

Abstract
The tumour suppressor PTEN is a negative regulator of the PI3K/AKT signalling pathway. Liver‐specific deletion of Pten in mice results in the hyper‐activation PI3K/AKT signalling accompanied by enhanced genome duplication (polyploidization), marked lipid accumulation (steatosis) and formation of hepatocellular carcinomas. However, it is unknown whether polyploidization in this model has an impact on the development of steatosis and the progression towards liver cancer. Here, we used a liver‐specific conditional knockout approach to delete Pten in combination with deletion of E2f7/8, known key inducers of polyploidization. As expected, Pten deletion caused severe steatosis and liver tumours accompanied by enhanced polyploidization. Additional deletion of E2f7/8 inhibited polyploidization, alleviated Pten‐induced steatosis without affecting lipid species composition and accelerated liver tumour progression. Global transcriptomic analysis showed that inhibition of polyploidization in Pten‐deficient livers resulted in reduced expression of genes involved in energy metabolism, including PPAR‐gamma signalling. However, we find no evidence that deregulated genes in Pten‐deficient livers are direct transcriptional targets of E2F7/8, supporting that reduction in steatosis and progression towards liver cancer are likely consequences of inhibiting polyploidization. Lastly, flow cytometry and image analysis on isolated primary wildtype mouse hepatocytes provided further support that polyploid cells can accumulate more lipid droplets than diploid hepatocytes. Collectively, we show that polyploidization promotes steatosis and function as an important barrier against liver tumour progression in Pten‐deficient livers.
Keywords: atypical E2Fs, conditional knockout mice, hepatocellular carcinoma, non‐alcoholic fatty liver disease, polyploidization, PTEN, steatosis
Key points.
Every fourth person in the world is overweight and develops fatty liver disease, which increases the risk for liver cancer. Importantly, increased number of polyploid liver cells, with 4 or more whole genome sets, have been detected in fatty liver disease. In a mouse model of fatty liver through knocking out Pten, we inhibited the formation of polyploid cells and discovered that polyploid cells promote lipid accumulation and prevent liver cancer.
1. INTRODUCTION
PTEN is a tumour suppressor gene mutated in less than 5% of diagnosed patients with hepatocellular carcinoma (HCC), and its decreased expression correlates with advanced disease stage and poor prognosis. 1 PTEN dictates several cellular functions such as proliferation, energy metabolism and survival via activation of PI3K/AKT signalling. Mice with Pten‐deficient livers are characterized by severe steatosis, increased hepatocyte polyploidization, also known as pathological polyploidization, and liver tumorigenesis. 2 , 3 , 4 , 5 Therefore, liver‐specific Pten‐deficient mice are a wildly used as a mouse model for non‐alcoholic fatty liver disease (NAFLD) progressing to HCC. 6 However, the mechanisms underlying the association between polyploidization and lipid accumulation and liver cancer are still incompletely understood.
During normal postnatal development, hepatocytes can undergo successive rounds of genome duplication in the absence of cytokinesis to become polyploid. 7 , 8 The percentage of polyploid hepatocytes in mammalian livers ranges from up to 90% in rodents to 30% in humans. 9 , 10 Programmed polyploidization occurs in the liver during postnatal development, starting at 3 weeks of age in mice. 11 This developmental hepatocyte polyploidization programme is at least in part driven by insulin signalling and involves transcriptional inhibition of cytokinesis genes as well as the PIDDosome components Casp2 and Pidd1 via the E2F7 and E2F8 repressors. 11 , 12 , 13 , 14 However, a substantial increase in hepatocyte polyploidization is frequently observed as a result of liver injury or cell stress, which is referred to as pathological polyploidization. 15 , 16 , 17 It has been proposed that pathological polyploidization may act as a mechanism to increase genetic diversity and stress‐resistant hepatocyte clones. 18 Other studies suggest that the increase in cell size that comes with polyploidization could rewire the energy metabolism of hepatocytes. For example, polyploidization shifted ATP synthesis from mitochondrial oxidative phosphorylation towards glycolysis. 19 Furthermore, liver cell polyploidization is associated with increased lipid accumulation, 4 and impairment of insulin signalling, which is commonly seen in metabolic disorders, reduces the formation of polyploid hepatocytes. 12
We and others have previously shown that physiological polyploidization in the mouse liver can be inhibited by deletion of atypical E2Fs. 11 , 13 Surprisingly, blocking hepatocyte polyploidization had no major impact on liver cell differentiation, apoptosis or regeneration. 11 Mice with livers composed of predominantly diploid hepatocytes had the same life expectancy compared to mice with normal polyploid livers. Interestingly, end of life analysis revealed that mice with reduced polyploidy had an increased incidence of liver tumours compared to mice with normal polyploid livers. 20 The fact that enhanced polyploidization is associated with NAFLD suggests that pathological polyploidization is induced by metabolic stress. However, it remains obscure whether polyploidization in hepatocytes with altered metabolic activity has an impact on NAFLD development and its progression towards liver cancer.
In the present study, we combined liver‐specific deletion of E2f7/8 to inhibit liver cell polyploidization with deletion of Pten. We evaluated long‐term consequences of abolishing polyploidization in this model on lipid accumulation and liver tumorigenesis. We show that polyploidy favours steatosis and inhibits liver tumour formation under metabolic stress conditions induced by overactivation of the PI3K/Akt pathway. Our data strongly suggest that polyploidization functions as a potent barrier against liver tumour formation in Pten‐deficient mice, despite facilitating lipid accumulation.
2. MATERIALS AND METHODS
2.1. Animal experiments
Animal experiments were approved by the Utrecht University Animal Ethics Committee (approval number: 2011.III.02.019) and performed according to institutional and national guidelines (experimental protocol: 103976–2). Mice were housed under standard conditions of temperature and housing. Breeding and generation of Albumin‐cre, Pten f/f and E2f7/8 f/f mice for experiments have been previously described. 11 , 21 Genotyping was performed using allele‐specific primers (Supplementary Table S1). Mouse lines were maintained on at least sixth generation of FVB background. Figures 1, 2, 3 were generated with liver‐specific knock‐out male and female mice harvested at the age of 16 weeks old. For the long‐term cross‐sectional study, male and female mice with liver‐specific loss of E2f7/8 (78 ∆/∆, n = 18), Pten (Pten ∆/∆, n = 18), and combination of the three genes (78Pten ∆/∆, n = 21) together with controls (78Pten f/f , n = 20) littermates were sacrificed at the age of 10 months and analysed for macroscopic and microscopic tumours.
FIGURE 1.
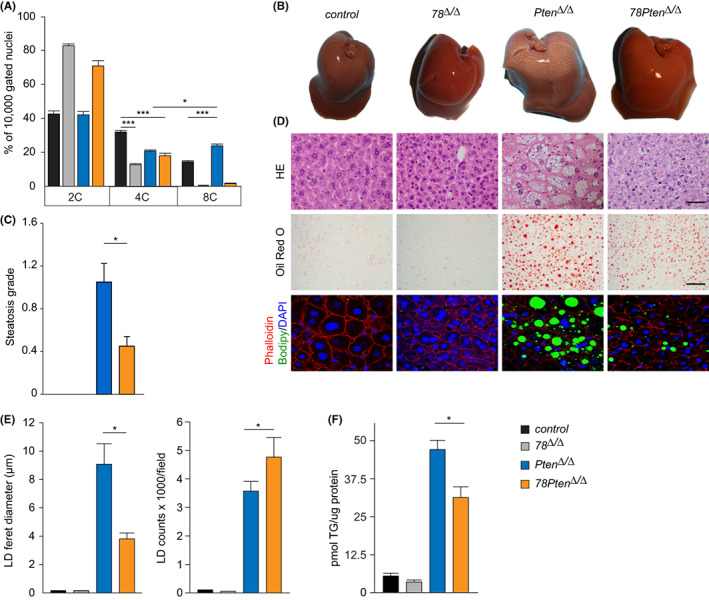
Loss of E2f7/8 prevents polyploidization and steatosis in Pten deficient livers. (A) Ploidy status of 16 weeks‐old livers from the indicated genotypes (control n = 10; 78 Δ/Δ n = 5; Pten Δ/Δ n = 8; 78Pten Δ/Δ n = 5 mice/genotype). Bar graphs represent average and standard error of the mean(SEM). (B) Representative macroscopic images of 16 weeks‐old livers from the indicated genotypes. (C) Steatosis grade of liver sections determined by a board‐certified veterinary pathologist (n = at least 5 mice/genotype). (D) Representative microscopic pictures of haematoxylin and eosin (HE) (Scale bar 20 μm), Oil Red O (lipid droplets, Scale bar 20 μm) and Bodipy (lipid droplets)/ Phalloidin(hepatocyte membrane)/ DAPI (nuclei, 6300x magnification) stained liver sections from 16 weeks‐old livers from the indicated genotypes. (E) Quantification of lipid droplet (LD) size and counts from 8–10 Oil Red O‐stained images per liver section from the indicated genotypes (n = 4 mice/genotype). (F) Quantification of triglyceride content in liver homogenates. Bar graphs represent average and SEM (n = 5). Data information: In (A), *p < .05, ***p < .001 (t‐test). In (C, E and F), *p < .05 (Kruskal Wallis one way analysis of variance on ranks and Dunn's post hoc correction).
FIGURE 2.
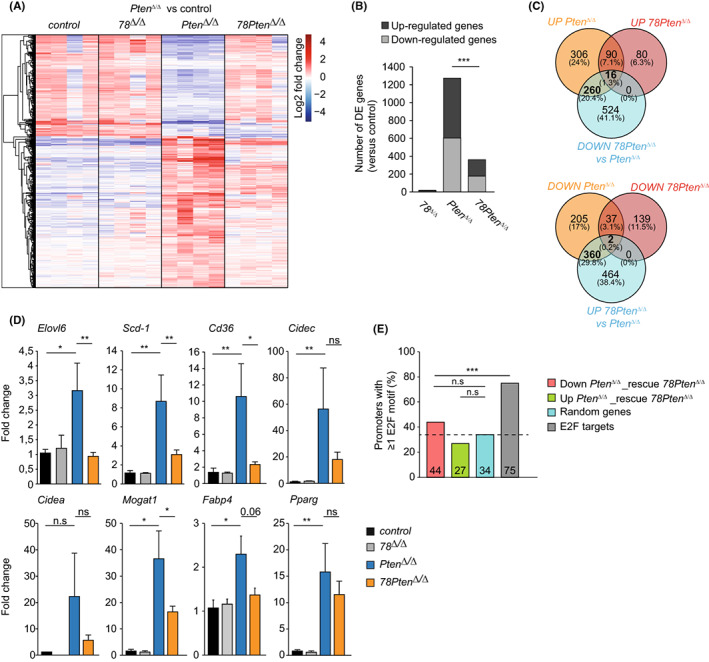
Polyploid hepatocytes promote lipid biosynthesis by enhancing PPAR gamma signalling. (A) Differential gene expression analysis of gene transcripts identified in RNA seq samples from the indicated genotypes (n = 4). Heatmap showing 2log‐fold change in expression of transcripts that were differentially expressed in Pten Δ/Δ compared to control livers. (B) Stack histogram showing the numbers of differentially expressed genes (up‐ or down‐regulated) in the indicated genotypes compared to control livers. ***p < .001 (chi‐square). (C) Venn diagram showing overlapping genes identified in RNAseq samples between the comparisons indicated. UP or DOWN comparisons are versus controls. (D) Transcript levels of PPARγ target gene expression in liver tissue of 16 weeks‐old mice. Fold changes were adjusted to average of controls and GAPDH and β‐Actin were used to normalize the expression. Data represent average ± SEM (n = 6 controls; n = 4 78 Δ/Δ, n = 9 Pten Δ/Δ , n = 8 78Pten Δ/Δ). **p < .05, **p < .01, ***p < .001; n.s non‐significant (Mann–Whitney rank sum test). (E) Histogram showing the overall percentage of promoters with at least 1 E2F consensus motif. ***p < .001; n.s non‐significant (Chi square).
FIGURE 3.
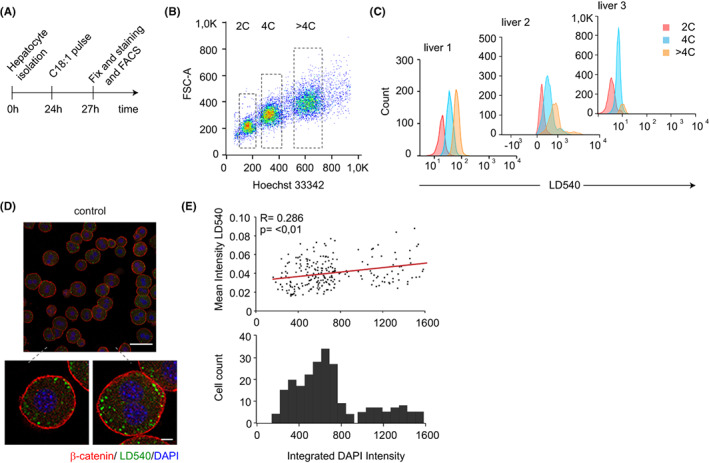
Polyploidy promotes lipid accumulation in mouse hepatocytes. (A) Experimental set‐up of primary hepatocytes isolation for immunofluorescent staining and flow cytometry analysis of lipid loading. (B) Representative flow cytometry plot of hepatocytes isolated from control mice. Hoechst 33342 and forward scatter (FSC) were used to determine the level of ploidy (n = 3 mice). (C) LD540 fluorescence was measured by FACS of 2C, 4C and >4C primary hepatocytes incubated for 3 h with oleic acid from control mice (n = 3 mice). (D) Representative staining of β‐catenin (red), the lipophilic dye LD540 (green) and DAPI (blue) of primary isolated hepatocytes from control mice cultured for 3 h with 200 μM oleic acid (C18:1). Scale bar: 10 μm. (E) Image quantification of the mean intensity of LD540 and integrated DAPI intensity per cell (top panel) and histogram of the integrated DAPI intensity per cell (bottom panel). Correlation R was calculated using the Pearson coefficient, statistical significance of the correlation was evaluated by a Welch's t‐test (n = 233 cells).
2.2. Flow cytometry
Determination of hepatocyte ploidy by propidium iodide staining was done as previously described. 11 Briefly, Pepsin (0.5 mg/mL 0.1 N in HCl) was used to generate the nuclei suspensions from frozen livers. Afterwards, the nuclei were washed twice with TBS and then stained with propidium iodide (20 μg/mL propidium iodide, 250 μg/mL RNase A and 0.1% bovine serum albumin). All samples were measured on a BD FACS Canto II (BD Biosciences) and further analysed using FlowJo software (FlowJo LLC). FACS on isolated hepatocytes (Figure 3) was done as the following. Hepatocytes were resuspended in prewarmed Hepatozyme (Invitrogen) containing 2% FCS, 1 mM Hepes, glutamax, penicillin/streptomycin, 15 μg/mL Hoeschst33342, 5 μM reserpine and 0.5 μg/mL LD540 dye at 1 million cells/mL. Cells were incubated for 30 min in a water bath at 37 degrees. Afterwards, the pellet was washed twice with HBSS and resuspended on cold HBSS containing 2%FCS, 1 mM Hepes, penicillin, streptomycin, and 5 μg/mL Sytox red for FACS.
2.3. Hepatocyte isolation, culture and IF staining of neutral lipids
Hepatocytes isolation and staining were done as previously described, 10 and cultured overnight in high glucose Dulbecco's modified Eagle's medium (DMEM) containing 2% Foetal bovine serum, 1 mM penicillin/streptomycin and 10 mM HEPES. Staining for imaging was done by plating the hepatocytes on coverslips overnight and incubated them for 24 h with exogenous lipids C18:1 (200 μM). After that period, hepatocytes were fixed with 1% paraformaldehyde, washed twice with PBS, blocked with 2%BSA and 0.2% Saponin in PBS for an hour and stained with LD540 (0.5 μg/mL), β‐catenin (1:1000 diluted on PBS containing 1% BSA and 0.2% saponin; AB6302, Abcam) and Hoechst 33342 (15 μg/mL, B2261, Sigma). Quantification of lipid droplets per cell was done using an open source cell profiler software available. 22
2.4. Neutral lipid staining in frozen liver tissue
Frozen liver samples from mice aged 4 months were cut at 8 μm thickness, air dried (1 min), fixed in 3% paraformaldehyde (10 min) and stained with filtered oil red O dye. Images were taken within 3 h after staining. All histological images were acquired using Labsens soft imaging software version 1.1 and DP25 camera mounted on Olympus BX45 microscope. For immunofluorescent staining, sections were incubated with blocking solution containing 10% goat serum, 0.2% BSA, 1 mM CaCl2 and MgCl2, and 0.1% Tween‐20 for 1 h at room temperature. After blocking, sections were incubated with staining solution containing Bodipy (1:50), Alexa‐fluor 568 phalloidin (1:400; A12380, Invitrogen) and DAPI (5 mg/mL; 1:4000; D1306 Invitrogen) for 30 min at room temperature.
2.5. Immunoblotting
Protein lysates were obtained with RIPA buffer (50 mM Tris–HCl pH 7,5, 1 mM EDTA, 150 mM NaCl, 0.25% deoxycholic acid, 1% Nonidet‐ P40), 1 mM NaF and NaV3O4 and protease inhibitor cocktail (11 873 580 001, Sigma Aldrich). After centrifugation at full speed (12 000 × g) for 10 min, supernatants were collected and proceed to a standard SDS‐PAGE Immunoblot. Antibodies used for western blots included pAKT‐T308 (9272, Cell Signalling), pAKT‐S473 (BD560378, BD Biosciences), anti‐AKT (9272S, Cell Signalling), p‐mTOR ser2448 (#5536 T, Cell Signalling), anti‐mTOR (#2983, Cell Signalling), p‐S6 ribosomal (#2211, Cell Signalling) and Total S6 ribosomal (#2217, Cell Signalling) and ƴ‐tubulin (GTU‐88; T6557, Sigma) at 1:1000 dilution.
2.6. Immunohistochemistry
Immunohistochemistry for Ki67 was conducted as previously described. 23 Briefly, tissues were embedded in paraffin and sectioned at 4 μm. After tissue deparaffinization in xylene and clearing using gadded ethanol, 10 mM Citrate buffer (pH 6) was used for heat‐induced antigen retrieval. Endogenous peroxidase activity was blocked with 1% H2O2. Rabbit anti‐Ki67 at 1:75 dilution in PBS (RM‐9106‐S0, Labvision/Neomarkers) was used for immunohistochemistry. Slides were counterstained with haematoxylin.
2.7. RNA isolation, cDNA and quantitative PCR
RNA isolation, cDNA synthesis and quantitative PCR were performed based on the manufacturer's instructions for QIAGEN (RNeasy Kits), Thermo Fisher Scientific (cDNA synthesis Kits) and Bio‐Rad (SYBR Green Master Mix) respectively. Reactions were performed in duplicate and the relative amount of cDNA was normalized to GAPDH and actin using the ΔΔCt method. qPCR primer sequences are provided in Supplementary Table S2.
2.8. RNA‐sequencing
Liver samples taken from 4 months old mice were used. Library preparation and RNA sequencing were done at the Utrecht Sequencing Facility according to standard procedures. Barcoded cDNA libraries were prepared with a cDNA TruSeq® Stranded mRNA poly‐A kit (Illumina). All 16 barcoded samples were mixed and sequenced simultaneously with a 1 × 75 bp run on a NextSeq500 sequencer (Illumina). All samples passed a quality control using FastQC v0.10.1. The sequencing reads were then mapped to the mouse genome (assembly GRCm38/mm10 using ENCODE's STAR software version 2.4.2a). The mapped reads were further analysed using the R packages EdgeR, 24 Deseq2 25 and Pheatmap. Differential expression analysis was done on raw counts using Deseq2, and an FDR‐corrected value of p < .05 was considered statistically significant. Gene set enrichment analysis was done using Enrichr. 26 A minimum of five genes and Benjamini–Hochberg‐corrected p < 0.05 were taken as cut‐off for significant enrichment. Raw and processed RNA‐sequencing data generated in this study are available on Gene Expression Omnibus under accession number GSE202546.
2.9. Pathological analysis: steatosis and tumours
Pathological analysis was performed by a board‐certified veterinary pathologist. Pathological changes were classified according to the nomenclature and diagnostic criteria for hepatobiliary lesions in rats and mice. 27
2.10. Statistics
The number of independent experiments, the number of mice and the type of statistical analysis for each figure are indicated in the legends. Differences between groups were compared with a one‐way analysis of variance using Tukey's test post hoc analysis. Where data were not normally distributed, groups were tested by Kruskal–Wallis tests with Dunn's post hoc correction. Asterisks indicate where significant differences were seen (p < .05). Where relevant for understanding the figure and individual comparisons, we indicate with “n.s.” that significance was not reached.
3. RESULTS
3.1. Inhibition of polyploidization via deletion of E2f7/8 prevents accumulation of large lipid droplets in Pten‐deficient livers
Lipid accumulation and pathological polyploidy are positively correlated during NAFLD in both humans and mice. 4 We first investigated whether inhibiting of polyploidization via ablation of E2F7 and −8 affects lipid accumulation in Pten‐deficient mice. To this end, we conditionally deleted E2f7, E2f8 and Pten (hereafter 78Pten Δ/Δ) in mouse livers using Cre/LoxP technology under the control of the hepatocyte‐specific albumin promoter (Alb‐Cre). We performed a cross‐sectional pathology study at 16 weeks of age with controls (Alb‐Cre negative), double E2f7 and −8 (78 Δ/Δ) mutant mice, single Pten knock‐out (Pten Δ/Δ) and triple knockout mice (78Pten Δ/Δ). In line with previous work, single deletion of Pten caused polyploidy, as seen by an increase in the percentage of 8C mononuclei compared to control livers (Figure 1A). We noted that E2f7 and E2f8 mRNA levels are elevated after Pten single deletion, indicating that pathological polyploidy could be mediated by these transcription factors (Figure S1A). Consistent with this, the liver polyploidy was strongly reduced in the absence of atypical E2Fs, leading to hepatocytes containing mainly 2C nuclei in 78 Δ/Δ as well as 78Pten Δ/Δ livers (Figure 1A and Figure S1B). In addition, Pten Δ/Δ livers showed a marked increase in liver weight and size characterized by pale, bright and grossly visible boundaries of the liver lobules, indicative of steatosis (Figure 1B and Figure S1C). Histological examination by a board‐certified veterinary pathologist confirmed severe hepatic steatosis in Pten Δ/Δ livers (previously described scoring system 28 ), which was substantially ameliorated in 78Pten Δ/Δ livers (Figure 1C,D). Liver weights of 78Pten Δ/Δ mice were not significantly reduced compared to Pten Δ/Δ livers, suggesting that increased cell density in 78Pten Δ/Δ might compensate for the presence of enlarged hepatocytes in Pten Δ/Δ livers (Figure S1C). Other parameters such as glycogen and lobular inflammation grade were neither significantly reduced in 78Pten Δ/Δ compared to Pten Δ/Δ livers at the age of 16 weeks old (Figure S1D–E).
Microscopic analysis confirmed that Pten Δ/Δ livers accumulated large lipid droplets in hepatocytes, as shown with haematoxylin & eosin, lipid‐specific oil red O and bodipy staining (Figure 1D). Morphometric analysis revealed that livers with additional deletion of E2f7 and E2f8 (78Pten Δ/Δ) carried substantially smaller lipid droplets compared to Pten Δ/Δ livers, although the total number of lipid droplets was increased (Figure 1E). This resulted in a significantly reduced triglyceride content in the 78Pten Δ/Δ compared to Pten Δ/Δ livers (Figure 1F). MS‐based lipidomic analysis on endogenous triacylglycerols (TAGs) and a subset of lipid species distribution confirmed the significant increase of TAGs in 78Pten Δ/Δ and Pten Δ/Δ livers (Figure S2A,B). However, there were no overall differences in lipid classes neither on the level of saturation of the triglyceride (Figure S2C,D), demonstrating that ploidy reduction did not affect lipid composition. Together, these findings show that steatosis and polyploidization can be alleviated in Pten‐deficient livers by additional deletion of atypical E2fs without altering lipid composition.
3.2. Polyploid hepatocytes promote lipid biosynthesis by enhancing PPAR gamma signalling
To gain comprehensive insights into how polyploidization promotes lipid accumulation in Pten Δ/Δ livers, we first analysed whether PI3K‐AKT–mTOR signalling was affected by inhibiting polyploidization through deletion of E2f7/8. We found that PTEN loss caused a strong increase in AKT and mTOR phosphorylation, which were not significantly decreased by additional deletion of atypical E2Fs (Figure S3A,B).
We then performed differential expression (DE) analysis on the liver samples of 16‐week‐old mice via RNA‐sequencing. Strikingly, we observed that the vast majority of expression changes caused by Pten deletion was partly or completely rescued by additional deletion of E2f7/8 (Figure 2A). In addition, far less genes were differentially expressed in 78Pten Δ/Δ versus control livers than in Pten Δ/Δ versus control livers (Figure 2B). Specifically, 276 transcripts that were upregulated through Pten deletion were rescued downwards upon additional deletion of E2f7/8. In addition, 362 transcripts that were downregulated in Pten Δ/Δ livers were rescued upwards when Pten was deleted together with atypical E2fs (Figure 2C). We did not identify any notable alterations in gene expression in livers with deletion of E2f7/8 compared to controls, suggesting that E2f7/8 do not interfere with the liver physiology of healthy adult mice (Figure 2A,B). In stress‐induced livers by Pten deletion, our data indicate that aberrant gene expression is, to a large extent, rescued by additional deletion of atypical E2fs.
Although the lists DE genes in Pten Δ/Δ versus control livers comprised hundreds of genes, these lists were not strongly enriched for specific pathways or molecular processes, indicating that these genes are involved in a wide variety of different processes (Figure S3C). The strongest functional enrichments among genes upregulated in Pten Δ/Δ livers were PPAR signalling and triglyceride metabolism (Figure S3C,D and Table S3). Previous studies demonstrated that PPARγ and its direct target are key inducers of steatosis in Pten‐deficient livers. 2 , 29 Given the importance of PPARγ and its direct targets in steatosis, we asked to which extent this pathway was normalized in 78Pten Δ/Δ livers. Indeed, additional deletion of atypical E2Fs in Pten‐deficient livers resulted in reduced expression of Pparg and a set of its target genes, which we validated by quantitative PCR analysis (Figure 2D and Figure S4A). Apart from this partial rescue in PPAR signalling, we did not find significant enrichment for any pathway or GO molecular process in the list of 276 genes upregulated in Pten Δ/Δ compared to control livers and rescued downwards in 78Pten Δ/Δ livers (Figure S3C,D). Of note, Hippo signalling and multiple processes linked to transcription regulation were the strongest enriched pathways among the downregulated genes in Pten Δ/Δ livers (Figure S3C,D). This is consistent with the role of Hippo in negative regulation of growth in mammalian livers. 30 However, when analysing genes that were downregulated in Pten Δ/Δ livers and significantly rescued upwards in 78Pten Δ/Δ livers we did not find evidence that Hippo signalling or transcription factor programmes were rescued. Instead, we found a rescue of gene products known to be involved in ribosome functions (Rplp1, Rps2 and Rps18; Figure S3C) as well as protein synthesis (Mrps24, Rps5, Pabpc4; Figure S3D). Notably, processes related to protein processing in ER and cellular response to ER stress were significantly downregulated in 78Pten Δ/Δ livers compared to controls, suggesting that polyploidization is used for cellular adaptation (Figure S3C,D). Together, these analyses show that additional deletion of atypical E2Fs in Pten‐deficient livers can partially rescue PPARγ signalling, as well as processes related to ribosomal function and protein synthesis.
Next, we aimed to investigate to what extent the observed gene transcription changes could be result of direct transcriptional repression or activation by atypical E2Fs. We hypothesized that the proximal promoters of the rescued genes should be enriched with E2F binding motifs compared to a randomly generated list of genes. Therefore, we searched for consensus E2F‐binding motifs (TTTSSCGC or highly similar) in the proximal promoters of the genes up‐ or down‐regulated in Pten Δ/Δ livers and rescued by additional deletion of atypical E2Fs. We analysed the lists of 362 rescued upwards and 276 genes rescued downwards in 78Pten Δ/Δ livers separately. A list of 250 randomly picked genes served to detect the background level of coincidental presence of motifs, and a list of 80 well‐known E2F targets genes from previous RNA‐ and ChIP‐sequencing data was used as positive control. 20 , 31 Unexpectedly, the lists of upward or downward rescued genes did not contain more genes with at least one E2F binding motif than the randomly generated gene list (Figure 2E). Furthermore, the list of known E2F7/8 target genes contained a markedly higher percentage of E2F‐motif‐containing genes than any of the other lists, showing that our approach is in principle feasible to pick up enrichment for E2F‐binding genes (Figure 2E). Notwithstanding these findings, we observed that some genes rescued by E2f7/8 deletion contained putative E2F binding motifs, and we asked if this limited set of genes could be primarily responsible for rescuing the PPARγ and steatosis phenotype in 78Pten Δ/Δ livers. To narrow down the lists of candidate genes, we screened which of the rescued genes containing putative E2F motifs were previously shown to be bound directly by E2F7 and/or E2F8 in ChIP‐sequencing experiments. 20 , 31 In total 48 transcripts rescued by E2f7/8 deletion were previously identified as E2F7/8 target genes with ChIP‐sequencing (Figure S4B; Table S4). We manually curated the functions of these target genes and we noticed that expression of Sik2 and Irs2, which are both involved in insulin signalling, was decreased in Pten Δ/Δ livers, and rescued in upward direction in 78Pten Δ/Δ livers by RNA sequencing. We hence sought to validate these genes in larger amount of livers by quantitative PCR. Neither of these genes showed noticeable upregulation in 78Pten Δ/Δ versus Pten Δ/Δ livers suggesting that Sik2 and Irs2 were not involved in the observed phenotypes (Figure S4C). Finally, we re‐analysed our previously published E2F7/8 ChIP‐sequening data to investigate whether the PPARγ target genes are bound by E2F7/8. However, proximal promoters of the PPARγ target gene promoters contained neither E2F7 nor E2F8 enrichment (data not shown).
Together our transcriptomic data suggest that atypical E2Fs do not alleviate steatosis in Pten‐mutant livers via direct binding to gene promoters related to lipid metabolism. Instead, this rescue effect is likely mediated by inhibition of polyploidization in 78Pten Δ/Δ hepatocytes.
3.3. Enhanced lipid accumulation in wild‐type polyploid versus diploid hepatocytes
Next, we explored whether the polyploid and polynuclear state would permit a higher level of lipid accumulation in hepatocytes isolated from young adult wild‐type mice after incubation with free fatty acids (Figure 3A). Flow cytometry analysis showed that the amount of neutral lipids, as reported by the lipophilic dye LD540, slightly increased with ploidy (Figure 3B,C and Figure S5A). In addition, we performed fluorescence microscopy and subsequent imaging analysis to segment and compare the isolated primary hepatocytes with different ploidy status on the single‐cell level. After the quantification of neutral lipid (LD540 signal) and DNA (DAPI signal) content of single‐cells, we observed again a mild positive correlation between those two parameters (Figure 3D,E), although no significant change was observed between polynuclear states (mono vs. binucleated hepatocytes) (Figure S5B,C). These data showed that polyploid cells with larger cell size facilitate lipid accumulation. Next, we evaluate whether the inactivation of atypical E2Fs could directly encounter for the reduction of lipid storage in hepatocytes. We isolated and FACS‐sorted diploid hepatocytes from control and 78 Δ/Δ livers based on Hoechst 33342 signal. Sorted hepatocytes had equal nuclear and cell size (Figure S5D,E). We observed that LD540 stained control 2C hepatocytes displayed similar LD540 fluorescence intensity compared to 2C 78 Δ/Δ hepatocytes (Figure S5F). Hepatocytes derived from control livers not stained with LD540 were used to detect background fluorescent. Altogether, these data suggest that the polyploid state and/or cell size may play an important role in facilitating the lipid storage capacity of hepatocytes, independent of E2F7/8.
3.4. Blocking polyploidization accelerates liver cancer formation in Pten‐deficient livers
NAFLD patients are at high risk to develop liver cancer. 32 Liver‐specific deletion of Pten is an established mouse model for NAFLD that progress to liver cancer. 2 To determine if inhibition of polyploidization has also impact on the progression from NAFLD towards liver cancer, we performed pathological analysis on livers from 10‐months‐old mice with the different genotypes. At this age, livers from Pten Δ/Δ mice were enlarged compared to controls (Figure 4A,B). However, we could detect only two mice with macroscopic tumour lesions. Strikingly, livers from 78Pten Δ/Δ mice presented evident macroscopic tumour lesions, although average liver mass was comparable to Pten Δ/Δ liver masses (Figure 4A–C). This suggests that enhanced neoplastic growth in 78Pten Δ/Δ liver and enhanced lipid loading in Pten Δ/Δ liver might compensate each other to result in similarly enlarged livers. Control and 78 Δ/Δ mice did not present any macroscopic tumours at this time point (Figure 4B,C). We next analysed these livers histologically and confirmed that Pten Δ/Δ mice presented mainly premalignant lesions, characterized as focal cellular alteration (FCA; Figure 4D). Importantly, hepatocellular carcinomas (HCCs) or cholangiocarcinomas (CCs) were only found on 78Pten Δ/Δ livers, suggesting that inhibition of polyploidization through additional deletion of E2f7/8 markedly accelerates liver cancer formation in Pten‐deficient livers. An alternative explanation would be that deletion of E2f7/8, a known cell cycle regulator, increases replication stress and genomic instability resulting in additional mutations on cancer‐related genes and thereby promoting tumorigenesis. Consistently, 78Pten Δ/Δ tumours showed increased proliferation, measured by Ki67‐IHC staining, compared to Pten Δ/Δ lesions (Figure 4E,F). These results demonstrate that polyploidization functions as an important barrier against liver tumour progression in a mouse model for NAFLD.
FIGURE 4.
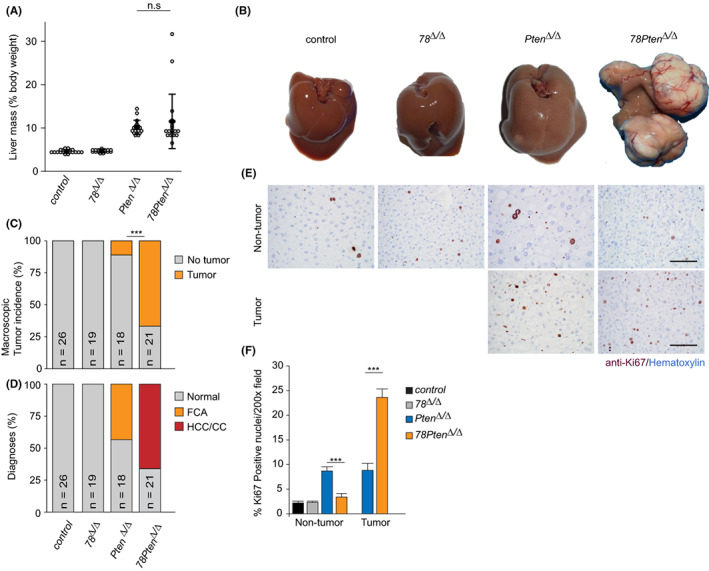
Blocking polyploidization enhances tumour formation in PTEN‐deficient livers. (A) Dot plot indicating liver mass/body weight percentage of 10 months old mice from the indicated genotypes. n = 18–26. (B) Representative macroscopic images of 10 months old livers from the indicated genotypes. (C) Macroscopic liver tumour incidence. ***p < .001 (chi‐square). (D) Pathology analysis of HE‐stained livers. FCA, Foci of cellular alteration; HCC, hepatocellular carcinoma; CC, Cholangiocarcinoma. (E) Representative immunohistochemistry pictures of Ki67‐stained 10 months‐old liver sections from the indicated regions (tumour vs non‐tumour). Scale bars: 50 μm. (F) Bar chart shows the quantification of Ki‐67‐positive hepatocytes in tumour and remote tissue of the indicated genotypes. Bars represent average and SEM (n = 5 pictures taken with 20x objective/condition and mouse; n = 5 mice/genotype; n = 2 Pten KO tumours).
4. DISCUSSION
The liver parenchyma displays changes in the polyploidy level of hepatocytes upon injury or stress. In particular, hepatocytes have the ability to acquire enhanced polyploid phenotype when exposed to increased lipid‐related stress. 4 In addition, physiological liver cell polyploidization was previously shown to function as a barrier against hepatocarcinogenesis. 33 , 34 However, the relevance of hepatocyte polyploidization in the pathophysiology of the liver is still largely unknown. The presence of mouse models that allow the manipulation of polyploidization in the liver facilitate further investigation of this issue. For instance, repressing cell cycle regulators (such as Cdk1, TP53 or RB) or inducing a metabolic overload (such as ob/ob mice or mice fed with high‐fat diet or MCD diet). In this study, we investigated the role of enhanced hepatocyte ploidy in Pten‐deficient mice on lipid accumulation by comparing controls and 78 ∆/∆ as well as Pten ∆/∆ and 78Pten ∆/∆ conditional knockout livers. On the one hand, liver‐specific loss of Pten results in severe steatosis via activated insulin signalling and is accompanied by increased hepatocyte polyploidization. 4 , 12 On the other hand, deletion of E2f7/8 blocks hepatocyte polyploidization and is an established and powerful in vivo model to explore the relevance of polyploidy in liver diseases. 11 It is tempting to speculate that our proposed model, which states that polyploidization favours lipid accumulation in the liver despite being a barrier for tumorigenesis, is applicable also to other mouse models. However, future experiments are required to address this question further.
We verified that Pten ∆/∆ hepatocytes showed increased hepatocyte ploidy and lipid accumulation, which is in agreement with previous findings. 2 , 4 But surprisingly, inhibition of polyploidization via deletion of E2f7/8 in Pten ∆/∆ deficient liver reduces steatosis and was accompanied by smaller lipid droplet formation. It has been shown that polyploid hepatocytes have relatively larger cell volumes compared to diploid hepatocytes. 35 Therefore, it is conceivable that the larger cell volume provides more space to increase lipid storage capacity of individual polyploid hepatocytes compared to smaller diploid hepatocytes. Indeed, comparison of murine wildtype hepatocytes of different ploidy have shown that polyploid hepatocytes with larger cell volume are capable of accommodating more lipid droplets signal per cell compared to diploid hepatocytes. These findings were consistent with previous observations showing that lipid droplets in polyploid hepatocytes can grow to larger sizes than in diploid hepatocytes. 4 , 36 Since deletion of E2f7/8 results in the formation of predominantly diploid hepatocytes, the presence of smaller lipids droplets could be related to the smaller cytoplasmic volume of these hepatocytes and consequently less lipid accumulation. In addition, the higher surface‐to‐volume ratio in small lipid droplets allows better accessibility for LD‐associated enzymes. However, the overall number of lipid droplets per area is increased in livers with predominantly diploid cells (78Pten ∆/∆) compared to livers with mostly polyploid cells (Pten ∆/∆). This difference can be explained by the relative increase in cellular density in E2f7/8 deficient liver. 11 Basically, lack of large polyploid cells is compensated by increasing the number of small diploid cells per liver unit.
Previous work also showed that an increase in cell‐size causes a strong increase in expression of the fatty acid transporter CD36, as well as triglyceride accumulation in liver. 19 In this line, we observed that Cd36 transcripts were upregulated in polyploid Pten ∆/∆ mutant livers and rescued in 78Pten ∆/∆ livers with predominately diploid hepatocytes. Interestingly, Cd36 is a PPARγ target gene, and we observed that many other PPARγ targets were also rescued in 78Pten ∆/∆ livers. It is therefore conceivable that the increase in cell volume caused by Pten ∆/∆‐induced polyploidization can affect PPARƴ signalling and CD36 expression, thus polyploid cells take up more fatty acids. However, further investigations are required to explore how polyploidization regulates PPAR signalling and CD36.
As E2F7/8 acts as transcriptional repressors, we initially expected to find novel target genes involved in steatosis. Previous work suggested that genes involved in de novo fatty acid synthesis are transcriptionally controlled by E2F8 and the activating E2F family member E2F1. 37 , 38 However, we did not observe that E2F7/8 loss caused de‐repression of these lipid synthesis genes in our transcriptome analyses (data not shown). Moreover, despite in‐depth gene expression analysis, we could not identify direct E2F7/8 target genes that could explain the changes in the steatosis or cancer phenotype. We did identify downregulation of Hippo signalling in RNA seq of Pten ∆/∆ mice, which was previously linked to enhanced hepatic steatosis by interacting with AKT via IRS2. 39 However, we did not detect significant changes in this signalling pathway either in RNA seq or qPCR of IRS2 by additional deletion of E2F7/8. Together, these results suggest that alternative variables, such as differences in the ploidy status of the livers are responsible for the phenotype changes we observe.
Lastly, PTEN and atypical E2Fs are both known tumour suppressors in the liver, but the mechanisms of action are completely different. PTEN prevents excessive insulin signalling, fat accumulation and polyploidization, whereas atypical E2Fs promote liver polyploidization. Physiological liver cell polyploidization was previously shown to function as a barrier against hepatocarcinogenesis. 33 , 34 By combining genetic ablation of PTEN and atypical E2Fs, our findings indicate that Pten‐induced polyploidization might be also an important barrier against liver tumour formation. Consistent with previous results, this occurs independent on the degree of steatosis. 33 This is counterintuitive, as steatosis is generally thought to drive carcinogenesis. We observed that inhibition of polyploidization reduced steatosis and PPARγ signalling in Pten‐mutant livers, although tumour formation was strongly accelerated. Our data are at first sight inconsistent with a previous study showing that inactivation of PPARγ signalling blocks tumorigenesis in Pten‐mutant mouse livers. 29 However, the rescue was only partial, and PPARγ may not be the exclusive tumour‐promoting pathway downstream of PI3K/AKT. For example, we observed that the tumour‐suppressive Hippo signalling pathway was downregulated in Pten‐mutant livers, and not rescued in E2f7/8/Pten triple mutant livers. We also detected upregulation of ribosome activity (e.g RPS2), previously shown to correlate with increased cell proliferation, 40 , 41 in E2f7/8/Pten Δ/Δ livers. So rather than being incompatible with the previous study, our data indicate that prevention of polyploidization overrides the potentially tumour‐suppressing effect of partly reducing PPARγ signalling on liver tumorigenesis.
Given that NAFLD is a spectrum of chronic liver diseases that involves several metabolic dysfunctions and other factors beyond metabolism such as liver inflammation, we believe that lipid loading per se does not directly compromise the severity of the progression of the disease. Instead, massive production of proinflammatory cytokines as observed in NASH or enrichment of diploid cells via ploidy reduction may contribute to progression towards liver cancer. Since NAFLD is not only caused by PTEN deficiency but can also be a consequence of continuous intake of diets with high fat or sugar content, future studies could explore whether polyploidization contributes also to steatosis in diet‐induced models of NAFLD.
CONFLICT OF INTEREST
The authors do not have any disclosures to report.
Supporting information
Data S1
Figure S1
ACKNOWLEDGEMENTS
We thank Wout Puijk and the rest of animal caretakers for excellent care of our mice.
Moreno E, Matondo AB, Bongiovanni L, et al. Inhibition of polyploidization in Pten‐deficient livers reduces steatosis. Liver Int. 2022;42:2442‐2452. doi: 10.1111/liv.15384
Eva Moreno and Augustine B. Matondo shared first authorship.
Handling Editor: Dr. Luca Valenti
REFERENCES
- 1. Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011;11(4):289‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Horie Y, Suzuki A, Kataoka E, et al. Hepatocyte‐specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest. 2004;113(12):1774‐1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Caviglia JM, Schwabe RF. Mouse models of liver cancer. Methods Mol Biol. 2015;1267:165‐183. [DOI] [PubMed] [Google Scholar]
- 4. Gentric G, Maillet V, Paradis V, et al. Oxidative stress promotes pathologic polyploidization in nonalcoholic fatty liver disease. J Clin Invest. 2015;125(3):981‐992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stiles B, Wang Y, Stahl A, et al. Liver‐specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity. PNAS. 2004;101(7):2082‐2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol. 2013;10(11):656‐665. [DOI] [PubMed] [Google Scholar]
- 7. Fox DT, Duronio RJ. Endoreplication and polyploidy: insights into development and disease. Development. 2013;140(1):3‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Edgar BA, Zielke N, Gutierrez C. Endocycles: a recurrent evolutionary innovation for post‐mitotic cell growth. Nat Rev Mol Cell Biol. 2014;15(3):197‐210. [DOI] [PubMed] [Google Scholar]
- 9. Celton‐Morizur S, Desdouets C. Polyploidization of liver cells. Adv Exp Med Biol. 2010;676:123‐135. [DOI] [PubMed] [Google Scholar]
- 10. Duncan AW, Taylor MH, Hickey RD, et al. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature (London). 2010;467(7316):707‐710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pandit S, Westendorp B, Nantasanti S, et al. E2F8 is essential for polyploidization in mammalian cells. Nat Cell Biol. 2012;14(11):1181‐1191. [DOI] [PubMed] [Google Scholar]
- 12. Celton‐Morizur S, Merlen G, Couton D, Margall‐Ducos G, Desdouets C. The insulin/Akt pathway controls a specific cell division program that leads to generation of binucleated tetraploid liver cells in rodents. J Clin Invest. 2009;119(7):1880‐1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen H, Ouseph MM, Li J, et al. Canonical and atypical E2Fs regulate the mammalian endocycle. Nat Cell Biol. 2012;14(11):1192‐1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sladky VC, Knapp K, Soratroi C, et al. E2F‐family members engage the PIDDosome to limit hepatocyte ploidy in liver development and regeneration. Dev Cell. 2020;52(3):335‐349.e7. [DOI] [PubMed] [Google Scholar]
- 15. Muramatsu Y, Yamada T, Moralejo DH, Mochizuki H, Sogawa K, Matsumoto K. Increased polyploid incidence is associated with abnormal copper accumulation in the liver of LEC mutant rat. Res Commun Mol Pathol Pharmacol. 2000;107(1–2):129‐136. [PubMed] [Google Scholar]
- 16. Lazzerini Denchi E, Celli G, de Lange T. Hepatocytes with extensive telomere deprotection and fusion remain viable and regenerate liver mass through endoreduplication. Genes Dev. 2006;20(19):2648‐2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Toyoda H, Bregerie O, Vallet A, et al. Changes to hepatocyte ploidy and binuclearity profiles during human chronic viral hepatitis. Gut. 2005;54(2):297‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Duncan AW, Hanlon Newell AE, Bi W, et al. Aneuploidy as a mechanism for stress‐induced liver adaptation. J Clin Invest. 2012;122(9):3307‐3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miettinen TP, Pessa HKJ, Caldez MJ, et al. Identification of transcriptional and metabolic programs related to mammalian cell size. Curr Biol. 2014;24(6):598‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kent L, Rakijas J, Pandit S, et al. E2f8 mediates tumor suppression in postnatal liver development. J Clin Invest. 2016;126(8):2955‐2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21(1):70‐71. [DOI] [PubMed] [Google Scholar]
- 22. Carpenter AE, Jones TR, Lamprecht MR, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7(10):R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matondo RB, Toussaint MJM, Govaert KM, et al. Surgical resection and radiofrequency ablation initiate cancer in cytokeratin‐19 + − liver cells deficient for p53 and Rb. Oncotarget. 2016;7(34):54662‐54675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xie Z, Bailey A, Kuleshov MV, et al. Gene set knowledge discovery with enrichr. Curr Protoc. 2021;1(3):e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thoolen B, Maronpot R, Harada T, et al. Proliferative and nonproliferative lesions of the rat and mouse hepatobiliary system. Toxicol Pathol. 2010;38(7 Suppl):5S‐81S. [DOI] [PubMed] [Google Scholar]
- 28. Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41(6):1313‐1321. [DOI] [PubMed] [Google Scholar]
- 29. Patitucci C, Couchy G, Bagattin A, et al. Hepatocyte nuclear factor 1α suppresses steatosis‐associated liver cancer by inhibiting PPARγ transcription. J Clin Invest. 2017;127(5):1873‐1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19(4):491‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Westendorp B, Mokry M, Koerkamp G, et al. E2F7 represses a network of oscillating cell cycle genes to control S‐phase progression. Nucleic Acids Res. 2012;40(8):3511‐3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nagaya T, Tanaka N, Komatsu M, et al. Development from simple steatosis to liver cirrhosis and hepatocellular carcinoma: a 27‐year follow‐up case. Clin J Gastroenterol. 2008;1(3):116‐121. [DOI] [PubMed] [Google Scholar]
- 33. Zhang S, Zhou K, Luo X, et al. The polyploid state plays a tumor suppressive role in the liver. Dev Cell. 2018;44(4):447‐459.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lin Y, Zhang S, Zhu M, et al. Mice with increased numbers of polyploid hepatocytes maintain regenerative capacity but develop fewer hepatocellular carcinomas following chronic liver injury. Gastroenterology. 2020;158(6):1698‐1712.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Watanabe T, Tanaka Y. Age‐related alterations in the size of human hepatocytes. A study of mononuclear and binucleate cells. Virchows Arch B Cell Pathol Incl Mol Pathol. 1982;39(1):9‐20. [DOI] [PubMed] [Google Scholar]
- 36. Xu W, Wu L, Yu M, et al. Differential roles of cell death‐inducing DNA fragmentation factor‐α‐like effector (CIDE) Proteins in promoting lipid droplet fusion and growth in subpopulations of hepatocytes. J Biol Chem. 2016;291(9):4282‐4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Denechaud P, Lopez‐Mejia IC, Giralt A, et al. E2F1 mediates sustained lipogenesis and contributes to hepatic steatosis. J Clin Invest. 2016;126(1):137‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shimada Y, Kuninaga S, Ariyoshi M, et al. E2F8 promotes hepatic steatosis through FABP3 expression in diet‐induced obesity in zebrafish. Nutr Metab (Lond). 2015;12:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jeong S, Kim H, Kim M, et al. Hippo‐mediated suppression of IRS2/AKT signaling prevents hepatic steatosis and liver cancer. J Clin Invest. 2018;128(3):1010‐1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kowalczyk P, Woszczyński M, Ostrowski J. Increased expression of ribosomal protein S2 in liver tumors, posthepactomized livers, and proliferating hepatocytes in vitro. Acta Biochim Pol. 2002;49(3):615‐624. [PubMed] [Google Scholar]
- 41. Boon K, Caron HN, van Asperen R, et al. N‐myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J. 2001;20(6):1383‐1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Figure S1