

Abstract
Duchenne muscular dystrophy (DMD) is the most common pediatric‐onset form of muscular dystrophy, occurring in 1 in 5,000 live male births. DMD is a multi‐system disease resulting in muscle weakness with progressive deterioration of skeletal, heart, and smooth muscle, and learning disabilities. Pathogenic/likely pathogenic (P/LP) variants in the DMD gene, which encodes dystrophin protein, cause dystrophinopathy. All males with a P/LP variant in the X‐linked DMD gene are expected to be affected. Two to 20% of female heterozygotes with a P/LP variant develop symptoms of dystrophinopathy ranging from mild muscle weakness to significant disability similar to Becker muscular dystrophy. Recently, with improvements in therapies and testing methodology, there is stronger evidence supporting newborn screening (NBS) for DMD for males and females because females may also develop symptoms. A consented pilot study to screen newborns for DMD was initiated in New York State (NYS) and conducted from 2019 to 2021. The identification of female carriers and the realization of the subsequent uncertainty of providers concerning follow‐up during the pilot led to the development of algorithms for screening and diagnosis of carrier females, including both NBS and cascade molecular testing of family members.
Keywords: duchenne muscular dystrophy, dystrophinopathy, female carrier, newborn screening
1. INTRODUCTION
Duchenne muscular dystrophy (DMD) is an X‐linked disorder and the most common pediatric‐onset form of muscular dystrophy, with an incidence of ~1 in 3,600–5,000 live male births (Kwon et al., 2016; Mah et al., 2014). DMD is a dystrophinopathy caused by pathogenic variants in the DMD gene, which encodes dystrophin protein (Cho & Choi, 2013). Dystrophin is localized on the cytoplasmic surface of the plasma membrane of skeletal and cardiac muscle cells (Hoffman, Brown Jr, & Kunkel, 1987). The disease results in muscle weakness and progressive deterioration of skeletal, cardiac, and smooth muscle, as well as an increased risk for learning disabilities. DMD and Becker muscular dystrophies (BMD) are both dystrophinopathies with similar signs and symptoms but caused by different pathogenic variants in the DMD gene. The two conditions differ in their severity, age of onset, and rate of progression, with BMD being less severe.
Because the DMD gene is X‐linked (Boyd et al., 1986; Greenstein et al., 1980), all males with a P/LP variant in the DMD gene are expected to be affected. A female with a single P/LP variant is a “carrier.” There is currently no consensus regarding proper nomenclature for unaffected and affected “female carriers” (Apkon et al., 2021). Female carriers of DMD can transmit pathogenic variants to their offspring and may not exhibit symptoms themselves. However, under certain circumstances, female carriers may also be symptomatic and are referred to as “manifesting carriers” or females with dystrophinopathy (Apkon et al., 2021). In this article, we will refer to females who are heterozygous for P/LP variants in the DMD gene as female carriers. We will refer to females who are heterozygous for a P/LP variant and symptomatic as “females with dystrophinopathy.”
In recent years, with improvements in therapies and testing methodology, including CK‐MM from dried blood spots, there is stronger evidence and feasibility to support NBS for DMD for males, but the utility for females is less clear. Currently in the United States, DMD is not included as a condition on the Recommended Uniform Screening Panel (RUSP), and no states are routinely screening for DMD. However, pilots are underway or recently completed in three states (Hartnett et al., 2022; Migliore et al., 2022; Parad, Sheldon, & Bhattacharjee, 2021), a RUSP nomination package has been submitted, and at least one state is considering adding DMD to their panel. This increase in support for DMD NBS is consistent with an overall movement to more broadly consider this disorder for NBS, recognizing benefits beyond medical treatment, which often includes avoiding a diagnostic odyssey, family planning, and enrollment in early childhood intervention services, as discussed by Chung et al. (2022). The addition of neuromuscular diseases to NBS is recent, with spinal muscular atrophy (SMA) added to the RUSP in the United States in 2018. The addition of SMA has developed a pathway for the inclusion of neuromuscular disorders to newborn screening panels.
NBS in the United States is universal and performed irrespective of sex. Although the data regarding female carriers of P/LP variants in the DMD gene are limited, female carriers identified through family history can have musculoskeletal and cardiac symptoms. Rarely, females have symptoms and disabilities similar to males with DMD/BMD. The prevalence of manifesting female carriers of DMD varies among studies. A recent nationwide epidemiological study from New Zealand conducted by Theadom et al. (2014) reported a prevalence of 0.35/100,000, while a study of hospital records in Norway over 21 years conducted by Müller, Ghelue, Lund, Jonsrud, and Arntzen (2021) reported prevalence as 0.8/100,000. Given that female DMD carriers may develop symptoms, it is important to screen both sexes early on and identify babies who are at risk for symptoms.
2. PHENOTYPE OF FEMALE CARRIERS
2.1. Neuromuscular phenotype
Female carriers can develop a broad range of symptoms of dystrophinopathy. The reported percentage of heterozygotes who are symptomatic varies widely, depending on the population evaluated, the method of evaluation, and the definition of symptomatic, with 2.5% to more than 20% of carriers being reported as symptomatic in past studies (Cho & Choi, 2013; Hoogerwaard, Bakker, et al., 1999; Hoogerwaard, van der Wouw, et al., 1999). Female carriers can manifest symptoms from muscle weakness and clumsiness in childhood, proximal muscle weakness, myalgia/cramps, unexplained abdominal or chest pain, pseudohypertrophy of the calf muscle, cardiac abnormalities to severe gait problems in young girls with the classic DMD clinical phenotype (Darras, Urion, & Ghosh, 2022) (Table 1). Some manifesting female carriers only present with myalgia or cramps without limb weakness (Ceulemans, Storm, Reyniers Jr, Callewaert, & Martin, 2008), camptocormia (Findlay, Lewis, Sahenk, & Flanigan, 2013), or developmental delays in childhood, while others may present with isolated cardiomyopathy without skeletal muscle weakness (Kinoshita et al., 1995; Mah et al., 2020; Mirabella et al., 1993; Walcher et al., 2010; Table 1). A recent evaluation of muscle MRI and muscle strength of adult carriers showed that 81% had reduced muscle strength or elevated fat fraction (Fornander et al., 2021). The age of muscle symptom onset is broad, with reports of onset from ages 1 to 50 years (Hoogerwaard, Bakker, et al., 1999; Silva et al., 2020). The muscle symptoms, including loss of strength, can be progressive over time (Silva et al., 2020).
TABLE 1.
Common symptoms reported in female carriers
| Female carriers commonly report: |
| A history of weakness and clumsiness in childhood (e.g., poor performance in sports) |
| Proximal muscle weakness (maybe asymmetric) leading to: |
| Exhaustion/tired all the time (TATT) |
| Difficulty getting up and down stairs |
| Difficulty getting up from sitting to standing position |
| Awkward gait |
| Myalgia (growing pains, cramps) |
| Unexplained abdominal or chest pain |
| Tachycardia of unknown origin |
| Pseudohypertrophy of calf muscle |
2.2. Cardiac manifestations
Female carriers of DMD P/LP variants have long been known to have an increased risk for cardiomyopathy in adulthood (Hoogerwaard, van der Wouw, et al., 1999). However, past research regarding its prevalence and severity has been inconsistent and dependent on the method of evaluation and definition of cardiac disease. Prevalence of dilated cardiomyopathy in carriers has been reported as 8%–18% (Adachi et al., 2018). Cases of female carriers with left ventricular noncompaction cardiomyopathy have also been observed (Parent et al., 2015). More recent studies using higher resolution imaging such as cardiovascular magnetic resonance imaging (cMRI) have shown a higher prevalence of female carriers with cardiac involvement. Mah et al. (2020) found that 49% of female carriers had cardiac fibrosis on cardiac MRI. Fibrosis and inflammation may alter conduction pathways and increase susceptibility to cardiac arrhythmias, with persistent labile sinus tachycardia being the most common (Cullom, Vo, & MD, 2021). Age and serum CK levels were also positively associated with an increased risk of identification of cardiac fibrosis. More than two‐thirds of female carriers in a recent small study had cardiac involvement, including fibrosis, abnormal ECG, abnormal Holter monitoring, or abnormal function on echocardiography (Solheim et al., 2021). Fibrosis was more frequent in female carriers of DMD‐associated variants than in female carriers with BMD‐associated variants. Florian et al. (2016) demonstrated similar findings with elevated CK and a DMD‐associated variant and a higher likelihood of abnormalities on cardiac MRI.
Cardiac disease may be subclinical or progressive, and in some cases, it may even require heart transplantation. It may precede or occur in the absence of muscle involvement. Pregnancy and delivery have been reported to exacerbate the development and progression of cardiac conditions, with multiple case reports of heart failure in the third trimester or pospartum (Cullom et al., 2021; Kondo et al., 2017). Early detection and prompt initiation of heart failure therapy can delay progression and may reverse remodeling (Cullom et al., 2021). Clinical guidelines recommend that female carriers have a cardiac evaluation in late adolescence or early adulthood with electrocardiogram and echocardiogram, and reevaluation at least every 5 years, starting at age 25 (American Academy of Pediatrics, 2005; Bushby et al., 2002; Cullom et al., 2021). Addition of speckle tracking echocardiography (STE) to detect subclinical myocardial dysfunction is also recommended. STE enables quantitative assessment of regional myocardial deformation for early detection of cardiac fibrosis due to cardiomyopathy in patients with dystrophinopathy (Adachi et al., 2018). However, since myocardial damage may be present even if echocardiogram findings are normal, it is recommended that cMRI with late gadolinium‐enhancement is also performed starting in the third decade (Apkon et al., 2021; Cullom et al., 2021). cMRI has demonstrated a similar pattern of myocardial fibrosis in DMD patients as it does in carrier females (Cullom et al., 2021).
2.3. Anesthesia
Because females with dystrophinopathy may develop skeletal muscle weakness and cardiomyopathy and may be at risk for cardiac arrhythmia, rhabdomyolysis, respiratory compromise, difficult airway, and coagulopathy, appropriate anesthetic management may be required (Cullom et al., 2021). Currently there are no guidelines specific to females with dystrophinopathy and the management is based on treatment of males with DMD and BMD. Cases of hyperkalemic cardiac arrest after exposure to volatile anesthetics and succinylcholine have been reported in individuals with dystrophinopathy.
3. MECHANISM OF SEVERE DISEASE IN FEMALES
Manifesting disease symptoms in females is likely related to the amount of dystrophin present. Genetic mechanisms causing severe symptoms in females include a large deletion involving Xp21.2, an X‐chromosome rearrangement involving Xp21.2, 45 X, uniparental disomy (UPD) of the X‐chromosome, two DMD pathogenic variants, and nonrandom X‐chromosome inactivation (Bakker et al., 1987; Martinez, Pignatelli, Belmont, Craigen, & Jefferies, 2011; Soltanzadeh et al., 2010). Genotype–phenotype correlations in males with dystrophinopathy are well‐described but predicting phenotype correlations in females with DMD variants is more complicated. In general, males with deletion/duplication variants that preserve the reading frame are more likely to have BMD, while males who have variants that disrupt the reading frame are more likely to have DMD. Additional studies have shown that the exon location of nonsense sequence variants can be predictive of phenotype (Torella et al., 2020). The cognitive phenotype may also be affected by variant location.
Within the DMD spectrum, some variants have been noted to result in faster or slower progression of neuromuscular disease. Variants that allow for increased expression, have a slower disease course, often with a later age of loss of ambulation (de Feraudy, 2021). For example, copy number variants amenable to exon skipping of exon 44 have been noted to result in “milder” disease with slower progression (van den Bergen, Ginjaar, Niks, Aartsma‐Rus, & Verschuuren, 2014), while deletions amenable to exon 51 skipping may result in more severe disease (Wang et al., 2018). Deletions flanking exon 44 appear to have endogenous exon‐skipping, resulting in some full‐length dystrophin production (Wang et al., 2018).
The Duchenne Registry (https://www.duchenneregistry.org/), a patient report registry supported by PPMD, includes verified genetic variants and self‐reported phenotypes on more than 200 female carriers (The Duchenne Registry, 2022). Female carriers report whether they consider themselves manifesting or asymptomatic. Within the database, manifesting carriers were more likely to have an out‐of‐frame deletion or duplication, 80% versus 62% of asymptomatic females (The Duchenne Registry, 2022).
4. THE NEW YORK STATE PILOT STUDY
On October 1, 2019, a consented pilot study to screen newborns for DMD was initiated in New York State (NYS). The pilot study was a collaboration between Parent Project Muscular Dystrophy (PPMD), New York State Newborn Screening (NYS NBS) program, Northwell Health Hospitals, New York‐Presbyterian Hospitals, the National Institutes of Health (NIH)‐supported Newborn Screening Translational Research Network (NBSTRN) housed at the American College of Medical Genetics and Genomics (ACMG), and funders. The NYS and the clinical sites' Institutional Review Boards (IRB) approved the study.
The NYS pilot followed a two‐tier screening program, with CK‐MM as the first‐tier assay, followed by DMD gene molecular testing as the second tier. Persistently elevated levels of the muscle isoform of creatine kinase (CK‐MM) are associated with neuromuscular diseases, with dystrophinopathies being the most common cause (Moat et al., 2017; Timonen et al., 2019). CK‐MM results were categorized as normal, borderline, or elevated. The cut‐offs selected for the assay were irrespective of gender and based on the age of the newborn at specimen collection (Park et al., 2022). A repeat sample was requested from babies with borderline CK‐MM measurements for repeat CK‐MM, while those with elevated levels were recommended to undergo DMD genetic testing. In the early stages of the pilot, genetic testing required an additional sample. In the later stages of the pilot, genetic testing could be performed on remaining NBS dried blood samples.
Elevations in CK‐MM are well‐described in Duchenne, Becker, and manifesting female carriers (Timonen et al., 2019; Moat et al., 2017). In December 2019, the FDA approved the first‐tier screen kit for DMD, which measures the level of creatine kinase‐MM in dried blood spots. The test kit is high throughput and provides a method for universal screening of DMD in newborns (Chien et al., 2022; Ke et al., 2017; Migliore et al., 2022; Moat et al., 2017; Parad, Sheldon, & Bhattacharjee, 2021; Timonen et al., 2019). Data from pilot studies indicate that the assay can also detect other muscular dystrophies (Ke et al., 2017; Timonen et al., 2019). Consequently, genetic testing for a panel of neuromuscular conditions was offered in cases where the baby had elevated CK‐MM and negative DMD genetic testing.
The NYS 2‐year Duchenne NBS pilot results support the implementation of NBS for DMD. During the duration of the pilot, 36,781 newborns were screened for CK‐MM. Forty‐two newborns (25 male, 17 female) were referred for second‐tier testing. Deletions or duplications in the DMD gene were detected in four male infants consistent with DMD or BMD. One female DMD carrier was identified. A second female baby had a borderline CK‐MM result requiring follow‐up. However, the mother, who later identified herself as a carrier, declined to submit another specimen for testing or follow‐up. CK‐MM screening in an earlier study also identified one carrier of a variant associated with BMD, while another carrier was missed (Timonen et al., 2019).
NBS for genetic disorders often results in the identification of other affected or at‐risk family members through cascade genetic testing. In the NYS pilot, babies identified through NBS were offered genetic counseling and testing of family members. While DMD is a relatively common genetic condition, primary care providers caring for families within the pilot struggled as to how to manage any females identified through NBS or cascade testing. Consequently, as the pilot proceeded, algorithms were developed to assist in the screening and diagnosis of carrier females.
5. IDENTIFICATION OF FEMALE CARRIERS: FEMALE INFANT WITH ABNORMAL CK‐MM (SCENARIO 1)
The level of serum CK in female carriers, and therefore CK‐MM, is dependent on the expression of dystrophin, which varies based on the DMD variant itself, X‐inactivation, and distribution of the DMD variant. Measuring CK‐MM levels may be a useful initial screen for symptomatic carriers. The majority of female carriers have elevated CK serum levels, but the levels are higher in those with muscle symptoms such as weakness. Fornander et al. (2021) found that 57% of DMD variant carriers had elevated CK, with carriers of variants associated with a DMD phenotype having a higher mean CK than those with variants associated with a BMD phenotype. This is not unexpected, as carriers of BMD‐associated variants are likely to produce more dystrophin and therefore have less muscle damage than carriers with a DMD‐associated variant. A higher CK level was also associated with a higher fat fraction in this study, supporting previous studies that have shown that female carriers with muscle symptoms are more likely to have elevated CK levels. Similarly, when Zhong et al. (2019) compared females with dystrophinopathy to asymptomatic carriers identified because of family history, females with dystrophinopathy had significantly higher serum CK levels. Serial CK levels may also be useful in assessing disease severity, as persistently elevated CK is seen in individuals with muscle damage (Apkon et al., 2021).
Evaluation (Figure 1 ): Female infants with an elevated serum CK‐MM were confirmed to have elevated CK‐MM on repeat CK‐MM measurement and then had molecular genetic testing to confirm the diagnosis of DMD‐BMD or other neuromuscular diseases. An adequate time for the counseling session was allowed to explain the potential likelihood and risks of male DMD/BMD, female dystrophinopathy, and the possibility of other types of non‐sex‐linked muscular dystrophies. Family history of symptoms of neurological and cardiac disease was elicited. If the diagnosis of DMD/BMD is confirmed by genetic testing (i.e., detection of P/LP variants in the DMD gene via sequencing or deletion/duplication analysis), consultation with the pediatric neuromuscular disease specialist or comprehensive muscular dystrophy clinic and subsequent continued developmental evaluation was recommended. Female carriers who have symptoms in the DMD/BMD spectrum are typically treated according to their symptoms (Apkon et al., 2021).
FIGURE 1.
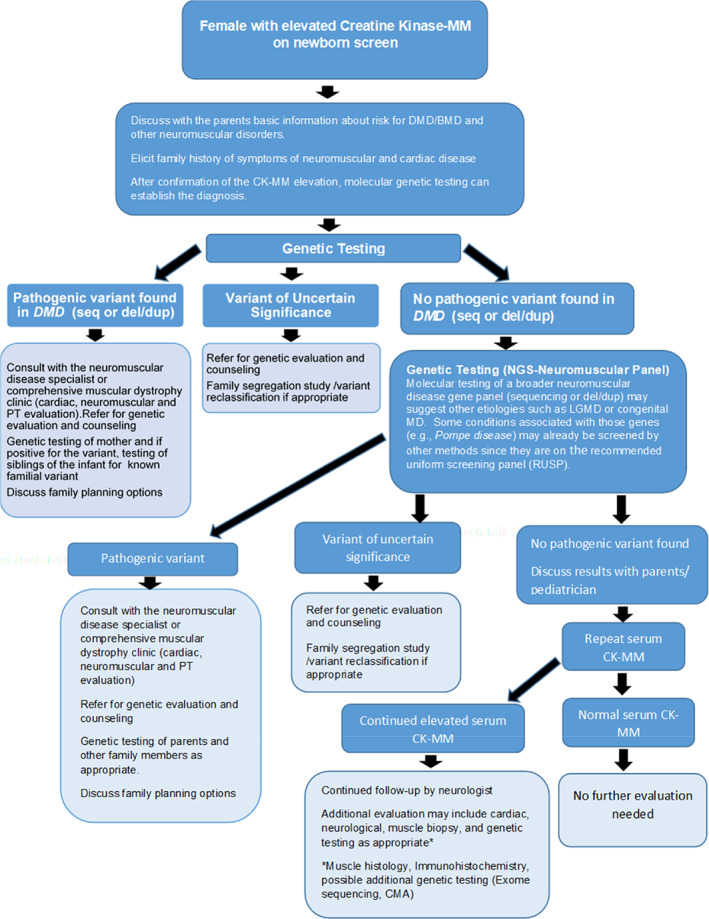
Identification of female carriers: Female Infant presenting with an Elevated Serum CK‐MM (Scenario 1)
In addition, once a pathogenic variant is identified in the family, testing of family members was offered. Carrier testing for at‐risk mothers and siblings, particularly brothers who have not yet been tested to determine their variant status, and prenatal testing or preimplantation genetic testing for future pregnancies were discussed.
6. IDENTIFICATION OF FEMALE CARRIERS: FAMILY MEMBERS (MALE OR FEMALE) WITH CONFIRMED GENETIC DIAGNOSES OF DMD OR BMD IDENTIFIED VIA AVENUES OTHER THAN NBS (SCENARIO 2)
Female carriers are most often identified later in life as part of family cascade testing triggered by a known P/LP variant in the DMD gene identified in a family member via genetic testing not associated with NBS. Identification of babies with results consistent with DMD/BMD allows for parental and family testing of family members at risk of inheriting the same variant and developing DMD/BMD.
If there is no family history of DMD/BMD, the DMD mutation is de novo in approximately 30% of boys with Duchenne; the other 70% of mothers are carriers. The risk to the sibling of an infant diagnosed with DMD/BMD depends on the genetic status of the mother. Heterozygous females have a 50% chance of transmitting the DMD/BMD pathogenic variant in each pregnancy. If an infant is identified with DMD/BMD through NBS, it is possible that older brothers also may be affected but are not yet recognized clinically and/or have not been diagnosed. It is also possible that sisters are carriers.
Carrier testing for at‐risk females and prenatal or preimplantation genetic testing for future pregnancies are possible when the DMD/BMD pathogenic variant in the family is known. Genetic counseling and additional testing are recommended for the mother in these cases to help the family understand the health risks to family members, the consequences for future children, and options available for family planning.
Evaluation (Figure 2): As in Scenario 1, once the diagnosis of DMD/BMD is confirmed by genetic testing and the female carrier is identified (i.e., P/LP variant was found in the DMD gene via sequencing or deletion/duplication analysis), consultation with the neuromuscular disease specialist or comprehensive muscular dystrophy clinic and subsequent continued developmental evaluation should be considered. Further evaluation for female carriers is presented in Figure 2.
FIGURE 2.
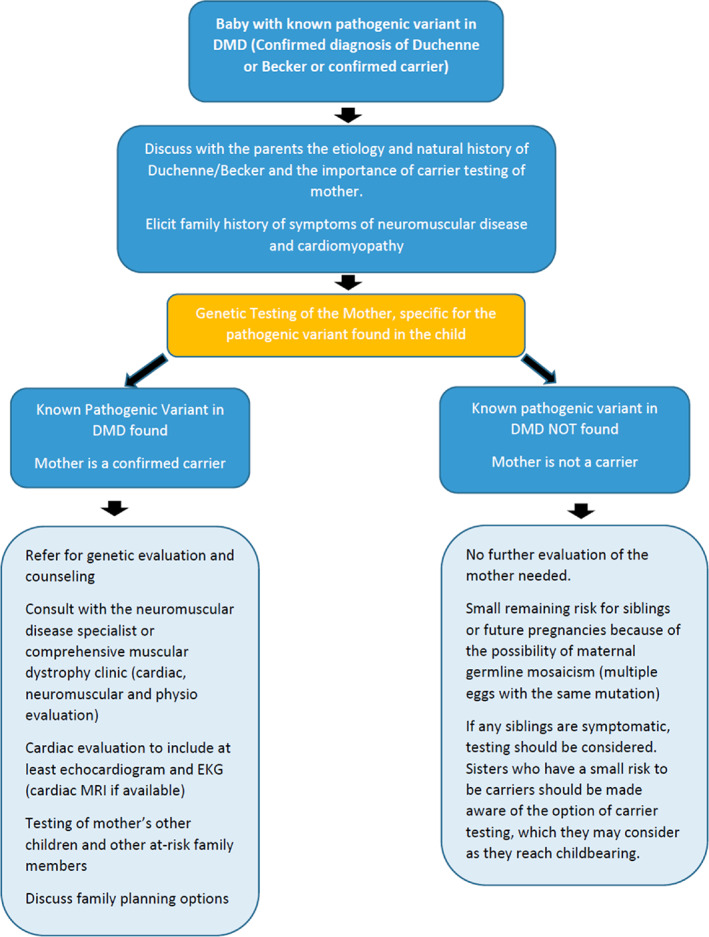
Identification of female carriers: Family members (Male or Female) with confirmed genetic diagnoses of DMD or BMD identified via avenue other than NBS (Scenario 2)
7. CLINICAL EVALUATION OF FEMALE CARRIERS
Female carriers may benefit from evaluations with a physical therapist, neurologist, and cardiologist. Assessing muscle strength and function and addressing issues that have been described as muscle cramps, exercise intolerance, and weakness can be done with a neurologist and physical therapist.
Generally, care depends upon the age of the carrier. In early adulthood, cardiac screening including echocardiogram, ECG, Holter and other methods of monitoring for arrhythmia, and cardiac MRI, can be utilized to look for underlying changes in cardiac structure and function.
Decreased health‐related quality of life and emotional distress among mothers of sons with muscular dystrophy have been described in a study conducted at Nationwide Children's Hospital (Jackson et al., 2021). When compared to sex and age group matched controls, mothers of sons with muscular dystrophy reported poorer health‐related quality of life across all domains of their health‐related quality of life, as well as higher levels of emotional distress (Jackson et al., 2021). This study was not able to differentiate whether these issues were related to being a carrier or being the mother of a child with degenerative and chronic condition. As in any other chronic condition, the need for psychological support for female carriers and their parents and siblings should also be addressed.
8. AVAILABILITY OF CURRENT THERAPEUTICS FOR DMD/BMD PATIENTS
Currently, there is no cure for DMD but there are treatments that slow disease progression. The advances in understanding the molecular pathways affected in DMD have led to the development of many therapeutic strategies. Two major strategies include restoration of dystrophin or the use of a surrogate molecule at the sarcolemma to improve the structural integrity of muscle fibers and address secondary symptoms of the condition and slow down disease progression (Fortunato, Rossi, Falzarano, & Ferlini, 2021; Falzarano, Scotton, Passarelli, & Ferlini, 2015). Corticosteroids are the only medication currently available for all patients, while exon skipping therapies are available for a subset of patients with particular genetic variants. Corticosteroids can slow the loss of skeletal muscle strength and function in DMD. For females with dystrophinopathy, it has been proposed that any female with symptoms consistent with DMD should be treated similarly, while those whose features are more consistent with BMD should be treated as BMD is treated (Apkon et al., 2021). This is especially relevant for any female with a DMD P/LP variant and 45, X karyotype, given the absence of the second DMD copy.
9. FUTURE STUDIES
While female carriers are relatively common and have been identified for decades, large long‐term studies are needed to understand the frequency and severity of muscle and cardiac disease. At this time, exact long‐term outcomes for infant female carriers identified by CK‐MM NBS are unknown. Additional studies following these babies' development, CK levels, and outcomes should be conducted to provide information to families identified through NBS. In addition, clinical trials that allow for the enrollment of females with dystrophinopathy are needed.
10. CONCLUSION
As females are at risk of developing dystrophinopathies, NBS for DMD/BMD should be made available to all infants. Female carriers may experience a variety of medical issues, including muscle weakness and cramps, exercise intolerance, myalgias, and cardiac disease. There is clinical heterogeneity with some women experiencing no signs or symptoms and others having progressive disease. A better understanding of the natural history in female carriers will support future evidence‐based clinical management. Identification of female carriers has the secondary benefit of identifying other affected males or carrier females in the family.
ACKNOWLEDGMENTS
The authors wish to thank the Duchenne Steering Committee for valuable input throughout the pilot study. This study was funded by Parent Project Muscular Dystrophy, PerkinElmer, Inc. (in‐kind support), Pfizer, Inc., PTC Therapeutics, Sarepta Therapeutics, Solid Biosciences, and Wave Life Sciences.
Gruber, D. , Lloyd‐Puryear, M. , Armstrong, N. , Scavina, M. , Tavakoli, N. P. , Brower, A. M. , Caggana, M. , & Chung, W. K. (2022). Newborn screening for Duchenne muscular dystrophy‐early detection and diagnostic algorithm for female carriers of Duchenne muscular dystrophy. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 190C:197–205. 10.1002/ajmg.c.32000
Funding information Wave Life Sciences; Solid Biosciences; Sarepta Therapeutics; PTC Therapeutics; Pfizer, Inc.; PerkinElmer, Inc.; Parent Project Muscular Dystrophy
REFERENCES
- Adachi, K. , Hashiguchi, S. , Saito, M. , Kashiwagi, S. , Miyazaki, T. , Kawai, H. , … Kimura, E. (2018). Detection and management of cardiomyopathy in female dystrophinopathy carriers. Journal of the Neurological Sciences, 386, 74–80. 10.1016/j.jns.2017.12.024 [DOI] [PubMed] [Google Scholar]
- American Academy of Pediatrics Section on Cardiology and Cardiac Surgery . (2005). Cardiovascular health supervision for individuals affected by Duchenne or Becker muscular dystrophy. Pediatrics, 116, 1569–1573. [DOI] [PubMed] [Google Scholar]
- Apkon, S. , Kinnett, K. , Cripe, L. , Duan, D. , Jackson, J. L. , Kornegay, J. N. , … Flanigan, K. M. (2021). Parent Project Muscular Dystrophy females with Dystrophinopathy conference, Orlando, Florida June 26–June 27, 2019. Journal of Neuromuscular Diseases, 8(2), 315–322. 10.3233/JND-200555 PMID: 33361607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker, E. , Van Broeckhoven, C. , Bonten, E. J. , van de Vooren, M. J. , Veenema, H. , Van Hul, W. , … Pearson, P. L. (1987). Germline mosaicism and Duchenne muscular dystrophy mutations. Nature, 329(6139), 554–556. 10.1038/329554a0 [DOI] [PubMed] [Google Scholar]
- Bushby, K. , Muntoni, F. , & Bourke, J. P. (2002). 107th ENMC international workshop: The management of cardiac involvement in muscular dystrophy and myotonic dystrophy. 7th‐9th June 2002, Naarden, The Netherlands. Neuromuscular Disorders, 2003(13), 166–172. [DOI] [PubMed] [Google Scholar]
- Boyd, Y. , Buckle, V. , Holt, S. , Munro, E. , Hunter, D. , & Craig, I. (1986). Muscular dystrophy in girls with X;autosome translocations. Journal of Medical Genetics, 23(6), 484–490. 10.1136/jmg.23.6.484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceulemans, B. P. , Storm, K. , Reyniers, E., Jr. , Callewaert, L. , & Martin, J. J. (2008). Muscle pain as the only presenting symptom in a girl with dystrophinopathy. Pediatric Neurology, 38(1), 64–66. 10.1016/j.pediatrneurol.2007.09.006 [DOI] [PubMed] [Google Scholar]
- Chien, Y. H. , Lee, N. C. , Weng, W. C. , Chen, L. C. , Huang, Y. H. , Wu, C. S. , & Hwu, W. L. (2022). Duchenne muscular dystrophy newborn screening: The first 50,000 newborns screened in Taiwan. Neurological Sciences, 43, 4563–4566. 10.1007/s10072-022-06128-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, Y. N. , & Choi, Y. C. (2013). Female carriers of Duchenne muscular dystrophy. Journal of Genetic Medicine, 10, 94–98 Published online December 31, 2013. 10.5734/JGM.2013.10.2.94 [DOI] [Google Scholar]
- Chung, W. K. , Berg, J. S. , Botkin, J. R. , Brenner, S. E. , Brosco, J. P. , Brothers, K. B. , … Hu, Z. (2022). Newborn screening for neurodevelopmental diseases: Are we there yet? American Journal of Medical Genetics. Part C, Seminars in Medical Genetics, 190(2), 222–230. 10.1002/ajmg.c.31988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullom, C. , Vo, V. , & MD, M. C. (2021). Orthotopic heart transplantation in manifesting carrier of Duchenne muscular dystrophy. Journal of Cardiothoracic and Vascular Anesthesia, 00, 1–7. 10.1053/j.jvca.2021.09.047 [DOI] [PubMed] [Google Scholar]
- Darras, B.T., Urion, D. K., & Ghosh, P. S. (2022). Dystrophinopathies. In M. P. Adam, D. B. Everman, G. M. Mirzaa, et al. (Eds.), GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle. [Google Scholar]
- van den Bergen, J. C. , Ginjaar, H. B. , Niks, E. H. , Aartsma‐Rus, A. , & Verschuuren, J. J. (2014). Prolonged ambulation in Duchenne patients with a mutation amenable to exon 44 skipping. Journal of Neuromuscular Diseases, 1(1), 91–94. [PubMed] [Google Scholar]
- Falzarano, M. S. , Scotton, C. , Passarelli, C. , & Ferlini, A. (2015). Duchenne muscular dystrophy: From diagnosis to therapy. Molecules, 20(10), 18168–18184. 10.3390/molecules201018168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Feraudy, Y. , Ben Yaou, R. , Wahbi, K. , Stalens, C. , Stantzou, A. , Laugel, V. , … Amthor, H. (2021. Feb). Very low residual dystrophin quantity is associated with milder Dystrophinopathy. Annals of Neurology, 89(2), 280–292. 10.1002/ana.25951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay, A. R. , Lewis, S. , Sahenk, Z. , & Flanigan, K. M. (2013. Jan). Camptocormia as a late presentation in a manifesting carrier of duchenne muscular dystrophy. Muscle & Nerve, 47(1), 124–127. 10.1002/mus.23497 [DOI] [PubMed] [Google Scholar]
- Florian, A. , Rösch, S. , Bietenbeck, M. , Engelen, M. , Stypmann, J. , Waltenberger, J. , … Yilmaz, A. (2016). Cardiac involvement in female Duchenne and Becker muscular dystrophy carriers in comparison to their first‐degree male relatives: A comparative cardiovascular magnetic resonance study. European Heart Journal Cardiovascular Imaging, 17(3), 326–333. 10.1093/ehjci/jev161 [DOI] [PubMed] [Google Scholar]
- Fornander, F. , Solheim, T. Å. , Eisum, A. V. , Poulsen, N. S. , Andersen, A. G. , Dahlqvist, J. R. , … Vissing, J. (2021). Quantitative muscle MRI and clinical findings in women with pathogenic dystrophin gene variants. Frontiers in Neurology, 12, 707837. 10.3389/fneur.2021.707837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortunato, F. , Rossi, R. , Falzarano, M. S. , & Ferlini, A. (2021). Innovative therapeutic approaches for Duchenne muscular dystrophy. Journal of Clinical Medicine, 10(4), 820. 10.3390/jcm10040820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenstein, R. M. , Reardon, M. P. , Chan, T. S. , Middleton, A. B. , Mulivor, R. A. , Greene, A. E. , & Coriell, L. L. (1980). An (X;11) translocation in a girl with Duchenne muscular dystrophy. Repository identification no. GM1695. Cytogenetics and Cell Genetics, 27(4), 268. 10.1159/000131496 [DOI] [PubMed] [Google Scholar]
- Hartnett, M. J. , Lloyd‐Puryear, M. A. , Tavakoli, N. P. , Wynn, J. , Koval‐Burt, C. L. , Gruber, D. , … Brower, A. M. (2022). Newborn screening for Duchenne muscular dystrophy: First year results of a population‐based pilot. International Journal of Neonatal Screening, 8, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman, E. P. , Brown, R. H., Jr. , & Kunkel, L. M. (1987). Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell, 51(6), 919–928. 10.1016/0092-8674(87)90579-4 [DOI] [PubMed] [Google Scholar]
- Hoogerwaard, E. M. , Bakker, E. , Ippel, P. F. , Oosterwijk, J. C. , Majoor‐Krakauer, D. F. , Leschot, N. J. , … de Visser, M. (1999). Signs and symptoms of Duchenne muscular dystrophy and Becker muscular dystrophy among carriers in The Netherlands: A cohort study. Lancet, 353(9170), 2116–2119. 10.1016/s0140-6736(98)10028-4 [DOI] [PubMed] [Google Scholar]
- Hoogerwaard, E. M. , van der Wouw, P. A. , Wilde, A. A. , Bakker, E. , Ippel, P. F. , Oosterwijk, J. C. , … de Visser, M. (1999). Cardiac involvement in carriers of Duchenne and Becker muscular dystrophy. Neuromuscular Disorders, 9(5), 347–351. 10.1016/s0960-8966(99)00018-8 [DOI] [PubMed] [Google Scholar]
- Jackson, J. L. , Korth, C. X. , Leslie, C. E. , Cotto, J. , Mah, M. L. , Hor, K. , … Mendell, J. R. (2021). Health‐related quality of life and emotional distress among mothers of sons with muscular dystrophy as compared to sex‐ and age group‐matched controls. Journal of Child Neurology, 36(3), 177–185. 10.1177/0883073820962927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita, H. , Goto, Y. , Ishikawa, M. , Uemura, T. , Matsumoto, K. , Hayashi, Y. K. , … Nonaka, I. (1995). A carrier of Duchenne muscular dystrophy with dilated cardiomyopathy but no skeletal muscle symptom. Brain & Development, 17(3), 202–205. 10.1016/0387-7604(95)00018-7 [DOI] [PubMed] [Google Scholar]
- Ke, Q. , Zhao, Z. Y. , Griggs, R. , Wiley, V. , Connolly, A. , Kwon, J. , … Gatheridge, M. (2017. Jun). Newborn screening for Duchenne muscular dystrophy in China: Follow‐up diagnosis and subsequent treatment. World Journal of Pediatrics, 13(3), 197–201. 10.1007/s12519-017-0036-3 [DOI] [PubMed] [Google Scholar]
- Kondo, T. , Okumura, T. , Takefuji, M. , Hiraiwa, H. , Sugiura, Y. , Watanabe, N. , … Murohara, T. (2017). Long‐term pathological follow‐up of myocardium in a carrier of Duchenne muscular dystrophy with dilated cardiomyopathy. Circulation. Heart Failure, 10(3), e003826. 10.1161/CIRCHEARTFAILURE.117.003826 [DOI] [PubMed] [Google Scholar]
- Kwon, J. M. , Abdel‐Hamid, H. Z. , Al‐Zaidy, S. A. , Mendell, J. R. , Kennedy, A. , Kinnett, K. , … Connolly, A. M. (2016). Clinical follow‐up for Duchenne muscular dystrophy newborn screening: A proposal. Muscle & Nerve, 54(2), 186–191. 10.1002/mus.25185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah, M. L. , Cripe, L. , Slawinski, M. K. , Al‐Zaidy, S. A. , Camino, E. , Lehman, K. J. , … Hor, K. N. (2020). Duchenne and Becker muscular dystrophy carriers: Evidence of cardiomyopathy by exercise and cardiac MRI testing. International Journal of Cardiology, 1(316), 257–265. 10.1016/j.ijcard.2020.05.052 [DOI] [PubMed] [Google Scholar]
- Mah, J. K., Korngut, L., Dykeman, J., Day, L., Pringsheim, T., & Jette, N. (2014). A systematic review and meta‐analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscular disorders: NMD, 24(6), 482–491. 10.1016/j.nmd.2014.03.008 [DOI] [PubMed] [Google Scholar]
- Martinez, H. R. , Pignatelli, R. , Belmont, J. W. , Craigen, W. J. , & Jefferies, J. L. (2011). Childhood onset of left ventricular dysfunction in a female manifesting carrier of muscular dystrophy. American Journal of Medical Genetics. Part A, 155A(12), 3025–3029. 10.1002/ajmg.a.33784 [DOI] [PubMed] [Google Scholar]
- Migliore, B. A. , Zhou, L. , Duparc, M. , Robles, V. R. , Rehder, C. W. , Peay, H. L. , & Kucera, K. S. (2022). Evaluation of the GSP Creatine kinase‐MM assay and assessment of CK‐MM stability in newborn, patient, and contrived dried blood spots for newborn screening for Duchenne muscular dystrophy. International Journal of Neonatal Screening, 8(1), 12. 10.3390/ijns8010012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabella, M. , Servidei, S. , Manfredi, G. , Ricci, E. , Frustaci, A. , Bertini, E. , … Tonali, P. (1993). Cardiomyopathy may be the only clinical manifestation in female carriers of Duchenne muscular dystrophy. Neurology, 43(11), 2342–2345. 10.1212/wnl.43.11.2342 [DOI] [PubMed] [Google Scholar]
- Moat, S. J. , Korpimäki, T. , Furu, P. , Hakala, H. , Polari, H. , Meriö, L. , … Weeks, I. (2017). Characterization of a blood spot Creatine kinase skeletal muscle isoform immunoassay for high‐throughput newborn screening of Duchenne muscular dystrophy. Clinical Chemistry, 63(4), 908–914. 10.1373/clinchem.2016.268425 [DOI] [PubMed] [Google Scholar]
- Müller, K. I. , Ghelue, M. V. , Lund, I. , Jonsrud, C. , & Arntzen, K. A. (2021. Jan). The prevalence of hereditary neuromuscular disorders in northern Norway. Brain and Behavior: A Cognitive Neuroscience Perspective, 11(1), e01948. 10.1002/brb3.1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parad, R. B. , Sheldon, Y. , & Bhattacharjee, A. (2021). Implementation of hospital‐based supplemental Duchenne muscular dystrophy newborn screening (sDMDNBS): A pathway to broadening adoption. International Journal of Neonatal Screening, 7(4), 77. 10.3390/ijns7040077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent, J. , Moore, R. , Taylor, M. D. , Towbin, J. A. , & Jefferies, J. L. (2015). Left ventricular noncompaction cardiomyopathy in Duchenne muscular dystrophy carriers. Journal of Cardiology Cases, 11(1), 7–9. 10.1016/j.jccase.2014.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S. , Maloney, B. , Caggana, M. , & Tavakoli, N. P. (2022). Creatine kinase‐MM concentration in dried blood spots from newborns and implications for newborn screening for Duchenne muscular dystrophy. Muscle & Nerve, 65, 652–658. 10.1002/mus.27533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva, T. , Anequini, I. P. , Fávero, F. M. , Voos, M. C. , Oliveira, A. , Telles, J. , & Caromano, F. A. (2020). Functional performance and muscular strength in symptomatic female carriers of Duchenne muscular dystrophy. Arquivos de Neuro‐Psiquiatria, 78(3), 143–148. 10.1590/0004-282X20190168 [DOI] [PubMed] [Google Scholar]
- Solheim, T. Å. , Fornander, F. , Raja, A. A. , Møgelvang, R. , Poulsen, N. S. , Dunø, M. , … Vissing, J. (2021). Cardiac involvement in women with pathogenic dystrophin gene variants. Frontiers in Neurology, 12, 707838. 10.3389/fneur.2021.707838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltanzadeh, P. , Friez, M. J. , Dunn, D. , von Niederhausern, A. , Gurvich, O. L. , Swoboda, K. J. , … Flanigan, K. M. (2010). Clinical and genetic characterization of manifesting carriers of DMD mutations. Neuromuscular Disorders, 20(8), 499–504. 10.1016/j.nmd.2010.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theadom, A. , Rodrigues, M. , Roxburgh, R. , Balalla, S. , Higgins, C. , Bhattacharjee, R. , … Feigin, V. (2014). Prevalence of muscular dystrophies: A systematic literature review. Neuroepidemiology, 43(3‐4), 259–268. [DOI] [PubMed] [Google Scholar]
- The Duchenne Registry . (2022). Genotype ‐Phenotype in carriers. [Unpublished raw data ]. Parent Project Muscular Dystrophy. Retrievd from https://www.duchenneregistry.org/
- Timonen, A. , Lloyd‐Puryear, M. , Hougaard, D. M. , Meriö, L. , Mäkinen, P. , Laitala, V. , … Korpimäki, T. (2019). Duchenne muscular dystrophy newborn screening: Evaluation of a new GSP® neonatal Creatine kinase‐MM kit in a US and Danish population. International Journal of Neonatal Screening, 5(3), 27. 10.3390/ijns5030027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torella, A. , Zanobio, M. , Zeuli, R. , Del Vecchio, B. F. , Savarese, M. , Giugliano, T. , … Nigro, V. (2020). The position of nonsense mutations can predict the phenotype severity: A survey on the DMD gene. PLoS One, 15(8), e0237803. 10.1371/journal.pone.0237803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walcher, T. , Kunze, M. , Steinbach, P. , Sperfeld, A. D. , Burgstahler, C. , Hombach, V. , & Torzewski, J. (2010). Cardiac involvement in a female carrier of Duchenne muscular dystrophy. International Journal of Cardiology, 138(3), 302–305. 10.1016/j.ijcard.2008.06.084 [DOI] [PubMed] [Google Scholar]
- Wang, R. T. , Barthelemy, F. , Martin, A. S. , Douine, E. D. , Eskin, A. , Lucas, A. , … Nelson, S. F. (2018). DMD genotype correlations from the Duchenne Registry: Endogenous exon skipping is a factor in prolonged ambulation for individuals with a defined mutation subtype. Human Mutation, 39(9), 1193–1202. 10.1002/humu.23561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, J. , Xie, Y. , Bhandari, V. , Chen, G. , Dang, Y. , Liao, H. , … Lan, D. (2019). Clinical and genetic characteristics of female dystrophinopathy carriers. Molecular Medicine Reports, 19(4), 3035–3044. 10.3892/mmr.2019.9982 [DOI] [PMC free article] [PubMed] [Google Scholar]