.161) |
| D3, n = 6 | 2.098 (0.257–15.275) | 2.915 (0.39–22.022) |
| D4(Dp140−), n = 6 | 1.957 (0.02–215.243) | 0.876 (0.009–82.252) |
| D5, n = 3 | 10.824 (1.314–98.235) c | 44.302 (2.075–2,530.511) c |
| D7, n = 4 | 0.196 (0.002–14.81) | 4.656 (0.121–190.056) |
| D9, n = 3 | 0.33 (0.023–4.046) | 0.047 (0.001–1.358) |
| D10, n = 3 | 0.09 (0.001–3.271) | 0.085 (0.001–3.62) |
| lncRNA, n = 55 | ||
| PPPP, n = 4 | Ref. | Ref. |
| PPNN, n = 45 | 9.835 (0.217–593.61) | 2.112 (0.062–79.874) |
| NNPP, n = 6 | 2.033 (0.024–206.376) | 0.807 (0.009–68.4) |
| Modifiers, n = 33 | ||
| SPP1 | 1.125 (0.135–8.59) | 1.213 (0.132–10.35) |
| LTBP4 | 0.576 (0.065–6.127) | 40.829 (1.996–1,707.37) c |
| ACTN3 Model 1 | 0.639 (0.109–3.733) | 0.486 (0.065–3.327) |
| ACTN3 Model 2 | 0.519 (0.151–1.53) | 0.733 (0.223–2.177) |
| Age | 1.086 (1.029–1.155) c | 1.072 (1.013–1.145) c |
Deletion 1 was used as a reference, in addition to the group containing the 4 lncRNAs. Age was considered as a cofounding variable for all models. Deletions and lncRNA pattern groups containing only one subject were excluded from the analysis (D6 and D8). Evidence for an effect was considered relevant when the 95% CI (confidence interval) did not include 1. The odds ratios and CIs are indicated for each comparison. In the lncRNA pattern, N = absence and P = presence.
Bayesian ordinal regression model.
Bayesian logistic regression model.
Statistically significant.
Results
Patient Clinical Features
Full clinical evaluation was carried out on 39 subjects, including 23 index and 16 secondary cases. Reliable data from 18 additional subjects, either deceased (n = 13) or unable to attend a face‐to‐face visit (n = 5), were obtained via telephone interviews and from clinical records. Supplementary Table 1 summarizes the clinical data of the 57 patients in this series and specifies the assigned deletion group according to the respective index case. The analysis of the 23 pedigrees demonstrated X‐linked transmission in all but 5 of the kindred.
Patients were divided into the following categories: isolated hyperCKemia (53% of cases), further subdivided into asymptomatic (n = 23) and paucisymptomatic (n = 7). The BMD phenotype accounted for 28% of cases (n = 16); isolated cardiopathy occurred in 19% of cases (n = 11). Cardiopathy was also present in 7 patients with the BMD phenotype. Cognitive and/or behavioral disturbances were detected in 10 subjects, 9 of pediatric age, and all manifested hyperCKemia. The adult patient also presented cardiopathy.
Figure 1 represents the age of patients at evaluation. The patients in the isolated hyperCKemia group were significantly younger than those in the BMD and isolated cardiopathy groups. The median age at onset in index patients was 12 years (range = 2–40 years). Muscle impairment in patients with BMD always occurred after the age of 20 years, but on most occasions (10/16) after the age of 40 years (late onset BMD). Approximately 44% (7/16) of patients with BMD lost ambulation, usually after 50 years of age.
FIGURE 1.
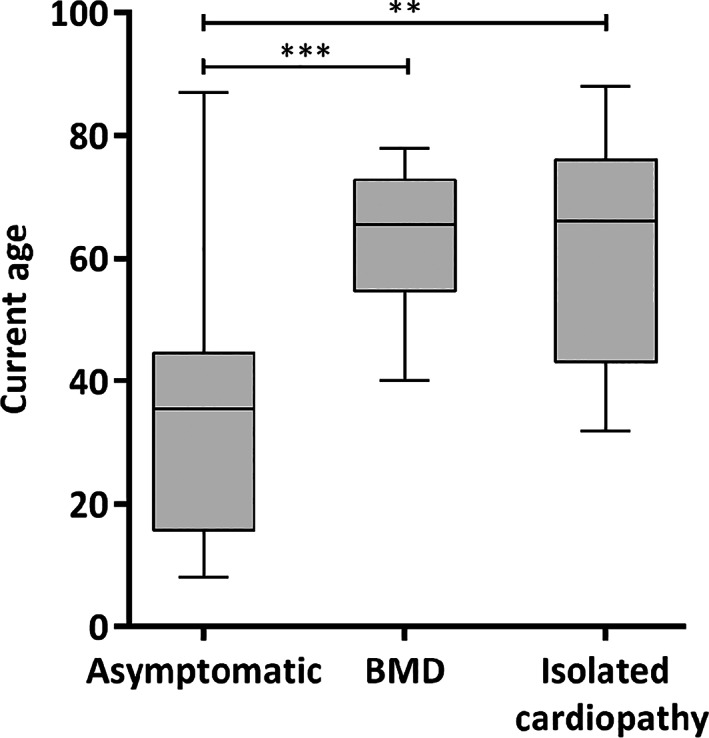
Graphical representation of age at evaluation of del45–55 cohort. Boxplot shows the median and range of assessment age of the 3 diagnostic categories. There is a significant difference between the asymptomatic/paucisymptomatic group (median = 35.5, range = 8–87) and the Becker muscular dystrophy (BMD; median = 65.5, range = 40–78) and isolated cardiopathy (median = 66, range = 32–88) groups (** p < 0.01, *** p < 0.001 according to Kruskal‐Wallis test).
Cardiopathy debut was always after the age of 20 years, with an increasing age‐related incidence rate (Fig 2A). Details of the available cardiological data are listed in Supplementary Table 3. Four cases presented atrioventricular block requiring emergency pacemaker implantation, one of them at age 29 years, as an inaugural symptom. Two others received an implantable cardioverter–defibrillator. In many instances, patients remained stable with medical therapy, but the cardiac transplant was indicated in 3 cases; one survived 9 years after the intervention, a second one died while on the waiting list, and the third one is still expecting the intervention.
FIGURE 2.
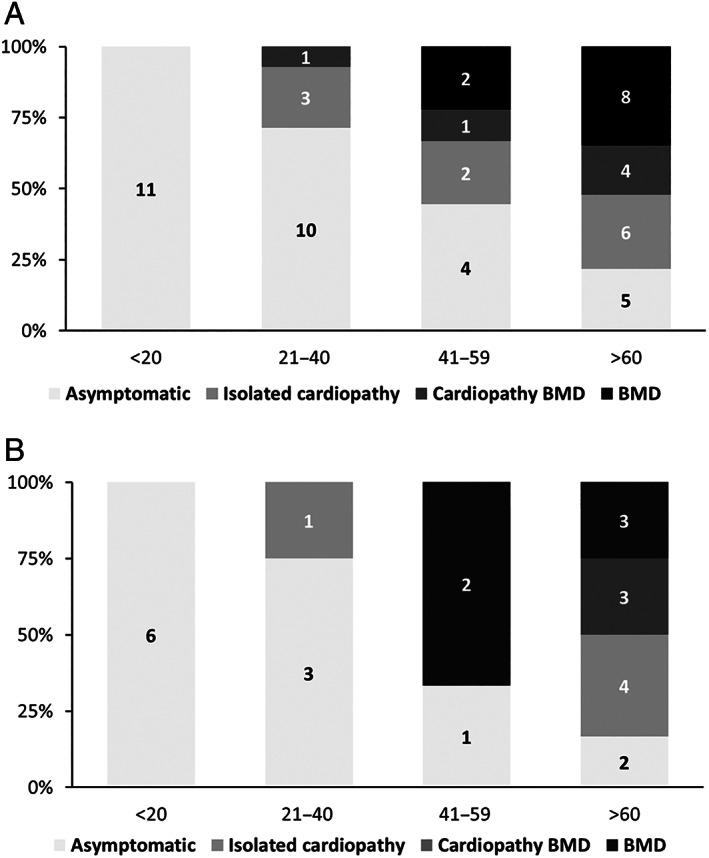
Prevalence of clinical manifestations across age of del45–55 series. (A) Global series (n = 57) prevalence of symptoms at 4 age periods. (B) Same representation in the Deletion 1 group (n = 25).
None of the patients required respiratory attention, and routine evaluation tests always showed an FVC of >50%.
The prevalence of symptoms across all ages of the whole series is shown in Figure 2A. Whereas the proportion of asymptomatic patients dropped from 100% at age 20 years to 22% after the age of 60 years, that of patients with symptomatic BMD rose to 52% in their 60s. The prevalence of cardiopathy reached 43.5% at the age of 60 years (26% of them isolated). The proportion of symptoms in the largest BP subgroup (D1) followed a similar trend to the whole series (see Fig 2B).
Supplementary Table 4 collects the results of the cognitive assessment. Nine of the 10 pediatric patients evaluated presented abnormalities, including neurodevelopmental features (80%), reduced intelligence quotient (IQ) score (40%), speech abnormalities at phonological (88%) or verbal processing (50%), executive function (55%), and emotional–behavioral features (80% internalization and 70% externalization). The adult patient profile showed a similar profile.
When the data were collected, 14 patients had died; cardiopathy was imputed as the causative factor in 6 subjects, 1 case presented as sudden death, and other medical or nonspecific causes were reported in the remaining 6 cases. The median age at death was 70 years (range = 52–88 years). Life expectancy in the Spanish male population has ranged from 76 to 79 over the past 20 years.
Intronic BP Analysis
Ten different deletions with specific intronic BPs were recognized in the 23 index cases (Fig 3). Four of them were shared by several index cases (deletion groups D1–D4), whereas the other 6 (D5–D10) appeared in a single case. Within the shared deletion, D1 was present in 9 index cases, D2 and D3 in 3 cases each, and D4 in 2 others.
FIGURE 3.
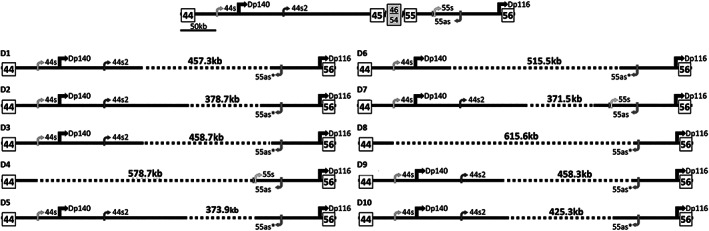
Schematic representation of the 10 specific deletion breakpoints of the del45–55 cohort. In the upper part, there is a schematic representation of the normal genomic structure spanning the del45–55 region. The promoter and first exons of the Dp140 and Dp116 isoforms are shown. The positions of the long noncoding RNAs (lncRNAs) 44s, 44s2, 55s, and 55as and their transcription directions are also represented. Each specific deletion is represented according to its intronic breakpoint position and labeled as D1 to D10. All deletions, except D4 and D7, disrupted the sequence of the lncRNA 55as containing the polyadenylation site, as indicated by asterisks. Dotted lines indicate the deleted segment.
The genomic location of each BP and the deletion length of all 10 specific groups are presented in Supplementary Table 5. D4 and D8 disrupted the promoter region of the dystrophin isoform Dp140 located in intron 44 (see Fig 3). In addition, Figure 3 and Supplementary Table 2 show the effects of the different deletions across intronic lncRNAs sequences. Only D7 preserved all lncRNA sequences, whereas most deletions partially disrupted these lncRNAs, leading to different preservation profiles. D8 was the only one that deleted all the lncRNA sequences.
Deletion junction characterization revealed the presence of sequences capable of inducing DNA double‐strand break (DSB) and other features, such as insertions and microhomologies. Repetitive elements were found near the BPs, with absence of extensive homology. Details are given in Supplementary Table 5. 32 , 33 The analysis of pseudoexon activation depicted the potential formation of cryptic exons in all the deletion junctions (Supplementary Table 6), whereas the analysis of the SREs showed that the deletions did not modify the exonic splicing enhancer/silencer ratio, which was close to 1 in all 10 (Supplementary Table 7).
Dystrophin Expression
Dystrophin immunostaining in 9 available muscle biopsies (from P1; P2; P4,2; P7; P8,1; P9; P17; P18; P22,1) showed a correct dystrophin sarcolemma expression, with a slight intensity reduction in some cases (Fig 4A). Quantification of dystrophin expression via Western blotting showed high levels in all muscle samples, ranging from 67 to 106% of a healthy control sample (see Fig 4B).
FIGURE 4.

Dystrophin protein analysis of the 9 del45–55 muscle biopsies; healthy (C) and Duchenne muscular dystrophy (DMD) samples. (A) Representative images of dystrophin immunostaining in transverse sections revealing a proper sarcolemma protein location with a slight or mild staining reduction in some patients (DYS2, upper image). Laminin immunostaining (lower image) was used to localize muscle sarcolemma. Scale bar = 50μm. (B) Dystrophin quantification by Western blotting of total protein extract. Note the del45–55 dystrophin with a lower molecular weight than control, and absent protein in the DMD sample. Dystrophin levels were normalized to α‐actinin signal and are represented as percent of healthy control. Data represent the mean of 3 technical replicates. Bars of the same shade indicate subjects from the same deletion group (D1 group = P1; P2; P4,2; P7; P8,1; P9. D4 group = P17. D5 group = P18. D9 group = P22,1).
The RT‐PCR analysis did not confirm the presence of pseudoexons (Supplementary Table 6). However, the sample from P18 (D5 group presenting a severe phenotype) showed a significant number of the replicates with an additional skipping of exons 56 and 57, which led to transcripts with an in‐frame deletion of exons 45 to 57 (del45–57; Fig 5A). A subsequent specific amplification of this transcript (using a primer targeting the 44–58 exon junction) revealed its presence in all the samples. The quantification of the del45–57 transcript using duplex ddPCR showed a residual expression, accounting for <0.1% in all cases (see Fig 5B, C).
FIGURE 5.
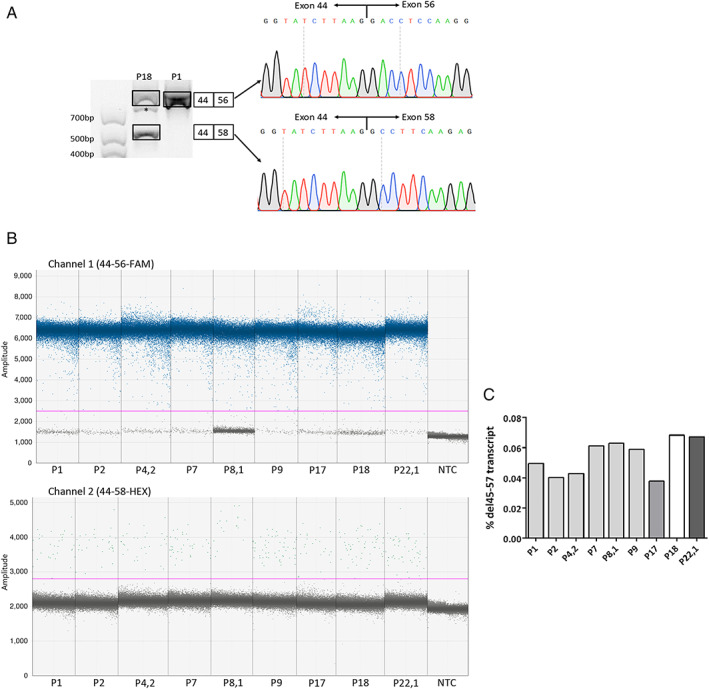
Dystrophin expression analysis of 9 muscle cDNA samples. (A) Presence of an alternative transcript in P18 (D5) muscle cDNA. Nested reverse transcription polymerase chain reaction products from P18 (D5) and P1 (D1) showed an upper band that corresponds to the expected del45–55 transcript. In addition, P18 showed a lower band, revealing an additional skipping of exons 56 and 57. The intermediate band in P18, marked with an asterisk, was considered a heteroduplex formation. The identities of all bands were confirmed by Sanger sequencing. (B) One‐dimensional fluorescence amplitude plots of the 2 ddPCR assays, quantifying the levels of the del45–55 transcript (upper part, FAM‐labeled probe targeting the exon 44–56 junction) and the del45–57 transcript (bottom part, HEX‐labeled probe targeting the exon 44–58 junction) in the nine samples. The horizontal lines indicate the threshold level established to discriminate the positive from the negative droplets (above and below, respectively). Three technical replicates per sample were run, and a no template control (NTC) was included as negative control. FAM= 6‐carboxyfluorescein fluorophore; HEX= 6‐carboxy‐2',4,4',5',7,7'‐hexaclorofluorescein fluorophore. (C) Low percentage of the del45–57 transcript in all samples, determined as [(copies/ul 44–58)/(copies/ul 44–58 + copies/ul 44–56)]*100. Percentages are shown as mean (n = 3). Bars of the same shade indicate subjects from the same deletion group (D1 group = P1; P2; P4,2; P7; P8,1; P9. D4 group = P17. D5 group = P18. D9 group = P22,1). [Color figure can be viewed at www.annalsofneurology.org]
Haplotype Analysis
Given that most of the subjects sharing the same deletions (D1–D4) were residing in the same geographical region (Supplementary Table 1), we performed a haplotype analysis to search for founder effects. All individuals within their respective D2, D3, and D4 groups harbored the same haplotype (Fig 6A). However, the D1 prototype contained 2 different subgroups of haplotypes, indicating the presence of 2 different founder events in the same specific deletion (see Fig 6B). Globally, we identified 5 distinct founder mutations.
FIGURE 6.
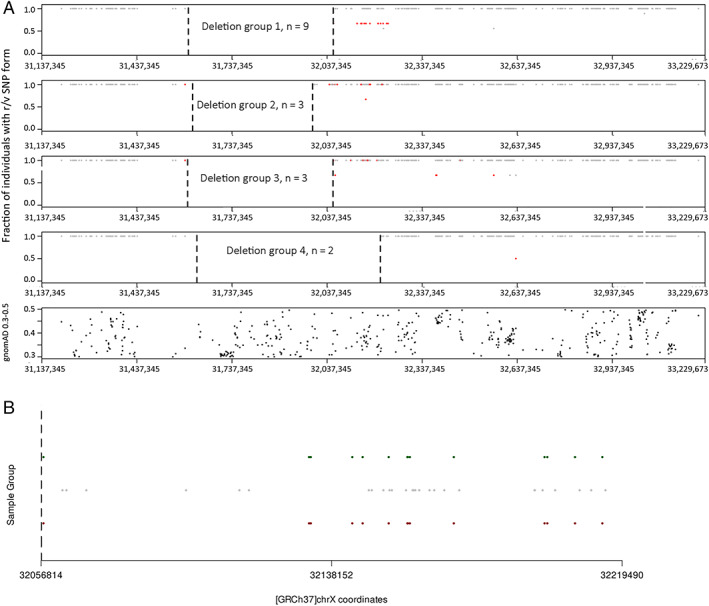
Haplotype analysis of subjects with the shared deletions D1 to D4. (A) Plot representing a selection of single nucleotide polymorphism (SNP) forms within the DMD sequenced region from subjects of the deletion groups D1 to D4. For each SNP, both reference (r) and variant (v) sequences are considered, and only the predominant form is plotted, as gray (r) or red dots (v) according to its proportion within the subjects of each group (the position in the y‐axis represents the proportion). Note that for all subjects from each D2 to D4 group, mostly a common SNP profile is depicted, except subjects from the D1 group showing 2 profiles. The position of the deletion breakpoints is represented as vertical dashed bars. The plot at the bottom represents SNPs reaching the selection criteria (allele frequency ≥ 0.3 < 0.5) along the entire DMD gene. (B) Magnification of the region of interest in the D1 group (chrX: 32,056,814–32,219,490), highlighting the 2 SNP profiles. The SNPs (r or v) common in the 9 subjects are represented in gray, whereas the subgroups of 6 and 3 subjects with distinct haplotypes are represented with green and red dots, respectively. The position of the dots along the y‐axis is arbitrary. [Color figure can be viewed at www.annalsofneurology.org]
BP Effect on Clinical Severity
Table1 shows the result of the analysis of the diverse variables on clinical severity. Only a significant effect of the D5 BP appeared on muscle severity impairment (odds ratio [OR] = 10.824, 95% confidence interval [CI] = 1.314–98.235) and the presence of cardiopathy (OR = 44.302, 95% CI = 2.075–2,530.511).
Effect of the Intronic Sequences
As represented in Table1, no significant effect was observed from the lncRNA profiles (presence, absence, or partial disruption) and the Dp140 disruption (deletions D4 and D8) over the severity of skeletal muscle involvement and development of cardiopathy. The connection between cognitive and behavioral impairment with Dp140 is depicted in Supplementary Table 4, where 8 of 10 cases showing cognitive abnormalities held a preserved promotor (Dp140+), whereas in the 2 others affected the promotor was absent (Dp140−).
Trans‐Modifying Factors
No statistically significant effect of the risky SNPs in SPP1, LTBP4, or ACTN3 was found on the clinical severity of myopathy in the assessed patients (n = 33). However, the absence of the LTBP4 IAAM/IAAM haplotype was associated with the presence of cardiopathy (OR = 40.829, 95% CI = 1.996–1,707.37; Table1). On the other hand, concurrent heterozygous mutations were present in 4 subjects (Supplementary Table 2), but no relationship with the symptoms was observed (ie, metabolic gene/pseudometabolic features).
Discussion
In this study, we analyzed a large cohort of patients with dystrophinopathy, harboring the deletion of DMD exons 45 to 55, wherein data were collected across different neuromuscular centers and evaluated using a systematic clinical protocol. This mutation is highly infrequent, representing <2% of the cases recorded in DMD registries. 7 To the best of our knowledge, no previous studies have provided a series of significant sample sizes enabling a rigorous clinical and genetic analysis. Data available on this condition are based on the reporting of small case series or anecdotic descriptions from which Echigoya et al 17 collected a total of 48 cases. As shown in Figure 7, our data differ to some extent from Echigoya's review in the proportion of phenotype incidence, particularly a higher rate of cardiopathy and a considerable proportion of cognitive impairment.
FIGURE 7.
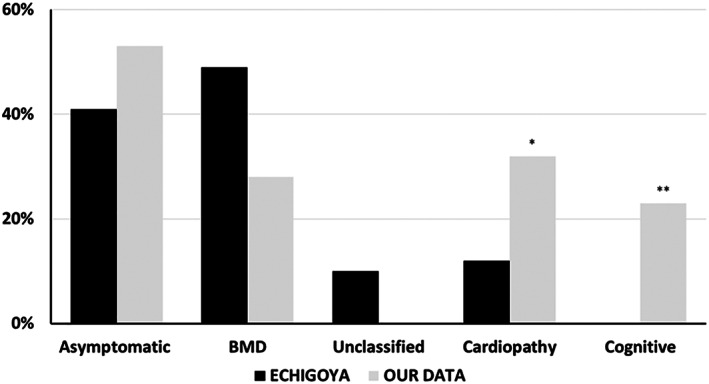
Comparison of the del45–55 clinical profile from the Echigoya et al 17 review series and our cohort. Note a higher proportion of asymptomatic/paucisymptomatic and cardiomyopathy cases (*19% isolated and 13% associated with Becker muscular dystrophy [BMD]) in our series. Cognitive impairment was always associated with hyperCKemia,** which was absent in Echigoya's work. The unclassified group of Echigoya's series correspond to 5 subjects with limited clinical information.
There are several clinical aspects to be highlighted in our series, which allowed us to draw up a natural history profile of the condition. As a rule, hyperCKemia and pseudometabolic features may be present since early childhood and can continue to be the only manifestations in a sizable number of cases even after the age of 60 years. Second, although the incidence of cardiopathy falls in the average of BMD reported series, 34 , 35 this mutation tends to present a high propensity to early cardiac impairment and isolated cardiopathy appearance. Third, this condition does not appear to be as benign as supposed, because up to 40% of patients with BMD suffered a loss of ambulation or reached a high degree of motor disability, whereas a significant number of cases evolved with severe cardiopathy demanding cardiac transplant or suffered from unexpected serious acute heart events and sudden death, indicative of threatening cardiac arrhythmias. Furthermore, data from affected deaths indicate a shorter life expectancy. Finally, learning and cognitive features are other remarkable issues scarcely evaluated in this deletion. Signs of brain impairment were recorded in 23% of the total series; however, the proportion rose to 90% in the pediatric population, possibly because this group was subjected to a more rigorous cognitive evaluation. This figure is the highest rate of those reported in the DMD and BMD series, 24 , 36 and it is worth mentioning that it has rarely been reported as an isolated manifestation. 2
The cognitive profile depicted by most of the evaluated del45–55 patients was on par with the signature reported in DMD studies. 37 The high rate of neurodevelopmental and behavioral features observed was also in consonance with the reported BMD series. 36 , 38 However, these data are discrepant with a study observing a distinct BMD profile with preservation of full scale IQ and higher impairment of performance scale over verbal scale. Differences in the battery of tests cannot explain the discrepancy, but differences in patient samples might; a proportion of patients reported in Young et al study were not genetically characterized.38
So far, several studies of BMD patient series have analyzed the clinical severity of different in‐frame deletions in the dystrophin rod domain spanning exons 44 to 55. Deletions presenting the most favorable phenotypes, such as del45–55 and those that are exon‐51 skippable, exhibit a dystrophin hybrid repeat conformation, preserving the filamentous protein structure. As opposed to this, there are other deletions (eg, del45–47 and del 45–49) causing fractional repeats associated with greater severity. 4 , 6 , 34 , 35 , 39 Moreover, studies of internal variability and potential modifying factors in a single BMD mutation have scarcely been addressed, such as the analysis of the impact of neuronal nitric oxide synthase mislocalization in del45–55 muscle 12 and the dystrophin amount in del45–47. 40 Regarding this issue, although the number of samples in our study was limited, they were representative of distinct groups and grades of severity and all cases showed high dystrophin expression, not accounting for the variable phenotypic severity.
A central objective of our study was to characterize intronic BPs and establish their implications in the phenotypic variability. For this purpose, we devised an NGS‐based targeted design complemented with Sanger sequencing for BP identification, which proved to be accurate and highly resolute compared to the classical approaches. 32 , 41 Third‐generation long‐read sequencing has also enabled the accurate identification of intronic BPs along DMD intron 44. 33 The analysis of the deletion junctions revealed the presence of diverse repetitive elements in the vicinity of the deletion BPs that may contribute to the genomic instability, but no extensive homology was found, eliminating the homologous recombination as the underlying mechanism. Nevertheless, we found a variety of elements associated with DSB formation in all the deletion junctions, indicating that del45–55 does not arise from a single mechanism but from the involvement of a variety of factors that induce DNA instability and DSB formation, as well as the involvement of different genomic repair pathways (Supplementary Table 5). 32 , 33
In keeping with another study that analyzed a del45–55 series, we did not find hotspots predisposing to BP recurrence. 42
The haplotype study in our series revealed that all index case groups harboring the same BP originated from distinct founder mutations, except for the D1 group, which turned out to be the result of 2 independent founder mutation events. The high rate of founder effects observed in our series contrasts with their rare occurrence in the BMD disease population, as only 2 similar founder effects occur have been reported. 43 , 44 In any case, this phenomenon is indicative of the preservation of fertility. On the other hand, this observation provided us with the chance to dispose of groups with sufficient size to enable the comparison of the effect of BP locations on the outcomes.
In our analysis, we found a significant influence of BP only in the D5 group with severe manifestations. In the first instance, the RNA analysis of a subject from this group revealed an alternative skipping of exons 56 and 57, which might be induced by the changes in the splicing sequences of this specific BP. However, later, the skipped transcript was found in all biopsies, raising concerns about its pathogenicity. Alternative exon skipping events have been detected in different pathological conditions 45 and are infrequent in healthy subjects. 46 Furthermore, its residual expression in all the samples suggests that is presumably a subproduct derived from multiexon‐skipping events triggered by post‐transcriptional introns. 47 Otherwise, no evidence of pseudoexon activation was found in any biopsies despite the positive in silico predictions.
We also performed an approximation of the disruption of the lncRNAs based on the BP location, and no clinical impact was observed regarding the different preservation patterns. Gargaun et al 42 reported the specific PNNN profile (it preserves only the lncRNA44s) as protective, but we could not confirm this, because it was present in only one of our subjects (P19, D6). Nevertheless, these speculations should be rigorously addressed at the RNA level to evaluate the role of lncRNAs in dystrophin transcription. 22 Similarly, we evaluated the effect of the BP location on the Dp140 promotor on the cognitive status in the properly evaluated patients, not observing a straightforward relationship with its preservation. However, this interpretation may be controversial, because the Dp140 translation should be altered by the deletion of the translation start site at exon 51. However, Bardoni et al 48 proposed the existence of alternative translation sites located between exon 52 and exon 56, although this suggestion has not been experimentally substantiated.
The validated DMD genetic modifiers, including the 3 investigated in this study, have not yet been assessed in BMD. Our exploratory analysis is limited by the small sample size (n = 33) that underpowers the statistical analysis. Nevertheless, we found an effect between the LTBP4 and the presence of cardiopathy, which is an interesting observation that needed to be confirmed. Similarly, although heterozygous concomitant mutations were observed in several subjects that might have acted as double trouble, 49 we did not find any aggravating or confusing manifestation over the phenotype in those cases.
Overall, our study confirms del45–55 as a model capable of segregating a high proportion of asymptomatic and benign cases responsible for founder effects. However, it is also causative of significant functional impairment, severe cardiac complications, cognitive alterations, and potential shortening of life expectancy, the mechanopathogenesis of which remains largely unrevealed by our extensive search for modifying factors. Solving these issues would consolidate this mutation as one of the best potential models for DMD gene therapy, comparable to monoexon skipping therapies, with the advantage of being applicable to a broader spectrum of mutations. It also preserves dystrophin functional domains, which is an issue of the minidystrophin gene transfer approach. 20 To achieve this goal, it is essential to overcome the hurdles derived from the usage of MES AO‐cocktails or to the development of gene‐editing therapies.
Author Contributions
J.P.‐G., P.M., and J.J.V. contributed to conception and design of the study. All authors contributed to acquisition and analysis of data. J.P‐G., P.M., A.L., B.R., and J.J.V. contributed to drafting a significant portion of the manuscript or figures.
Potential Conflicts of Interest
J.P.‐G. has received a PhD grant from Fundación Isabel Gemio. J.J.V. has received grants from Fundación Isabel Gemio and ISCIII and has been a consultant to BioMarin, Genzyme Therapeutics, and PTC Therapeutics. I.P. has received grants from Duchenne Parent Project España (DPPE) and has received speaker honoraria from Genzyme Therapeutics and PTC Therapeutics. J.D‐M. is an external advisor for Sanofi‐Genzyme Spain and has participated in advisory meetings for Sanofi‐Genzyme, Amicus, Audentes, Sarepta, and Lupin. He has received funding for research from Sanofi‐Genzyme, Amicus, Spark Therapeutics, and Boehringer‐Ingelheim. He has also received grants from DPPE, Fundación Isabel Gemio, ISCIII, CIBERER, and AFM. The other authors declare no conflicts of interest.
Supporting information
SUPPLEMENTARY TABLE 1. Summary of Clinical Data from the 57 Patients of the Series
SUPPLEMENTARY TABLE 2. Modifying Factors Genotype, Presence of Concurrent Mutations, and Long Noncoding RNA Preservation Profile in the 23 Index Cases Analyzed by Next Generation Sequencing and 10 Secondary Cases Analyzed by Sanger
SUPPLEMENTARY TABLE 3. Cardiac Assessment
SUPPLEMENTARY TABLE 4. Cognitive Assessment
SUPPLEMENTARY TABLE 5. Genomic Characterization of the 10 Deletion Breakpoints
SUPPLEMENTARY TABLE 6. In Silico Prediction of Pseudoexon Activation
SUPPLEMENTARY TABLE 7. Analysis of the Exonic Splicing Enhancer/Silencer Ratio at the Breakpoint Flanking Regions
Acknowledgments
We acknowledge Fundación Isabel Gemio for the financial support of this project and for the PhD grant to J.P.‐G. (2018/0200). A.L. is a recipient of the GVA fellowship APOSTD2021/212. N.M. and J.J.V. are funded by PI16/00316 supported by Instituto de Salud Carlos III (ISCIII) Madrid and by PROMETEO/2019075 supported by Conselleria de Sanitat, Generalitat Valenciana. I.P. and M.D. are funded by Duchenne Parent Project Spain (2018/0541). P.G. and L.G.‐Q. are funded by PI18/01585 supported by ISCIII. T.S. is funded by PROMETEO/2018/135 (Conselleria de Sanitat, Generalitat Valenciana) and by PI19/01178 (ISCIII).
We thank all the patients and family members for their cooperation. We acknowledge the Data Science Unit of IIS La Fe, Valencia for statistical analysis and the Genomic and Translational Genetics Unit of CIPF, Valencia for sequencing support. We are grateful to the collaboration of “Càtedra de Malalties rares,” University of Barcelona. We also thank Editage (Cactus Communications) for editing support.
Data Availability
The data that support the findings of this study are available from the corresponding author upon request.
References
- 1. Darras BT, Urion DK, Ghosh PS. Dystrophinopathies. GeneReviews® Last Update: January 20, 2022 [Internet]. University of Washington, Seattle; 1993‐2022.
- 2. North KN, Miller G, Iannaccone ST, et al. Cognitive dysfunction as the major presenting feature of Becker's muscular dystrophy. Neurology 1996;46:461–464. [DOI] [PubMed] [Google Scholar]
- 3. Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol 2003;2:731–740. [DOI] [PubMed] [Google Scholar]
- 4. Nicolas A, Raguénès‐Nicol C, Ben Yaou R, et al. Becker muscular dystrophy severity is linked to the structure of dystrophin. Hum Mol Genet 2015;24:1267–1279. [DOI] [PubMed] [Google Scholar]
- 5. Bello L, Pegoraro E. The “usual suspects”: genes for inflammation, fibrosis, regeneration, and muscle strength modify Duchenne muscular dystrophy. J Clin Med 2019;8:649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nakamura A, Shiba N, Miyazaki D, et al. Comparison of the phenotypes of patients harboring in‐frame deletions starting at exon 45 in the Duchenne muscular dystrophy gene indicates potential for the development of exon skipping therapy. J Hum Genet 2017;62:459–463. [DOI] [PubMed] [Google Scholar]
- 7. Béroud C, Tuffery‐Giraud S, Matsuo M, et al. Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Hum Mutat 2007;28:196–202. [DOI] [PubMed] [Google Scholar]
- 8. Flanigan KM, Dunn DM, von Niederhausern A, et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat 2009;30:1657–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nakamura A, Yoshida K, Fukushima K, et al. Follow‐up of three patients with a large in‐frame deletion of exons 45–55 in the Duchenne muscular dystrophy (DMD) gene. J Clin Neurosci 2008;15:757–763. [DOI] [PubMed] [Google Scholar]
- 10. Ferreiro V, Giliberto F, Noelia Muñiz GM, et al. Asymptomatic Becker muscular dystrophy in a family with a multiexon deletion. Muscle Nerve 2009;39:239–243. [DOI] [PubMed] [Google Scholar]
- 11. Taglia A, Petillo R, D'Ambrosio P, et al. Clinical features of patients with dystrophinopathy sharing the 45–55 exon deletion of DMD gene. Acta Myol 2015;34:9–13. [PMC free article] [PubMed] [Google Scholar]
- 12. Gentil C, Leturcq F, Ben Yaou R, et al. Variable phenotype of del45–55 Becker patients correlated with nNOSμ mislocalization and RYR1 hypernitrosylation. Hum Mol Genet 2012;21:3449–3460. [DOI] [PubMed] [Google Scholar]
- 13. Anthony K, Cirak S, Torelli S, et al. Dystrophin quantification and clinical correlations in Becker muscular dystrophy: implications for clinical trials. Brain 2011;134:3547–3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Echigoya Y, Aoki Y, Miskew B, et al. Long‐term efficacy of systemic multiexon skipping targeting dystrophin exons 45–55 with a cocktail of vivo‐morpholinos in mdx52 mice. Mol Ther Nucleic Acids 2015;4:e225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee J, Echigoya Y, Duddy W, et al. Antisense PMO cocktails effectively skip dystrophin exons 45–55 in myotubes transdifferentiated from DMD patient fibroblasts. PLoS One 2018;13:e0197084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aoki Y, Yokota T, Nagata T, et al. Bodywide skipping of exons 45–55 in dystrophic mdx52 mice by systemic antisense delivery. Proc Natl Acad Sci U S A 2012;109:13763–13768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Echigoya Y, Lim KRQ, Melo D, et al. Exons 45–55 skipping using mutation‐tailored cocktails of antisense morpholinos in the DMD gene. Mol Ther 2019;27:2005–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Young CS, Hicks MR, Ermolova NV, et al. A single CRISPR‐Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC‐derived muscle cells. Cell Stem Cell 2016;18:533–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ousterout DG, Kabadi AM, Thakore PI, et al. Multiplex CRISPR/Cas9‐based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat Commun 2015;6:6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Happi Mbakam C, Lamothe G, Tremblay JP. Therapeutic strategies for dystrophin replacement in Duchenne muscular dystrophy. Front Med 2022;9:774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Felisari G, Boneschi FM, Bardoni A, et al. Loss of Dp140 dystrophin isoform and intellectual impairment in Duchenne dystrophy. Neurology 2000;55:559–564. [DOI] [PubMed] [Google Scholar]
- 22. Bovolenta M, Erriquez D, Valli E, et al. The DMD locus harbours multiple long non‐coding RNAs which orchestrate and control transcription of muscle dystrophin mRNA isoforms. PLoS One 2012;7:e45328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tuffery‐Giraud S, Miro J, Koenig M, Claustres M. Normal and altered pre‐mRNA processing in the DMD gene. Hum Genet 2017;136:1155–1172. [DOI] [PubMed] [Google Scholar]
- 24. Ricotti V, Mandy WPL, Scoto M, et al. Neurodevelopmental, emotional, and behavioural problems in Duchenne muscular dystrophy in relation to underlying dystrophin gene mutations. Dev Med Child Neurol 2016;58:77–84. [DOI] [PubMed] [Google Scholar]
- 25. Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J 2016;37:2129–2200. [DOI] [PubMed] [Google Scholar]
- 26. Bonne G, Rivier F, Hamroun D. The 2018 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscul Disord 2017;27:1152–1183. [DOI] [PubMed] [Google Scholar]
- 27. Soblechero‐Martín P, Albiasu‐Arteta E, Anton‐Martinez A, et al. Duchenne muscular dystrophy cell culture models created by CRISPR/Cas9 gene editing and their application in drug screening. Sci Rep 2021;11:18188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Verheul RC, van Deutekom JCT, Datson NA. Digital droplet PCR for the absolute quantification of exon skipping induced by antisense oligonucleotides in (pre‐)clinical development for Duchenne muscular dystrophy. PLoS One 2016;11:e0162467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Anthony K, Arechavala‐Gomeza V, Taylor LE, et al. Dystrophin quantification: biological and translational research implications. Neurology 2014;83:2062–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liquori A, Vaché C, Baux D, et al. Whole USH2A gene sequencing identifies several new deep intronic mutations. Hum Mutat 2016;37:184–193. [DOI] [PubMed] [Google Scholar]
- 31. Poplin R, Ruano‐Rubio V, DePristo MA, et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2017;201178. [Google Scholar]
- 32. Miyazaki D, Yoshida K, Fukushima K, et al. Characterization of deletion breakpoints in patients with dystrophinopathy carrying a deletion of exons 45–55 of the Duchenne muscular dystrophy (DMD) gene. J Hum Genet 2009;54:127–130. [DOI] [PubMed] [Google Scholar]
- 33. Geng C, Tong Y, Zhang S, et al. Sequence and structure characteristics of 22 deletion breakpoints in intron 44 of the DMD gene based on long‐read sequencing. Front Genet 2021;12:638220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kaspar RW, Allen HD, Ray WC, et al. Analysis of dystrophin deletion mutations predicts age of cardiomyopathy onset in Becker muscular dystrophy. Circ Cardiovasc Genet 2009;2:544–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bello L, Campadello P, Barp A, et al. Functional changes in Becker muscular dystrophy: implications for clinical trials in dystrophinopathies. Sci Rep 2016;6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lambert JT, Darmahkasih AJ, Horn PS, et al. Neurodevelopmental, behavioral, and emotional symptoms in Becker muscular dystrophy. Muscle Nerve 2020;61:156–162. [DOI] [PubMed] [Google Scholar]
- 37. Hinton V, Fee RJ, Goldstein EM, De Vivo DC. Verbal and memory skills in males with Duchenne muscular dystrophy. Dev Med Child Neurol 2007;49:123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Young HK, Barton BA, Waisbren S, et al. Cognitive and psychological profile of males with Becker muscular dystrophy. J Child Neurol 2008;23:155–162. [DOI] [PubMed] [Google Scholar]
- 39. Findlay AR, Wein N, Kaminoh Y, et al. Clinical phenotypes as predictors of the outcome of skipping around DMD exon 45. Ann Neurol 2015;77:668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Van Den Bergen JC, Wokke BH, Janson AA, et al. Dystrophin levels and clinical severity in Becker muscular dystrophy patients. J Neurol Neurosurg Psychiatry 2014;85:747–753. [DOI] [PubMed] [Google Scholar]
- 41. Ishmukhametova A, Van Kien PK, Méchin D, et al. Comprehensive oligonucleotide array‐comparative genomic hybridization analysis: new insights into the molecular pathology of the DMD gene. Eur J Hum Genet 2012;20:1096–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gargaun E, Falcone S, Solé G, et al. The lncRNA 44s2 study applicability to the design of 45–55 exon skipping therapeutic strategy for DMD. Biomedicine 2021;9:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pons R, Kekou K, Gkika A, et al. Single amino acid loss in the dystrophin protein associated with a mild clinical phenotype. Muscle Nerve 2017;55:46–50. [DOI] [PubMed] [Google Scholar]
- 44. Gurvich OL, Maiti B, Weiss RB, et al. DMD exon 1 truncating point mutations: amelioration of phenotype by alternative translation initiation in exon 6. Hum Mutat 2009;30:633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sironi M, Cagliani R, Comi GP, et al. Trans‐acting factors may cause dystrophin splicing misregulation in BMD skeletal muscles. FEBS Lett 2003;537:30–34. [DOI] [PubMed] [Google Scholar]
- 46. Bougé A‐L, Murauer E, Beyne E, et al. Targeted RNA‐seq profiling of splicing pattern in the DMD gene: exons are mostly constitutively spliced in human skeletal muscle. Sci Rep 2017;7:39094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Suzuki H, Aoki Y, Kameyama T, et al. Endogenous multiple exon skipping and back‐splicing at the DMD mutation hotspot. Int J Mol Sci 2016;17:1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bardoni A, Sironi M, Felisari G, et al. Absence of brain Dp140 isoform and cognitive impairment in Becker muscular dystrophy. Lancet 1999;353:897–898. [DOI] [PubMed] [Google Scholar]
- 49. Kerst B, Mennerich D, Schuelke M, et al. Heterozygous myogenic factor 6 mutation associated with myopathy and severe course of Becker muscular dystrophy. Neuromuscul Disord 2000;10:572–577. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY TABLE 1. Summary of Clinical Data from the 57 Patients of the Series
SUPPLEMENTARY TABLE 2. Modifying Factors Genotype, Presence of Concurrent Mutations, and Long Noncoding RNA Preservation Profile in the 23 Index Cases Analyzed by Next Generation Sequencing and 10 Secondary Cases Analyzed by Sanger
SUPPLEMENTARY TABLE 3. Cardiac Assessment
SUPPLEMENTARY TABLE 4. Cognitive Assessment
SUPPLEMENTARY TABLE 5. Genomic Characterization of the 10 Deletion Breakpoints
SUPPLEMENTARY TABLE 6. In Silico Prediction of Pseudoexon Activation
SUPPLEMENTARY TABLE 7. Analysis of the Exonic Splicing Enhancer/Silencer Ratio at the Breakpoint Flanking Regions
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.