

Summary
This analysis is the largest population‐based study to date to provide contemporary and comprehensive epidemiological estimates of all third edition of the International Classification of Diseases for Oncology (ICD‐O‐3) coded Langerhans cell histiocytosis (LCH) from England. People of all ages were identified from the National Cancer Registration Dataset using ICD‐O‐3 morphologies 9751–9754 for neoplasms diagnosed in 2013–2019. A total of 658 patients were identified, of whom 324 (49%) were children aged <15 years. The age‐standardised incidence rate was 4.46 (95% confidence interval [CI] 3.99–4.98) per million children and 1.06 (95% CI 0.94–1.18) per million adults aged ≥15 years. Prevalence of LCH was 9.95 (95% CI 9.14–10.81) per million persons at the end of 2019. The 1‐year overall survival (OS) was 99% (95% CI 97%–100%) for children and 90% (95% CI 87%–93%) for adults. Those aged ≥60 years had poorer OS than those aged <15 years (hazard ratio [HR] 22.12, 95% CI 7.10–68.94; p < 0.001). People in deprived areas had lower OS than those in the least deprived areas (HR 5.36, 95% CI 1.16–24.87; p = 0.03). There will inevitably be other environmental factors and associations yet to be identified, and the continued standardised data collection will allow further evaluation of data over time. This will be increasingly important with developments in LCH management following the large collaborative international trials such as LCH IV.
Keywords: cancer, childhood haematological malignancies, epidemiology, Langerhans cell histiocytosis (LCH), survival
INTRODUCTION
Eosinophilic granuloma, Hand–Schüller–Christian disease and Letterer–Siwe disease were first recognised in 1953 as diverse manifestations of the same disease, initially termed histiocytosis X 1 but since 1987 referred to as Langerhans cell histiocytosis (LCH). 2 X‐linked inactivation first suggested LCH to be a neoplastic disorder, 3 but it was the subsequent presence of the B‐Raf proto‐oncogene, serine/threonine kinase (BRAF) V600E mutation in ~50% of cases, followed by the finding of other somatic mutations activating the mitogen‐activated protein kinase (MAPK) signalling pathway that confirmed LCH was a clonal myeloid neoplasia. 4 , 5
This delay in the full recognition of LCH as a common neoplastic process is reflected in the history of its cancer registry coding worldwide and leads to our poor understanding of LCH epidemiology. For example, the most severe form, Letterer–Siwe disease, was first included in the ‘Neoplasms’ chapter of International Statistical Classification of Diseases and Related Health Problems ninth Revision (ICD‐9) in 1978 and the first edition of International Classification of Diseases for Oncology (ICD‐O) in 1976, but the other forms did not appear in ICD‐O until the third edition (ICD‐O‐3) and were only assigned malignant behaviour codes in the first revision of ICD‐O‐3 (ICD‐O‐3.1). 6 This historical piecemeal coding explains the paucity of population‐based information on the incidence and outcome of LCH with a historical bias in cases to the more severe end of the disease spectrum. However, following the introduction of ICD‐O‐3.1 coding in 2013, the entire spectrum of LCH finally fell under the remit of population‐based cancer registries.
The only published population‐based reports on the epidemiology of LCH that have included all forms of the disease have been studies of children in European countries, 7 , 8 , 9 , 10 , 11 , 12 , 13 which suggested that incidence among children was between two and nine per million per year. These numbers are most probably an underestimate, 14 and the high proportion of multisystem LCH in studies with the lowest incidence rates 9 , 10 is consistent with under‐ascertainment of single‐system disease. Two reports from the United States Surveillance, Epidemiology, and End Results (SEER) Program cancer registries covered only disseminated LCH in children 15 and adults. 16 The incidence rate in the childhood study was below one per million. 15 Another study of childhood histiocytosis from the SEER Program combined LCH with other class I histiocytosis 17 but the incidence rates were similarly low, presumably as the great majority of cases were of disseminated LCH. Other studies originated from referral centres with an inevitable bias towards severe cases. 11 , 18 The total annual incidence of LCH across all age groups has been suggested closer to 11–12 per million, 19 but this is especially uncertain in the almost complete absence of population‐based data on LCH among adults. Except for the French study of outcomes in children diagnosed during 1983–2012, 8 the period for all of the population‐based studies ended >10 years ago. The SEER studies also included ethnicity as a factor in their analyses. 15 , 16 , 17 Lower socioeconomic status has not yet been considered as a risk factor in LCH, but for other cancers it is associated with increased incidence and inferior survival. 20 , 21
We report a nationwide population‐based study and provide contemporary and comprehensive epidemiological estimates of all ICD‐O‐3 coded LCH from England, for the first time looking at incidence, prevalence and survival associated with deprivation.
METHODS
Data sources
The most recently released data from the National Cancer Registration Dataset (NCRD) was used. The NCRD holds the population‐based national cancer registry for England and is linked to other datasets for analysis purposes. Cancer data collection from all health institutions is a statutory requirement. Henson et al. 22 provides a detailed description of the NCRD.
Study population
People of all ages were identified using ICD‐O‐3 morphologies 9751–9754 (detailed description is in the Table S1) for neoplasms diagnosed between 1 January 2013 and 31 December 2019. There are cases of single‐site, single‐system and multisystem LCH. Multisystem disease at LCH diagnosis present many coding difficulties, 6 and consequently the coding within the routine data of the NCRD lacked detailed topographical recording of all sites affected by LCH in each patient, so we were unable to clearly delineate between cases of single‐site, single‐system and multisystem LCH. Restricting to start date since 2013 and via ICD‐O‐3 contemporary coding was to ensure that all cases of LCH were registered prospectively, minimising any bias towards the more severe forms of the disease.
Study end‐points and statistical analysis
Numbers and frequencies by patient characteristics were described. Two‐tailed hypothesis test was used to assess difference between groups. All LCH end‐points were reported for the overall population, children (aged <15 years at diagnosis), and adults (aged ≥15 years at diagnosis) based on previous studies showing higher incidence and different outcomes among children compared to adults, 7 , 8 , 9 , 10 , 11 , 12 , 13 , 15 , 16 , 17 , 18 as well as sex, deprivation (quintiles 1–5, 1 the least deprived and 5 the most deprived) and region. Children were further divided into ages of <1 and 1–14 years, so our results could be compared to extant literature. 7 , 9 , 10 For adults, the International Cancer Survival Standards 23 divides them into ages of 15–44, 45–54, 55–64, and ≥65 years. However, the frequency of LCH by age is different from that of common cancer: the International Registry of the Histiocyte Society has reported that LCH is more common in young adults (aged <30 years). 24 It would be more informative to subdivide those aged 15–44 years into 15–19, 20–29 and 30–44 years. Our study was large in the context of LCH, but LCH remains a rare disease and small numbers limit further splits of age‐bands such as 5‐ or 10‐year in the analyses. Therefore, for those aged ≥15 years we categorised them into three 15‐year bands 15–29, 30–44 and 45–59 years, and the ≥60 years group. Deprivation was the Index of Multiple Deprivation (IMD), which is the official measure of relative deprivation for small areas in England. 25 Each individual in the NCRD has their IMD assigned from their postcode of residence at diagnosis as do the entire population denominator for England. The quintiles are calculated by ranking the 32 844 neighbourhoods of Lower‐layer Super Output Areas (LSOAs) in England from most deprived to least deprived and dividing them into five quintiles. The population sizes are not identical for each LSOA nationally, but each quintile percentage varies between 19.4% and 20.6%. Region‐level geographies were based on the postcode of the residence of the patient at the time of the diagnosis. The latest IMD 2019 26 reveals concentrations of deprivation in areas that have historically had large heavy industry manufacturing and/or mining sectors (such as the West Midlands, the East Midlands, and the North East), coastal towns (such as the North West or the South East), and parts of east London. There are also pockets of deprivation surrounded by less deprived places in every region of England. Survival was also reported by ethnicity. Incidence and prevalence by ethnicity were not calculated because the Office for National Statistics (ONS) 27 no longer publishes approved populations by ethnicity.
Incidence
Crude and directly age‐standardised incidence rate (ASR) per million persons per year were calculated. The ASR was age‐adjusted to the 2013 European Standard Population. 28 The total at‐risk population (the denominator) was the ONS mid‐year population estimates for 2013–2019. Incidence‐rate ratios (IRRs) were calculated to compare incidence between groups. To estimate if there was a trend in incidence associated with deprivation a Poisson regression model was fitted using deprivation quintile as a linear term adjusted for age, sex and region. This model was compared to the model with deprivation quintile as a categorical variable, using a likelihood ratio test to assess evidence of departure from a linear trend.
Prevalence
Using a method described in Maddams et al, 29 a 7‐year limited‐duration LCH prevalence per million persons was calculated. The survivors (the numerator) were the number of people that were diagnosed with LCH between 1 January 2013 and 31 December 2019 and were still alive on 31 December 2019. The total at‐risk population (the denominator) was the ONS mid‐year population estimates for 2019.
Survival
A check on the vital status of each patient with cancer is made annually through the Personal Demographics Service (PDS) 30 and the last trace for death in the most recently released data from the NCRD was made on 5 January 2021. Survival time measured the time, in years, from the date of being diagnosed with LCH to the earliest of the patient's date of death, fifth anniversary of diagnosis with LCH, date of embarkation or loss to follow‐up at the PDS, or the censor date on 5 January 2021. Overall survival (OS) and hazard ratio (HR) were calculated. The Kaplan–Meier method was used to estimate OS. 31 A Cox proportional hazards regression model 32 (limited to the first year after diagnosis when all patients had complete follow‐up) and the Efron method of tie handling were used to assess relative risk (hazard) of death, adjusting for age, sex, deprivation, region, and ethnicity. Cox proportional hazards regression model assumption tests were based on Schoenfeld residuals.
Statistical software
All statistical analyses were performed using Stata, version 15.1 (StataCorp LLC).
RESULTS
Patient characteristics
We identified 658 patients with LCH diagnosed in 2013–2019. Almost half (324 [49%]) were children, of whom 53 (8%) were infants aged <1 year. Over half (370 [56%]) were male and 523 (79%) from White ethnic groups. Significantly more (315 [48%]) resided in the 40% deprived areas (deprivation quintiles 4 and 5), compared to 229 (35%) in the 40% non‐deprived areas (quintiles 1 and 2) (p < 0.001). Overall, 165 (25%) arose from bone, 152 (23%) bone marrow, 73 (11%) skin, and 62 (9%) bronchus/lung. In children, 128 (40%) arose from bone, 73 (23%) bone marrow and two (1%) bronchus/lung; whereas in adults 37 (11%) arose from bone, 79 (24%) bone marrow and 60 (18%) bronchus/lung (Table 1; patient characteristics by age <1, 1–14, 15–29, 30–44, 45–59, or ≥60 years are in Table S2; detailed site codes and description are in Table S3). Of the 62 cases that arose in the bronchus/lung, a significantly higher proportion resided in the most deprived areas (20 [32%]) than in the least deprived areas (seven [11%]) (p = 0.02).
TABLE 1.
Patients’ characteristics for Langerhans cell histiocytosis in England, 2013–2019
| Characteristic | N (%) a |
|---|---|
| Age at diagnosis, years | |
| Infants (<1) | 53 (8) |
| 1–14 | 271 (41) |
| 15–29 | 72 (11) |
| 30–44 | 80 (12) |
| 45–59 | 100 (15) |
| ≥60 | 82 (12) |
| Children (<15) | 324 (49) |
| Adults (≥15) | 334 (51) |
| Overall | 658 (100) |
| Sex | |
| Female | 288 (44) |
| Male | 370 (56) |
| Ethnicity | |
| White | 523 (79) |
| Asian | 44 (7) |
| Other b | 34 (5) |
| Unknown | 57 (9) |
| Deprivation quintile | |
| 1 – least deprived | 116 (18) |
| 2 | 113 (17) |
| 3 | 114 (17) |
| 4 | 156 (24) |
| 5 – most deprived | 159 (24) |
| Region | |
| North East | 34 (5) |
| North West | 75 (11) |
| Yorkshire/Humber | 60 (9) |
| East Midlands | 46 (7) |
| West Midlands | 85 (13) |
| East of England | 61 (9) |
| London | 97 (15) |
| South East | 119 (18) |
| South West | 81 (12) |
| Site of disease c | |
| Bone | 165 (25) |
| Bone marrow | 152 (23) |
| Skin | 73 (11) |
| Bronchus/lung | 62 (9) |
| Other and unknown | 206 (31) |
Percentages might not total 100% due to approximation.
Combined all other known ethnic groups due to low numbers.
Detailed site codes and description are in Table S3.
Incidence
The ASR was 1.60 per million persons per year overall, 4.46 for children and 1.06 for adults. Children had significantly higher incidence than adults (IRR 4.35, 95% confidence interval [CI] 3.73–5.07; p < 0.001). Incidence for males was significantly higher than that for females, with a male ASR of 1.79, compared to a female ASR of 1.41 (IRR 1.28, 95% CI 1.10–1.49; p < 0.01). There was substantial variation in the incidence with age: the highest crude incidence was seen in infants with 11.56 (95% CI 8.66–15.13) per million, compared to the lowest 0.91 (95% CI 0.73–1.13) for those aged ≥60 years. The ASR was 1.50 per million for those who resided in the least deprived areas, compared to 1.86 for those who resided in the most deprived areas (IRR 1.27, 95% CI 0.99–1.62; p = 0.06). The estimated increase in relative incidence per deprivation quintile (deprivation quintile fitted as a continuous variable), assuming a linear trend, was 6% (IRR 1.06, 95% CI 1.01–1.13; p = 0.03, adjusted for age, sex and region). When compared to the model with deprivation quintile fitted as a categorical variable, there was no evidence of departure from a linear trend (p = 0.31). There was variation in incidence with region, but small numbers meant there was greater uncertainty in the estimates thus preventing further assessment (Table 2).
TABLE 2.
Incidence per million persons for Langerhans cell histiocytosis in England, 2013–2019
| Crude rate (95% CI) a | ASR b (95% CI) | IRR c (95% CI) | p | |
|---|---|---|---|---|
| Age at diagnosis, years | ||||
| Infants (<1) | 11.56 (8.66–15.13) | |||
| 1–14 | 4.18 (3.70–4.71) | |||
| 15–29 | 0.98 (0.77–1.23) | |||
| 30–44 | 1.05 (0.83–1.31) | |||
| 45–59 | 1.29 (1.05–1.57) | |||
| ≥60 | 0.91 (0.73–1.13) | |||
| Children (<15) | 4.67 (4.18–5.21) | 4.46 (3.99–4.98) | 4.35 (3.73–5.07) | <0.001 |
| Adults (≥15) | 1.05 (0.94–1.17) | 1.06 (0.94–1.18) | 1 [Reference] | |
| Overall | 1.70 (1.58–1.84) | 1.60 (1.48–1.73) | ||
| Sex | ||||
| Female | 1.47 (1.31–1.65) | 1.41 (1.25–1.59) | 1 [Reference] | |
| Male | 1.94 (1.75–2.15) | 1.79 (1.61–1.98) | 1.28 (1.10–1.49) | <0.01 |
| Deprivation quintile | ||||
| 1 – least deprived | 1.54 (1.28–1.85) | 1.50 (1.31–1.70) | 1 [Reference] | |
| 2 | 1.48 (1.22–1.78) | 1.45 (1.27–1.65) | 0.98 (0.76–1.28) | 0.90 |
| 3 | 1.46 (1.20–1.75) | 1.41 (1.23–1.60) | 0.96 (0.74–1.25) | 0.78 |
| 4 | 1.97 (1.67–2.30) | 1.84 (1.63–2.06) | 1.29 (1.01–1.64) | 0.04 |
| 5 – most deprived | 2.06 (1.75–2.41) | 1.86 (1.65–2.09) | 1.27 (0.99–1.62) | 0.06 |
| Region | ||||
| North East | 1.84 (1.28–2.57) | 1.78 (1.23–2.49) | 1 [Reference] | |
| North West | 1.48 (1.17–1.86) | 1.39 (1.09–1.74) | 0.79 (0.53–1.18) | 0.26 |
| Yorkshire/Humber | 1.58 (1.21–2.04) | 1.49 (1.13–1.92) | 0.85 (0.56–1.29) | 0.47 |
| East Midlands | 1.39 (1.02–1.86) | 1.30 (0.95–1.73) | 0.78 (0.50–1.21) | 0.28 |
| West Midlands | 2.09 (1.67–2.59) | 1.98 (1.58–2.45) | 1.10 (0.74–1.64) | 0.60 |
| East of England | 1.43 (1.09–1.83) | 1.37 (1.04–1.76) | 0.80 (0.53–1.23) | 0.34 |
| London | 1.59 (1.29–1.94) | 1.49 (1.19–1.83) | 0.83 (0.56–1.23) | 0.35 |
| South East | 1.89 (1.56–2.26) | 1.78 (1.47–2.13) | 1.08 (0.74–1.59) | 0.68 |
| South West | 2.10 (1.67–2.61) | 2.08 (1.65–2.59) | 1.21 (0.81–1.82) | 0.32 |
95% CI, 95% confidence interval.
ASR, directly age‐standardised incidence.
IRR, incidence‐rate ratios, adjusted for children/adults, sex, deprivation, and region.
Prevalence
There were 560 people diagnosed with LCH in 2013–2019 and still alive at the end of 2019. This equated to 9.95 (95% CI 9.14–10.81) per million persons overall, 27.67 (95% CI 24.53–31.09) for children and 6.03 (95% CI 5.34–6.78) for adults. The highest prevalence was seen in those aged 1–14 years, males, and in the most deprived areas (Table 3).
TABLE 3.
Prevalence per million persons of Langerhans cell histiocytosis in England on 31 December 2019
| N (%) a of people diagnosed with LCH in 2013–2019 and still alive at the end of 2019 | Prevalence (95% CI) | |
|---|---|---|
| Age on 31 December 2019, years | ||
| Infants (<1) | 2 (0) | 3.23 (0.39–11.67) |
| 1–14 | 280 (50) | 29.25 (25.92–32.88) |
| 15–29 | 81 (14) | 7.80 (6.20–9.70) |
| 30–44 | 66 (12) | 6.02 (4.66–7.66) |
| 45–59 | 79 (14) | 6.99 (5.54–8.72) |
| ≥60 | 52 (9) | 3.86 (2.88–5.06) |
| Children (<15) | 282 (50) | 27.67 (24.53–31.09) |
| Adults (≥15) | 278 (50) | 6.03 (5.34–6.78) |
| Overall | 560 (100) | 9.95 (9.14–10.81) |
| Sex | ||
| Female | 245 (44) | 8.61 (7.56–9.76) |
| Male | 315 (56) | 11.32 (10.10–12.64) |
| Deprivation quintile | ||
| 1 – least deprived | 101 (18) | 9.27 (7.55–11.26) |
| 2 | 94 (17) | 8.46 (6.83–10.35) |
| 3 | 98 (18) | 8.58 (6.96–10.45) |
| 4 | 130 (23) | 11.23 (9.38–13.33) |
| 5 – most deprived | 137 (24) | 12.16 (10.21–14.37) |
| Region | ||
| North East | 26 (5) | 9.74 (6.36–14.27) |
| North West | 62 (11) | 8.45 (6.48–10.83) |
| Yorkshire/Humber | 53 (9) | 9.63 (7.21–12.60) |
| East Midlands | 37 (7) | 7.65 (5.39–10.55) |
| West Midlands | 77 (14) | 12.98 (10.24–16.22) |
| East of England | 50 (9) | 8.02 (5.95–10.57) |
| London | 80 (14) | 8.93 (7.08–11.11) |
| South East | 104 (19) | 11.33 (9.26–13.73) |
| South West | 71 (13) | 12.62 (9.86–15.92) |
Abbreviations: CI, confidence interval; LCH, Langerhans cell histiocytosis. a :Percentages might not total 100% due to approximation.
Survival
Two adults had uncertain date of death and were excluded from survival analyses; therefore, 656 patients were included in the survival analyses. Survival was >50% at the last time point (the fifth year) for age groups <60 years hence the median OS was undefined overall. The 1‐year OS was 95% (95% CI 92%–96%) overall, 99% (95% CI 97%–100%) for children and 90% (95% CI 87%–93%) for adults. The 5‐year OS was 86% (95% CI 82%–88%) overall, the same as the 1‐year OS for children as there was no death beyond the first‐year time point, and 72% (95% CI 65%–77%) for adults. OS varied substantially by age: 1‐ and 5‐year OS was 99% (95% CI 97%–100%) for the youngest group (aged <15 years), compared to 1‐year OS of 77% (95% CI 66%–85%) and 5‐year OS of 33% (95% CI 20%–46%) for the oldest group (aged ≥60 years). For females the 1‐year OS was 95% (95% CI 92%–97%) and the 5‐year OS was 86% (95% CI 81%–90%), compared to males with a 1‐year OS of 94% (95% CI 91%–96%) and 5‐year OS of 85% (95% CI 80%–89%); males had poorer survival compared to females, although admittedly the significant level was at borderline 0.05 (HR 2.0, 95% CI 0.99–4.09; p = 0.054). The best OS was seen for those in the least deprived areas, with a 1‐year OS of 98% (95% CI 93%–100%) and 5‐year OS of 93% (95% CI 84%–97%). There was no clear variation in OS by region or ethnicity, with uncertainty in the estimates from small numbers (Figure 1; Table S4). The Cox proportional hazards regression assumption was met (p = 0.97). Those aged ≥60 years had poorer OS compared to those aged <15 years (HR 22.12, 95% CI 7.10–68.94; p < 0.001). People in deprived areas had lower OS than those in the least deprived areas (HR 5.36, 95% CI 1.16–24.87; p = 0.03) (Table 4).
FIGURE 1.
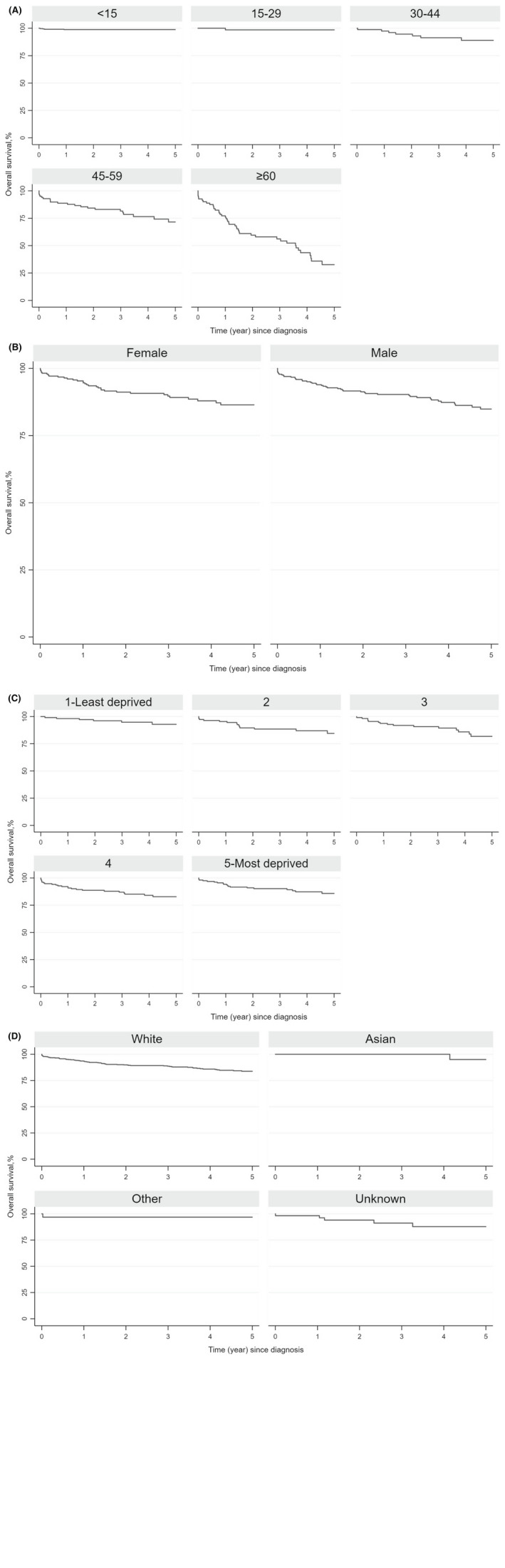
The 5‐year overall survival (OS) of patients with Langerhans cell histiocytosis in England, 2013–2019. (A) OS, by age (year) at diagnosis. (B) OS, by sex. (C) OS, by deprivation quintile. (D) OS, by ethnicity.
TABLE 4.
Multivariate analysis of baseline factors associated with overall survival for Langerhans cell histiocytosis in England, 2013–2019
| Hazard ratio (95% CI) a | p | |
|---|---|---|
| Age at diagnosis, years | ||
| Children (<15) | 1 [Reference] | |
| 15–29 | [no death] | |
| 30–44 | 2.32 (0.42–12.93) | 0.34 |
| 45–59 | 11.04 (3.40–35.88) | <0.001 |
| ≥60 | 22.12 (7.10–68.94) | <0.001 |
| Sex | ||
| Female | 1 [Reference] | |
| Male | 2.01 (0.99–4.09) | 0.054 |
| Deprivation quintile | ||
| 1 – least deprived | 1 [Reference] | |
| 2 | 2.45 (0.45–13.24) | 0.30 |
| 3 | 3.13 (0.64–15.35) | 0.16 |
| 4 | 5.36 (1.16–24.87) | 0.03 |
| 5 – most deprived | 3.24 (0.66–15.87) | 0.15 |
| Region | ||
| North East | 1 [Reference] | |
| North West | 0.61 (0.14–2.62) | 0.50 |
| Yorkshire/Humber | 0.63 (0.10–3.94) | 0.62 |
| East Midlands | 0.62 (0.12–3.18) | 0.56 |
| West Midlands | 0.63 (0.14–2.90) | 0.55 |
| East of England | 1.14 (0.25–5.21) | 0.87 |
| London | 0.17 (0.02–1.19) | 0.07 |
| South East | 0.69 (0.16–3.03) | 0.63 |
| South West | 0.61 (0.13–2.78) | 0.52 |
| Ethnicity | ||
| White | 1 [Reference] | |
| Asian | [no death] | |
| Other | 2.38 (0.26–22.07) | 0.44 |
| Unknown | 0.30 (0.04–2.28) | 0.25 |
Abbreviation: CI, confidence interval. a :Adjusted for patient age, sex, deprivation, region, and ethnicity.
DISCUSSION
Over the 7‐year study period, 658 patients diagnosed with LCH were identified via ICD‐O‐3 contemporary coding in England. Our data confirms LCH to be a disease more common in children and to have a modest male predominance, which has been described in some case series but not all. 18 , 24 , 33 We found that children had significantly higher incidence than adults. OS was excellent in children but decreased with increasing age. Older age at diagnosis and residence in more deprived areas were associated with poorer OS.
Our data showed that significantly more patients with LCH resided in deprived areas and a significantly higher proportion with LCH in the bronchus/lung resided in the most deprived areas. For the first time we showed that ASR was 24% higher for people in the most deprived areas compared to those in the least deprived areas and the estimated increase in relative incidence per deprivation quintile was 6%. As nearly all cases of primary pulmonary histiocytosis are associated with a history of current or prior cigarette smoking and 27% of adults living in England's most deprived areas were smokers compared to just 8% of adults in the least deprived neighbourhoods, 34 this difference is to be expected. Our data suggested that this had accounted for some of the observed increased incidence of LCH in more deprived areas: the difference in the ASR between the least and most deprived reduced to 11% (from 24%) when excluding lung/bronchus cases in the calculation. Notably, of the 53 infants, 15 (28%) were in the most deprived areas whereas only three (6%) were from the least deprived areas. Influences, in children, such as maternal smoking in pregnancy, or an alternate in utero exposure yet to be discovered are possible explanations for the deprivation gradient. It seems possible that the deprivation gradient, if confirmed in other cohorts, will be related to an interaction between maternal exposures, early childhood exposures and the BRAF mutation but all of this deserves further investigation.
In comparison to other studies, we found a much higher ASR of 1.06 (95% CI 0.94–1.18) per million for those aged ≥15 years, compared to the previously reported ASR of 0.07 (95% CI 0.05–0.10) for those aged >18 years, although the prior study only considered cases of disseminated LCH. 16 In contrast, for children aged <15 years we found an ASR of 4.46 (95% CI 3.99–4.98) per million, compared to the previously reported ASR of 4.12 (95% CI 4.11–4.13) in the UK and Republic of Ireland in 2003–2005, 10 whilst there was a reported ASR of 5.0 in France in 2000–2004. 7 Some of these differences are due to ASRs age‐adjusted to different standard populations (to compare incidence for children from the extant literature, we re‐calculated ASRs age adjusted to different standard populations in Table S5). When childhood was further stratified, we still showed a slightly higher crude incidence of 11.56 (95% CI 8.66–15.13) per million for those aged <1 year and 3.39 (95% CI 2.66–4.26) for those aged 10–14 years, compared to previously reported 9.0 for <1 year and 0.7 for 10–14 years. 9
Previous studies that used the SEER dataset from the USA reported a lower incidence of disseminated LCH in patients of Black ethnicity for those aged 0–19 years 15 and adults, 16 and a higher incidence among patients of Asian and/or Pacific Islander or Native American ethnicity compared to patients of White ethnicity. 17 A further paediatric paper based on data from the Texas Cancer Registry in 1995–2011 suggested an increased risk of LCH for children whose parents were Hispanic. 35 Comparisons by ethnicity with the United States studies are very difficult to do and interpret as the United States ‘Asian and/or Pacific Islander or Native American’ group is heterogeneous in a quite different way from the UK ‘Asian’ category, and the United States definition of ‘Black’ includes mixed‐race. In addition, the necessary underlying populations to calculate incidence by ethnicity in our study were not available. However, we were able to demonstrate there was no reduced survival in patients reporting ethnicities other than White compared to those reporting White.
For survival in children, the comparable population‐based studies from France reported 1‐year OS of 99% (95% CI 97%–100%) for those aged <18 years and diagnosed during 2000–2004 7 and 5‐year OS of 98.7% for those aged <18 years and diagnosed during 1998–2012. 8 These were consistent with our 1‐ and 5‐year OS of 99% (95% CI 97%–100%) for those aged <15 years. For adults, a French multicentre study of vinblastine treatment reported 10‐year OS of 86.2% (95% CI 71.8%–100%) in 35 adult patients (median [interquartile range] age 33 [28–42] years). 36 Although all the patients in that study required active therapy, the results appear consistent with our 5‐year OS of 89% (95% CI 78%–95%) for patients aged 30–44 years.
LIMITATIONS
This study was large in the context of LCH, but LCH remains a rare disease and small numbers still meant there was greater uncertainty in the estimates (e.g., by region and ethnicity) thus preventing further assessment. In addition, it is possible that a few LCH cases that arose in the bronchus/lung may not have been picked up by the NCRD as those patients with characteristic clinical presentation, a history of smoking and CT imaging may not undergo diagnostic biopsy. While a UK registry of pulmonary LCH 37 reports that only 60% of cases had a biopsy, our data showed that among bronchus/lung cases 87% were identified via histology or cytology. Another limitation was that we were unable to clearly delineate between cases of single‐site, single‐system and multisystem LCH, and consequently our results could not be directly compared with those of the Histiocyte Society LCH‐III trial 18 in which substantial numbers of UK children were entered during 2001–2008. However, this will improve over time as work is currently underway to collect all involved sites as a separate data item for LCH within the Cancer Outcomes and Services Dataset – the national standard for reporting cancer in England. 22 Because ICD‐O‐3 coded LCH data were only available since 2013, the length of follow‐up is necessarily short. This limited our ability to assess the association of LCH with other malignancies, but, as all patients within the cancer registry are on indefinite passive follow‐up, the methods from our study will provide a valuable framework to examine these associations in the future. This is important as LCH diagnosis can be both preceded and followed by associations with other malignancies. 38 , 39 , 40
CONCLUSIONS
This analysis is the largest population‐based study to date to provide contemporary and comprehensive epidemiological estimates of all ICD‐O‐3 coded LCH. It shows that a higher incidence is associated with younger age and male sex. Survival is excellent in children, but older age is associated with poorer OS. For the first time this study shows that a higher incidence and lower survival are associated with deprivation. There will inevitably be other environmental factors and associations yet to be identified, and the continued standardised data collection described in this study will allow further evaluation of data over time. This will be increasingly important with developments in LCH management following the large collaborative international trials such as LCH IV. 41
AUTHOR CONTRIBUTIONS
Joe West (principal investigator), Mary Bythell, Judith Rankin, Tim R. Card, Colin J. Crooks, Lu Ban, Vasanta Nanduri, Lucy Elliss‐Brookes, Mark Bishton, Peter Lanyon, and Johann Visser wrote the grant application and obtained funding for the study. Joe West and Hanhua Liu designed the study. Hanhua Liu extracted the data from the National Cancer Registration Dataset for England. Joe West, Brian Rous, and Charles A. Stiller verified the data. Hanhua Liu and Joe West planned and conducted the analysis. Hanhua Liu, Joe West, Charles A. Stiller, Mark Bishton, and Colin J. Crooks interpreted data. All authors were involved in writing, reviewing, and editing drafts of the paper, and approving the manuscript for submission.
FUNDING INFORMATION
This paper was independent research funded by the Histio UK (award number: HistioUK/2019/07/01).
CONFLICT OF INTEREST
Dr Lanyon is recipient of a research grant from Vifor Pharma. Vifor Pharma had no influence on the design, conduct, or interpretation of this study. All other authors declare no competing interests.
Supporting information
Table S1
Table S2
Table S3
Table S4
Table S5
ACKNOWLEDGEMENTS
This paper uses data that has been provided by patients and collected by the NHS as part of their care and support. The data is collated, maintained and quality assured by the National Cancer Registration and Analysis Service, which is part of NHS Digital. We would like to acknowledge the contributions of the late Dr Johann Visser who was a co‐applicant on the grant funding this work. Joe West had single‐system multifocal LCH aged 9 years, diagnosed and managed at Great Ormond Street Hospital by the late Dr Jon Pritchard – he would like to acknowledge the care and inspiration he received.
Liu H, Stiller CA, Crooks CJ, Rous B, Bythell M, Broggio J, et al. Incidence, prevalence and survival in patients with Langerhans cell histiocytosis: A national registry study from England, 2013–2019. Br J Haematol. 2022;199:728–738. 10.1111/bjh.18459
DATA AVAILABILITY STATEMENT
The study data was obtained from the National Cancer Registration Dataset for England and belongs to the National Cancer Registration and Analysis Service (NCRAS), which is part of the National Disease Registration Service, NHS Digital. We do not own these data and hence are not permitted to share them in the original form. All data included in this study are stored in the Cancer Analysis System database hosted by NCRAS. Any request for access to cancer registration data needs to go through the NHS Digital Data Access Request Service via data.application@nhsdigital.net
REFERENCES
- 1. Lichtenstein L. Histiocytosis X; integration of eosinophilic granuloma of bone, letterer‐Siwe disease, and Schüller‐Christian disease as related manifestations of a single nosologic entity. AMA Arch Pathol. 1953;56(1):84–102. [PubMed] [Google Scholar]
- 2. Writing Group of the Histiocyte Society . Histiocytosis syndromes in children. Lancet. 1987;1(8526):208–9. [PubMed] [Google Scholar]
- 3. Willman CL, Busque L, Griffith BB, Favara BE, McClain KL, Duncan MH, et al. Langerhans'‐cell histiocytosis (histiocytosis X)—a clonal proliferative disease. N Engl J Med. 1994;331(3):154–60. [DOI] [PubMed] [Google Scholar]
- 4. Badalian‐Very G, Vergilio JA, Degar BA, MacConaill L, Brandner B, Calicchio ML, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116(11):1919–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Emile JF, Cohen‐Aubart F, Collin M, Fraitag S, Idbaih A, Abdel‐Wahab O, et al. Histiocytosis. Lancet. 2021;398(10295):157–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fritz A, Percy C, Jack A, Shanmugaratnam K, Sobin L, Parkin DM, et al., editors. International classification of diseases for oncology, 3rd edition, first revision. Geneva: WHO Press; 2013. [Google Scholar]
- 7. Guyot‐Goubin A, Donadieu J, Barkaoui M, Bellec S, Thomas C, Clavel J. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000‐2004. Pediatr Blood Cancer. 2008;51(1):71–5. [DOI] [PubMed] [Google Scholar]
- 8. Rigaud C, Barkaoui MA, Thomas C, Bertrand Y, Lambilliotte A, Miron J, et al. Langerhans cell histiocytosis: therapeutic strategy and outcome in a 30‐year nationwide cohort of 1478 patients under 18 years of age. Br J Haematol. 2016;174(6):887–98. [DOI] [PubMed] [Google Scholar]
- 9. Alston RD, Tatevossian RG, McNally RJ, Kelsey A, Birch JM, Eden TO. Incidence and survival of childhood Langerhans cell histiocytosis in Northwest England from 1954 to 1998. Pediatr Blood Cancer. 2007;48(5):555–60. [DOI] [PubMed] [Google Scholar]
- 10. Salotti JA, Nanduri V, Pearce MS, Parker L, Lynn R, Windebank KP. Incidence and clinical features of Langerhans cell histiocytosis in the UK and Ireland. Arch Dis Child. 2009;94(5):376–80. [DOI] [PubMed] [Google Scholar]
- 11. Müller J, Garami M, Hauser P, Schuler D, Csóka M, Kovács G, et al. Hungarian experience with Langerhans cell histiocytosis in childhood. Pediatr Hematol Oncol. 2006;23(2):135–42. [DOI] [PubMed] [Google Scholar]
- 12. Stålemark H, Laurencikas E, Karis J, Gavhed D, Fadeel B, Henter JI. Incidence of Langerhans cell histiocytosis in children: a population‐based study. Pediatr Blood Cancer. 2008;51(1):76–81. [DOI] [PubMed] [Google Scholar]
- 13. Carstensen H, Ornvold K. The epidemiology of Langerhans cell histiocytosis in children in Denmark, 1975‐1989. Med Pediatr Oncol. 1993;21:387–8. [Google Scholar]
- 14. Stine K, Abla O, Egeler RM, Jubran R. Langerhans cell histiocytosis: straight to the point of care. BMJ Best Practice. 2022; https://bestpractice.bmj.com/topics/en‐gb/585/pdf/585/Langerhans%20cell%20histiocytosis.pdf. Accessed 8 Mar 2022. [Google Scholar]
- 15. Ribeiro KB, Degar B, Antoneli CB, Rollins B, Rodriguez‐Galindo C. Ethnicity, race, and socioeconomic status influence incidence of Langerhans cell histiocytosis. Pediatr Blood Cancer. 2015;62(6):982–7. [DOI] [PubMed] [Google Scholar]
- 16. Goyal G, Shah MV, Hook CC, Wolanskyj AP, Call TG, Rech KL, et al. Adult disseminated Langerhans cell histiocytosis: incidence, racial disparities and long‐term outcomes. Br J Haematol. 2018;182(4):579–81. [DOI] [PubMed] [Google Scholar]
- 17. Golpanian S, Tashiro J, Gerth DJ, Thaller SR. Pediatric histiocytoses in the United States: incidence and outcomes. J Surg Res. 2014;190(1):221–9. [DOI] [PubMed] [Google Scholar]
- 18. Gadner H, Minkov M, Grois N, Pötschger U, Thiem E, Aricò M, et al. Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis. Blood. 2013;121(25):5006–14. [DOI] [PubMed] [Google Scholar]
- 19. Berry DH, Becton DL. Natural history of histiocytosis X. Hematol Oncol Clin North Am. 1987;1(1):23–34. [PubMed] [Google Scholar]
- 20. Kogevinas M, Porta M. Socioeconomic differences in cancer survival: a review of the evidence. IARC Sci Publ. 1997;138:177–206. [PubMed] [Google Scholar]
- 21. Mackillop WJ, Zhang‐Salomons J, Boyd CJ, Groome PA. Associations between community income and cancer incidence in Canada and the United States. Cancer. 2000;89(4):901–12. [DOI] [PubMed] [Google Scholar]
- 22. Henson KE, Elliss‐Brookes L, Coupland VH, Payne E, Vernon S, Rous B, et al. Data resource profile: National Cancer Registration Dataset in England. Int J Epidemiol. 2020;49(1):16–16h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Corazziari I, Quinn M, Capocaccia R. Standard cancer patient population for age standardising survival ratios. Eur J Cancer. 2004;40:2307–16. [DOI] [PubMed] [Google Scholar]
- 24. Aricò M, Girschikofsky M, Généreau T, Klersy C, McClain K, Grois N, et al. Langerhans cell histiocytosis in adults. Report from the international registry of the histiocyte society. Eur J Cancer. 2003;39(16):2341–8. [DOI] [PubMed] [Google Scholar]
- 25. Ministry of Housing, Communities & Local Government . The English Indices of Deprivation 2019: Frequently Asked Questions (FAQs). https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/853811/IoD2019_FAQ_v4.pdf. Accessed 8 Mar 2022.
- 26. Ministry of Housing, Communities & Local Government . The English Indices of Deprivation 2019 (IoD 2019). https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/835115/IoD2019_Statistical_Release.pdf. Accessed 10 Aug 2022.
- 27. Office for National Statistics (ONS) . Population estimates for the UK, England and Wales, Scotland and Northern Ireland: mid‐2020. National and subnational mid‐year population estimates for the UK and its constituent countries by administrative area, age and sex. Release date: June 25, 2021. https://www.ons.gov.uk/peoplepopulationandcommunity/populationandmigration/populationestimates/bulletins/annualmidyearpopulationestimates/mid2020. Accessed 8 Mar 2022.
- 28. Eurostat Methodologies and Working papers . Revision of the European Standard Population: Report of Eurostat's Task Force. 2013th ed. Luxembourg: Publications Office of the European Union; 2013. https://ec.europa.eu/eurostat/documents/3859598/5926869/KS‐RA‐13‐028‐EN.PDF.pdf/e713fa79‐1add‐44e8‐b23d‐5e8fa09b3f8f?t=1414782757000 [Google Scholar]
- 29. Maddams J, Brewster D, Gavin A, Steward J, Elliott J, Utley M, et al. Cancer prevalence in the United Kingdom: estimates for 2008. Br J Cancer. 2009;101(3):541–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. NHS Digital . The Personal Demographics Service (PDS). https://digital.nhs.uk/services/demographics. Accessed 8 Mar 2022.
- 31. Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53(282):457–81. [Google Scholar]
- 32. Cox DR. Regression models and life tables (with discussion). J R Stat Soc Series B Stat Methodol. 1972;34(2):187–202. [Google Scholar]
- 33. Islinger RB, Kuklo TR, Owens BD, Horan PJ, Choma TJ, Murphey MD, et al. Langerhans' cell histiocytosis in patients older than 21 years. Clin Orthop Relat Res. 2000;379:231–5. [DOI] [PubMed] [Google Scholar]
- 34. Office for National Statistics . Likelihood of smoking four times higher in England's most deprived areas than least deprived, 14 March 2018. https://www.ons.gov.uk/peoplepopulationandcommunity/healthandsocialcare/drugusealcoholandsmoking/articles/likelihoodofsmokingfourtimeshigherinenglandsmostdeprivedareasthanleastdeprived/2018‐03‐14#:~:text=Around%206.3%20million%20people%20aged,in%20the%20least%20deprived%20neighbourhoods. Accessed 8 Mar 2022.
- 35. Peckham‐Gregory EC, McClain KL, Allen CE, Scheurer ME, Lupo PJ. The role of parental and perinatal characteristics on Langerhans cell histiocytosis: characterizing increased risk among Hispanics. Ann Epidemiol. 2018;28(8):521–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tazi A, Lorillon G, Haroche J, Neel A, Dominique S, Aouba A, et al. Vinblastine chemotherapy in adult patients with langerhans cell histiocytosis: a multicenter retrospective study. Orphanet J Rare Dis. 2017;12(1):95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mason RH, Foley NM, Branley HM, Adamali HI, Hetzel M, Maher TM, et al. Pulmonary Langerhans cell histiocytosis (PLCH): a new UK register. Thorax. 2014;69(8):766–7. [DOI] [PubMed] [Google Scholar]
- 38. Cohen‐Barak E, Sonnenscien D, Ziv M, Shani‐Adir A, Rozenman D. Kaposi's sarcoma in a patient with pemphigus vulgaris. Int J Dermatol. 2016;55(1):85–8. [DOI] [PubMed] [Google Scholar]
- 39. Egeler RM, Neglia JP, Aricò M, Favara BE, Heitger A, Nesbit ME. Acute leukemia in association with Langerhans cell histiocytosis. Med Pediatr Oncol. 1994;23(2):81–5. [DOI] [PubMed] [Google Scholar]
- 40. Haupt R, Minkov M, Astigarraga I, Schäfer E, Nanduri V, Jubran R, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work‐up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60(2):175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. LCH‐IV, International collaborative treatment protocol for children and adolescents with Langerhans Cell Histiocytosis. https://clinicaltrials.gov/ct2/show/NCT02205762.Accessed 8 Mar 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2
Table S3
Table S4
Table S5
Data Availability Statement
The study data was obtained from the National Cancer Registration Dataset for England and belongs to the National Cancer Registration and Analysis Service (NCRAS), which is part of the National Disease Registration Service, NHS Digital. We do not own these data and hence are not permitted to share them in the original form. All data included in this study are stored in the Cancer Analysis System database hosted by NCRAS. Any request for access to cancer registration data needs to go through the NHS Digital Data Access Request Service via data.application@nhsdigital.net