

Abstract
While the range of accessible borylenes has significantly broadened over the last decade, applications remain limited. Herein, we present tricoordinate oxy‐borylenes as potent photoreductants that can be readily activated by visible light. Facile oxidation of CAAC stabilized oxy‐borylenes (CAAC)(IPr2Me2)BOR (R=TMS, CH2CH2C6H5, CH2CH2(4‐F)C6H4) to their corresponding radical cations is achieved with mildly oxidizing ferrocenium ion. Cyclovoltammetric studies reveal ground‐state redox potentials of up to −1.90 V vs. Fc+/0 for such oxy‐borylenes placing them among the strongest organic super electron donors. Their ability as photoreductants is further supported by theoretical studies and showcased by the application as stoichiometric reagents for the photochemical hydrodehalogenation of aryl chlorides, aryl bromides and unactivated alkyl bromides as well as the detosylation of anilines.
Keywords: Boron Compounds, EPR Spectroscopy, Photochemistry, Radical Reactions, SET Reduction
The synthesis of a series of oxy‐borylenes and their corresponding radical cations is reported. Cyclovoltammetric studies reveal ground‐state redox potentials of up to −1.90 V vs. Fc+/0 for those highly reducing oxy‐borylenes. Their ability as photoreductants is then showcased by the detosylation of tosylated anilines and the dehalogenation of electron‐rich aryl as well as alkyl halides under visible light irradiation.

Introduction
The chemistry of borylenes, in which boron adopts the +1 oxidation state, has attracted increasing attention over the last decades. [1] Free monocoordinate borylenes, bearing a lone pair and only one substituent at boron, are highly electron rich, transient species, which have not yet been isolated, but could be studied spectroscopically and be traced by trapping experiments.[ 2 , 3 ] In two seminal studies, Braunschweig showed that borylenes may be stabilized and isolated, either in a terminal or bridged position, by coordination to an electron rich transition metal. [4] Inspired by a report from Robinson, who showed that the reduction of (NHC)BBr3 (NHC=N‐heterocyclic carbene) led to a stable diborene, [5] a formal dimer of the dicoordinate borylene, in 2011 Bertrand and co‐workers were able to isolate the first metal‐free borylene (CAAC)2BH (1) via stabilization with two cyclic (alkyl)(amino)carbenes (CAACs), thus delocalizing the free lone pair at boron (Scheme 1a). [6] CAACs were shown to be more nucleophilic than NHCs therefore acting as stronger σ donors to the empty p orbitals at boron while simultaneously being better π acceptors to decrease the Lewis basicity of the boron atom. [7] Subsequently, the application of CAAC as ligand in combination with an amino‐substituent on boron, resulting in a push‐pull effect, allowed for the isolation of dicoordinate borylene (CAAC)BN(TMS)2, whose electrophilicity under one atmosphere of CO gave rise to the corresponding mixed base stabilized tricoordinate borylene (CAAC)(L)BR with L=CO and R=N(TMS)2. [8] Moreover, the reduction of (CAAC)BBr2CN in the presence of an excess of PEt3, enabled the synthesis of the first phosphine ligated borylene (CAAC)(PEt3)BCN. [9] This strategy was further applied for the synthesis of several mixed Lewis‐base tricoordinate halo‐ and pseudohalo‐borylenes.[ 10 , 11 ]
Scheme 1.

a) One electron oxidation of borylene 1 to radical cation 2. b) Organic electron donors 3–5. Dipp=2,6‐diisopropylphenyl.
Their meanwhile good availability allowed to better explore the general reactivity of such tricoordinate borylenes. The nucleophilic nature of boron in borylene 1 was documented by protonation using triflic acid. [6] Moreover, Kinjo and co‐workers demonstrated that bis(oxazol‐2‐ylidine)‐stabilized borylenes can act as σ donors which coordinate to Lewis acids and transition metals like gold and chromium. As an application, Au‐borylene complexes were successfully used as catalysts for the hydroamination of alkynes in the presence of KBArF 4. [12] For tricoordinate (CAAC)(CO)BDur (Dur=2,3,5,6‐tetramethylphenyl) it was shown that under UV light irradiation, CO is readily cleaved off and the free dicoordinate borylene can either be trapped intermolecularly by a Lewis base or undergo intramolecular C−H insertion. [13] Furthermore, the one electron oxidation of borylene 1 to the corresponding boryl radical cation 2 was realized by adding mildly oxidizing GaCl3 (Scheme 1a). Cyclovoltammetric studies revealed a reversible oxidation at −0.940 V vs. Fc+/0. [6] Reversible chemical redox operation was achieved for pseudohalo‐borylenes (CAAC)(IiPr)BCN (IiPr=1,3‐diisopropylimidazol‐2‐ylidene) and (CAAC)(IiPr)BSCN by one electron oxidation using AgOTf and subsequent reduction with strongly reducing KC8. [11b] Braunschweig and co‐workers showed that the absorption maxima (350 nm–530 nm) and also the ground state redox potential for chloro‐borylenes (CAAC)(L)BCl, ranging from −0.95 V (L=PEt3) to −1.26 V (L=IMe4) vs. Fc+/0 depend on the ligand L. [10a]
These potentials are comparable to those of organic electron donors like tetrakis(dimethylamino)ethene (3, TDAE), that was successfully used for the reduction of electron‐poor perfluoro alkenes. [14] Along with 3, a wide range of organic electron donors based on electron‐rich alkenes were developed since, which enable the reduction of alkyl halides and arenesulfonamides among other substrates. [15]
Importantly, upon UV light irradiation of electron donor 4, the reduction of even more challenging substrates such as aryl chlorides could be achieved. [16] Converting the energy of light, especially visible light, into chemical redox potential, i.e. photoinduced electron transfer (PET), has attracted great attention over the last decades. [17] Especially the design of new concepts and photosystems for difficult reductions, e.g. of aryl/alkyl halides, is of major interest. [18] In this regard the activation of a “super electron donor” by visible light irradiation was demonstrated for boronate 5. With a ground‐state redox potential of −1.11 V vs. Fc+/0, irradiation at 400 nm resulted in an excited state potential of −3.87 V vs. Fc+/0 enabling the challenging reduction of electron‐rich aryl chlorides. [19]
Based on the reported ground‐state redox potentials and absorption properties of tricoordinate borylenes, we assumed that such borylenes should be very strong reductants in their excited states. Moreover, we reasoned that installing an electron‐donating substituent on boron could further increase the borylene redox potential. We therefore initiated a program towards the synthesis of oxy‐borylenes (CAAC)(IPr2Me2)BOR. In addition, their physical properties and first applications as photoreductants in synthesis were studied.
Results and Discussion
Room temperature reduction of (CAACCy)Cl2BOTMS (6, CAACCy=1‐(2,6‐diisopropylphenyl)‐3,3,5,5‐tetramethylpyrrolidin‐2‐ylidene, for its preparation see the Supporting Information) with 2.5 equivalents KC8 in the presence of IPr2Me2 (1,3‐diisopropyl‐4,5‐dimethylimidazol‐2‐ylidene) in toluene provided the tricoordinate siloxy‐borylenes Z‐7 and E‐7 as a 91 : 9 diastereoisomeric mixture in 76 % yield (Scheme 2). NMR analysis revealed characteristic 11B NMR resonances at 13.8 ppm (major isomer) and 17.6 ppm (minor isomer) for these two compounds. Of note, depending on the batch the isomer ratio slightly varied (see Supporting Information). Slow evaporation of the dark red toluene solution yielded single crystals of Z‐7 suitable for X‐ray crystallographic analysis, which allowed the unambiguous assignment of the relative configuration of the major isomer with respect to the C=B double bond (Figure 1a). [20] Interestingly, irradiation of a Z/E‐isomeric mixture of 7 in C6D6 at room temperature using a 390 nm LED resulted in full isomerization to the E‐isomer. [11a] Full isomerization to the E‐borylene could also be achieved by heating of a C6D6 solution of Z‐7/E‐7 at 80 °C for six days, showing that E‐7 is the thermodynamic product whereas reduction of 6 leads mainly to the kinetic product Z‐7. This is confirmed by DFT calculations which indicate that E‐ 7 is more stable (ΔG298(Z‐ 7→E‐ 7)=−2.3 kcal mol−1, see Supporting Information). The structure of E‐7 was also confirmed by X‐ray analysis (Figure 1b).
Scheme 2.

Synthesis and isomerization of tricoordinate siloxyborylenes Z‐7 and E‐ 7.
Figure 1.
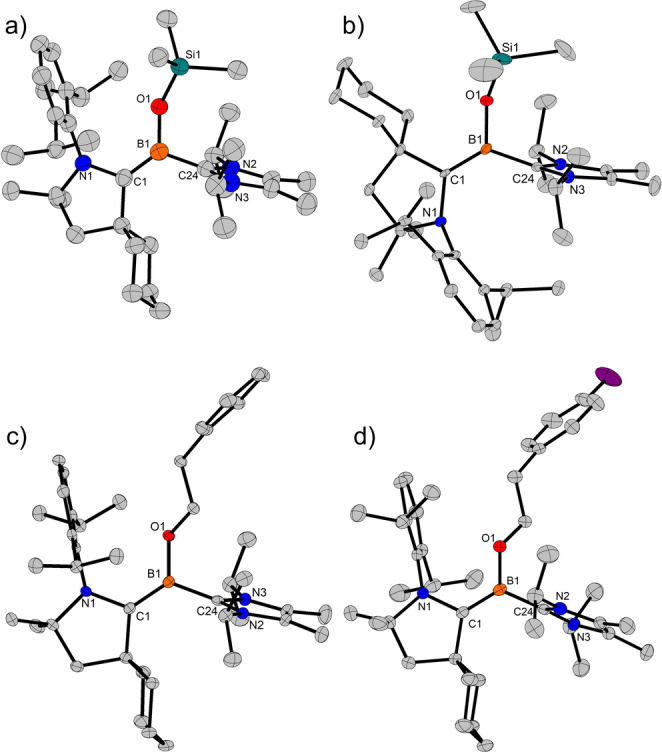
Molecular structures of a) Z‐7 (only the independent molecule A is depicted), b) E‐7, c) Z‐10Cy , d) Z‐11Cy . Hydrogen atoms are omitted for clarity and thermal ellipsoids are set at 50 % probability. Selected bond lengths [Å] and angles for Z‐7 (molecule A): B1−O1 1.457(5), B1−C1 1.444(6), B1−C24 1.601(6), O1−B1−C24−N3 ca. −71°. For E‐7: B1−O1 1.440(3), B1−C1 1.446(4), B1−C24 1.614(3), O1−B1−C24−N3 ca. −80°. For Z‐10Cy : B1−O1 1.448(2), B1−C1 1.453(3), B1−C24 1.615(3), O1−B1−C24−N3 ca. −65°. For Z‐11Cy : B1−O1 1.445(2), B1−C1 1.451(2), B1−C24 1.609(2), O1−B1−C24−N2 ca. 62°.
To evaluate the influence of the nature of the O‐substituent on the properties and structure of the oxy‐borylenes, the reduction of phenethyl‐ and 4‐fluorophenethyl‐dichloro precursors 8 and 9 was performed, aiming at evaluating their steric as well as electronic effect as compared to the parent trimethylsilyl‐substituent (Scheme 3). Reduction of 8–9 yielded the corresponding borylenes 10–11 in all cases as dark purple powders in good yields (74–76 %). While borylenes 10Et , 11Et and 11Cy were obtained with complete Z‐selectivity, 10Cy was formed as a 92 : 8 Z/E‐mixture of isomers favoring the Z‐isomer. In contrast to Z‐7, photoisomerization of the Z/E‐mixture of 10Cy at 390 nm for 24 h could not be achieved.
Scheme 3.

Synthesis of phenethyl‐ and 4‐fluorophenethyl‐substituted borylenes 10 and 11.
Comparison of the solid‐state structures shows, that there are only slight differences between the oxy‐borylenes Z‐7, E‐7, Z‐10Cy and Z‐11Cy . They all show a trigonal planar boron center, connected to CAAC, that is only slightly tilted away from the borylene plane (torsion angle O1−B1−C1−N1=ca. 2°–9°). The B−CCAAC bond (1.44–1.46 Å) is shortened, typical for a B=C double bond, [21] indicating a strong π backdonation from the boron center to the CAAC ligand. In contrast, the IPr2Me2 ligand is turned almost perpendicular to the borylene plane (torsion angle O1−B1−C24−N2(3)=ca. 61°–80°), therefore only acting as σ donor to boron with negligible π overlap. This is further supported by the B−CIPr2Me2 bond length of 1.60–1.62 Å in line with a B−C single bond. [21] The B−O bond length of 1.44–1.46 Å, typical for a B−O single bond, [21] also indicates, that there is no significant π character involved. Comparison of these X‐ray data shows, that neither the substituent on oxygen nor the relative orientation (Z or E) or choice of CAAC (CAACEt or CAACCy) have a major influence on the core‐solid‐state structure of such borylenes.
To further explore their properties, cyclovoltammetry experiments of all oxy‐borylenes were performed. For the TMS‐O‐substituted borylene Z‐7, a fully reversible oxidation at E 1/2=−1.88 V vs. Fc+/0 and a second partially reversible oxidation at E 1/2=−0.65 V vs. Fc+/0 could be observed. This corresponds to an increased redox potential of −0.6 V as compared to the most strongly reducing chloroborylenes reported, [10a] which shows that the donating ability of the oxygen‐substituent on boron indeed has a major impact on the borylene redox potential. For the E‐isomer of 7 the reversible oxidation was found at E 1/2=−1.66 V vs. Fc+/0. For the phenethyl‐O‐substituted borylenes 10 and 11, a first reversible oxidation at E 1/2=−1.84 V to −1.90 V vs. Fc+/0 and a second oxidation event at E 1/2=−0.61 V to −0.68 V vs. Fc+/0 was measured, which depending on the substrate could be rendered reversible by an increase of the scan rate. This indicates, that the nature of the oxygen‐bound substituent does not have a significant impact on the borylenes redox potential.
In contrast, the oxy‐borylenes exhibited rather different absorption spectra depending on the oxygen‐bound substituent as well as the E/Z geometrical structure of the CAAC (Figure 2a). In THF, while both E‐7 and Z‐7 exhibited broad absorption bands with a maximum wavelength (λ abs) of around 430 nm, the molar absorption coefficient (ϵ) of E‐7 (2.28×103 M−1cm−1) is more than twice as large as that of Z‐7 (0.928×103 M−1cm−1). Furthermore, a significant red‐shift relative to oxy‐borylene 7 could be observed for the phenethyl‐substituted 10Et and 11Et (λ abs=505 nm, for both compounds), whereas the substituent on the phenyl group does neither affect the absorption maximum nor the ϵ value. The λ abs values of 10Et and 11Et are even larger relative to that of a CAAC‐substituted chloro‐borylene bearing an orthogonally‐oriented IMe4 group reported by Braunschweig and co‐workers. [10a]
Figure 2.

a) UV/Vis absorption spectra of E‐7, Z‐7, 10 Et and 11 Et in THF and b) pictorial drawings of HOMO and LUMO+2 for 10 Et calculated at the CAM‐B3LYP/6‐31G(d) level of theory.
To gain in‐depth insights into these differences, we conducted time‐dependent (TD) DFT calculations on the four compounds E‐7, Z‐7, 10Et and 11Et at the CAM‐B3LYP/6‐31G(d) level of theory including THF as a solvent using the polarizable continuum model (PCM). The calculated absorption spectra well reproduced the experimental results in terms of the spectral shape (Figure S27). Notably, the absorption bands around 430 nm observed for E‐7 and Z‐7 are not assigned to the S0→S1 electronic transition, but the S0→S2 and S0→S3 transition, respectively. The TD‐DFT calculations indicate that their S0→S1 transitions, corresponding to the HOMO→LUMO transitions, for both E‐7 and Z‐7 have forbidden character with small oscillator strengths, while the transition energies are comparable or even smaller relative to those of 10Et and 11Et . In contrast, the S0→S1 transitions for 10Et and 11Et , mainly attributed to the HOMO→LUMO+2 and HOMO→LUMO+1 transitions, respectively, have larger oscillator strengths than those of 7, which should be responsible for their larger molar absorption coefficients. For all four compounds, the HOMO is predominantly localized on the borylene B−C double bond, while the unoccupied MOs relevant to the electronic transitions are mainly localized on the orthogonally‐oriented NHC moiety (Figure 2b). In a comparison between E‐7 and Z‐7, the HOMO of Z‐7 is higher by 0.23 eV than that of E‐7 (Figure S27), consistent with the cyclic voltammetry results, demonstrating the impact of the E/Z geometrical structures on the redox properties.
In accordance with the CV measurements the preparative one‐electron oxidation of oxy‐borylenes proved to be feasible upon treatment with FeCp2GaCl4 [22] in toluene at room temperature (Scheme 4). The reaction could be readily monitored by the naked eye by a color change from dark red/purple solutions to yellow suspensions. Since the resulting boryl radical cations did not show any solubility in toluene, filtration, subsequent extraction of the greyish residue with acetonitrile and removal of solvent afforded the clean persistent boryl radical cations 12Cy , 13Et , 13Cy , 14Et and 14Cy as orange crystals in moderate to good yields (48–70 %). X‐ray crystal structure analysis of 12Cy and 14Cy showed that the boron center remains trigonal planar, but the reduced electron density at boron leads to a lengthening of the B−CCAAC bond (1.51–1.52 Å), now only showing partial double bond character (Figure 3). Consequently, a slightly shortened B−O‐bond (1.39–1.52 Å) can be observed, as a result of π donation from the OR‐substituent to the boron center.
Scheme 4.

Synthesis of boryl radical cations 12–14 by one electron oxidation.
Figure 3.

a) Molecular structures of 12Cy (left) and 14Cy (right). Hydrogen atoms are omitted for clarity and thermal ellipsoids are set at 50 % probability. Selected bond lengths [Å] and angles for 12Cy : B1−O1 1.385(3), B1−C1 1.518(4), B1−C24 1.612(4), O1−B1−C24−N3 ca. −80°. For 14Cy : B1−O1 1.383(7), B1−C1 1.515(8), B1−C24 1.603(8), O1−B1−C24−N2 ca. −77°. b) Spin density isosurface plots ((ρα−ρβ), 5×10−3 a.u.), obtained with PW6B95/def2‐TZVP c) Liquid‐state CW EPR spectra of the radical 12Cy (left) and 14Cy (right) recorded in ACN at X‐band and at rt. The black curve represents the experimental spectrum and the red curve is the best‐fit simulation.
The liquid‐state CW EPR spectra for 12Cy and 14Cy in Figure 3 show g‐factors around 2.0035 (Table S2) and magnetic hyperfine coupling to one 14N nucleus (I=1, AN ≈6.7 G) and one 11B nucleus (I=3/2, AB ≈1.4 to 1.7G), which is in a good agreement with the spin density calculations (Figure 3b), which indicate a spin delocalization over all four atoms (N−C−B−O). The spin distribution of 13Et shows the same form (see Supporting Information). Consequently, a quartet of triplets is theoretically expected, however, the individual components are only partially resolved due to relatively strong line broadening effects as shown by the simulations displayed in Figure S10. The AN values are similar to, whereas the AB values are significantly lower than those published on similar boryl radicals. [10a]
To evaluate the applicability of the novel borylenes as photoreductants, the reductive dehalogenation of aryl halides was studied. We were pleased to find, that all three tested borylenes 7, 10Et and 11Et (1.2 equivalents) were able to hydrodehalogenate 4‐chloroanisole (15 a) upon 405 nm LED light irradiation in dimethylacetamide (DMA) as solvent at room temperature. The TMS‐O‐substituted borylene 7 (used as a mixture of isomers) afforded anisole in 83 % yield, whereas a lower efficiency was noted for the reduction with 10Et and 11Et (49 %, 57 %). Control experiments revealed the necessity of light as well as the borylene for the reduction.
Based on these initial results, all following experiments were conducted with the most efficient reductant 7. A number of electron‐rich and electron‐poor aryl bromides and aryl chlorides 15–16 were subjected to the photochemical reduction and the corresponding hydrodehalogenated products were obtained in moderate to excellent yields (57–95 %, Scheme 5). Of note, ester substituents and boronic ester moieties are tolerated (see 15 e and 15 d). The dehalogenation of aryl halide 15 f afforded dihydrobenzofurane 18 (51 %) resulting from a 5‐exo‐cyclization of the intermediate aryl radical with subsequent reduction. This result supports the radical nature of the dehalogenation. Next the reduction of alkyl bromides was attempted and we were pleased to find that the tertiary alkyl bromide 17 b was smoothly reduced in 88 % yield to adamantane. It was further shown that the reaction of the unactivated primary alkyl bromide 17 a to propyl benzene worked, albeit a lower yield was obtained in this case (55 %).
Scheme 5.

Photoreduction of various aryl and alkyl halides and tosylated amines with borylene 7. Reactions were performed at a 0.1 mmol scale. Yields were determined by GC‐FID measurements using mesitylene as internal standard. a Byproduct which partially decomposed during the reaction. b Yield determined by 1H NMR measurements using CH2Br2 as internal standard. c vs. SCE. [23]
Finally, the reductive N‐deprotection of tosylated anilines was studied. Pleasingly, applying the same reaction conditions, detosylation of the Ts‐indole 19 a was achieved in 85 % yield. An even better yield of 91 % was obtained upon switching to dimethylformamide (DMF) as solvent. Under these optimized conditions, tosylated anilines 19 b and 19 c could be converted to the free anilines in 88 % and 60 % yield, respectively.
To get insights into the mechanism of the photo‐mediated reduction we first performed the dehalogenation of the aryl bromide 16 b in fully deuterated DMF in place of DMA. Analysis by GC‐MS (see Supporting Information) revealed that the desired tert‐butylbenzene was partially deuterated, suggesting that the solvent as well as the oxidized boron species act as H‐atom donors in the reduction of the intermediate aryl radical, which is generated upon single electron reduction and mesolysis of the C−Br bond of the starting halide. In addition, we analyzed the fate of the boron species by using EPR and GC measurements. In the crude reaction mixture of the reduction of chloroanisole 15 a with borylene 7, we could identify the boryl radical cation, as expected byproduct (see Supporting Information for experimental and simulated data). However, this boryl radical cation was found not to be stable under the reaction conditions (see Supporting Information for GC‐trace), which is in accordance with its likely role as H‐atom donor and the fact, that none of our attempts to render the reaction catalytic was successful.
Conclusion
In summary, we have shown that oxy‐borylenes under visible light irradiation are efficient single electron transfer reductants. Structures of the oxy‐borylenes and also their oxidized persistent radical cations were characterized by X‐ray diffraction. For the latter, EPR studies were conducted. All compounds were additionally analyzed by cyclovoltammetry revealing a reversible first oxidation of the oxy‐borylenes. We have found that the installation of an electron‐donating siloxy or alkoxy substituent on the boron atom leads to a significant increase of the ground state redox potential to up to −1.90 vs. Fc+/0 as compared to literature known tricoordinate borylenes. Their absorption properties were examined and further investigated by theoretical studies. The ability of oxy‐borylenes to compete with the most efficient organic SET‐reductants has been documented by the application of oxy‐borylene 7 as efficient photoreductant for the hydrodehalogenation of aryl and alkyl halides as well as for the deprotection of N‐tosylated anilines.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—SFB 858 and GRK 2678‐437785492—for financial support. This work was further supported by KAKENHI grant 18H05261 from the Japan Society for the Promotion of Science (JSPS). Open Access funding enabled and organized by Projekt DEAL.
Dedicated to Professor Mukund P. Sibi on the occasion of his 70th birthday.
P. Lenz, R. Oshimizu, S. Klabunde, C. G. Daniliuc, C. Mück-Lichtenfeld, J. C. Tendyck, T. Mori, W. Uhl, M. R. Hansen, H. Eckert, S. Yamaguchi, A. Studer, Angew. Chem. Int. Ed. 2022, 61, e202209391; Angew. Chem. 2022, 134, e202209391.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Soleilhavoup M., Bertrand G., Angew. Chem. Int. Ed. 2017, 56, 10282–10292; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10416–10426. [Google Scholar]
- 2.selected spectroscopic studies:
- 2a. Timms P. L., J. Am. Chem. Soc. 1967, 89, 1629–1632; [Google Scholar]
- 2b. Timms P. L., Acc. Chem. Res. 1973, 6, 118–123; [Google Scholar]
- 2c. Nomoto M., Okabayashi T., Klaus T., Tanimoto M., J. Mol. Struct. 1997, 413–414, 471–476; [Google Scholar]
- 2d. Andrews L., Hassanzadeh P., Martin J. M. L., Taylor P. R., J. Phys. Chem. 1993, 97, 5839–5847; [Google Scholar]
- 2e. Thompson C. A., Andrews L., Martin J. M. L., El-Yazal J., J. Phys. Chem. 1995, 99, 13839–13849; [Google Scholar]
- 2f. Bettinger H. F., J. Am. Chem. Soc. 2006, 128, 2534–2535. [DOI] [PubMed] [Google Scholar]
- 3.selected trapping experiments:
- 3a. Pachaly B., West R., Angew. Chem. Int. Ed. Engl. 1984, 23, 454–455; [Google Scholar]; Angew. Chem. 1984, 96, 444–445; [Google Scholar]
- 3b. Meller A., Seebold U., Maringgele W., Noltemeyer M., Sheldrick G. M., J. Am. Chem. Soc. 1989, 111, 8299–8300; [Google Scholar]
- 3c. Grigsby W. J., Power P. P., J. Am. Chem. Soc. 1996, 118, 7981–7988. [Google Scholar]
- 4.
- 4a. Braunschweig H., Wagner T., Angew. Chem. Int. Ed. Engl. 1995, 34, 825–826; [Google Scholar]; Angew. Chem. 1995, 107, 904–905; [Google Scholar]
- 4b. Braunschweig H., Kollann C., Englert U., Angew. Chem. Int. Ed. 1998, 37, 3179–3180; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 3355–3357. [Google Scholar]
- 5. Wang Y., Quillian B., Wei P., Wannere C. S., Xie Y., King R. B., Schaefer H. F., Schleyer P. V. R., Robinson G. H., J. Am. Chem. Soc. 2007, 129, 12412–12413. [DOI] [PubMed] [Google Scholar]
- 6. Kinjo R., Donnadieu B., Celik M. A., Frenking G., Bertrand G., Science 2011, 333, 610–613. [DOI] [PubMed] [Google Scholar]
- 7. Soleilhavoup M., Bertrand G., Acc. Chem. Res. 2015, 48, 256–266. [DOI] [PubMed] [Google Scholar]
- 8. Dahcheh F., Martin D., Stephan D. W., Bertrand G., Angew. Chem. Int. Ed. 2014, 53, 13159–13163; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13375–13379. [Google Scholar]
- 9. Arrowsmith M., Auerhammer D., Bertermann R., Braunschweig H., Bringmann G., Celik M. A., Dewhurst R. D., Finze M., Grüne M., Hailmann M., Hertle T., Krummenacher I., Angew. Chem. Int. Ed. 2016, 55, 14464–14468; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14680–14684. [Google Scholar]
- 10.
- 10a. Arrowsmith M., Schweizer J. I., Heinz M., Härterich M., Krummenacher I., Holthausen M. C., Braunschweig H., Chem. Sci. 2019, 10, 5095–5103; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Sarkar S. K., Siddiqui M. M., Kundu S., Ghosh M., Kretsch J., Stollberg P., Herbst-Irmer R., Stalke D., Stückl A. C., Schwederski B., Kaim W., Ghorai S., Jemmis E. D., Roesky H. W., Dalton Trans. 2019, 48, 8551–8555. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Hagspiel S., Arrowsmith M., Fantuzzi F., Vargas A., Rempel A., Hermann A., Brückner T., Braunschweig H., Angew. Chem. Int. Ed. 2021, 60, 6446–6450; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 6519–6524; [Google Scholar]
- 11b. Hagspiel S., Elezi D., Arrowsmith M., Fantuzzi F., Vargas A., Rempel A., Härterich M., Krummenacher I., Braunschweig H., Chem. Sci. 2021, 12, 7937–7942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Kong L., Li Y., Ganguly R., Vidovic D., Kinjo R., Angew. Chem. Int. Ed. 2014, 53, 9280–9283; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9434–9437; [Google Scholar]
- 12b. Kong L., Ganguly R., Li Y., Kinjo R., Chem. Sci. 2015, 6, 2893–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Braunschweig H., Kurmmenacher I., Légaré M.-A., Matler A., Radacki K., Ye Q., J. Am. Chem. Soc. 2017, 139, 1802–1805. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Pruett R. L., Barr J. T., Rapp K. E., Bahner C. T., Gibson J. D., R. H. Lafferty Jr , J. Am. Chem. Soc. 1950, 72, 3646–3650; [Google Scholar]
- 14b. Briscoe M. W., Chambers R. D., Mullins S. J., Nakamura T., Vaughan J. F. S., Drakesmith F. G., J. Chem. Soc. Perkin Trans. 1 1994, 3115–3118. [Google Scholar]
- 15.for reviews:
- 15a. Broggi J., Terme T., Vanelle P., Angew. Chem. Int. Ed. 2014, 53, 384–413; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 392–423; [Google Scholar]
- 15b. Murphy J. A., J. Org. Chem. 2014, 79, 3731–3746; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Doni E., Murphy J. A., Chem. Commun. 2014, 50, 6073–6087. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Cahard E., Schoenebeck F., Garnier J., Cutulic S. P. Y., Zhou S., Murphy J. A., Angew. Chem. Int. Ed. 2012, 51, 3673–3676; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3733–3736; [Google Scholar]
- 16b. Doni E., O'Sullivan S., Murphy J. A., Angew. Chem. Int. Ed. 2013, 52, 2239–2242; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2295–2298; [Google Scholar]
- 16c. O′Sullivan S., Doni E., Tuttle T., Murphy J. A., Angew. Chem. Int. Ed. 2014, 53, 474–478; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 484–488. [Google Scholar]
- 17.
- 17a. Prier C. K., Rankic D. A., MacMillan D. W. C., Chem. Rev. 2013, 113, 5322–5363; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Romero N. A., Nicewicz D. A., Chem. Rev. 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
- 18.selected examples:
- 18a. Cybularczyk-Cecotka M., Szczepanik J., Giedyk M., Nat. Catal. 2020, 3, 872–886; [Google Scholar]
- 18b. Ghosh I., Ghosgh T., Bardagi J. I., König B., Science 2014, 346, 725–728; [DOI] [PubMed] [Google Scholar]
- 18c. Ghosh I., Marzo L., Das A., Shaikh R., König B., Acc. Chem. Res. 2016, 49, 1566–1577; [DOI] [PubMed] [Google Scholar]
- 18d. Ghosh I., König B., Angew. Chem. Int. Ed. 2016, 55, 7676–7679; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7806–7810; [Google Scholar]
- 18e. Singh-Rachford T. N., Castellano F. N., Coord. Chem. Rev. 2010, 254, 2560–2573; [Google Scholar]
- 18f. Ghosh I., Shaikh R. S., König B., Angew. Chem. Int. Ed. 2017, 56, 8544–8549; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 8664–8669; [Google Scholar]
- 18g. Kim H., Kim H., Lambert T. H., Lin S., J. Am. Chem. Soc. 2020, 142, 2087–2092; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18h. Cowper N. G. W., Chernowsky C. P., Williams O. P., Wickens Z. K., J. Am. Chem. Soc. 2020, 142, 2093–2099; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18i. Yin H., Jin Y., Hertzog J. E., Mullane K. C., Carroll P. J., Manor B. C., Anna J. M., Schelter E. J., J. Am. Chem. Soc. 2016, 138, 16266–16273; [DOI] [PubMed] [Google Scholar]
- 18j. Mackenzie I. A., Wang L., Onuska N. O. R., Williams O. F., Begam K., Moran A. M., Dunietz B. D., Nicewicz D. A., Nature 2020, 580, 76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Zhang L., Jiao L., Chem. Sci. 2018, 9, 2711–2722; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19b. Zhang L., Jiao L., J. Am. Chem. Soc. 2019, 141, 9124–9128. [DOI] [PubMed] [Google Scholar]
- 20.Deposition Numbers 2181414 (for 6), 2181417 (for Z-7), 2181418 (for E-7), 2181415 (for 8 Et ), 2181416 (for 9 Et ), 2181419 (for 10 Cy ), 2181420 (for 10 Et ), 2181421 (for 11 Cy ), 2181422 (for 12 Cy ), 2181423 (for 13 Et ) and 2181424 (for 14 Cy ) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 21. Straub D. K., J. Chem. Educ. 1995, 72, 494–497. [Google Scholar]
- 22.This oxidant was chosen due to the counter ion GaCl4 −, to enable facile crystallization and X-ray characterization.
- 23. Chmiel A. F., Williams O. P., Chernowsky C. P., Yeung C. S., Wickens Z. K., J. Am. Chem. Soc. 2021, 143, 10882–10889. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.