

Abstract
Objective
The aim of this study was to evaluate the effect of a novel, oral, modified‐release formulation of the lipase inhibitor orlistat and the glucosidase/amylase inhibitor acarbose (denoted EMP16) on relative body weight after 26 weeks compared with placebo.
Methods
The randomized, double‐blind, placebo‐controlled trial had a 26‐week treatment period, with dose escalation up to 6 weeks. Participants, adults between ages 18 and 75 years, with BMI ≥30 kg/m2 or ≥28 kg/m2 with risk factors, were randomly assigned to EMP16 120‐mg orlistat/40‐mg acarbose (EMP16‐120/40), EMP16‐150/50, or placebo. The primary end point was relative weight loss from baseline to week 26 assessed in participants with at least one post‐baseline weight measurement.
Results
Of 156 randomized participants, 149 constituted the intention‐to‐treat population. The mean (95% CI) estimated treatment difference to placebo in relative weight loss after 26 weeks in the intention‐to‐treat population was −4.70% (−6.16% to −3.24%; p < 0.0001) with EMP16‐120/40 and −5.42% (−6.60% to −4.24%; p < 0.0001) with EMP16‐150/50.
Conclusions
This trial indicates that orlistat and acarbose can be successfully combined in a modified‐release formulation to provide efficacious weight loss with no unexpected safety issues. EMP16 may be a promising candidate among other medications for improved weight management.
Study Importance.
1. What is already known?
As obesity is a chronic disease, lifestyle modification often needs to be supplemented by weight‐loss medication. Compared with other chronic diseases, the availability of effective drugs with proven long‐term safety is limited.
What does this study add?
In this 6‐month randomized trial in 156 adults with obesity, participants taking the new modified‐release combination (orlistat plus acarbose) product EMP16 lost ~5% more weight compared with the placebo group. Quality of life improved in the intervention groups and withdrawal due to adverse events was low; no serious events occurred.
How might these results change the direction of research or the focus of clinical practice?
This trial supports that orlistat and acarbose can be successfully combined as a promising potential candidate for improved weight management and can be part of the expanding treatment portfolio for obesity.
INTRODUCTION
Obesity is a multidimensional health problem worldwide and a major risk factor for additional health concerns such as cardiovascular diseases, diabetes, musculoskeletal disorders, and some cancers [1, 2]. Although lifestyle changes are seen as a crucial part of the treatment of obesity, most patients need supplementary pharmaceutical intervention to achieve a clinically meaningful weight loss [2, 3]. As obesity is a chronic disease [3], drug products need to be both efficacious and safe for long‐term use. Currently, only orlistat and liraglutide have been assessed for more than 2 years [4, 5]. Orlistat reversibly inhibits dietary lipid digestion in the gastrointestinal (GI) lumen, leading to a modest weight loss, and it has been used for decades with no serious side effects [6]. Although orlistat's efficacy and attrition are similar to those of other oral weight‐loss products [2], its conventional oral dosage forms are associated with frequent (~20% of patients) GI side effects, such as liquid and oily stools [6]. Moreover, particularly unfortunate side effects associated with orlistat in its conventional dosage form include accelerated gastric emptying and increased appetite [7].
In the present proof‐of‐concept trial, a fixed‐dose combination of orlistat and the antidiabetic drug acarbose (Precose) was tested in a population of adults with obesity. The novel, oral, modified‐release formulation, denoted EMP16, was designed to improve efficacy and tolerability compared with the conventional dosage forms. Acarbose, like orlistat, acts locally in the GI tract to delay carbohydrate digestion and reduce the subsequent intestinal glucose absorption rate [8]. The conventional oral dosage form of acarbose is associated with frequent (~3% to 30% of patients) GI side effects, mainly flatulence and sometimes soft stools or abdominal discomfort [8]. However, acarbose decreases the gastric emptying rate and increases the secretion of glucagon‐like peptide‐1 (GLP‐1) [9]. EMP16 was designed to deliver orlistat and acarbose with distinct release rates in different GI compartments. We hypothesized that the GI side effects would be reduced and that the two drugs would work synergistically to improve efficiency (see Background in online Supporting Information). In a previous pilot trial, EMP16 reduced appetite and improved both glucose metabolism and tolerability, with a similar safety profile compared with orlistat in its conventional dosage form [10, 11].
The primary objective of the present trial was to evaluate the effect of EMP16 on relative body weight loss after a 26‐week period of oral treatment as compared with placebo. We also evaluated the effects of EMP16 on absolute body weight, anthropometric characteristics, satiety and other components of appetite, and biomarkers of glucose and lipid metabolism, as well as inflammation, patient‐reported quality of life, and safety and tolerability. We also wanted to investigate higher doses of EMP16 as the pilot study indicated that higher doses could increase efficacy without causing problematic side effects [10, 11].
METHODS
Study design
This was a randomized, double‐blind, placebo‐controlled phase 2a trial. The trial was conducted by an independent clinical research organization, Clinical Trial Consultants AB, Uppsala, Sweden, in Linköping and in Uppsala. The protocol was approved by the local ethics committee in Stockholm (Approval# Dnr 2020–00835). The trial was performed in accordance with the Declaration of Helsinki and International Conference on Harmonisation (ICH) Good Clinical Practice, as well as the Consolidated Standards of Reporting Trials (CONSORT) reporting guideline, and was registered in the European Union Drug Regulating Authorities Clinical Trials Database (EudraCT 2019‐004545‐32). The trial included a screening period of up to 4 weeks, a 26‐week treatment period, and a 2‐week safety follow‐up visit. A protocol amendment was added during the trial, wherein participants were asked to return to the research sites 6 months after their last treatment dose, and measurements of acarbose plasma concentrations at week 26 were added. Data from this extension period will be presented separately.
Participants
Participants were recruited using the clinical research organization database and advertisements in social media platforms and radio, mainly from the local area. The trial population consisted of women and men aged between 18 and 75 years with body mass index (BMI) of at least 30 kg/m2 or at least 28 kg/m2 in combination with other risk factors such as hypertension, glucose dysregulation (impaired glucose tolerance or type 2 diabetes mellitus [T2DM]), and/or dyslipidemia based on interview. Main exclusion criteria were T2DM treated with medication, a medical history that might affect the safety of the enrolled individual or the interpretation of trial results, and clinically significant findings in the physical examination, as well as clinically abnormal vital signs, electrocardiogram (ECG), or laboratory values at the time of screening as judged by the investigator. Full inclusion and exclusion criteria are listed in online Supporting Information. Written informed consent was obtained from all individuals prior to initiation of any trial‐related activities.
Procedures
The procedure is described in detail in online Supporting Information. In brief, the trial consisted of six visits at the research units. Participants arrived at the research clinic in the morning of the first dosing day (day 1, visit 2), and a reevaluation of eligibility, including a brief physical examination, check of vital signs, and assessment of body weight, was conducted before randomization (randomization and masking are described in the online Supporting Information). Blood sampling (fasting) and anthropometric measurements were performed. Participants were asked to complete a satiety and craving questionnaire before consuming a standardized breakfast at the clinic and then once every hour for 4 hours until lunch, which was eaten at home. Participants randomized to the two EMP16 arms started with a titration period of 6 weeks during which the dose was sequentially increased to allow gradual adaptation to the acarbose dose in case of GI adverse effects (Supporting Information Table S1). Placebo treatment consisted of matching oral capsules containing only cellulose granules. Lifestyle instructions were limited to instructing the patient to adhere to the Nordic Nutrition Recommendations [12], with several trial‐specific clarifications (online Supporting Information). Participants returned to the clinic at visits 3 (week 7), 4 (week 14), and 5 (week 26) for efficacy assessments of body weight and body composition and safety assessments, including reporting of adverse events (AEs) and fasting blood sampling. A safety follow‐up visit took place in week 28.
Outcomes
The primary outcome variable was weight loss assessed as the relative change from baseline in body weight (in percentage) after 26 weeks of treatment with EMP16 120‐mg orlistat/40‐mg acarbose (EMP16‐120/40) compared with placebo. Weight loss was also assessed after 14 and 26 weeks, including the absolute change from baseline in body weight after treatment with EMP16‐120/40 versus placebo and EMP16‐150/50 versus placebo, the relative and absolute changes from baseline in body weight, and the proportion of participants losing at least 5% or 10% of their baseline body weight.
Additional secondary outcome variables were BMI, waist circumference sagittal diameter, and body fat percentage; lipid and glucose variables such as total cholesterol, high‐density lipoprotein (HDL) cholesterol, low‐density lipoprotein (LDL) cholesterol, triglycerides, glucose, insulin, and glycosylated hemoglobin A1c (HbA1c); inflammation and liver safety markers such as high‐sensitivity C‐reactive protein, albumin, alanine aminotransferase, aspartate aminotransferase, alkaline phosphatase, and gamma‐glutamyl transferase; and proportion of participants with T2DM, defined as fasting glucose ≥ 126 mg/dL (≥ 7.0 mmol/L), and prediabetes, defined as fasting glucose ≥ 110 to 126 mg/dL (≥ 6.1 to < 7.0 mmol/L). Several patient‐related outcome measures were assessed using questionnaires related to satiety and craving [13], meal pattern [14], quality of life after 26 (RAND‐36) [15], and the gastrointestinal symptoms rating scale (GSRS) [16]. AEs were collected at each visit (online Supporting Information) and evaluated by the investigator in terms of severity and relationship to the investigated medical product. The grading of AE severity followed the Common Terminology Criteria for AEs (CTCAE) v5.0. To assess the relationship between participant withdrawal and tolerability, the rate of participant withdrawal from the trial (overall and due to GI‐related AEs) following treatment with EMP16‐120/40 or EMP16‐150/50 was evaluated versus placebo after 26 weeks. In addition, safety laboratory parameters and vital signs including blood pressure and pulse were assessed, as well as treatment compliance.
Statistical analysis
The statistical analysis plan (SAP) was written and signed before locking the database. The primary hypothesis was that individuals treated with EMP16‐120/40 would experience a greater relative weight loss as compared with placebo. The estimated treatment difference was set to 5% with a standard deviation (SD) of 8% [17], assuming a statistical power of 80% and significance level of 5% based on two‐sided hypothesis testing. Using PASS (Power Analysis & Sample Size Software) version 16.0 (NCSS), the number of evaluable individuals needed per treatment arm was calculated to be 41. Assuming a dropout rate of 20%, a total of 156 individuals needed to be randomized. Efficacy analyses were done on a modified intention‐to‐treat (ITT) population, which included all randomized individuals who were exposed to at least one dose of EMP and who had at least one post‐baseline body weight assessment. In addition, a per‐protocol (PP) population included participants without any major protocol deviations (online Supporting Information). The safety population included all randomized individuals who were exposed to at least one dose of IEMP. In the SAP, the last‐observation‐carried‐forward imputation method was prespecified. Based on interactions with regulatory advisory boards and guidelines [18] after finalizing the SAP, we included an additional post hoc imputation method when analyzing the body weight data. A placebo‐based imputation method assumed that individuals discontinuing the trial followed the trajectory of compliant participants in the placebo group (online Supporting Information) [18].
Mean data were analyzed using ANCOVA or ANOVA. Treatment was included as an independent variable and body weight at baseline as a covariate. Categorical data were analyzed using a χ2 test without continuity correction. Ordinal data were analyzed using the Wilcoxon rank sum test. Test of normal distribution was not done as according to the central limit theorem [19]. Statistical analyses were performed using SAS version 9.4 (SAS Institute Inc.), and the level of significance was set at an α level of 0.05. Analyses of secondary outcomes were performed without any imputation. No adjustment was made for any p values of secondary outcomes; therefore, these need to be interpreted cautiously.
RESULTS
Participants were recruited between May and August 2020, and the final participant visit was in March 2021. Of the 156 randomized participants, 149 constituted the modified ITT population and were assessed for the primary end point, and 135 completed the 28‐week trial period (Figure 1). The PP population was composed of 122 participants across treatment groups. In total, 111 women and 45 men were randomized in the trial (Supporting Information Table S2). There were no important differences between the three groups at baseline (Supporting Information Tables S2 and S3).
FIGURE 1.
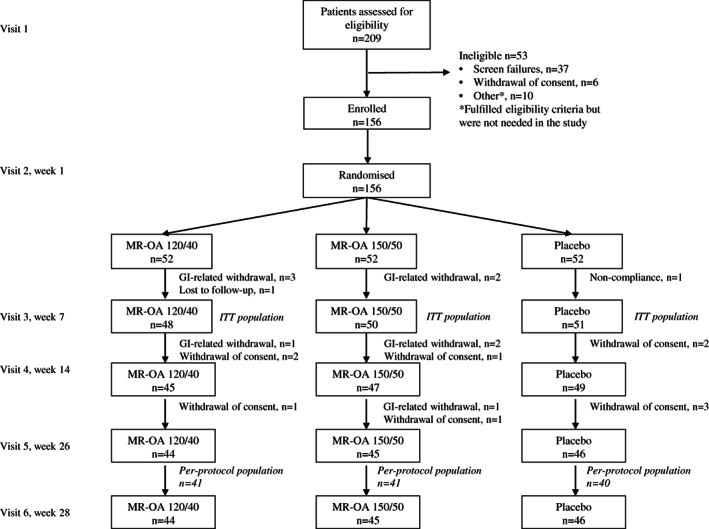
Trial profile. GI, gastrointestinal; ITT, intention‐to‐treat; MR‐OA, modified release ‐ orlistat acarbose
Primary outcomes
As illustrated in Figure 2A, participants treated with both doses of EMP16 for 26 weeks lost more weight than those treated with placebo (p < 0.0001). Mean relative weight loss in the ITT population was −5.53% and − 6.25% after 26 weeks of treatment with both EMP16‐120/40 and EMP16‐150/50, respectively, as compared with −0.83% in the placebo group at week 26 (Table 1). More participants in the active treatment groups lost at least 5% and 10% of their baseline body weight at week 26 (Figure 2B). Similar weight losses were seen in the PP population or using the more conservative imputation method (Supporting Information Table S4).
FIGURE 2.
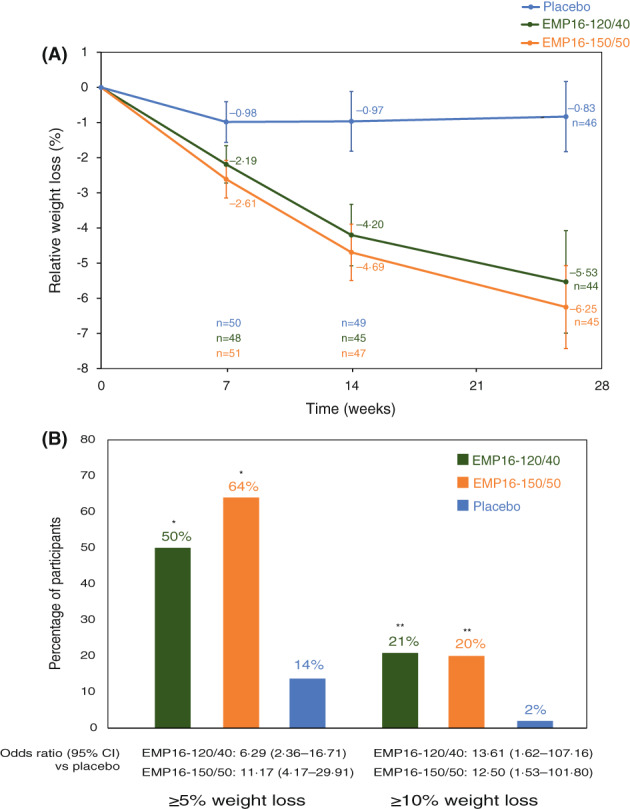
Change in body weight. (A) Data are mean percent weight loss from baseline (95% CI) (ANCOVA estimate) for the intention‐to‐treat population with the last‐observation‐carried‐forward imputation. (B) Percentage of individuals who lost more than 5% and more than 10% of baseline weight at week 26 (intention‐to‐treat population with the last‐observation‐carried‐forward imputation). *p < 0.0001 vs. placebo. **p ≤ 0.001 vs. placebo [Color figure can be viewed at wileyonlinelibrary.com]
TABLE 1.
Primary and secondary outcome variables (changes from baseline to week 26)
| EMP16‐120/40 | Estimated difference (95% CI), EMP16‐120/40 vs. placebo | p value, EMP16‐120/40 vs. placebo | EMP16‐150/50 | Estimated difference (95% CI), EMP16‐150/50 vs. placebo | p value, EMP16‐150/50 vs. placebo | Placebo | |
|---|---|---|---|---|---|---|---|
| Primary outcome variable a | |||||||
| Absolute weight change (kg) from baseline to week 26 | −6.03 (5.35) | −4.56 (−6.04 to −3.09) | < 0.001 | −6.40 (4.80) | −5.36 (−6.59 to −4.12) | < 0.001 | −0.82 (3.88) |
| Relative weight change (%) from baseline to week 26 | −5.53 (5.15) | −4.70 (−6.16 to −3.24) | < 0.001 | −6.25 (4.26) | −5.42 (−6.60 to −4.24) | < 0.001 | −0.83 (3.64) |
| Secondary outcome variables b | |||||||
| BMI (kg/m2) | −2.07 (1.97) | −1.80 (−2.50 to −1.10) | < 0.001 | −2.22 (1.54) | −1.95 (−2.55 to −1.35) | < 0.001 | −0.27 (1.33) |
| Body fat (%) | −1.16 (3.83) | −1.11 (−2.56 to 0.34) | 0.0676 | −1.94 (2.69) | −1.89 (−3.09 to −0.69) | 0.004 | −0.05 (3.05) |
| Waist circumference (cm) | −6.30 (5.70) | −3.15 (−5.44 to −0.86) | 0.009 | −7.17 (5.21) | −4.02 (−6.21 to −1.83) | 0.005 | −3.15 (5.26) |
| Sagittal abdominal diameter (cm) | −1.3 (2.2) | −0.90 (−1.80 to 0.00) | 0.052 | −1.8 (2.0) | −1.40 (−2.26 to −0.54) | 0.002 | −0.4 (2.1) |
| Systolic blood pressure (mmHg) | −2.7 (12.0) | −0.90 (−5.75 to 3.95) | 0.577 | −4.9 (13.6) | −3.10 (−8.30 to 2.10) | 0.182 | −1.8 (11.2) |
| Diastolic blood pressure (mmHg) | −3.0 (7.0) | −1.50 (−4.49 to 1.49) | 0.377 | −3.0 (8.1) | −1.50 (−4.72 to 1.72) | 0.384 | −1.5 (7.3) |
| Pulse (beats/min) | 0.6 (6.9) | −0.70 (−3.96 to 2.56) | 0.684 | −1.2 (9.1) | −2.50 (−6.20 to 1.20) | 0.184 | 1.3 (8.6) |
| Fasting glucose (mg/dL) | −4.9 (9.0) | −2.0 (−5.8 to 1.8) | 0.342 | −4.7 (0.65) | −0.10 (−11.7 to 2.5) | 0.365 | −2.9 (8.8) |
| HbA1c (mmol/mol) | −1.2 (2.3) | −0.60 (−1.54 to 0.34) | 0.291 | −1.1 (2.4) | −0.50 (−1.46 to 0.46) | 0.342 | −0.6 (2.2) |
| Fasting insulin (mIU/L) | −3.62 (9.71) | −2.11 (−5.90 to 1.68) | 0.397 | 0.08 (18.10) | 1.59 (−4.30 to 7.48) | 0.591 | −1.51 (8.41) |
| Fasting triglycerides (mmol/L) | −7.97 (56.58) | 13.29 (−13.29 to 39.86) | 0.439 | −2.66 (55.80) | 18.60 (−7.97 to 45.17) | 0.191 | −21.26 (69.08) |
| Total cholesterol (mg/dL) | −20.50 (28.23) | −15.47 (−26.30 to −4.64) | 0.007 | −17.40 (29.39) | −12.37 (−23.20 to −1.55) | 0.030 | −5.03 (22.82) |
| LDL (mg/dL) | −13.15 (20.11) | −13.53 (−22.04 to −5.03) | 0.002 | −10.05 (23.59) | −10.44 (−19.72 to −1.16) | 0.020 | 0.39 (21.27) |
| HDL (mg/dL) | −4.25 (6.19) | −3.48 (−6.19 to −0.77) | 0.025 | −6.19 (7.35) | −5.41 (−8.51 to −2.32) | <0.001 | −0.77 (6.96) |
Note: For the primary outcome variable, weight loss, mean (SD) data and estimated difference (95% CI) from the ITT analysis set with LOCF imputation.
Abbreviations: HbA1c, glycosylated hemoglobin A1C; HDL, high‐density lipoprotein; ITT, intention‐to‐treat population, everyone with at least one post–first dose measurement; LDL, low‐density lipoprotein; LOCF, last‐observation‐carried‐forward.
For EMP16‐120/40 n = 48, for EMP16‐150/50 n = 50, and for placebo n = 51. For secondary outcome variables, observed mean data (SD) and estimated difference (95% CI) are presented for the ITT analysis set without imputations. ANCOVA was performed with imputations using LOCF.
For EMP16‐120/40 n = 44, for EMP16‐150/50 n = 45, and for placebo n = 46. Changes at weeks 7 and 14 during the trial are presented in online Supporting Information.
Secondary outcomes
Statistically significant absolute mean reductions in BMI and waist circumference were observed for participants treated with both EMP16 doses for 26 weeks as compared with placebo (Table 1). The absolute mean sagittal diameter and body composition in terms of percentage body fat were significantly reduced in participants treated with EMP16‐150/50 as compared with placebo, whereas the reductions in the EMP16‐120/40 treatment group were not statistically significant.
There were no significant treatment differences in absolute mean changes from baseline in glucose metabolism markers (fasting glucose, insulin, HbA1c) or vital signs at week 26 (Table 1). The lipid metabolism markers (LDL and HDL cholesterol and total cholesterol, but not triglycerides) exhibited small but statistically significant reductions compared with the placebo group at week 26. There were no significant treatment differences in changes from baseline in T2DM or prediabetes status at week 26 (Supporting Information Table S7).
In general, there were no differences between the active treatment groups and the placebo group in the total scores for satiety and craving at week 26 (Table 2).
TABLE 2.
Questionnaires (absolute changes from baseline to week 26)
| EMP16‐120/40 (n = 44) | Estimated difference (95% CI), EMP16‐120/40 vs. placebo | p value, EMP16‐120/40 vs. placebo | EMP16‐150/50 (n = 44) | Estimated difference (95% CI), EMP16‐150/50 vs. placebo | p value, EMP16‐150/50 vs. placebo | Placebo (n = 46) | |
|---|---|---|---|---|---|---|---|
| Satiety and craving total score | 8.1 (35.4) | 9.00 (−5.74 to 23.74) | 0.229 | −0.8 (29.7) | 0.10 (−13.50 to 13.70) | 0.919 | −0.9 (35.2) |
| RAND‐36 | |||||||
| Physical functioning | 9.0 (14.9) | 7.00 (1.48 to 12.52) | 0.008 | 11.5 (15.3) | 9.50 (3.88 to 15.12) | 0.002 | 2.0 (11.3) |
| Role functioning/physical | 7.7 (36.0) | 7.70 (−5.95 to 21.35) | 0.181 | 12.2 (35.8) | 12.20 (−1.40 to 25.80) | 0.075 | 0.0 (29.0) |
| Pain (bodily pain) | 1.3 (17.4) | 1.50 (−5.68 to 8.68) | 0.877 | 11.8 (21.0) | 12.00 (4.02 to 19.98) | 0.013 | −0.2 (17.0) |
| General health | 3.3 (17.1) | 8.00 (1.68 to 14.32) | 0.017 | 6.0 (12.6) | 10.70 (5.38 to 16.02) | < 0.001 | −4.7 (±12.9) |
| Energy/fatigue (vitality) | −1.4 (19.9) | 1.70 (−6.01 to 9.41 | 0.437 | 7.2 (20.0) | 10.30 (2.57 to 18.03) | 0.022 | −3.1 (16.9) |
| Social functioning | 1.2 (20.3) | 3.90 (−4.37 to 12.17) | 0.468 | 6.4 (26.8) | 9.10 (−0.65 to 18.85) | 0.089 | −2.7 (19.3) |
| Role functioning/emotional | −2.4 (38.5) | 8.90 (−5.75 to 23.55) | 0.286 | 3.3 (43.3 | 14.60 (−1.17 to 30.37) | 0.145 | −11.3 (31.3) |
| Emotional well‐being (mental) | −3.8 (16.6) | 0.50 (−5.41 to 6.41) | 0.302 | 1.7 (17.8) | 6.00 (−0.21 to 12.21) | 0.036 | −4.3 (11.2) |
| Health transition score | 18.5 (29.8) | 12.60 (2.05 to 23.15) | 0.006 | 16.5 (20.6) | 10.60 (2.18 to 19.02) | 0.011 | 5.9 (19.7) |
| GSRS | |||||||
| Diarrhea syndrome | 1.3 (1.5) | 1.00 (0.50 to 1.50) | < 0.001 | 1.8 (1.4) | 1.50 (1.02 to 1.98) | < 0.001 | 0.3 (0.8) |
| Indigestion syndrome | 0.8 (1.0) | 0.70 (0.34 to 1.06) | < 0.001 | 1.0 (1.1) | 0.90 (0.52 to 1.28) | < 0.001 | 0.1 (0.7) |
| Constipation syndrome | 0.2 (1.0) | 0.10 (−0.23 to 0.43) | 0.831 | 0.3 (0.6) | 0.20 (−0.03 to 0.43) | 0.404 | 0.1 (0.5) |
| Abdominal pain syndrome | 0.2 (0.7) | 0.30 (0.03 to 0.57) | 0.079 | 0.1 (0.8) | 0.20 (−0.10 to 0.50) | 0.284 | −0.1 (0.6) |
| Reflux syndrome | 0.0 (0.9) | −0.20 (−0.50 to 0.10) | 0.717 | −0.1 (0.4) | −0.30 (−0.49 to −0.11) | 0.110 | 0.2 (0.5) |
Note: Observed mean data (SD) and estimated difference (95% CI) are presented for the intention‐to‐treat analysis set without imputations. ANCOVA was performed with imputations using last‐observation‐carried‐forward. Changes at weeks 7 and 14 during the trial are presented in online Supporting Information.
Abbreviations: GSRS, gastrointestinal symptoms rating scale, where higher scores indicate increased intensity; RAND‐36, 36‐item short form health survey where higher score indicates better quality of life.
Similarly, most participants appeared to follow the recommendations for healthy eating habits at baseline, and no treatment differences in overall meal patterns were observed (Supporting Information Table S10). Eating habits in relation to sweet food (cookies, chocolate, sweets, chips, soft drinks) and breakfast were improved in the EMP16‐150/50 group but not the EMP16‐120/40 group, as compared with the placebo group at week 26 (Supporting Information Tables S11 and S12).
Quality of life, based on the RAND‐36 health survey, improved more in both active treatment groups compared with the placebo group between baseline and week 26 with respect to physical functioning, general health, and the overall health transition score (Table 3). In addition, participants in the EMP16‐150/50 group improved more than those in the placebo group in terms of bodily pain, energy/fatigue, and emotional well‐being. The larger increase in rated quality of life was somewhat unexpected, and in an exploratory posthoc analysis (Supporting Information Figure S1), relative weight loss was plotted against the GSRS domain health transition, and no clear correlation was observed. There were no differences in activity and sleep habits between the study groups during the trial (Supporting Information Table S13).
TABLE 3.
Participant withdrawal and AEs reported by ≥ 5% of participants in any group
| EMP16‐120/40 (n = 52) | EMP16‐150/50 (n = 52) | Placebo (n = 52) | |
|---|---|---|---|
| Overall withdrawal rate | 8 (15%) | 7 (14%) | 6 (12%) |
| Any AE | 32 (62%) | 39 (75%) | 30 (58%) |
| Any AE leading to withdrawal | 4 (8%) | 6 (12%) | 0 |
| Most frequent AEs by MedDRA PT | |||
| Nasopharyngitis | 4 (8%); 4 | 10 (19%); 10 | 13(25%); 15 |
| Diarrhea | 8 (15%); 9 | 8 (15%); 9 | 0 |
| Headache | 4 (8%); 5 | 2 (4%); 2 | 3 (6%); 4 |
| Flatulence | 4 (8%); 4 | 3 (6%); 4 | 1 (2%); 1 |
| COVID‐19 | 0 | 6 (12%); 6 | 2 (4%); 2 |
| Abdominal distension | 4 (8%); 4 | 1 (2%); 1 | 0 |
| Causality | |||
| Unlikely | 22 (42%) | 28 (54%) | 27 (52%) |
| Possibly | 7 (13%) | 9 (17%) | 6 (12%) |
| Probably | 13 (25%) | 12 (23%) | 2 (4%) |
| Severity | |||
| Mild | 21 (40%) | 24 (46%) | 22 (42%) |
| Moderate | 11 (21%) | 13 (25%) | 11 (21%) |
| Severe | 14 (27%) | 12 (23%) | 3 (6%) |
Note: Data are shown as number of participants (percentage of treatment arm); number of events. Data are from the safety population (all participants who were randomized and exposed to at least one treatment dose). Full list in online Supporting Information.
Abbreviations: AE, adverse event; MedDRA, medical dictionary for regulatory activities; PT, preferred term.
The mean scores in the GSRS diarrhea and indigestion syndromes increased to a significantly greater extent in both active treatment groups compared with the placebo group at week 26 (Table 2). Most participants rated their symptoms as mild or moderate. There were no treatment differences observed in the other parts of the GSRS. In Supporting Information Figure S3, the percentage of participants in the three groups reporting minor or major discomfort in the GSRS question “Have you been bothered by DIARRHEA during the past week? (Diarrhea refers to a too frequent emptying of the bowels.)” during the trial is reported. A minority reported major discomfort, and the highest level of discomfort were observed at week 8.
A total of 191 AEs were reported by 101 (65%) of the 156 randomized participants with the three most common events being nasopharyngitis, diarrhea, and headache (Table 3). Diarrhea was reported only in the active treatment groups; 4 of 52 participants (8%) in the EMP16‐120/40 group and 5 of 52 participants (10%) in the EMP16‐150/50 group withdrew early from the trial because of GI‐related AEs. In addition, one participant in the EMP‐150/50 group withdrew consent to remain in the trial because of a COVID‐19 infection. No participants in the placebo group withdrew because of AEs, whereas the overall withdrawal rate was comparable between the active treatment groups. Most AEs were mild or moderate in intensity. No deaths or serious AEs occurred during the trial. Compliance was high, and no difference between the treatment groups was observed (Supporting Information Table S17).
There were no clinically noteworthy changes in liver enzymes during the trial (Supporting Information Table S8). A few individuals had a transient increase; however, these were not judged to be related to the investigated medical product according to the investigator, and the participants continued in the trial. There were no clinically relevant or statistically significant changes or differences in safety laboratory parameters or ECG during the trial (data at emprospharma.com). Additional data are presented in the online Supporting Information.
DISCUSSION
In this trial, treatment with EMP16 for 26 weeks led to a continuous and clinically relevant weight loss. More than 50% of the participants in both active treatment groups lost at least 5% of their baseline weight, and more than 20% lost at least 10%, compared with 14% and 2% of participants, respectively, in the placebo group. Other anthropometric measurements such as BMI, waist circumference, sagittal abdominal diameter, and percentage of body fat showed similar treatment effects, albeit with larger effects for the higher EMP16‐150/50 dose. Patient‐reported quality of life showed improvements in both intervention groups, notably in physical functioning, general health, and the overall health transition score. Blood pressure, glucose metabolism markers, and blood lipids were not notably affected, and no treatment differences were observed in the ratings of satiety and craving, although minor improvements in meal patterns in relation to sweet food and breakfast were seen in the EMP16‐150/50 intervention group. EMP16 was generally well tolerated, and no safety concerns were noted.
This trial corroborates the findings from the previous pilot trial with EMP16, which demonstrated that both the efficacy and tolerability of orlistat and acarbose were increased by employing a modified‐release intervention [10, 11]. Orlistat and acarbose treatment in their conventional dosage forms typically provide an approximate relative weight loss of 2% to 3% [17] and less than 0.5%, respectively [20], whereas the observed mean placebo‐adjusted weight losses in the EMP16 arms were approximately 5%. The efficacy of EMP16 seems equivalent to most currently approved oral weight‐loss drugs and liraglutide [1, 2]. Furthermore, a combination of orlistat and acarbose in their conventional dosage forms would likely cause tolerability problems and potentially augment their associated GI side effects such as flatulence with or without discharge [6, 8]. EMP16 was designed to encompass three contributing factors: (i) the mechanisms of action of orlistat and acarbose in their conventional dosage form on energy uptake; (ii) employment of a modified‐release pattern to ensure that food‐derived ligands are delivered to various appetite regulating checkpoints in the GI tract in an appropriate manner; and (iii) improved tolerability by releasing acarbose and orlistat at selected part of the GI tract (online Supporting Information).
Despite the weight loss achieved with EMP16, there were positive albeit limited effects on blood pressure, glucose metabolism, and blood lipids. The decrease in HDL is typically seen initially during weight loss but is not of clinical concern [21]. However, as evident from the baseline characteristics, this was a fairly healthy population of individuals with obesity. The prevalence of hypertension at baseline was approximately 30%, whereas a prevalence of about 60% has been observed in patients with obesity in other studies [22]. Furthermore, baseline blood lipid levels were generally low in the current study, with a small proportion of individuals (7%) on blood lipid medication, and most participants having a baseline HbA1c value below 40 mmol/mol. The observed lack of clinically relevant changes in metabolic risk markers is in line with similar weight‐loss studies in patients with obesity without diabetes using liraglutide [23] or semaglutide [24].
Patient‐reported quality of life was clearly improved in the intervention groups, especially in the EMP16‐150/50 group, with distinct clinically relevant differences in several of the RAND‐36 domains. The differences recorded were larger than seen in other weight‐loss studies using orlistat in its conventional dosage form [25] or liraglutide [23].
The larger increase in rated quality of life was somewhat unexpected, and in an exploratory post hoc analysis, relative weight loss was plotted against health transition. The explained variance was low; additional factors apart from weight loss seem to have been involved in the increased quality of life ratings.
There were no significant treatment differences in the ratings of satiety and craving. In the pilot study, satiety was greater in the EMP16 groups compared with conventional orlistat [11]. As stated earlier, orlistat administered in a conventional dosage form is associated with an increased appetite compared with placebo, partly by its effect on satiety sensing cells in the duodenum [7]. With the modified release preparation employed in EMP16, the orlistat‐mediated effect on appetite seems to be reduced.
No large treatment differences were observed in reported meal patterns, although participants in the EMP16‐150/50 group reported increased breakfast intake and decreased intake of sweets and cakes. This could possibly be a “nudging” effect [26] of EMP16, as side effects are worsened if sweet, high‐fat meal items are consumed, and the “cost” of not eating correctly appeared higher in the EMP16‐150/50 group. Alternatively, EMP16 may have triggered an incretin effect, which may have affected the participants’ preference for sweets and cakes [27]. We did not observe any effects on GLP‐1 in the previous 14‐day pilot trial [11]. However, it possibly takes longer and/or higher doses of ligands to elicit an incretin response in participants with obesity [28].
Orlistat and acarbose in their conventional dosage forms are associated with frequent GI side effects [6, 8], which limit their popularity. In the present trial, 15% of the participants receiving EMP16 reported diarrhea of severe intensity, whereas no participants in the placebo group did so, and more subjects (6%–8%) in the EMP16 groups reported flatulence of severe intensity compared with the placebo group (2%). Because GI events were recorded as AEs in the trial only if they were judged by the investigator as being severe or leading to withdrawal, cases of minor or moderate GI events were not registered. The results from the GSRS corroborate that the frequency of diarrhea with EMP16 was greater than that in the placebo group, but the mean diarrhea syndrome scores in both intervention groups were around 3 (mild discomfort), and only a small minority reported major discomfort. Fecal incontinence is perhaps the most problematic side effect associated with conventional orlistat, but this seemed not to be an issue in the present trial with only one reported event of severe intensity in the EMP16‐150/50 group. In a similar 6‐month trial, 5% of participants in the conventional orlistat arm reported fecal incontinence [29], whereas approximately 18% of participants reported such events in trials of longer duration [6]. Lastly, only one participant reported one event of nausea of severe intensity, and none reported vomiting, which is in contrast to studies using liraglutide [23].
This was a proof‐of‐concept trial with limited interaction between trial sites and participants; there were few visits with limited activities. Moreover, the participants did not receive any lifestyle instructions, only limited information regarding dietary choices. In contrast to many weight‐loss studies [23, 29], a run‐in participant selection procedure before randomization was not used. One reason for this “lean” design was to mimic a real‐life situation and better understand the efficacy of EMP16 in a setting more closely resembling a clinical situation, somewhat analogous to a phase IV trial [30]. The chosen design might explain the limited placebo effect and ensured that the trial could be conducted during the COVID‐19 pandemic with few major interruptions. Only a few participants had their last visit postponed for more than a week.
One limitation of the trial was the chosen imputation method. The last‐observation‐carried‐forward imputation method was prespecified in the protocol and has previously been recommended by regulatory authorities, but it is no longer regarded as optimal [18]. A more conservative imputation method was added post hoc [18], and comparable results were obtained.
Another weakness was the potential problem of maintaining masking, not unique to EMP16. One of the known side effects of conventional orlistat is oily stools. Participants guessing that they had received placebo may have had a lower motivation to fulfill the additional lifestyle instructions, which could have led to an increased difference in outcomes between the active treatments and placebo. However, there were no differences in compliance between the treatment groups and the withdrawal rate for all groups was low (≤ 15%).
Overall, this trial supports that orlistat and acarbose can be successfully combined as a promising potential candidate for improved weight management. The magnitude of the weight loss may have been less than that achieved with semaglutide and tirzepatide [1, 2, 31], but it was similar to other oral weight‐loss products, as well as liraglutide [32]. As stated in the Obesity Canada guidelines, “The individual response to obesity management pharmacotherapy is heterogeneous; the response to medications can differ from patient to patient” [3]; thus, more tools in the toolbox can only benefit the individuals with obesity. Furthermore, obesity is a chronic disease [3] that may require long‐term treatment. Both orlistat and acarbose in their conventional dosage forms have already demonstrated long‐term safety [6, 8, 20]. No safety issues were observed in the present trial with EMP16, and in general, the safety and tolerability of the modified‐release drug product appeared to be improved compared with the conventional products [10]. The efficacy and safety of EMP16 need to be evaluated in a study of longer duration in a more diverse population.
AUTHOR CONTRIBUTIONS
All authors took part in designing the study outline. Helena Litorp was the signatory principal investigator of the clinical trial and responsible for site 1 (Uppsala, Sweden). Daniel Willhems was principal investigator for site 2 (Linköping, Sweden). Sandra Kuusk was the principal writer of the clinical trial protocol and the clinical trial report. Stefan Grudén and Göran Alderborn were responsible for the pharmaceutical development of EMP16. Ulf Holmbäck and Sandra Kuusk wrote the first draft of the manuscript. Statistical analyses were performed by the clinical research organization. All authors have taken part in writing, reviewing, and finalizing the manuscript. All authors have had access to the data and guarantee the validity of the presented data.
FUNDING INFORMATION
Empros Pharma AB funded the trial and provided the clinical research organization (CRO) with the trial synopsis. The funder and the CRO discussed the design of the trial, and the CRO wrote the protocol and the statistical analysis plan, collected the data, and performed the statistical analyses. The funder had access to the data after database lock. The funder and the authors interpreted the data and wrote the manuscript collectively; everyone had access to the data.
CONFLICT OF INTEREST
Ulf Holmbäck, Stefan Grudén, Arvid Söderhäll, Göran Alderborn, and Anders Forslund have equity interests in Empros Pharma AB and have acted as employees or consultants for the company. Ulf Holmbäck, Stefan Grudén, Göran Alderborn, and Anders Forslund hold the patent for the studied weight‐loss product. Helena Litorp, Daniel Willhems, and Sandra Kuusk declared no conflict of interest.
CLINICAL TRIAL REGISTRATION
European Union Drug Regulating Authorities Clinical Trials Database (EudraCT) 2019‐004545‐32.
Supporting information
Appendix S1 Supporting Information
ACKNOWLEDGMENTS
The authors express their great appreciation for the hard work and commitment of both trial participants and study staff during a difficult period. The authors also want to acknowledge medical writer Angela Stocks, PhD (Alpha Script, Denmark), who assisted with insightful comments, further editing, and proofreading.
Holmbäck U, Grudén S, Litorp H, et al. Effects of a novel weight‐loss combination product containing orlistat and acarbose on obesity: A randomized, placebo‐controlled trial. Obesity (Silver Spring). 2022;30(11):2222‐2232. doi: 10.1002/oby.23557
Funding information Empros Pharma AB
DATA AVAILABILITY STATEMENT
Beginning 3 months after publication without any deadline, individual participant data that underlie the results reported in this article, after deidentification (text, tables, figures, and appendices), will be available to anyone who provides a methodologically sound proposal. Information on how to apply and whom to contact will be given at emprospharma.com. The following documents will be freely available to anyone upon publication at emprospharma.com: Study Protocol, Statistical Analysis Plan, Informed Consent Form (in Swedish), and Clinical Study Report.
REFERENCES
- 1. Müller TD, Blüher M, Tschöp MH, DiMarchi RD. Anti‐obesity drug discovery: advances and challenges. Nat Rev Drug Discov. 2022;21:201‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shi Q, Wang Y, Hao Q, et al. Pharmacotherapy for adults with overweight and obesity: a systematic review and network meta‐analysis of randomised controlled trials. Lancet. 2022;399:259‐269. [DOI] [PubMed] [Google Scholar]
- 3. Obesity Canada. About obesity. Accessed June 1, 2022. https://obesitycanada.ca/about-obesity/
- 4. Torgerson JS, Hauptman J, Boldrin MN, Sjöström L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care. 2004;27:155‐161. [DOI] [PubMed] [Google Scholar]
- 5. le Roux CW, Astrup A, Fujioka K, et al. 3 years of liraglutide versus placebo for type 2 diabetes risk reduction and weight management in individuals with prediabetes: a randomised, double‐blind trial. Lancet. 2017;389:1399‐1409. [DOI] [PubMed] [Google Scholar]
- 6. McClendon KS, Riche DM, Uwaifo GI. Orlistat: current status in clinical therapeutics. Expert Opin Drug Saf. 2009;8:727‐744. [DOI] [PubMed] [Google Scholar]
- 7. Ellrichmann M, Kapelle M, Ritter PR, et al. Orlistat inhibition of intestinal lipase acutely increases appetite and attenuates postprandial glucagon‐like peptide‐1‐(7–36)‐amide‐1, cholecystokinin, and peptide YY concentrations. J Clin Endocrinol Metabol. 2008;93:3995‐3998. [DOI] [PubMed] [Google Scholar]
- 8. Laube H. Acarbose. Clin Drug Investig. 2002;22:141‐156. [Google Scholar]
- 9. Enç FY, Imeryüz N, Akin L, et al. Inhibition of gastric emptying by acarbose is correlated with GLP‐1 response and accompanied by CCK release. Am J Physiol Gastrointest Liver Physiol. 2001;281:G752‐G763. [DOI] [PubMed] [Google Scholar]
- 10. Grudén S, Forslund A, Alderborn G, Söderhäll A, Hellström PM, Holmbäck U. Safety of a novel weight loss combination product containing orlistat and acarbose. Clin Pharmacol Drug Dev. 2021;10:1242‐1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Holmbäck U, Forslund A, Grudén S, et al. Effects of a novel combination of orlistat and acarbose on tolerability, appetite, and glucose metabolism in persons with obesity. Obes Sci Pract. 2020;6:313‐323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nordic Council . Nordic Nutrition Recommendations 2012. Published November 3, 2014. Accessed May 22, 2022. https://www.norden.org/en/publication/nordic-nutrition-recommendations-2012
- 13. Blundell J, de Graaf C, Hulshof T, et al. Appetite control: methodological aspects of the evaluation of foods. Obes Rev. 2010;11:251‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. National Board of Health and Welfare. Enkla råd för bra matvanor. National Board of Health and Welfare. Published June 24, 2014. Accessed May 22, 2022. https://www.socialstyrelsen.se/globalassets/sharepoint‐dokument/artikelkatalog/kunskapsstod/2014‐6‐24.pdf
- 15. Hays RD, Morales LS. The RAND‐36 measure of health‐related quality of life. Ann Med. 2001;33:350‐357. [DOI] [PubMed] [Google Scholar]
- 16. Revicki DA, Wood M, Wiklund I, Crawley J. Reliability and validity of the gastrointestinal symptom rating scale in patients with gastroesophageal reflux disease. Qual Life Res. 1998;7:75‐83. [DOI] [PubMed] [Google Scholar]
- 17. Hutton B, Fergusson D. Changes in body weight and serum lipid profile in obese patients treated with orlistat in addition to a hypocaloric diet: a systematic review of randomized clinical trials. Am J Clin Nutr. 2004;80:1461‐1468. [DOI] [PubMed] [Google Scholar]
- 18. McEvoy BW. Missing data in clinical trials for weight management. J Biopharm Stat. 2016;26:30‐36. [DOI] [PubMed] [Google Scholar]
- 19. Kwak SG, Kim JH. Central limit theorem: the cornerstone of modern statistics. Korean J Anesthesiol. 2017;70:144‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li Y, Tong Y, Zhang Y, Huang L, Wu T, Tong N. Acarbose monotherapy and weight loss in Eastern and Western populations with hyperglycaemia: an ethnicity‐specific meta‐analysis. Int J Clin Pract. 2014;68:1318‐1332. [DOI] [PubMed] [Google Scholar]
- 21. Santos HO, Lavie CJ. Weight loss and its influence on high‐density lipoprotein cholesterol (HDL‐C) concentrations: a noble clinical hesitation. Clin Nutr ESPEN. 2021;42:90‐92. [DOI] [PubMed] [Google Scholar]
- 22. Czernichow S, Castetbon K, Salanave B, et al. Determinants of blood pressure treatment and control in obese people: evidence from the general population. J Hypertens. 2012;30:2338‐2344. [DOI] [PubMed] [Google Scholar]
- 23. Astrup A, Rössner S, Van Gaal L, et al. Effects of liraglutide in the treatment of obesity: a randomised, double‐blind, placebo‐controlled study. Lancet. 2009;374:1606‐1616. [DOI] [PubMed] [Google Scholar]
- 24. Wilding JPH, Batterham RL, Calanna S, et al. Once‐weekly semaglutide in adults with overweight or obesity. N Engl J Med. 2021;384:989‐1002. [DOI] [PubMed] [Google Scholar]
- 25. Swinburn BA, Carey D, Hills AP, et al. Effect of orlistat on cardiovascular disease risk in obese adults. Diabetes Obes Metab. 2005;7:254‐262. [DOI] [PubMed] [Google Scholar]
- 26. Arno A, Thomas S. The efficacy of nudge theory strategies in influencing adult dietary behaviour: a systematic review and meta‐analysis. BMC Public Health. 2016;16:676. doi: 10.1186/s12889-016-3272-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jensterle M, DeVries JH, Battelino T, Battelino S, Yildiz B, Janez A. Glucagon‐like peptide‐1, a matter of taste? Rev Endocr Metab Disord. 2021;22:763‐775. [DOI] [PubMed] [Google Scholar]
- 28. Ranganath LR, Beety JM, Morgan LM, Wright JW, Howland R, Marks V. Attenuated GLP‐1 secretion in obesity: cause or consequence? Gut. 1996;38:916‐919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Van Gaal LF, Broom JI, Enzi G, Toplak H. Efficacy and tolerability of orlistat in the treatment of obesity: a 6‐month dose‐ranging study. Orlistat dose‐ranging study group. Eur J Clin Pharmacol. 1998;54:125‐132. [DOI] [PubMed] [Google Scholar]
- 30. Gursoy A, Erdogan MF, Cin MO, Cesur M, Baskal N. Comparison of orlistat and sibutramine in an obesity management program: efficacy, compliance, and weight regain after noncompliance. Eat Weight Disord. 2006;11:e127‐e132. [DOI] [PubMed] [Google Scholar]
- 31. Jastreboff AM, Aronne LJ, Ahmad NN, et al. SURMOUNT‐1 Investigators. Tirzepatide once weekly for the treatment of obesity. N Engl J Med. 2022;387:205‐216. [DOI] [PubMed] [Google Scholar]
- 32. Khera R, Murad MH, Chandar AK, et al. Association of pharmacological treatments for obesity with weight loss and adverse events: a systematic review and meta‐analysis. JAMA. 2016;315:2424‐2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information
Data Availability Statement
Beginning 3 months after publication without any deadline, individual participant data that underlie the results reported in this article, after deidentification (text, tables, figures, and appendices), will be available to anyone who provides a methodologically sound proposal. Information on how to apply and whom to contact will be given at emprospharma.com. The following documents will be freely available to anyone upon publication at emprospharma.com: Study Protocol, Statistical Analysis Plan, Informed Consent Form (in Swedish), and Clinical Study Report.