

SUMMARY
Vegetative propagation (VP) is an important practice for production in many horticultural plants. Sugar supply constitutes the basis of VP in bulb flowers, but the underlying molecular basis remains elusive. By performing a combined sequencing technologies coupled with ultra‐high performance liquid chromatography‐quadrupole time‐of‐flight mass spectrometry approach for metabolic analyses, we compared two Lycoris species with contrasting regeneration rates: high‐regeneration Lycoris sprengeri and low‐regeneration Lycoris aurea. A comprehensive multi‐omics analyses identified both expected processes involving carbohydrate metabolism and transcription factor networks, as well as the metabolic characteristics for each developmental stage. A higher abundance of the differentially expressed genes including those encoding ethylene responsive factors was detected at bulblet initiation stage compared to the late stage of bulblet development. High hexose‐to‐sucrose ratio correlated to bulblet formation across all the species examined, indicating its role in the VP process in Lycoris bulb. Importantly, a clear difference between cell wall invertase (CWIN)‐catalyzed sucrose unloading in high‐regeneration species and the sucrose synthase‐catalyzed pathway in low‐regeneration species was observed at the bulblet initiation stage, which was supported by findings from carboxyfluorescein tracing and quantitative real‐time PCR analyses. Collectively, the findings indicate a sugar‐mediated model of the regulation of VP in which high CWIN expression or activity may promote bulblet initiation via enhancing apoplasmic unloading of sucrose or sugar signals, whereas the subsequent high ratio of hexose‐to‐sucrose likely supports cell division characterized in the next phase of bulblet formation.
Keywords: vegetative propagation, bulb yield, sugar dynamics, sucrose phloem unloading, invertase, full‐length sequencing, Lycoris
Significance Statement
At a time of unprecedented climate change and losses in biodiversity, vegetative propagation (VP) is a particularly practical strategy for the survival and yield realization of many plant species. Assisted by horticultural techniques such as cross‐cutting, VP contributes significantly to the commercial production and global horticultural industry as seen in the production of flower bulbs. However, the molecular mechanism orchestrating vegetative regeneration capacities remains largely unknown. Here, we addressed this issue in Lycoris bulbs using multi‐omics approaches. The analyses identified CWIN‐mediated sucrose‐to‐hexose conversion as a metabolic marker for bulblet initiation in Lycoris and a potential molecular target for improving bulb production.
INTRODUCTION
Sessile plants have evolved strategies to adjust their growth according to ever‐changing environments (Huby et al., 2020) by optimizing their metabolism and shaping their architecture (Davies, 2010; Lopez‐Salmeron et al., 2019; Tanaka et al., 2006; van Es et al., 2020). As opposed to the limited capacity to repair or regenerate damaged tissues in animals, plants exhibit much more diverse modes of regeneration depending on the developmental and/or environmental context (Birnbaum & Sanchez Alvarado, 2008; Ikeuchi et al., 2016; Xu & Huang, 2014), including a process known as vegetative propagation (VP). Assisted by manual techniques (e.g. scaling, cross‐cutting, cooping), VP has been widely used in horticultural practice to fulfill the need for constant and accelerated reproduction of flower bulbs of plants with long juvenile phases and high genetic variability (Kamenetsky & Okubo, 2012; Moreno‐Pachón, 2017), making VP pivotal in practice for improving and optimizing crop yield and quality in a sustainable manner.
Lycoris is an important member of the Amaryllidaceae family, a bulbous plant valued for its both ornamental features and medicinal ingredients, especially those used to combat infection and Alzheimer's disease (He et al., 2011; Jin & Yao, 2019; Ren, Lin, et al., 2021; Wang et al., 2016; Zhou et al., 2020). However, the regeneration efficiency of Lycoris species is very low (Ren, Xia, et al., 2017; Xu et al., 2020), only one or two axillary buds could sprout and formed bulblets afterwards each year under natural conditions. Artificial VP in Lycoris bulbs is induced by the traditional cross‐cutting method for formation of new individuals (bulblets) from cut sites, which is a unique feature of plant plasticity (Hartmann et al., 2011). Upon loss of apical meristems, new axillary buds (AxB) are formed from the scale abaxial side at the basal region connecting the basal plate (or stem) (Lv et al., 2020; Ren, Xia, et al., 2017). At the histological and subcellular levels, we previously reported comparisons between two different regenerated species, namely high‐regeneration species Lycoris sprengeri and low‐regeneration species Lycoris aurea. Lycoris sprengeri usually develops more AxBs in the inner and middle sacles, which ensures a higher propagation coefficient than L. aurea (Ren, Xia, et al., 2017). Differences in the number and size of regenerated bulblets between Lycoris species raise the question of whether different species share the similar or different developmental and metabolic processes and how these features may modulate early VP leading to their differential regenerative capacities.
The plant kingdom is exceptionally rich in metabolic diversity, harboring in excess of 200 000 structurally distinct metabolites (Wurtzel & Kutchan, 2016). A principal feature of plant metabolism is the flexibility to accommodate developmental changes and to respond to the changing environment (Butelli et al., 2008; Chen et al., 2016; Fernie et al., 2020; Lloyd & Zakhleniuk, 2004). Metabolomics is defined, by analogy to transcriptomics and proteomics, as the analysis of the metabolic complement of an organism (Wishart et al., 2007). Over the past decade, the integration of metabolic profiling with other omics tools has proven to be highly effective for functional gene identification and pathway elucidation in plant primary and secondary metabolism (Kusano et al., 2011; Matsuda et al., 2010; Zhu et al., 2018). In recent years, metabolite profiles of some Lycoris species have been analyzed regarding galantamine biosynthesis (Park et al., 2019), floral volatile organic compounds (Shi et al., 2019) and petal color development (Yang et al., 2021). However, details about the metabolic landscape associated with bulblet formation and development are essentially unknown.
Sucrose play a pivotal role in integrating developmental phases and environmental cues to fine‐tune plant yield (Fernie et al., 2020; Liao et al., 2020; Ruan, 1993, 2014; Wang et al., 2019, 2020; Wang & Ruan, 2016). Upon being delivered from source to sinks, sucrose is unloaded either symplastically or apoplastically (Palmer et al., 2015; Pugh et al., 2010; Ruan, 2014). In the former pathway, sucrose is reversibly degraded into UDP‐glucose and fructose by SUS (EC 2.4.1.13), whereas, in the latter pathway, sucrose is irreversibly hydrolyzed by CWIN (EC 3.2.1.26) into glucose and fructose (Chen et al., 2012; Fernie et al., 2020; Ruan, 2014; Ruan et al., 2008; Thomas, 2013; Wu et al., 2021). Increasing studies have revealed that sucrose and starch metabolism constitutes the predominant metabolic pathway during VP in various bulbous plants. It is generally agreed that higher expression of genes in the sucrose degrading pathway, such as those encoding SUS and invertase (INV), is beneficial for the early formation of regenerated propagules (Gao et al., 2018; Li et al., 2014; Xu et al., 2020), whereas increased expression of genes involved in the starch synthetic direction, including those encoding soluble starch synthase, starch branching enzyme, ADP‐glucose pyrophosphorylase and granule‐bound starch synthase, proceeds with filling of storage compounds (Li et al., 2014; Wu et al., 2021; Xu et al., 2020). Our recent study revealed that early sucrose degradation and the dominant sucrose cleavage pattern influence L. sprengeri bulblet regeneration in vitro (Ren, Xu, et al., 2021). However, the molecular mechanisms underlying early sucrose metabolism that may affect regeneration capacities during VP still remain to be explored. Furthermore, although the process of VP based on temporal variations has been analyzed in some flower bulbs (Li et al., 2014; Xu et al., 2020), developmental characteristics between high‐ and low‐regeneration species have not been compared.
To explore the molecular pathways underlying bulblet formation in Lycoris, we generated a large dataset in the present study, spanning the transcriptome and metabolome over the early VP process among species with different bulb‐regeneration capacities. The VP process was morphologically divided into three developmental stages. It is a bottom‐up strategy, which combined second generation sequencing (next‐generation sequencing) technology (Illumina RNA‐Seq) and single‐molecule, real‐time (SMRT) sequencing technology (Pacbio Iso‐seq) analyses to maximize the exclusion of species‐specific and genetic background orientated differences for L. sprengeri and L. aurea. By taking advantage of the ultra‐high performance liquid chromatography‐quadrupole time‐of‐flight mass spectrometry (UHPLC‐Q‐TOF MS) approach in the separation of sugars and sugar‐related compounds (Romo‐Pérez et al., 2020), the present study aimed to provide (i) a systematic basis to investigate the VP process; (ii) a comprehensive transcriptome and metabolitome map of bulblet formation and development; and (iii) an overview of the temporal differences in metabolic pathways and sugar dynamics. The present study may provide useful hints for the systematic analysis in improving bulb yield in the future.
RESULTS
VP in bulbs of Lycoris
A proposed schematic graph was established to describe the artificial VP in Lycoris (Figure 1a). Upon cross‐cutting, regeneration usually occurs at the abaxial side and the most proximal end of scales where they are attached to the basal plate in Lycoris (Ren, Xia, et al., 2017), reflecting a high architectural similarity to branching in non‐bulbous plant species, being different from those of the randomly formed‐bulblets in lily (Moreno‐Pachón, 2017; Xu et al., 2020). Using this method, multiple genetically identical bulblets could be obtained from the quartered‐bulb sections within 2 months (Figure 1a, A–D). Only three to four more years will be needed for these bulblets to flower (mature bulbs) compared to 5–7 years for those derived from sexual reproduction and seed germination.
Figure 1.
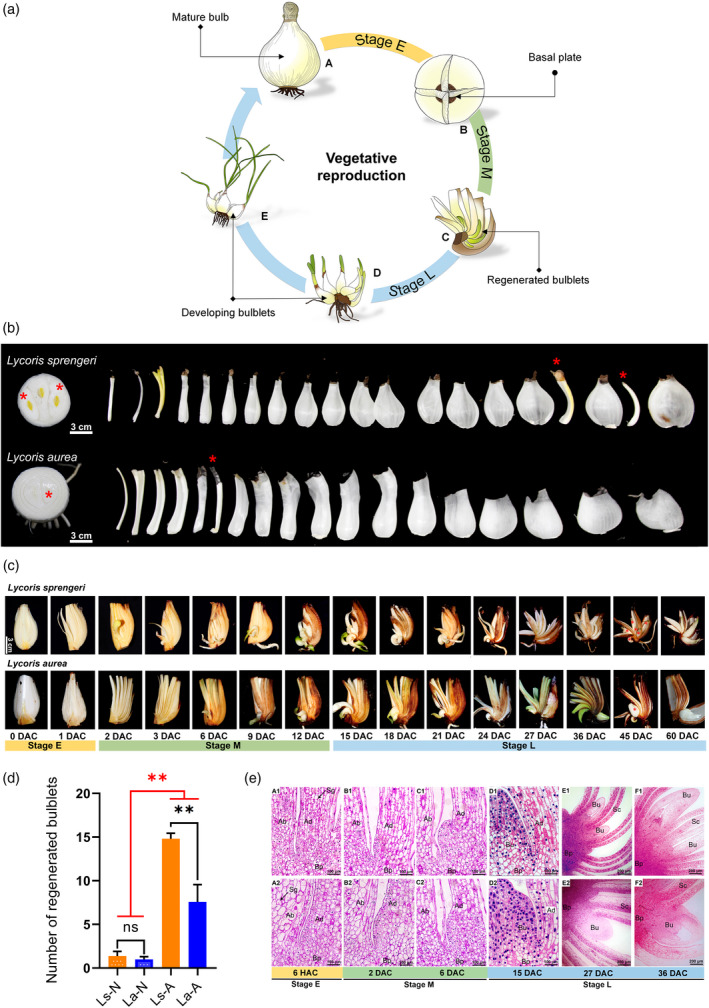
Morphological aspects and regenerative analysis of vegetative propagation (VP) in bulbs of Lycoris.
(a) A schematic model of artificially VP in Lycoris. Lycoris sprengeri (Ls) is shown as an example. (A) mature bulb. (B) Cross‐cutting method (wound stress). (C) Regenerated bulblets formed from the scale axils. (D, E) Development of regenerated bulblets.
(b) Native VP process. Asterisk, vegetative buds formed in one growing period.
(c) Samples under artificial conditions were temporally collected from the meristematic region around scale basal from 0 to 60 days after cutting (DAC) and termed stage E (0–1 DAC), stage M (2–12 DAC) and stage L (15–60 DAC) based on the morphological changes. Asterisk, vegetative buds formed during the culturing period.
(d) The number of regenerated bulblets under native (‐N) and artificial conditions (‐A). Values are represented as the mean ± SE of three biological replicates. Asterisks indicate significant differences between different propagation conditions (red) or between species (black) (Duncan's multiple range test, **P < 0.01).
(e) Comparative histological characteristics of VP under a dissecting microscope between Ls (A1–F1) and L. aurea (La) (A2–F2). Ad, adaxial side; Ab, abaxial side; Bp, basal plate; Sg, starch granule; Is, inner scales; Os, outer scales; Bu, bulblet. [Colour figure can be viewed at wileyonlinelibrary.com]
Artificial VP not only played a pivotal role in shortening the breeding cycle of Lycoris bulbs, but also significantly promoted the number of proliferating bulblets. Morphological and statistical comparisons were performed between native (Figure 1b) and artificial VP (Figure 1c) to assess potential effects of artificial VP in promoting regeneration. Specifically, artificial VP significantly increased the number of regenerated bulblets derived from a given bulb compared to natural VP (Figure 1d). Notably, no significant inter‐species differences were observed under natural conditions, whereas the number of regenerated bulblets of L. sprengeri was significantly higher than that of L. aurea via artificial VP (Figure 1d). Hereafter, we specifically refer to artificial VP as VP.
Inter‐species comparisons of the spatial arrangement of the scale cells indicated a high consistency in histological developmental processes during artificial VP between the two species, which could be represented by three main stages (Figure 1e). To compensate for the changes before bulblet morphogenesis, samples were initially collected before any observed morphological signs, which was termed the early stage [stage E, 0–6 h after cutting (HAC)] (Figure 1e, A1,A2). Compared to the loosely packed cells on the adaxial side of the scales, layers of closely packed cells were distributed along the abaxial side, where the appearance of cell clusters with denser nuclear staining was observed during the bulblet formation stage [middle stage, stage M, 2–6 days after cutting (DAC)] (Figure 1e, B1–C1,B2–C2). The bulblet development stage (later stage, stage L, 15e36 DAC) (Figure 1e, D1–F1,D2–F2) was characterized by a significant increase in the size of regenerated bulblets (Figure 1e, F1,F2). Notably, we found that the inner and middle scales of L. sprengeri produced more bulblets than that of L. aurea (Figure 1e, E1,E2), contributing to the prominent advantage in proliferation rate of L. sprengeri.
Generation of a high‐resolution temporal transcriptomic map of VP
As a result of the lack of genome information resources, we took advantage of both the Illumina HiSeq X ten platform (Illumina Inc., San Diego, CA, USA) and Pacbio RS II platform (Pacbio, Menlo Park, CA, USA) to obtain a high‐resolution transcriptome map of Lycoris (Figure S1). Tissues with three biological replicates at seven time points (42 samples in total), representing major events occurring within the bulb during VP, were collected from each species to construct a comparative transcript database. In total, 486 423 193 and 526 792 197 cleaned Illumina reads were obtained for L. sprengeri and L. aurea, respectively (Table S1). A mixed pool of equal amounts of RNA from seven stages in L. sprengeri was used to generate 10.39 Gb data from the PacBio sequencing, yielding 392 500 circular consensus reads, 213 561 (54.41%) of which were full‐length non‐chimeric (FLNC) reads (Table S1). Followed by error correction via Illumina reads, the uniquely mapped FLNC reads were merged into 285 130 non‐redundant transcripts. The transcriptome generated by incorporating PacBio collapsed isoforms and the Illumina final assembly was used as a reference for Lycoris, with an N50 of 2091 bp (Figure S1), which was eight times longer than that reported by Li, Yang, et al. (2017) who identified 86 243 unigenes (N50 = 256 bp) from mixed samples of bulbs and leaves of L. sprengeri, and approximately two times longer than that reported by Yang et al. (2021) who identified 34 336 total genes with an average length of 1037.74 bp from the petals of L. sprengeri.
Lycoris sprengeri and L. aurea are two genetically related wild species. To maximize the exclusion of species‐specific and genetic background orientated differences, these final cleaned Illumina reads were aligned to the reference transcriptome, with mapping rates of 76.76% and 68.35% in L. sprengeri and L. aurea, respectively. Only the uniquely mapped reads were further used to calculate the normalized gene expression defined by fragments per kilobase of transcript per million mapped reads (FPKM). Notably, the global heatmap revealed that extensive inter‐species and intra‐species differential gene expression occurs immediately in response to cross‐cutting in both species (Figure S2), indicating the key regulatory role of the early stage.
Global comparison uncovers stage‐specific regulation during VP
Principal component analysis (PCA) clearly separated the samples by morphological development stages in both species (Figure 2a). We investigated gene expression at different stages using k‐means clustering (Figure S3). In total, we identified 14 437 (70.52%, L. sprengeri) and 6851 (68.05%, L. aurea) genes of all differentially expressed genes (DEGs) that showed specific expression at a particular stage. Notably, more than half of the DEGs as well as transcription factors (TFs) and transcriptional regulators (TRs) were enriched in stage E (Figure 2b). Enrichment analysis of Kyoto Encyclopedia of Genes and Genomes (KEGG) (https://www.genome.jp/kegg) pathways and Gene Ontology (GO) (http://geneontology.org) terms in both species revealed high similarities at any given developmental stage of VP (Figure S4). By selecting genes highly activated at only one stage for assignment to metabolic categories at each stage, we analyzed the top 30 most enriched stage‐specific KEGG pathways in both species (Figure 2c; Table S2). Stage E was characterized by carbohydrate metabolism‐related pathways. Secondary metabolism‐related pathways contributed to the separation of stage M from other stages but made minor contributions to separate the two species. DNA replication pathway was characterized at stage L, which was consistent with the rapid development of the bulblets. Notably, the KEGG enrichment analysis demonstrated that, except at the early stage, the mechanisms underlying bulblet formation and development might be highly conserved between the two species.
Figure 2.
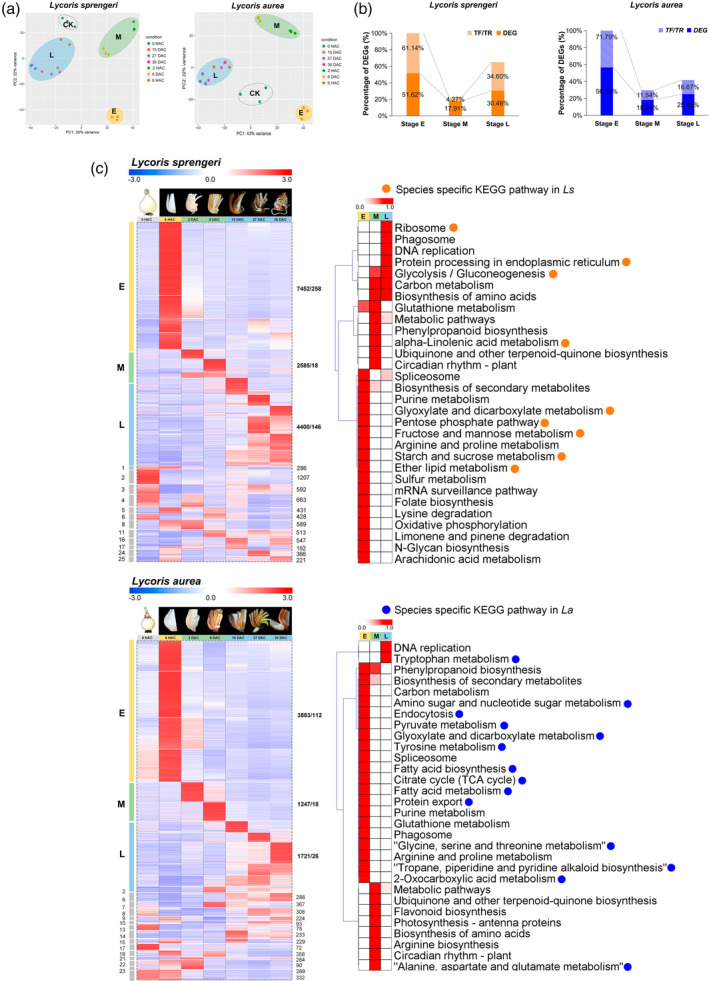
The landscape of transcriptional changes during vegetative propagation (VP) in bulbs of Lycoris.
(a) Principal component analysis (PCA) of the Lycoris transcriptome of 21 independent samples collected from Lycoris sprengeri (Ls) and Lycoris sprengeri (La) bulbs at CK [0 h after cutting (HAC)], E (6 HAC), M [48 HAC and 6 days after cutting (DAC)] and L stage (15, 27 and 36 DAC), respectively. Dots with the same colour represent three biological replicates.
(b) Percentage of stage specific differentially expressed genes (DEGs, including TFs/TRs) in Ls and La.
(c) Expression patterns of genes (cv > 0.5) in 25 different co‐expression modules using the k‐means clustering algorithum. Expression data per row were Z‐score standardized to −3 to 3. For each module, the numbers of DEGs, TF/TR are shown. Top 30 KEGG metabolism pathways enriched in stage‐specific co‐expression modules are shown on the right. [Colour figure can be viewed at wileyonlinelibrary.com]
Correlations between the transcriptome and metabolome during VP
PCA showed clear separation of the metabolite profiles between early stage (stage E) and bulblet formation and development stages (stage M and L) (Figure 3a). Large scale metabolite analysis identified 230 metabolites during VP in bulbs of Lycoris, which could be divided into 47 subclasses (Figure S3), represented by three main metabolites groups, including carbohydrates and carbohydrate conjugates (34), amino acids, peptides and analogues (31), and fatty acids and conjugates (14) (Figure 3b). In total, 153 metabolites were significantly changed during VP via overlap analysis, representing 87.4% and 84.1% of the total changed metabolites in L. sprengeri and L. aurea, respectively (Table S3). Different cross‐cutting (wounding) responses could be seen from changed metabolites belonging to stage E between two species, with limited effects of the wounding on main metabolite accumulation observed in L. sprengeri compared to those within 6 HAC in L. aurea (Figure S5). However, higher network connectedness could be seen for L. sprengeri compared to L. aurea for the analyzed metabolites represented by network topology (Figure S5).
Figure 3.
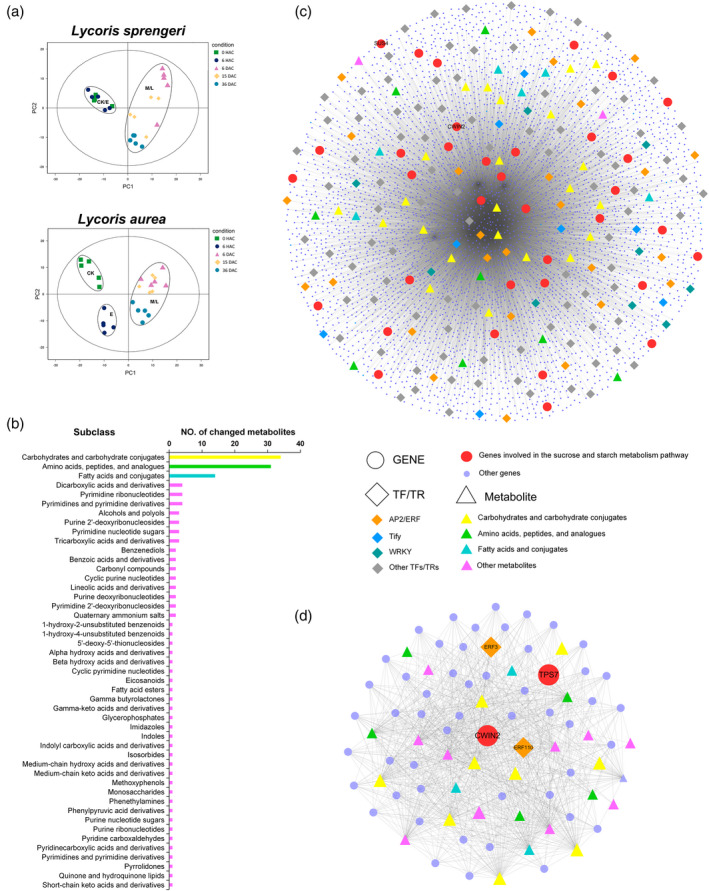
Generation of metabolisome and integration of Multi‐omics data.
(a) PCA of the Lycoris metabolome of 25 independent samples collected from Lycoris sprengeri (Ls) and L. aurea (La) bulbs at control [0 h after cutting (HAC)], E (6 HAC), M [6 days after cutting (DAC)] and L stage (15 and 36 DAC), respectively. Dots with the same colour represent five biological replicates.
(b) Histogram of differentially changed metabolites based on subclass‐level classification.
(c) Network built on all correlations among structural genes, transcription factors (TFs) and transcription regulators (TRs) and metabolites of Ls during stage E. Metabolites are shown as big triangles colored as in (b). The top three TFs/TRs in number are shown as large diamonds with a distinct color per family. Genes involved in the starch and sucrose metabolism pathway are shown as big red dots. Other genes are shown as small blue dots. The LsCWIN2 and LsSUS4 genes are denoted.
(d) Network built on genes specifically associated with metabolites. Each gene in this network is associated with at least one metabolite. The structural genes and TFs/TRs are denoted. [Colour figure can be viewed at wileyonlinelibrary.com]
To further correlate gene expression patterns with parallel metabolic profiling, we also applied co‐expression analysis to our transcriptome and metabolome data. In total, 408 390 expression metabolite correlations involving 28 chemicals and 7475 genes were identified in the multi‐omics network (Figure 3c). After removing genes that were not highly correlated with any of the metabolites, we further conducted visulization analysis for the remaining 803 expression metabolite correlations (Figure 3d). This network output facilitates both candidate gene identification and metabolic pathway elucidation. For example, we found that cell wall invertase 2 (CWIN2) and trehalose‐phosphatase/synthase 7 (TPS7) were the only two genes belonging to the starch and sucrose metabolic pathways that were highly associated with metabolites during VP in L. sprengeri (Figure 3d). Meanwhile, APETALA2/ethylene‐responsive factor (AP2/ERF) is the unique type of TF that is specifically expressed at stage E and is highly associated with metabolites (Figure 3d).
Analyses of sucrose and starch metabolism revealed different sucrose cleavage strategies during early VP
Sucrose and starch metabolism has been reported to play an important role during the VP of flower bulbs (Li et al., 2014; Wu et al., 2021; Xu et al., 2020; Yang et al., 2017). The stage‐specific patterns of 17 enzyme genes and seven metabolites in the sucrose and starch metabolism pathway that were differentially changed are visualized in Figure 4(a). The whole VP was characterized by active expression of DEGs involved in the metabolism of sucrose, UDP‐glucose and cellobiose in both species, whereas stage specific up‐regulation of DEGs regulating starch degradation and synthesis was observed in stage E and stage L, respectively (Figure 4a). Consistent with the pivotal role of CWIN2 in the coexpression network (Figure 3d), we found that CWIN, which is mainly involved in unloading of sucrose via apoplastic pathway, was significantly upregulated in L. sprengeri, but not in L. aurea, at stage E, whereas SUS, which associates the unloading of sucrose via symplastic pathway, was significantly upregulated in L. aurea only. Overall, the heatmap indicated a species‐specific sucrose cleavage pattern during the early stage (Figure 4b).
Figure 4.
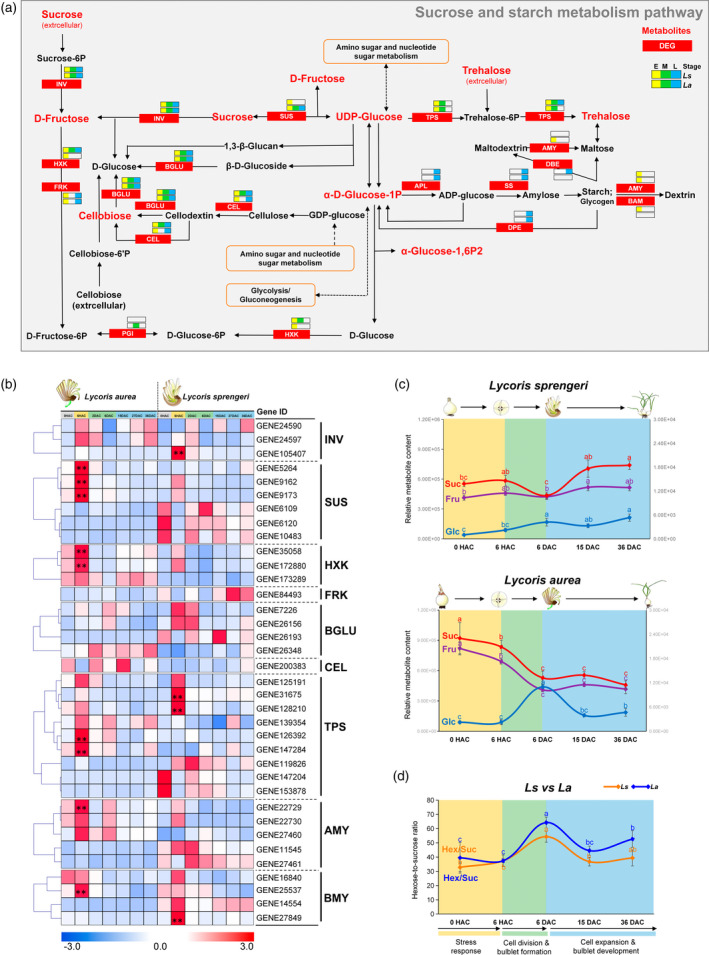
Stage specific expression patterns of differentially expressed genes (DEGs) and changes of related metabolites involved in the sucrose and starch metabolism pathway.
(a) Summarized gene and metabolite changes during vegetative propagation (VP) in both Lycoris species. Stage specific expression patterns of DEGs involved in the sucrose and starch metabolism pathway are colored as labeled. Metabolites that were significantly changed during VP within each species are colored in red.
(b) Expression patterns of DEGs involved in the sucrose and starch metabolism pathway in both Lycoris species. Expression data were Z‐score standardized to −3 to 3 per gene.
(c) Related sugar content changes via UHPLC‐Q‐TOF MS approach. Suc, sucrose; Fru, fructose; Glc, UDP‐glucose.
(d) The ratios of hexose‐to‐sucrose during VP in both Lycoris species. Different letters indicate significant differences at P < 0.05 within each species according to Duncan's multiple range test. [Colour figure can be viewed at wileyonlinelibrary.com]
To gain a basic understanding on the dynamics of sucrose metabolism, changing patterns of sucrose, fructose and UDP‐glucose were visualized across the VP process in both species (Figure 4c). The changing trend of fructose was highly consistent with that of sucrose in both species. The content of UDP‐glucose remained relatively stable during the early stage in both species. By contrast, a significant increase of UDP‐glucose was observed in both L. sprengeri and L. aurea during the middle stage, with a greater increase observed in L. aurea than that in L. sprengeri (Figure 4c). Notably, similar changing patterns were observed in the hexose‐to‐sucrose ratio during the VP process in both species (Figure 4d). The hexose‐to‐sucrose ratio did not show rapid accumulation at the early stage in response to wound stress (Figure 4d). Entering the middle stage, active cell divisions contribute to the bulblet formation accompanied with an evident increase in the ratio of hexose‐to‐sucrose. The ratio peaked at 6 DAC in both species with more rapid increasement from 6 HAC to 6 DAC in L. aurea than that of L. sprengeri. The hexose‐to‐sucrose ratio dramatically decreased to the same level of that for early stage at the beginning of bulblet development stage (Figure 4d).
Phylogenetic analyses of the CWINs and SUSs
To examine phylogenetic relationships among Lycoris CWIN proteins, we constructed a phylogenetic tree based on 147 INV proteins from dicots including Arabidopsis thaliana, Vitis vinifera and Solanum lycopersicum and monocots, including Oryza sativa, Zea mays, L. sprengeri and L. aurea, using two Cyanobacteria (Nostoc sp. PCC7120) and Volvox carteri as outgroups (Table S4). According to previous phylogenetic analyses, INV proteins were divided into three major clades, including CWIN, VIN and CIN (Figure 5a). In total, seven, nine and 10 INV genes, distributed into the CWIN, VIN and CIN subfamilies, respectively, were identified in the two Lycoris species. The LsCWIN2 gene (GENE105407), which was significantly increased during stage E, had the highest similarity to its ortholog in Arabidopsis (AtCWIN2), with the mRNA level increased by 356‐fold and 6‐fold in L. sprengeri and L. aurea, respectively. By contrast, all the expressed NEUTRAL/ALKALINE INVERTASES (CINs) were evenly clustered into the same subgroups (subgroups α and β) for both monocots and dicots (Figure S6).
Figure 5.
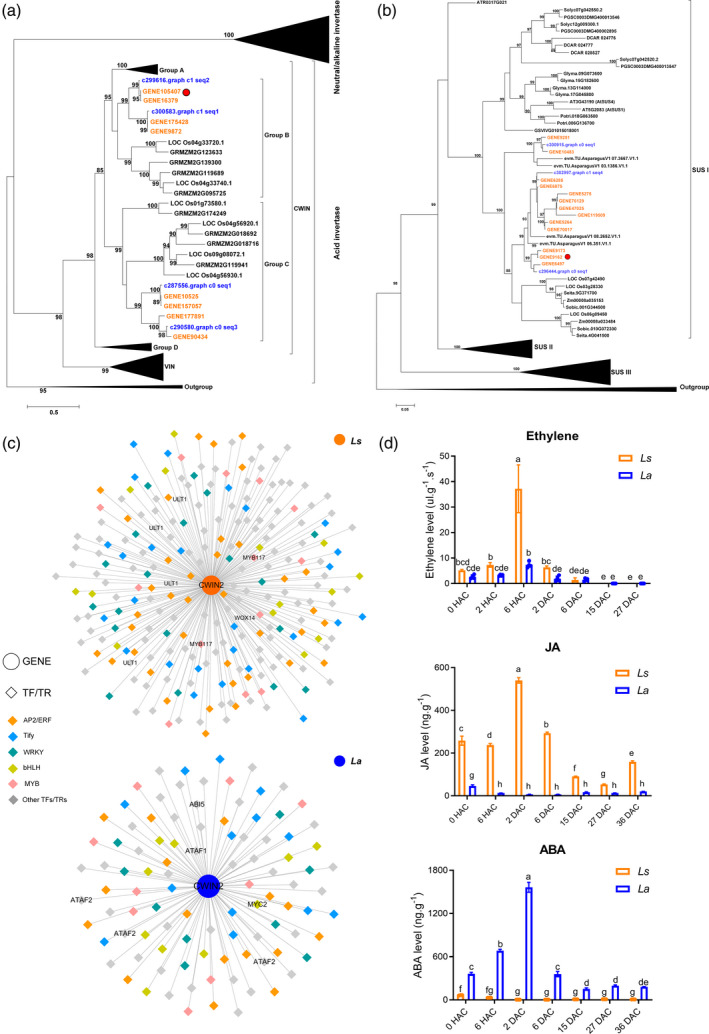
Comparative analysis regarding sucrose cleavage pathways during vegetative propagation (VP) in bulbs of Lycoris.
Phylogenetic analyses of invertase (a) and sucrose synthases (b) in Lycoris and 12 and 13 representative plants, respectively. Font colors of orange and blue indicate the Lycoris transcripts from Pacbio and Illumina platform, respectively.
(c) Networks built from correlations among CWIN2 and transcription factors (TFs)/transcription regulators (TRs) in Ls and La, respectively. Pearson correlation coefficient values were calculated for each pair of genes, and r ≥ 0.9 (P ≤ 0.05) was used to filter the TFs/TRs that were significantly correlated with each gene.
(d) Changes of stress response hormone levels during VP in both Lycoris species. Different letters indicate significant difference at P < 0.05 according to Duncan's multiple range test. [Colour figure can be viewed at wileyonlinelibrary.com]
To identify phylogenetic inferences for the SUSs among the representative species, 110 SUS sequences from six monocots, nine dicots and two outgroup species were used to build a phylogenetic tree (Table S5). According to previous phylogenetic analysis, the constructed phylogenetic tree clearly divided all the SUSs into three groups (Figure 5b). The majority of the Lycoris SUS proteins were classified into Group I, which contained AtSUS1 and AtSUS4. Consistent with the increased levels of UDP‐glucose and starch in SUS‐overexpressing potatotubers (Baroja‐Fernández et al., 2009), we noted significant upregulation of UDP‐glucose during stage M in L. aurea, in which species‐specific upregulation of LsSUS4 (orthologues of AtSUS4) was observed. Only two expressed homologues of SUS proteins were clustered into Group II, whereas none of the expressed SUSs were found in Group III, which contained AtSUS5 and AtSUS6 (Figure S7).
Coexpression networks reveal TFs and TRs associated with hormonal response
Co‐expression network analyses were conducted to uncover the full range of TFs and TRs associated with CWIN2 and SUS4 in L. sprengeri and L. aurea during VP (Figure 5c; Table S6). We found that 238 and 100 TFs/TRs were co‐expressed with CWIN2 in L. sprengeri and L. aurea, respectively, with ERF, Tify, bHLH, MYB, MYB‐related and WRKY representing the largest proportions (r ≥ 0.9) (Table S6). Mining the relationships associated with SUS4 during VP in both species, the most notable of these TFs/TRs were ERFs, being the same as that observed from the networks of CWIN2 (Table S6). The accumulation pattern of LsCWIN2 co‐expressed TFs/TRs (WOX14, MYB117, ULT1) involved in the meristem initiation and maintenance fits well with the active meristematic activities observed in the high regeneration species. For example, LsWOX14, a homolog of the Arabidopsis WUSCHEL‐related homeobox 14, was significantly upregulated in response to wounding and was co‐expressed with CWIN2 in L. sprengeri. WOX14 functions in the shoot meristem organizing center to maintain the stem cells in an undifferentiated state and has been reported to function in enhancing storage organ production (Fernie et al., 2020; Matsumoto‐Kitano et al., 2008).
High proportions of co‐expressed TFs/TRs genes related to hormonal responses were identified during VP with genes regulating sucrose cleavage patterns. This includes TFs/TRs associated with stress responses hormones, e.g. ethylene, jasmonic acid (JA) and ABA. Among the co‐expressed TFs/TRs of CWIN2, ethylene response factor 110 and TIFY domain family protein, which account for the largest proportion, caught our attention. This finding is in line with a previous study attempting to correlate regeneration capacity of lily bulb scales with ERFs expression level after wounding (Moreno‐Pachón, 2017). Previous studies identified ETHYLENE INSENSITIVE 3 (EIN3)/EIN3‐LIKE 1 as a key integration node whose activation requires both JA and ethylene signaling (Zhu et al., 2011). In the present study, species‐specific upregulation of LsEIL1 was only observed in L. sprengeri, along with the synergistically increase of ethylene and JA contents (Figure 5d), which was similar to the previous reports in A. thaliana (Zhu et al., 2011) and pear (Ni et al., 2020), suggesting that JA might enhance ethylene signal transduction in the high‐regeneration bulbs under stress conditions. However, we cannot draw any conclusions based on our observations that ERF and Tify play roles as triggering signal for bulblet regeneration in Lycoris. Intriguingly, during the early VP, contrasting changes in ABA content was observed (Figure 5d). This was evident from the significant increasing trend in L. aurea and decreasing trend in L. sprengeri at 6 HAC and 2 DAC (Figure 5d). ABA has been reported to inhibit the activities of soluble acid cell wall invertase in maize (Zhang et al., 2017), which hints toward the probable role of ABA in the regulation of early sucrose cleavage pattern regarding bulblet formation in Lycoris.
Observations of CF unloading during VP
To test the possibility that apoplasmic and symplasmic pathways may operate specifically during early VP in different regeneration bulbs, we detected the fluorescence signals of a symplasmic tracer, CF, from transverse sections of scales via CFDA labeling (Figure 6a). Upon loading into scale cells, the membrane‐permeable fluorescent CFDA is degraded into membrane‐impermeable fluorescent CF (Ruan et al., 2001; Ruan & Patrick, 1995). Notably, no CF was found in any of the vascular systems after CF transporting at 6 HAC (Figure 6b, A1–C1) and 2 DAC (data not shown) in L. sprengeri. By contrast, CF had already been unloaded from the upper site of the phloem to the lower site next to the basal plate by 6 HAC (Figure 6b, A2–C2) and extensive spread of CF to the surrounding storage parenchyma elements was evident by 6 DAC in L. aurea (Figure 6b, A4–C4).
Figure 6.
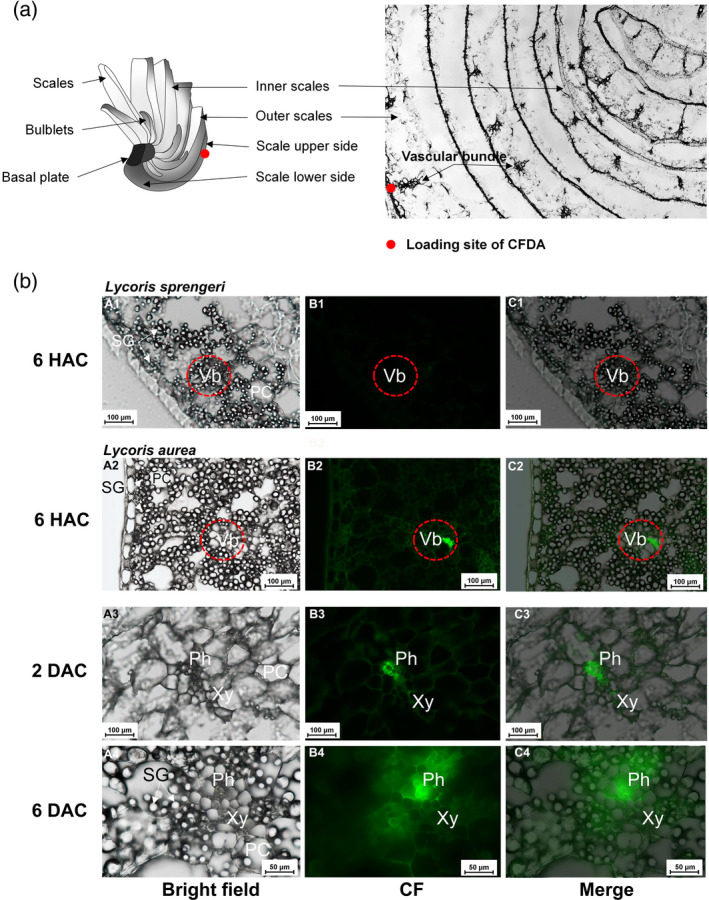
Carboxyfluorescein (CF) fluorescence tracing in the early vegetative propagation (VP).
(a) A diagrammatic drawing of the Lycoris bulbs, showing the loading site of non‐fluorescent 5(6)‐carboxyfluorescein diacetate (CFDA). A transection of the bulblet showing the sampling site, and the anatomy of scales and the distribution of the vascular bundles.
(b) Samples from Lycoris sprengeri (Ls) and Lycoris aurea (La) were collected at 6 h after cutting (HAC) (A1‐C1, A2‐C2) 2 HAC (A3‐C3), 6 days after cutting (DAC) (A4‐C4) and thereafter. Three explants were used each time for each species. Fluorescence was almost quenched after 6 DAC. Red dashed circles indicate vascular bundles. Xy, xylem; Ph, phloem; IS, inner scales; OS, outer scales; SG, starch granules; PC, parenchymal cell. BF, bright field. CFP, cyan fluorescent protein field. [Colour figure can be viewed at wileyonlinelibrary.com]
Metabolic and quantitative real‐time PCR (qRT‐PCR) validation in Lycoris species
To check whether our observations on the gene expression‐metabolite network are conserved in Lycoris for VP, we investigated this issue in other Lycoris species (Figure 7). Different bulblet regeneration abilities were observed in the four common Lycoris species under the same VP condition induced by artificial cross‐cutting (Figure 7a). Lycoris longituba and L. radiata, representing the most significant regeneration differences (Figure 7b), were selected for gene expression analyses and sugar metabolites profiling. Clearly, the expression differences are in line with regeneration differences between species. Specifically, DEGs encoding CWIN2 were upregulated in the early VP in both species. However, the up‐regulation of CWIN2 in L. radiata with a higher number of regenerated bulblets was significantly stronger than in L. longituba with less bulblets (Figure 7d). Meanwhile, expression of SUS4 was inhibited during the early stage in L. radiata compared to the high expression pattern of SUS4 in L. longituba (Figure 7d). Collectively, the relative expression patterns of both CWIN2 and SUS4 in a qRT‐PCR of the selected Lycoris species matched the results of RNA‐Seq analysis, providing evidence for the early sucrose cleavage pattern manipulated VP in Lycoris species. Notably, the changing ratio of hexose‐to‐sucrose during the VP of L. longituba and L radiata was similar to that of L. sprengeri and L. aurea (Figure 7e). The ratio of hexose‐to‐sucrose increased along with the formation of bulblet, and decreased thereafter with the development of bulblets. Notably, compared with the gentle increasement in the ratio of hexose‐to‐sucrose in L. radiata, more significant fluctuations of the ratio were observed during the early and middle stage in L. longituba. Overall, the relationship between hexose and sucrose hints towards its probable role as a growth‐specific marker for the VP process in Lycoris, which is worthy of further investigation.
Figure 7.
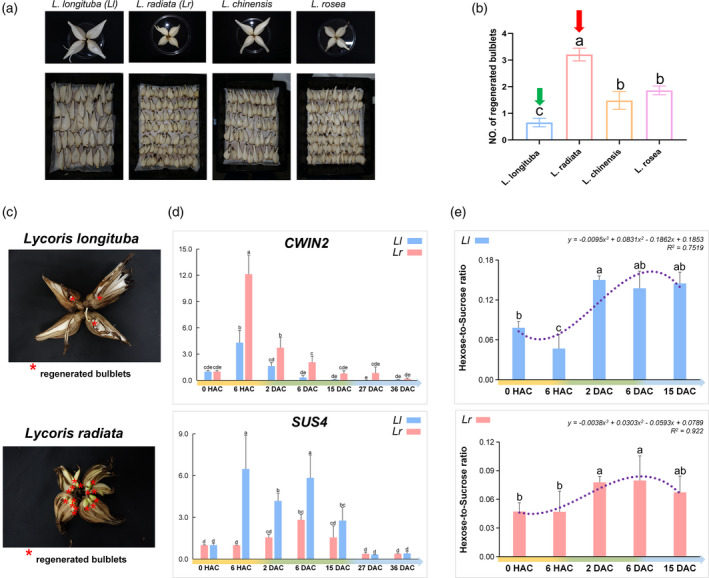
Expression patterns of CWIN and SUS genes and changes of related sugar levels in Lycoris species with high‐ and low‐regeneration capacities. (a) Vegetative propagation (VP) of L. longituba (Ll), L. radiata (Lr), L. chinensis and L. rosea via cross‐cutting under artificial conditions. (b) The number of regenerated bulblets per bulb section in the randomly selected Lycoris species under artificial conditions. Values are the mean ± SEM (n = 30 biological replicates). (c) Regenerated bulblets in Ll(low‐regeneration species) and Lr (high‐regeneration species). Asterisks indicate regenerated bulblets. (d) Expression patterns of CWIN2 and SUS4 in Lr and Ll. (e) Changes of the ratios of hexose‐tsucrose in Lr and Ll. Values are the mean ± SEM of three biological replicates. Different letters indicate significant differences between species at P < 0.05 according to Duncan's multiple range test. [Colour figure can be viewed at wileyonlinelibrary.com]
DISCUSSION
In the present study, we analyzed the VP process in Lycoris, focusing on the capacity of the bulbs to form bulblets (regeneration) in response to wounding stress (cutting). The vegetative regeneration ability plays a vital role in horticultural practice for realizing yield and for survival under harsh environments. Despite its central importance, whether and how sugar metabolism exerts a potential role in regulating regeneration during early VP is poorly understood. Here, we explored this issue by performing a comprehensive transcriptomic analysis and parallel metabolic profiling in bulbs of two Lycoris species with contrasting regenerative abilities.
Temporal transcriptome profiling uncovered molecular components potentially responsible for bulblet initiation during VP
Previous research on VP has largely focused on the biomass trait during bulblet development. By contrast, much less is known about the mechanisms influencing quantitative traits during the early initiation stage. As a result of the significant transcriptome differences during the early stage of VP via both inter‐ and intra‐species comparisons, emphasis was further given on exploring the underlying molecular mechanisms of this process within 6 HAC. This approach is consistent with a previous report on the pivotal role of early development in the formation of sink organs during sexual reproduction (e.g. seed and fruit development) when their final size and yield potential are determined (Ruan et al., 2012).
With the next‐generation sequencing technologies being developed, molecular technologies have become attractive tools for analyzing important biological processes in non‐model species (Leeggangers et al., 2013). To this end, Shahin (2012) provided the transcriptome dataset of Lilium and Tulipa by sequencing of ESTs with Roche 454 technology. In recent years, technical improvements such as Illumina paired‐end sequencing and outputs from different platforms (e.g. PacBio) have been incorporated to avoid the need to decipher complete genome sequences, which, in the case of tulip, is 200 times (Leeggangers et al., 2013) and, for Lycoris, 180 times (Ren, Lin, et al., 2021) the size of the Arabidopsis genome. RNA‐Seq experiments have helped to elucidate specific biological processes in flower bulbs, including Lilium (He et al., 2018; Lazare et al., 2019; Li, Zhang, et al., 2017; Li et al., 2014; Singh & Desgagne‐Penix, 2017; Wang, Su, et al., 2018; Xu et al., 2017; Yang et al., 2017; Zhang et al., 2015), Tulipa (Moreno‐Pachón et al., 2018; Shahin, 2012; Sun et al., 2022), Narcissus (He et al., 2020; Ren, Yang, et al., 2017; Singh & Desgagne‐Penix, 2017), Muscari (Lou et al., 2014) and Lycoris (Chang et al., 2013; Park et al., 2019; Wang et al., 2013, 2016; Xu et al., 2020; Yang et al., 2021), amongst others. However, the short reads resulting from this approach could rarely cover full‐length transcripts, especially in the absence of genomic information. We conducted SMRT sequencing to construct a full‐length reference transcriptome of a member of the Amaryllidaceae family. The length distribution has been significantly refined based on FLNC reads (Figure S1), highlighting the potential of this approach for generation of full‐length structures of each splicing isoform.
Through combined transcriptome assembly, we unified the reference background to conduct the inter‐species comparison between two Lycoris species with different regeneration rates, which helped to reduce the noise from the Illumina platform because of genotype differences to a certain extent, making up for deficiencies of lacking the mutants in most flower bulbs. We characterized transcriptomes in tightly defined temporal stages during VP so that the underlying regulatory networks could be precisely investigated. The high temporal‐resolution transcriptome profiling data generated here allowed us to identify genes specifically expressed in particular stages of VP in bulbs (Figure 2c; Table S2), which is highly informative for inferring gene function and metabolic regulation of the regeneration transition, thereby improving plant yield.
Sugar dynamics might be good indicators of the metabolic processes that lead to phase transition toward regeneration of bulblets
A principal feature of plant metabolism is the flexibility to accommodate developmental changes and to respond to the changing environment (Fernie et al., 2020; Lloyd & Zakhleniuk, 2004). In the present study, we observed a short time‐scale response to wound stress via metabolic regulation in bulbs of Lycoris. Overall, the detection of 230 metabolites by 7‐Q‐TOF MS provided a good overview into different types of metabolites in the storage organs of Lycoris and insights into metabolic processes throughout VP. Given the specific advantage of the UHPLC‐Q‐TOF MS approach for the separation of sugars and sugar‐related compounds (Romo‐Pérez et al., 2020), we identified 34 carbohydrates and carbohydrate conjugates, of which 25 (L. sprengeri) and 29 (L. aurea) changed significantly in at least one stage of VP in bulbs of Lycoris (Table S3). In sum, this report providing detailed compositional data about Lycoris bulbs including an assessment of the metabolic changes during wound‐induced VP based on data obtained by untargeted methods.
The carbohydrate status has been reported to be one of the regulatory signals in signaling networks that allow plants to perceive and respond to a changing environment and to developmental changes (Rolland et al., 2002; Smeekens et al., 2010). In the Lycoris species with higher regeneration capacity, VP via wound induction repressed those LsSUSs likely catalysing the reversible cleavage of sucrose but enhanced the expression of LsCWIN genes for apoplastic phloem unloading and sink induction during the early stage (Figures 4d and 7d). Intriguingly, although the major sucrose cleavage pathway differs between species during the early stage, similar trends in the changes of hexose‐to‐sucrose ratio was observed during the morphological development of bulblets in both species (Figure 4d). Importantly, a similar result was confirmed in another randomly selected pair of Lycoris species with contrasting regeneration capacities via quantitative detection of sugar metabolite (Figure 7e). Based on the analyses above, we proposed a sugar‐mediated model of the regulation of bulblet development in which high CWIN expression or activity may promote bulblet initiation via enhancing apoplasmic unloading of sucrose or sugar signals, whereas the subsequent high ratio of hexose‐to‐sucrose likely supports cell division characterized in the next phase of bulblet formation (Figure 8). The role of CWIN‐mediated sugar signaling has been shown to be essential for ovule initiation from floral primordia in Arabidopsis (Liao et al., 2020). On the other hand, a high hexose‐to‐sucrose ratio stimulates cell and nuclear division in the embryo and endosperm during early seed development (Ruan, 2014, 2022).
Figure 8.
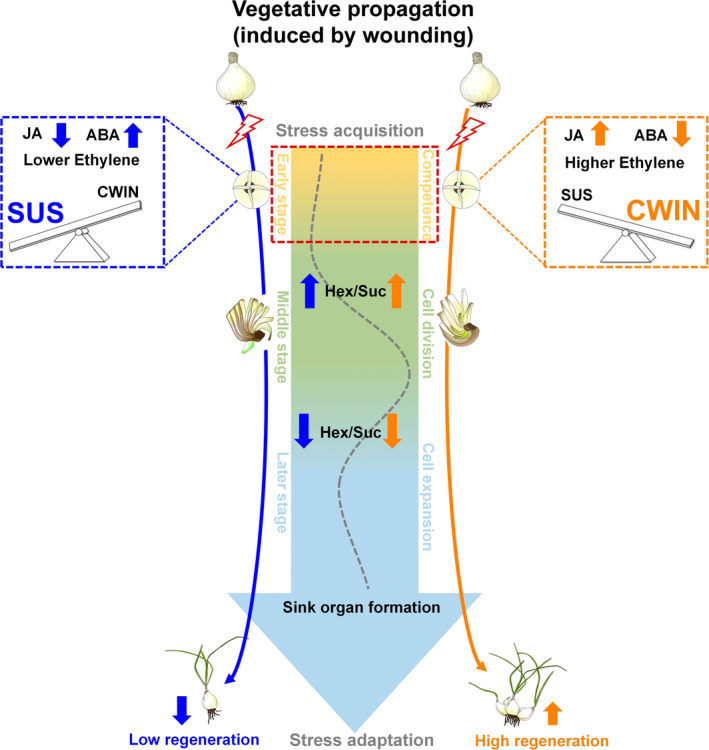
Schematic representation of how the vegetative propagation (VP) is affected from a sugar dynamics manipulation perspective. The rapid increase in the hexose‐to‐sucrose ratio, as driven by high expression levels of sucrose cleavage enzyme genes, is required during the early VP. The dominant sucrose cleavage pathway is therefore pivotal for activating the regeneration potential in bulbs. The ratio of hexose‐to‐sucrose increases during bulblet formation and decreases during bulblet development. The ratio of hexose‐to‐sucrose might be a good indicator of the sugar metabolic processes that leads to phase transition and regeneration. Stress response hormones with differential affects along with early sucrose cleavage is also predicted. CWIN, cell wall invertase; JA, jasmonic acid; SUS, sucrose synthase. [Colour figure can be viewed at wileyonlinelibrary.com]
Apart from CWIN, our co‐expression analysis also identified TPS as another gene associated with metabolite dynamics during VP in L. sprengeri (Figure 3d). TPS catalyzes the synthesis of trehalose‐6‐phosphate, which has been implicated in sugar signalling in some cases. Here, the TPS genes exhibited a similar increment in their expression levels in L. sprengeri and L. aurea at the bulblet initiation stage, although from different gene members (Figure 4b) with no significant changes in trehalose content over the whole period of VP in L. sprengeri (Table S3). Thus, TPS does not appear to play a major role in bulblet initiation.
Early sucrose cleavage pattern correlates variations in regeneration capacities during VP
Regeneration is a common strategy for plant recovery and survival following wound stress (Ikeuchi et al., 2016). During the process of wound‐induced regeneration in Lycoris, decapitation of the apical bud released the outgrowth of AxBs. More importantly, by wounding stimulation, certain endogenous environments permissive for de novo vegetative meristem formation and development could be established, which allows the realization of their regeneration potentials. Plant reproduction depends greatly on an adequate import of photoassimilates, which, for most plant species, is mainly in the form of sucrose (Ruan et al., 2012). During bulblet formation, sucrose breakdown into soluble sugars was observed. This was evident from the higher expression of CWIN and SUS at the early stage of VP, resulting in the release of hexose. A higher ratio of hexose‐to‐sucrose in L. radiata and L. longituba during early VP validated this observation (Figure 7e). According to our data, the efficient conversion of sucrose to hexose could be facilitated by CWIN or SUS, suggesting the role of these enzymes in the establishment of sugar homeostasis during early VP in bulbs.
Phloem‐unloading strategies have evolved to suit the requirements of photoassimilate distribution, for growth and development (Braun et al., 2014; Ma, Sun, et al., 2019; Patrick, 1997). Plants possess the ability to autonomously choose the most suitable strategies for sucrose phloem loading and unloading, with either symplastic or apoplastic pathways being dominant, depending on different structures, development stages and species (Ma, Sun, et al., 2019; Ruan, 2014). The regulation of phloem unloading in sinks is not understood as well as phloem loading in sources because, compared with source leaves, sinks vary in form, including meristems, seeds, fruits, bulbs and tubers, amongst others (Fernie et al., 2020; Ma, Sun, et al., 2019), resulting in more diverse unloading routes. It was difficult to distinguish which unloading pathway was superior because each has pros and cons (Ma, Li, et al., 2019; Ma, Sun, et al., 2019; Sonnewald & Fernie, 2018; Viola et al., 2001). For SUS‐mediated symplastic unloading, the requirement of diffusion and bulk flow at the two ends of the plasmodesma greatly limits the multi‐directional transport of assimilates, which might result in the limited formation of vegetative meristems as observed in L. aurea. Sucrose catabolism via CWINs requires more energy than via SUSs (Bologa et al., 2003) and often is functionally coupled with sugar transporters (Ruan, 2022). This active unloading mode without the prerequisite of a high concentration difference of sucrose between the phloem and surrounding recipient cells might be more efficient in the delivery of sugars for rapid cell or organ regeneration. The significant upregulation of LsCWIN2 during the early stage of VP in the high‐regeneration Lycoris species is consistent with the reported positive role of CWIN in sexual reproduction, such as to mediate ovule initiation (Liao et al., 2020) and to stimulate endosperm nuclear division after double fertilization (Ruan et al., 2008; Wang & Ruan, 2012). Further studies using approaches such as transgenic knockout are needed to determine the casualty between CWIN and bulblet initiation.
We also observed that bulblet regeneration in Lycoris was accompanied by a synergistically burst of ethylene and JA production induced by wounding in the high‐regeneration species (Figure 5d), consistent with the obtained large abundance of co‐expressed ERF and Tify family genes with LsCWIN2 and LsSUS4, respectively (Figure 5c; Table S6). This finding corresponded to a study reporting that wounding activated an extremely fast regeneration response probably mediated by AP2/ERF via mediating polar auxin re‐distribution, cell proliferation and cell dedifferentiation during de novo shoot meristem regeneration in Lilium (Moreno‐Pachón, 2017). Moreover, the opposite changing trend of ABA during the early VP between high and low‐regeneration species attracted our attention (Figure 5d). Interaction of CWIN with hormones has been indicated from past studies (Albacete et al., 2014; French et al., 2014; Zhang et al., 2017). In maize, an inhibitory effect was noted on the activities of CWIN for ABA application at the early post‐pollination stage (Zhang et al., 2017). In addition, overexpression of CWIN in cassava indicated its role in plant growth and stress response (Yan et al., 2019). ABA is typically accumulated in plants under stress conditions. It is not known whether ABA is involved in bulblet formation. Further investigations are required to assess whether these stress response hormones directly impacts enzyme activities of CWIN or sucrose, thereby regulating sucrose cleavage for early VP.
EXPERIMENTAL PROCEDURES
Plant materials and sample collection
Bulbs of L. sprengeri and L. aurea were harvested in late May from the Bulb and Perennial Root Germplasm Resources Garden at Zhejiang University, Hangzhou, China (E 120°11′, N 30°29′). Bulbs of uniform circumferences were longitudinally cut into quartered‐bulb sections via cross‐cutting. The sterilized bulb sections were placed on a plastic tray within a moist chamber (Yiheng Technical Co., Ltd, Shanghai, China) and monitored under the aerial cultivation conditions with a light intensity of ~90 μmol m−2 sec−1 and a 14 : 10 h light/dark photocycle at 28 ± 2°C. Sections were sprayed with sterilized water twice per day to maintain the humidity at 90–95% until new bulblets formed (approximately 1 cm in diameter).
RNA samples from seven time points after cross‐cutting (0 HAC, 6 HAC, 2 DAC, 6 DAC, 15 DAC, 27 DAC and 36 DAC) were collected from the basal regions of the scales connecting the stem to monitor the early VP process (Figure S1a). Meristem‐enriched tissues were excised from each bulb section, including tissues (1–2 cm) connecting scales and the basal plate. Five randomly selected samples from different bulbs were pooled to form one replicate, and each time point contained three biological replicates. Tissues were frozen immediately in liquid nitrogen and stored at −80°C before processing. Total RNA samples were isolated using the EASYspin Plus Plant RNA kit (RN38; Aidlab Bio, Beijing, China) in accordance with the manufacturer's instructions. Gel electrophoresis (1.0% agarose gel) and a Nanodrop® spectrophotometer (ND‐2000; Thermo Scientific, Waltham, MA, USA) were used to analyze and confirm the quality and quantity of the total RNA from each sample.
For the untargeted metabolomics analysis, five time points (0 HAC, 6 HAC, 6 DAC, 15 DAC and 36 DAC) selected based on transcriptome samples, representing four main developmental stages, were utilized. Five randomly selected samples from different bulbs were pooled to form one replicate, and each time point contained five biological replicates. Tissues were frozen immediately in liquid nitrogen, well ground to fine powder and kept frozen at –80°C until UHPLC‐Q‐TOF MS analysis.
Morphology and microscopy analysis
The number of regenerated bulblets from each bulb via native VP (without wound treatment) and each quartered‐bulb section via the artificial method (with cross‐cutting treatment) were analyzed and compared between two species. Comparative observations were conducted using the Periodic acid–Schiff reagent method of Mowry (1963) as reported in Ren, Xia, et al. (2017). Briefly, the sampled tissues were fixed in formaldehyde:glacial acetic acid:ethanol (1:1:18, v/v) fixation buffer for 24 h before being embedded in paraffin. Then, 12‐μm semi‐thin sections were cut with a microtome (Leica 2016; Leica Microsystems, Heidelberg, Germany). The sections were oxidized in periodic acid for 10 min and then treated with Schiff's reagent for 15 min, followed by stained in hematoxylin for 2 min. The sections of scales were examined under a Nikon ECLIPSE CI microscope (Nikon Corp., Tokyo, Japan) and photographed with a DS‐U3 digital camera (Nikon Corp.) connected to the microscope.
Library preparation and sequencing
The Iso‐Seq libraries were constructed using 1 μg of equally pooled RNAs from the seven time points as specified in Figure S1(b). First‐strand cDNA was synthesized using the SMARTerTM PCR cDNA Synthesis Kit (Clontech Laboratories, Mountain View, CA, USA), followed by amplification of double‐stranded cDNA via long‐distance PCR. The 1–2‐, 2–3‐ and 3–6‐kb cDNA fractions were generated with the BluePippin™ size selection system (Sage Science, Beverly, MA, USA). The three libraries were constructed with a SMRTbell Template Prep Kit 1.0 (http://www.pacb.com) and subsequently sequenced on the Pacific Biosciences RS II platform with a total of five SMRT cells: the 1–2‐ and 2–3‐kb libraries were sequencing on two SMRT cells, respectively, whereas the 3–6‐kb library was sequenced on one SMRT cell. Strand‐specific RNA‐Seq libraries were constructed with 3 μg of RNA per sample using the RNA‐Seq Library Prep Kit (Illumina Inc.) in accordance with the manufacturer's instructions and sequenced on the Illumina HiSeq Xten platform in paired‐end mode, each with three replicates. RNA Integrity Number (RIN) values were checked for each RNA sample using an Agilent Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA) (RIN ≥ 7.0 and ≥8.0 for RNA‐Seq and Iso‐Seq, respectively). Library preparation and sequencing were both performed at Biomarker Technologies (Beijing, China) using standard protocols.
RNA‐Seq data processing and transcript assembly
Raw RNA‐Seq reads were processed to remove adaptor and low‐quality sequences using trimmomatic (Bolger et al., 2014). Then, bowtie (Quast et al., 2013) was used to remove ribosomal RNA and virus reads. The resulting high‐quality clean reads were de novo assembled into transcripts using rnaspades (Bankevich et al., 2012). Redundancies of the assembled transcripts were removed using cd‐hit (Huang et al., 2010), and transcripts expressed at low levels (FPKM < 0.1) were excluded from downstream analyses (Figure S1c).
PacBio Iso‐Seq data analysis
PacBio subreads were analyzed according to the Iso‐Seq software in smrt® analysis, version 3.0 (https://github.com/ben‐lerch/IsoSeq‐3.0). The pipeline was mainly performed in three stages: the generation of circular consensus sequences (CCSs), classification and clustering. The CCSs generated from the subreads were the key inputs for classification, after which FLNC reads were obtained. High‐quality consensus isoforms (HQs) and low‐quality consensus isoforms (LQs) were obtained after clustering, and then the LQs were further corrected with cleaned RNA‐Seq reads using lordec (Guindon & Gascuel, 2003). HQs and corrected LQs were collapsed by cogent (https://github.com/Magdoll/Cogent) to form the collapsed PacBio isoforms (Figure S1d).
Merging RNA‐Seq assembly and Iso‐Seq isoforms
The assembled transcripts from the RNA‐Seq data and Iso‐Seq isoforms were combined. cd‐hit (Huang et al., 2010) was then used to cluster the transcripts and to remove redundancies in the combined transcripts. The resulting non‐redundant transcripts were processed using seqclean (https://sourceforge.net/projects/seqclean/files) to remove rRNA contaminants or those from non‐eukaryotic organisms. The longest transcript in each cluster from the final transcriptome assembly was selected as a representative transcript for downstream gene expression analysis (Figure S1e). The final representative transcripts were annotated using blast2go (Conesa et al., 2005) to obtain GO terms and then blastkoala (https://www.kegg.jp/blastkoala/) was used to assign KEGG Orthology terms. TFs and transcriptional regulators were identified from the transcripts using itak (Zheng et al., 2016). Furthermore, the representative transcripts were compared against the Arabidopsis (version Araport11), rice (v7.0) and maize (Zm‐B73‐REFERENCE‐GRAMENE‐4.0) protein databases (https://phytozome‐next.jgi.doe.gov/) to obtain their closest homologues using blastx (E‐value cut‐off ≤1 × 10−5) (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
DEG identification and classification
Cleaned reads were mapped to the final representative transcripts using bowtie2 (Langmead & Salzberg, 2012). Based on the alignments, raw counts for each gene were obtained using rsem (https://deweylab.github.io/RSEM), which were then fed into impulsede2 (Fischer et al., 2018) to identify DEGs (adjusted P ≤ 0.01). FPKM values were calculated from raw counts and used to measure the gene expression levels. The Pearson correlation coefficients between biological replicates with normalized expression levels (FPKM) were calculated. PCA was performed using r software 2013 (R Foundation for Statistical Computing, Vienna, Austria) with default settings. DEG analysis between species was performed with timesvector (Jung et al., 2017) to identify genes with distinctive expression patterns in the time series data. Hierarchical clustering of gene expression profiles was performed using the HCL method in mev, version 4.9 (http://www.tm4.org/mev.html) to select stage‐specific DEGs.
Identification and characterization of the metabolome
The metabolome analysis was conducted as previously described (Pi et al., 2018; Thomas, 2013), with minor modifications. The frozen powder (approximately 100 mg) was suspended in 1.0 ml of a mixture of pre‐cooled methanol:acetonitrile:water (2:2:1, v/v/v) and then briefly vortexed before being sonicated for 30 min (4°C). The vortex‐sonication step was repeated twice. The samples were incubated at −20°C for 1 h, and then centrifuged at 14 000 g for 20 min (4°C). The supernatant (2 μl) was analyzed using an UHPLC instrument coupled to an ion‐trap mass spectrometer equipped with an electrospray ionization (ESI) source (Triple TOF 6600; Applied Protein Technology Co., Ltd, Shanghai, China). The ESI sources were set according to the method described by Pi et al. (2018). The raw data was converted into mzxml format using proteowizard(https://proteowizard.sourceforge.io/) and then xcms(Smith et al., 2006) was used to align the peaks, calibrate the retention time and integrate the peaks. The identity of each ion was searched against the METLIN database (https://metlin.scripps.edu/index.php) for molecular annotation. Differential metabolites were defined as those with P < 0.05 via Student's t‐test.
Network building for transcripts and metabolites
Coexpression analysis was applied to correlate gene expression patterns with metabolite accumulation (Giovannoni, 2018; Serin et al., 2016). A rigorous multiple test correction (r ≥ 0.7) was used to filter the structural genes, TFs and TRs that were significantly correlated with each metabolite (Li et al., 2020). All the associations among structural genes, TFs and TRs and metabolites were visualized using cytoscape (version 3.7.2; Kohl et al., 2011).
Hormone measurements
Endogenous concentrations of JA and ABA were determined using UPLC‐ESI‐MS/MS as described previously by Liu et al. (2020). Briefly, the freeze‐dried samples (50 mg) were ground in 500 μl of a pre‐cooled mixture of isopropanol:H2O:formic acid (2:1:0.002, v/v/v). The extract was incubated at −20°C for 20 min before being sonicated in an ice bath for 30 min, followed by incubation at −20°C for 20 min (mixed with 1 ml of chloroform) and sonication in an ice bath for 5 min. The samples were vortexed for 1 min before being centrifuged at 10 000 g for 5 min (4°C). The supernatant (900 μl) was transferred to an LC‐MS vial and dried before being resuspended in 400 ml of a mixture of pre‐cooled methanol:H2O (4:1, v:v), followed by 1 min of sonication in the ice bath. Chromatography was carried out on a Poroshell 120, EC‐C18 (100 × 3 mm, 2.7 μm) (Agilent Technologies). The mobile phase and gradient elution procedures were set according to the method described by Liu et al., 2020. Peak areas were calculated using analyst, version 1.6.2 (AB Sciex, Toronto, ON, Canada). Standards for JA (CAS#77026‐92‐7; Yuanye Biochemical Company, Shaghai, China) and ABA (CAS#14375‐45‐2; Yuanye Biochemical Company) were used to quantify the concentrations.
Ethylene production in the bulbs of Lycoris was analyzed by placing five quartered‐bulb sections into glass vials during VP. The sealed vials were incubated in the growth chamber for 2 h before measurement as described in Wu et al. (2011). Gas (1 ml) was withdrawn from the airspace of each vial using a gas‐tight syringe (Focus GC; Thermo Scientific) and injected into a gas chromatograph (Focus GC; Thermo Scientific) equipped with a capillary column (CP‐carboPLOT P7; Varian, Palo Alto, CA, USA) and flameionization detector for ethylene determination. ET production was calculated on the basis of the fresh weight of root samples.
Carboxyfluorescein diacetate labeling
To determine the phloem unloading pathway during early VP in bulbs of two Lycoris species, the application and visualization of the symplastic fluorescent dye CF were performed as previously described in Ruan and Patrick (1995), with some modifications. CFDA (#C4995; APExBIO, Houston, TX, USA) was prepared as a 1.0 mg ml−1 stock solution in acetone. Direct loading of approximately 120 μl of CFDA solution into the wedge‐shaped block in the upper part of abaxial surface of the outer bulb scales was performed to expose the vascular bundle (Figure 6a). The loading site was immediately fixed with polythene film and aluminum foil to avoid dye loss and fluorescence quenching under light conditions. The CFDA‐fed materials from the two species were incubated back to the original growth condition to allow for CF transport for at least 24 h before monitoring as described in Wu et al. (2021). CFDA feeding was conducted at 6 HAC, 2 DAC and 6 DAC, respectively, to cover the early VP stages. A frozen slicer (SHARDON ORYTOME FE; Thermo Scientific) was used to slice the explants into 35‐μm thin sections, after which a fluorescence microscope (Eclipse Ni; Nikon Corp.) was used to monitor the movement of the CF fluorescence under blue light (488 nm).
Quantitative detection of sugar metabolite
A model 7890B gas chromatograph (Agilent Technologies) coupled to a 7000D mass spectrometer with a DB‐5MS column (30 m length × 0.25 mm i.d. × 0.25 μm film thickness; J&W Scientific, Flosom, CA, USA) was employed for gas chromatography‐MS analysis. Sugar contents in dried bulb samples of L. longituba and L. radiata were detected using metware (http://www.metware.cn) based on the Agilent 7890B‐7000D platform (for details regarding the protocol, refer to Methods S1).
Validation by qRT‐PCR
RNA samples from seven time points after cross‐cutting collected from Ll and Lr during the early VP process were applied for a qRT‐PCR. A PrimScript RT reagent Kit with gDNA Eraser (Takara, Otsu, Japan) was used for cDNA synthesis. The products were diluted 20‐fold with sterile water and used as templates, with primers designed using beacon designer, version 7.9 (Table S7) (Li et al., 2022). qRT‐PCRs were performed with a TB Green Premix Ex Taq Kit (RR420A; TaKaRa, Tokyo, Japan) in a Connect™ Optics Module (Bio‐Rad, Hercules, CA, USA). The expression levels of the tested transcripts were calculated by the 2−∆∆Ct method (Livak & Schmittgen, 2001). The values are expressed as the mean of three biological replicates.
Statistical analysis
All morphological traits consisted of at least ten biological replicates, and all other measurements consisted of three biological replicates unless indicated otherwise. Statistical analyses were conducted using one‐way analysis of variance in spss, version 20.0 (IBM Corp., Armonk, NY, USA) with Duncan's multiple range test. Illustrations were drawn using prism, version 8 (GraphPad Software Inc., San Diego, CA, USA) and powerpoint (Microsoft Corp., Redmond, WA, USA).
AUTHOR CONTRIBUTIONS
Z‐MR and Y‐PX conceived and designed the study. Z‐MR, DZ, YW and Y‐FL collected plant materials and conducted the experiments. Z‐MR, D‐QL, DZ, CG and Y‐LR analyzed the data with the help of CJ and X‐YW. Z‐MR, Y‐PX and Y‐LR wrote the original manuscript. Z‐MR and Y‐LR revised the manuscript. All authors read and approved the final version of the manuscript submitted for publication.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Figure S1. Flowchart of the experimental design, combined sequencing and bioinformatics pipeline.
Figure S2. Global heatmaps of inter‐species and intra‐species comparisons throughout VP.
Figure S3. k‐means clustering algorithum of DEGs in two Lycoris species.
Figure S4. KEGG and GO analysis of stage‐specific DEGs.
Figure S5. Analysis of the metabolome during VP in bulbs of Lycoris.
Figure S6. Phylogenetic tree of invertase (INV) in Lycoris and representative plants.
Figure S7. Phylogenetic tree of sucrose synthase (SUS) in Lycoris and representative plants.
Data S1. Quantitative detection of sugar metabolites.
Table S1. Summary of combined sequencing.
Table S2. GO and KEGG enrichment analysis of stage‐specific DEGs in both Lycoris species.
Table S3. Summary of metabolomics.
Table S4. Amino acid sequences of invertase genes.
Table S5. Amino acid sequences of sucrose synthase genes.
Table S6. Summary of co‐expression analysis.
Table S7. Primers used in a qRT‐PCR.
ACKNOWLEDGEMENTS
We are grateful to Zhangjun Fei (Boyce Thompson Institute), Liangsheng Zhang (Zhejiang University) and William Bill Miller (Cornell University) for technical support and critical advice. This work was supported by funds from National Natural Science Foundation of China (Grant No. 32101571), Zhejiang Science and Technology Major Program on Agricultural New Variety Breeding (Grant No. 2021C02071‐6), Zhejiang Sci‐Tech University Start‐up Fund (Grant No. 22052138‐Y) and the Australian Research Council (DP180103834) to Y‐LR.
Contributor Information
Yong‐Ling Ruan, Email: yong-ling.ruan@anu.edu.au.
Yi‐Ping Xia, Email: ypxia@zju.edu.cn.
DATA AVAILABILITY STATEMENT
The Illumina RNA‐Seq and PacBio Iso‐Seq raw data have been submitted to the Short Read Archive (SRA) data library under accession numbers PRJNA574869 and PRJNA579847.
REFERENCES
- Albacete, A. , Cantero‐Navarro, E. , Balibrea, M.E. , Grosskinsky, D.K. , de la Cruz Gonzalez, M. , Martinez‐Andujar, C. et al. (2014) Hormonal and metabolic regulation of tomato fruit sink activity and yield under salinity. Journal of Experimental Botany, 65(20), 6081–6095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, P.J. (Ed.) (2010) The plant hormones: their nature, occurrence, and functions. In: Plant Hormones. Dordrecht:Springer, pp. 1–15. [Google Scholar]
- Bankevich, A. , Nurk, S. , Antipov, D. , Gurevich, A.A. , Dvorkin, M. , Kulikov, A.S. et al. (2012) SPAdes: a new genome assembly algorithm and its applications to single‐cell sequencing. Journal of Computational Biology, 19, 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroja‐Fernández, E. , Muñoz, F.J. , Montero, M. , Etxeberria, E. , Sesma, M.T. , Ovecka, M. et al. (2009) Enhancing sucrose synthase activity in transgenic potato (Solanum tuberosum L.) tubers results in increased levels of starch, ADP glucose and UDP glucose and total yield. Plant & Cell Physiology, 50(9), 1651–1662. [DOI] [PubMed] [Google Scholar]
- Birnbaum, K.D. & Sanchez Alvarado, A. (2008) Slicing across kingdoms: regeneration in plants and animals. Cell, 132, 697–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. & Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bologa, K.L. , Fernie, A.R. , Leisse, A. , Ehlers Loureiro, M. & Geigenberger, P. (2003) A bypass of sucrose synthase leads to low internal oxygen and impaired metabolic performance in growing potato tubers. Plant Physiology, 132, 2058–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun, D.M. , Wang, L. & Ruan, Y.L. (2014) Understanding and manipulating sucrose phloem loading, unloading, metabolism, and signalling to enhance crop yield and food security. Journal of Experimental Botany, 65, 1713–1735. [DOI] [PubMed] [Google Scholar]
- Butelli, E. , Titta, L. , Giorgio, M. , Mock, H.P. , Matros, A. , Peterek, S. et al. (2008) Enrichment of tomato fruit with health‐promoting anthocyanins by expression of select transcription factors. Nature Biotechnology, 26, 1301–1308. [DOI] [PubMed] [Google Scholar]
- Chang, L. , Xiao, Y.M. , She, L.F. & Xia, Y.P. (2013) Analysis of gene expression and enzyme activities related to starch metabolism in Lycoris sprengeri bulbs of different sizes. Scientia Horticulturae, 161, 118–124. [Google Scholar]
- Chen, L.Q. , Qu, X.Q. , Hou, B.H. , Sosso, D. , Osorio, S. , Fernie, A.R. et al. (2012) Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science, 335, 207–211. [DOI] [PubMed] [Google Scholar]
- Chen, W. , Wang, W. , Peng, M. , Gong, L. , Gao, Y. , Wan, J. et al. (2016) Comparative and parallel genome‐wide association studies for metabolic and agronomic traits in cereals. Nature Communications, 7, 12767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa, A. , Gotz, S. , Garcia‐Gomez, J.M. , Terol, J. , Talon, M. & Robles, M. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics, 21, 3674–3676. [DOI] [PubMed] [Google Scholar]
- Fernie, A.R. , Bachem, C.W.B. , Helariutta, Y. , Neuhaus, H.E. , Prat, S. , Ruan, Y.L. et al. (2020) Synchronization of developmental, molecular and metabolic aspects of source‐sink interactions. Nature Plants, 6, 55–66. [DOI] [PubMed] [Google Scholar]
- Fischer, D.S. , Theis, F.J. & Yosef, N. (2018) Impulse model‐based differential expression analysis of time course sequencing data. Nucleic Acids Research, 46, e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French, S.R. , Abu‐Zaitoon, Y. , Uddin, M.M. , Bennett, K. & Nonhebel, H.M. (2014) Auxin and cell wall invertase related signaling during rice grain development. Plants, 3(1), 95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, S. , Zhu, Y. , Zhou, L. , Fu, X. , Lei, T. , Chen, Q. et al. (2018) Sucrose signaling function on the formation and swelling of bulblets of Lilium sargentiae E.H. Wilson. Plant Cell, Tissue and Organ Culture, 135, 143–153. [Google Scholar]
- Giovannoni, J. (2018) Tomato multiomics reveals consequences of crop domestication and improvement. Cell, 172, 6–8. [DOI] [PubMed] [Google Scholar]
- Guindon, S. & Gascuel, O. (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology, 52, 696–704. [DOI] [PubMed] [Google Scholar]
- Hartmann, A. , Senning, M. , Hedden, P. , Sonnewald, U. & Sonnewald, S. (2011) Reactivation of meristem activity and sprout growth in potato tubers require both cytokinin and gibberellin. Plant Physiology, 155, 776–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, Q. , Shen, Y. , Wang, M. , Huang, M. , Yang, R. , Zhu, S. et al. (2011) Natural variation in petal color in Lycoris longituba revealed by anthocyanin components. PLoS One, 6, e22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, X. , Shenkute, A.G. , Wang, W. & Xu, S. (2018) Characterization of conserved and novel microRNAs in Lilium lancifolium Thunb. by high‐throughput sequencing. Scientific Reports, 8, 2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, Y. , Xu, M. & Chen, X. (2020) De novo transcriptomics analysis of the floral scent of Chinese Narcissus . Tropical Plant Biology, 13, 172–188. [Google Scholar]
- Huang, Y. , Niu, B. , Gao, Y. , Fu, L. & Li, W. (2010) CD‐HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics, 26, 680–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huby, E. , Napier, J.A. , Baillieul, F. , Michaelson, L.V. & Dhondt‐Cordelier, S. (2020) Sphingolipids: towards an integrated view of metabolism during the plant stress response. The New Phytologist, 225, 659–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeuchi, M. , Ogawa, Y. , Iwase, A. & Sugimoto, K. (2016) Plant regeneration: cellular origins and molecular mechanisms. Development, 143, 1442–1451. [DOI] [PubMed] [Google Scholar]
- Jin, Z. & Yao, G. (2019) Amaryllidaceae and sceletium alkaloids. Natural Product Reports, 36, 1462–1488. [DOI] [PubMed] [Google Scholar]
- Jung, I. , Jo, K. , Kang, H. , Ahn, H. , Yu, Y. & Kim, S. (2017) TimesVector: a vectorized clustering approach to the analysis of time series transcriptome data from multiple phenotypes. Bioinformatics (Oxford, England), 33, 3827–3835. [DOI] [PubMed] [Google Scholar]
- Kamenetsky, R. & Okubo, H. (2012) Ornamental geophytes: from basic science to sustainable production. Boca Raton:CRC Press. [Google Scholar]
- Kohl, M. , Wiese, S. & Warscheid, B. (2011) Cytoscape: software for visualization and analysis of biological networks. Methods in Molecular Biology, 696, 291–303. [DOI] [PubMed] [Google Scholar]
- Kusano, M. , Tabuchi, M. , Fukushima, A. , Funayama, K. , Diaz, C. , Kobayashi, M. et al. (2011) Metabolomics data reveal a crucial role of cytosolic glutamine synthetase 1;1 in coordinating metabolic balance in rice. The Plant Journal, 66, 456–466. [DOI] [PubMed] [Google Scholar]
- Langmead, B. & Salzberg, S.L. (2012) Fast gapped‐read alignment with Bowtie 2. Nature Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazare, S. , Bechar, D. , Fernie, A.R. , Brotman, Y. & Zaccai, M. (2019) The proof is in the bulb: glycerol influences key stages of lily development. The Plant Journal, 97, 321–340. [DOI] [PubMed] [Google Scholar]
- Leeggangers, H.A. , Moreno‐Pachon, N. , Gude, H. & Immink, R.G. (2013) Transfer of knowledge about flowering and vegetative propagation from model species to bulbous plants. The International Journal of Developmental Biology, 57, 611–620. [DOI] [PubMed] [Google Scholar]
- Li, D. , Shao, L. , Zhang, J. , Wang, X. , Zhang, D. , Horvath, D.P. , et al. (2022) MADS-box transcription factors determine the duration of temporary winter dormancy in closely related evergreen and deciduous Iris spp. Journal of experimental botany, 73, 1429–1449. [DOI] [PubMed] [Google Scholar]
- Li, Q. , Yang, L. , Sun, Y. & Li, X. (2017) Transcriptome analysis of Lycoris sprengeri based on RNA‐seq technology. Molecular Plant Breeding, 15, 5044–5051 (in Chinese). [Google Scholar]
- Li, X. , Wang, C. , Cheng, J. , Zhang, J. , da Silva, J.A. , Liu, X. et al. (2014) Transcriptome analysis of carbohydrate metabolism during bulblet formation and development in Lilium davidii var. unicolor. BMC Plant Biology, 14, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Chen, Y. , Zhou, L. , You, S. , Deng, H. , Chen, Y. et al. (2020) MicroTom metabolic network: rewiring tomato metabolic regulatory network throughout the growth cycle. Molecular Plant, 13(8), 1203–1218. [DOI] [PubMed] [Google Scholar]
- Li, Y. , Zhang, M. , Zhang, M. & Jia, G. (2017) Analysis of global gene expression profiles during the flowering initiation process of Lilium × formolongi . Plant Molecular Biology, 94, 361–379. [DOI] [PubMed] [Google Scholar]
- Liao, S. , Wang, L. , Li, J. & Ruan, Y. (2020) Cell wall invertase is essential for ovule development through sugar signaling rather than provision of carbon nutrients. Plant Physiology, 183, 1126–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, B. , Wang, X. , Cao, Y. , Arora, R. , Zhou, H. & Xia, Y.P. (2020) Factors affecting freezing tolerance: a comparative transcriptomics study between field and artificial cold acclimations in overwintering evergreens. The Plant Journal, 103(6), 2279–2300. [DOI] [PubMed] [Google Scholar]
- Livak, K.J. & Schmittgen, T.D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2–DDCT method. Methods, 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Lloyd, J.C. & Zakhleniuk, O.V. (2004) Responses of primary and secondary metabolism to sugar accumulation revealed by microarray expression analysis of the Arabidopsis mutant, pho3. Journal of Experimental Botany, 55, 1221–1230. [DOI] [PubMed] [Google Scholar]
- Lopez‐Salmeron, V. , Cho, H. , Tonn, N. & Greb, T. (2019) The phloem as a mediator of plant growth plasticity. Current Biology, 29, R173–R181. [DOI] [PubMed] [Google Scholar]
- Lou, Q. , Liu, Y. , Qi, Y. , Jiao, S. , Tian, F. , Jiang, L. et al. (2014) Transcriptome sequencing and metabolite analysis reveals the role of delphinidin metabolism in flower colour in grape hyacinth. Journal of Experimental Botany, 65, 3157–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv, X. , Zhang, D. , Min, R. , Li, S. , Li, Z. , Ren, Z. et al. (2020) Effects of exogenous sucrose on bulblet formation of Lycoris sprengeri in vitro. Acta Horticulturae, 47(8), 1475–1489 (in Chinese). [Google Scholar]
- Ma, S. , Li, Y.X. , Li, X. , Sui, X.L. & Zhang, Z.X. (2019) Phloem unloading strategies and mechanisms in crop fruits. Journal of Plant Growth Regulation, 38, 494–500. [Google Scholar]
- Ma, S. , Sun, L. , Sui, X. , Li, Y. , Chang, Y. , Fan, J. et al. (2019) Phloem loading in cucumber: combined symplastic and apoplastic strategies. The Plant Journal, 98, 391–404. [DOI] [PubMed] [Google Scholar]
- Matsuda, F. , Hirai, M.Y. , Sasaki, E. , Akiyama, K. , Yonekura‐Sakakibara, K. , Provart, N.J. et al. (2010) AtMetExpress development: a phytochemical atlas of Arabidopsis development. Plant Physiology, 152, 566–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto‐Kitano, M. , Kusumoto, T. , Tarkowski, P. , Kinoshita‐Tsujimura, K. , Vaclavikova, K. , Miyawaki, K. et al. (2008) Cytokinins are central regulators of cambial activity. Proceedings of the National Academy of Sciences of the United States of America, 105, 20027–20031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno‐Pachón, N.M. (2017) Mechanisms of vegetative propagation in bulbs a molecular approach. Wageningen University, Doctoral Thesis.
- Moreno‐Pachón, N.M. , Mutimawurugo, M.C. , Heynen, E. , Sergeeva, L. , Benders, A. , Blilou, I. et al. (2018) Role of Tulipa gesneriana TEOSINTE BRANCHED1 (TgTB1) in the control of axillary bud outgrowth in bulbs. Plant Reproduction, 31, 145–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowry, R.W. (1963) The special value of methods that color both acidic and vicinal hydroxyl groups in the histochemical study of mucins. With revised directions for the colloidal iron stain, the use of alcian blue g8x and their combinations with the periodic acid‐Schiff reaction. Annals of the New York Academy of Sciences, 106, 402–423. [Google Scholar]
- Ni, J. , Zhao, Y. , Tao, R. , Yin, L. , Gao, L. , Strid, A. et al. (2020) Ethylene mediates the branching of the jasmonate‐induced flavonoid biosynthesis pathway by suppressing anthocyanin biosynthesis in red Chinese pear fruits. Plant Biotechnology Journal, 18(5), 1223–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer, W.M. , Ru, L. , Jin, Y. , Patrick, J.W. & Ruan, Y.‐L. (2015) Tomato ovary‐to‐fruit transition is characterized by a spatial shift of mRNAs for cell wall invertase and its inhibitor with the encoded proteins localized to sieve elements. Molecular Plant, 8, 315–328. [DOI] [PubMed] [Google Scholar]
- Park, C.H. , Yeo, H.J. , Park, Y.E. , Baek, S.A. , Kim, J.K. & Park, S.U. (2019) Transcriptome analysis and metabolic profiling of Lycoris Radiata . Biology (Basel), 8, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick, J.W. (1997) Phloem unloading: sieve element unloading and post-sieve element transport. Annual Review of Plant Physiology and Plant Molecular Biology, 48, 191–222. [DOI] [PubMed] [Google Scholar]
- Pi, E. , Zhu, C. , Fan, W. , Huang, Y. , Qu, L. , Li, Y. et al. (2018) Quantitative phosphoproteomic and metabolomic analyses reveal GmMYB173 optimizes flavonoid metabolism in soybean under salt stress. Molecular & Cellular Proteomics, 17, 1209–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh, D.A. , Offler, C.E. , Talbot, M.J. & Ruan, Y.‐L. (2010) Evidence for the role of transfer cells in the evolutionary increase of seed and fibre biomass yield in Cotton. Molecular Plant, 3, 1075–1086. [DOI] [PubMed] [Google Scholar]
- Quast, C. , Pruesse, E. , Yilmaz, P. , Gerken, J. , Schweer, T. , Yarza, P. et al. (2013) The SILVA ribosomal RNA gene database project: improved data processing and web‐based tools. Nucleic Acids Research, 41, D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, Y. , Yang, J. , Lu, B. , Jiang, Y. , Chen, H. , Hong, Y. et al. (2017) Structure of pigment metabolic pathways and their contributions to white tepal color formation of Chinese Narcissus tazetta var. chinensis cv Jinzhanyintai. International Journal of Molecular Sciences, 18, 1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, Z. , Lin, Y. , Lv, X. , Zhang, J. , Zhang, D. , Gao, C. et al. (2021) Clonal bulblet regeneration and endophytic communities profiling of Lycoris sprengeri, an economically valuable bulbous plant of pharmaceutical and ornamental value. Scientia Horticulturae, 279, 109856. [Google Scholar]
- Ren, Z. , Xia, Y. , Zhang, D. , Li, Y. & Wu, Y. (2017) Cytological analysis of the bulblet initiation and development in Lycoris species. Scientia Horticulturae, 218, 72–79. [Google Scholar]
- Ren, Z. , Xu, Y. , Lvy, X. , Zhang, D. , Gao, C. , Lin, Y. et al. (2021) Early sucrose degradation and the dominant sucrose cleavage pattern influence Lycoris sprengeri bulblet regeneration in vitro . International Journal of Molecular Sciences, 22, 11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland, F. , Moore, B. & Sheen, J. (2002) Sugar sensing and signaling in plants. Plant Cell, 14(Suppl), S185–S205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romo‐Pérez, M.L. , Weinert, C.H. , Häußler, M. , Egert, B. , Frechen, M.A. , Trierweiler, B. et al. (2020) Metabolite profiling of onion landraces and the cold storage effect. Plant Physiology and Biochemistry, 146, 428–437. [DOI] [PubMed] [Google Scholar]
- Ruan, Y.‐L. & Patrick, J.W. (1995) The cellular pathway of postphloem sugar‐transport in developing tomato fruit. Planta, 196, 434–444. [Google Scholar]
- Ruan, Y.‐L. (1993) Fruit set, young fruit and leaf growth of Citrus unshiu in relation to assimilate supply. Scientia Horticulturae, 53, 99–107. [Google Scholar]
- Ruan, Y.‐L. (2014) Sucrose metabolism: gateway to diverse carbon use and sugar signaling. Annual Review of Plant Biology, 65, 33–67. [DOI] [PubMed] [Google Scholar]
- Ruan, Y.‐L. (2022) CWIN‐sugar transporter nexus is a key component for reproductive success. Journal of Plant Physiology, 268, 153572. [DOI] [PubMed] [Google Scholar]
- Ruan, Y.-L. , Llewellyn, D.J. & Furbank, R.T. (2001) The control of single-celled cotton fibre elongation by developmentally reversible gating of plasmodesmata and coordinated expression of sucrose and K+ transporters and expansin. Plant Cell, 13, 47–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan, Y.‐L. , Llewellyn, D.J. , Liu, Q. , Xu, S. , Wu, L. , Wang, L. et al. (2008) Expression of sucrose synthase in the developing endosperm is essential for early seed development in cotton. Functional Plant Biology, 35, 382–393. [DOI] [PubMed] [Google Scholar]
- Ruan, Y.‐L. , Patrick, J.W. , Bouzayen, M. , Osorio, S. & Fernie, A.R. (2012) Molecular regulation of seed and fruit set. Trends in Plant Science, 17, 656–665. [DOI] [PubMed] [Google Scholar]
- Serin, E.A.R. , Nijveen, H. , Hilhorst, H.W.M. & Ligterink, W. (2016) Learning from co‐expression networks: possibilities and challenges. Frontiers in Plant Science, 7, 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahin, A. (2012) Generation and analysis of expressed sequence tags in the extreme large genomes Lilium and Tulipa . BMC Genomics, 13, 640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, T. , Yue, Y. , Shi, M. , Chen, M. , Yang, X. & Wang, L. (2019) Exploration of floral volatile organic compounds in six typical Lycoris taxa by GC‐MS. Plants (Basel), 8, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, A. & Desgagne‐Penix, I. (2017) Transcriptome and metabolome profiling of Narcissus pseudonarcissus ‘King Alfred’ reveal components of Amaryllidaceae alkaloid metabolism. Scientific Reports, 7, 17356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeekens, S. , Ma, J. , Hanson, J. & Rolland, F. (2010) Sugar signals and molecular networks controlling plant growth. Current Opinion in Plant Biology, 13, 274–279. [DOI] [PubMed] [Google Scholar]
- Smith, C.A. , Want, E.J. , O'Maille, G. , Abagyan, R. & Siuzdak, G. (2006) XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Analytical chemistry, 78, 779–787. [DOI] [PubMed] [Google Scholar]
- Sonnewald, U. & Fernie, A.R. (2018) Next‐generation strategies for understanding and influencing source‐sink relations in crop plants. Current Opinion in Plant Biology, 43, 63–70. [DOI] [PubMed] [Google Scholar]
- Sun, Q. , Zhang, B. , Yang, C. , Wang, W. , Xiang, L. , Wang, Y. et al. (2022) Jasmonic acid biosynthetic genes TgLOX4 and TgLOX5 are involved in daughter bulb development in tulip (Tulipa gesneriana). Horticultural Research, 9, uhac006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, H. , Dhonukshe, P. , Brewer, P.B. & Friml, J. (2006) Spatiotemporal asymmetric auxin distribution: a means to coordinate plant development. Cellular and Molecular Life Sciences, 63, 2738–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, H. (2013) Senescence, ageing and death of the whole plant. The New Phytologist, 197, 696–711. [DOI] [PubMed] [Google Scholar]
- van Es, S.W. , Muñoz‐Gasca, A. , Romero‐Campero, F.J. , González‐Grandío, E. , de los Reyes, P. , Tarancón, C. et al. (2020) A gene regulatory network critical for axillary bud dormancy directly controlled by Arabidopsis BRANCHED1. bioRxiv ,12, 394403. [DOI] [PubMed] [Google Scholar]
- Viola, R. , Roberts, A.G. , Haupt, S. , Gazzani, S. , Hancock, R.D. , Marmiroli, N. et al. (2001) Tuberization in potato involves a switch from apoplastic to symplastic phloem unloading. Plant Cell, 13, 385–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. & Ruan, Y. (2016) Critical roles of vacuolar invertase in floral organ development and male and female fertilities are revealed through characterization of GhVIN1‐RNAi cotton plants. Plant Physiology, 171, 405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. & Ruan, Y.L. (2012) New insights into roles of cell wall invertase in early seed development revealed by comprehensive spatial and temporal expression patterns of GhCWIN1 in cotton. Plant Physiology, 160, 777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Kartika, D. & Ruan, Y. (2020) Looking into ‘hair tonics’ for cotton fiber initiation. The New Phytologist, 229(4), 1844–1851. [DOI] [PubMed] [Google Scholar]
- Wang, R. , Xu, S. , Jiang, Y. , Jiang, J. , Li, X. , Liang, L. et al. (2013) De novo sequence assembly and characterization of Lycoris aurea transcriptome using GS FLX titanium platform of 454 pyrosequencing. PLoS One, 8, e60449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, R. , Xu, S. , Wang, N. , Xia, B. , Jiang, Y. & Wang, R. (2016) Transcriptome analysis of secondary metabolism pathway, transcription factors, and transporters in response to methyl jasmonate in Lycoris aurea . Frontiers in Plant Science, 7, 1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Yokosho, K. , Guo, R. , Whelan, J. , Ruan, Y. , Ma, J. et al. (2019) The soybean sugar transporter GmSWEET15 mediates sucrose export from the endosperm to early embryo. Plant Physiology, 180, 2133–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. , Su, X. , Tian, Z. , Liu, Y. , Zhou, Y. & He, M. (2018) Transcriptome profiling provides insights into dormancy release during cold storage of Lilium pumilum . BMC Genomics, 19, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart, D.S. , Tzur, D. , Knox, C. , Eisner, R. , Guo, A.C. , Young, N. et al. (2007) HMDB: the human metabolome database. Nucleic Acids Research, 35, D521–D526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J. , Wang, C. , Zheng, L. , Wang, L. , Chen, Y. , Whelan, J. et al. (2011) Ethylene is involved in the regulation of iron homeostasis by regulating the expression of iron‐acquisition‐related genes in Oryza sativa . Journal of Experimental Botany, 62, 667–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Y. , Ren, Z. , Gao, C. , Sun, M. , Li, S. , Min, R. et al. (2021) Change in sucrose cleavage pattern and rapid starch accumulation govern lily shoot‐to‐bulblet transition in vitro. Frontiers in Plant Science, 11, 564713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtzel, E.T. & Kutchan, T.M. (2016) Plant metabolism, the diverse chemistry set of the future. Science, 353, 1232–1236. [DOI] [PubMed] [Google Scholar]
- Xu, J. , Li, Q. , Yang, L. , Li, X. , Wang, Z. & Zhang, Y. (2020) Changes in carbohydrate metabolism and endogenous hormone regulation during bulblet initiation and development in Lycoris radiata . BMC Plant Biology, 20, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, L. & Huang, H. (2014) Genetic and epigenetic controls of plant regeneration. Current Topics in Developmental Biology, 108, 1–33. [DOI] [PubMed] [Google Scholar]
- Xu, L. , Yang, P. , Feng, Y. , Xu, H. , Cao, Y. , Tang, Y. et al. (2017) Spatiotemporal transcriptome analysis provides insights into bicolor tepal development in Lilium “Tiny Padhye”. Frontiers in Plant Science, 8, 398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, W. , Wu, X. , Li, Y. , Liu, G. , Cui, Z. , Jiang, T. et al. (2019) Cell wall invertase 3 affects cassava productivity via regulating sugar allocation from source to sink. Frontiers in Plant Science, 10, 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, F. , Li, C.‐h. , Das, D. , Zheng, Y. , Song, T. , Wang, L. et al. (2021) Comprehensive transcriptome and metabolic profiling of petal color development in Lycoris sprengeri . Frontiers in Plant Science, 12, 747131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, P. , Xu, L. , Xu, H. , Tang, Y. , He, G. , Cao, Y. et al. (2017) Histological and transcriptomic analysis during bulbil formation in Lilium lancifolium . Frontiers in Plant Science, 8, 1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Li, X.H. , Gao, Z. , Shen, S. , Liang, X.G. , Zhao, X. et al. (2017) Regulation of maize kernel weight and carbohydrate metabolism by abscisic acid applied at the early and middle post‐pollination stages in vitro . Journal of Plant Physiology, 216, 1–10. [DOI] [PubMed] [Google Scholar]
- Zhang, M. , Jiang, L. , Zhang, D. & Jia, G. (2015) De novo transcriptome characterization of Lilium ‘Sorbonne’ and key enzymes related to the flavonoid biosynthesis. Molecular Genetics and Genomics, 290, 399–412. [DOI] [PubMed] [Google Scholar]
- Zheng, Y. , Jiao, C. , Sun, H. , Rosli, H.G. , Pombo, M.A. , Zhang, P. et al. (2016) iTAK: a program for genome‐wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Molecular Plant, 9, 1667–1670. [DOI] [PubMed] [Google Scholar]
- Zhou, J. , Liu, Z. , Wang, S. , Li, J. , Li, Y. , Chen, W.K. et al. (2020) Fungal endophytes promote the accumulation of Amaryllidaceae alkaloids in Lycoris radiata . Environmental Microbiology, 22, 1421–1434. [DOI] [PubMed] [Google Scholar]
- Zhu, G. , Wang, S. , Huang, Z. , Zhang, S. , Liao, Q. , Zhang, C. et al. (2018) Rewiring of the fruit metabolome in tomato breeding. Cell, 172, 249–261. [DOI] [PubMed] [Google Scholar]
- Zhu, Z. , An, F. , Feng, Y. , Li, P. , Xue, L.A.M. , Jiang, Z. et al. (2011) Derepression of ethylene‐stabilized transcription factors (EIN3/EIL1) mediates jasmonate and ethylene signaling synergy in Arabidopsis. Proceedings of the National Academy of Sciences of the United States of America, 108, 12539–12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Flowchart of the experimental design, combined sequencing and bioinformatics pipeline.
Figure S2. Global heatmaps of inter‐species and intra‐species comparisons throughout VP.
Figure S3. k‐means clustering algorithum of DEGs in two Lycoris species.
Figure S4. KEGG and GO analysis of stage‐specific DEGs.
Figure S5. Analysis of the metabolome during VP in bulbs of Lycoris.
Figure S6. Phylogenetic tree of invertase (INV) in Lycoris and representative plants.
Figure S7. Phylogenetic tree of sucrose synthase (SUS) in Lycoris and representative plants.
Data S1. Quantitative detection of sugar metabolites.
Table S1. Summary of combined sequencing.
Table S2. GO and KEGG enrichment analysis of stage‐specific DEGs in both Lycoris species.
Table S3. Summary of metabolomics.
Table S4. Amino acid sequences of invertase genes.
Table S5. Amino acid sequences of sucrose synthase genes.
Table S6. Summary of co‐expression analysis.
Table S7. Primers used in a qRT‐PCR.
Data Availability Statement
The Illumina RNA‐Seq and PacBio Iso‐Seq raw data have been submitted to the Short Read Archive (SRA) data library under accession numbers PRJNA574869 and PRJNA579847.