

Abstract
We report an intramolecular conjugate addition/Truce‐Smiles/E1cb cascade of 2‐nitrobenzenesulfonamide‐functionalized cyclohexenones as a new entry to the core scaffold of monoterpene indole alkaloids. The method was applied to the asymmetric total synthesis of (−)‐limaspermidine, (−)‐kopsinilam, and (−)‐kopsinine, as well as the framework of the kopsifoline alkaloids, thus highlighting its complementarity to existing approaches involving the use of indole‐based starting materials or the interrupted Fischer indole synthesis. Furthermore, we show that the cascade tolerates various substituents on the nitroarene, opening the way to other natural products as well as non‐natural analogues.
Keywords: Alkaloids, Asymmetric Synthesis, Domino Reactions, Total Synthesis
2‐Nitrobenzenesulfonamides can be exploited as functional protective groups by their use in a long‐range nitroarene transfer cascade reaction. Using a suitably functionalized precursor, this process was used as the key step in the enantioselective total synthesis of the monoterpene indole alkaloids (−)‐limaspermidine, (−)‐kopsinilam, and (−)‐kopsinine as well as the unnatural alkaloid (−)‐tetra‐hydrokopsifoline D.

Monoterpene indole alkaloids (MIAs) constitute a very large class of natural products isolated from a wide range of flowering plants, most notably from the Kopsia, Aspidosperma, and Strychnos genera (Figure 1). MIAs often feature complex polycyclic molecular architectures as well as potent and diverse bioactivity, making them popular targets for total synthesis, dating back to Woodward's landmark synthesis of (±)‐strychnine. [1] (+)‐Limaspermidine (2) features the typical framework of Aspidosperma alkaloids which curiously have the opposite absolute configuration of the pentacyclic core compared to Strychnos and Kopsia alkaloids. Limaspermidine was first isolated in 1979 from A. rhombeosignatum by Di Genova, [2] but was already unwittingly synthesized by Ban in 1976 in their efforts towards aspidofractinine. [3] Since then, numerous elegant strategies have been reported. [4] (−)‐Kopsinine (3), first isolated in 1954 from K. longiflora by Michael, has demonstrated high antitussive properties. [5] Its complex cage‐like structure is characteristic for the Kopsia alkaloids. Although the first enantioselective synthesis by Magnus dates back to 1985, [6] its synthesis gained renewed interest in the past decade. [7] The kopsifolines [8] (including (−)‐kopsifoline D, 4) feature an alternatively bridged framework. [9]
Figure 1.

Selected Apocynaceae alkaloids, with the characteristic pentacyclic scaffold of the Kopsia and Aspidosperma alkaloids shown in green.
Biosynthetically, all MIAs are ultimately derived from strictosidine, the Pictet–Spengler product of tryptamine and the terpenoid aldehyde secologanin. [10] It is therefore hardly surprising that most synthetic approaches toward MIAs employ tryptamine (or a related indole derivative) as a starting point, often featuring a dearomative spirocyclization of the indole moiety as a key step. [11]
However, the intrinsic nucleophilicity of indoles may be incompatible with other key steps in the synthesis, leading to the development of various methods for the late‐stage introduction of the indole or indoline moiety, including the (interrupted) Fischer indole synthesis and the reductive cyclization of nitroarenes. Although the former approach presents a direct and frequently efficient method for late‐stage indolization, [12] it may suffer from regioselectivity issues or competing indole formation. The reductive cyclization of nitroarenes presents an interesting alternative to Fischer indolization.[ 4g , 13 ] However, also in this approach, the regioselective introduction of the nitroaryl moiety is far from trivial. Given our interest in the synthesis of indole alkaloids [14] and related compounds, [15] we became interested in new methods for the regioselective introduction of nitroaryl moieties to provide controlled access to polycyclic indole and indolenine frameworks.
In 2016, Canesi and co‐workers reported an intriguing cascade reaction of tyramine‐derived cyclohexadienones 5 bearing a 2‐nitrobenzenesulfonamide moiety (Scheme 1a). [16] This domino process involves conjugate addition of the sulfonamide moiety to the enone and subsequent Truce‐Smiles rearrangement of the resulting enolate to give nitrobenzyl ketone 7, which rapidly undergoes proton transfer, E1cb elimination, and conjugate addition on the opposite side of the ring to afford (racemic) tetrahydroindole derivatives 10. The latter reaction can be considered undesirable, as it prevents further elaboration of the cascade products to MIAs. We envisioned that the use of enone 11 as an alternative substrate would allow the cascade to be interrupted at the stage of aza‐decalin 12 or the ring‐opened product 13 (Scheme 1b). Moreover, the use of a chiral substrate in the key cascade reaction would allow control over the absolute stereochemistry in a unified approach to Kopsia and Aspidosperma alkaloids.
Scheme 1.

Conjugate addition/Truce‐Smiles cascade reaction.
Pentacycle 14 was considered as a key common intermediate in our retrosynthetic approach (Scheme 2). Construction of the final B and E rings was projected by reduction/condensation of the cascade product 15, followed by nucleophilic attack of the indole C3 position on the tethered chloroacetamide. Similar cyclization sequences have shown to be fully diastereoselective, making the formation of the C4 stereocenter critical in our enantioselective synthesis.[ 4b , 4e , 17 ] Instalment of this all‐carbon quaternary stereocenter was envisioned by Pd‐catalyzed enantioselective decarboxylative allylation as pioneered by Stoltz. [18] The resulting product 17 would be readily converted to the desired cascade precursor 16 by reduction, nosylation and Stork‐Danheiser transposition. [19] In turn, ketoester 18 can be readily derived from 1,3‐cyclohexadione (19).
Scheme 2.
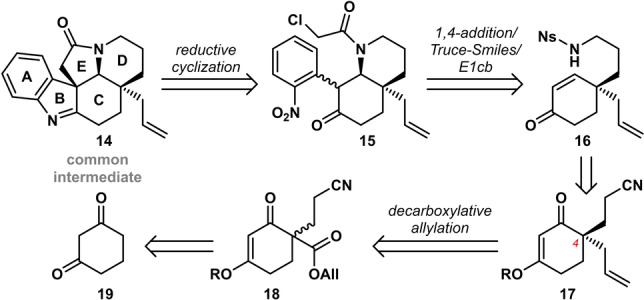
Retrosynthetic analysis of Kopsia and Aspidosperma alkaloids by a nitroaryl transfer cascade strategy. Ns=2‐nitrobenzenesulfonyl, All=allyl.
We started our synthetic approach with the protection of 19 under Dean–Stark conditions to provide the enol ether in near quantitative yield (Scheme 3). Acylation with allyl chloroformate, followed by Michael addition of the resulting ketoester to acrylonitrile furnished 18 in 78 % yield over three steps. Deracemization was accomplished by a palladium‐catalyzed decarboxylative allylation. [18] A brief optimization (Supporting Information Table S1) revealed the optimal conditions [2 mol% Pd(dba)2, 4 mol% (S)‐t‐BuPHOX (L3), Et2O, RT] furnishing 17 in 85 % yield with 91 % ee, even on >15 g scale. Conversion of optically enriched 17 into cascade precursor 16 proved to be challenging: simultaneous reduction of the ketone and nitrile moieties with LiAlH4 resulted in the undesired 1,4‐reduction of the α,β‐unsaturated ketone, contrary to earlier reports. [17a] Instead, DIBAL‐H proved suitable for selective 1,2‐reduction of the ketone moiety. Unfortunately, this procedure did not allow simultaneous reduction of the nitrile group. Thus, reduction of the ketone with DIBAL‐H followed by reduction of the nitrile with LiAlH4 gave us the desired alcohol and amine functionalities in product 20. Immediate nosylation of the amine with nosyl chloride under basic conditions and hydrolysis of the β‐hydroxyenol ether with aqueous HCl gave the cascade precursor 16 in 69 % yield.
Scheme 3.

Synthesis of cascade precursor 16. THF=tetrahydrofuran, dba=dibenzylideneacetone, Alloc=allyloxycarbonyl, DIBAL‐H=di‐isobutyl‐aluminum hydride.
With the cascade substrate in hand, we subjected it to the conditions reported by Canesi. [16] Disappointingly, refluxing 16 in acetonitrile in the presence of Cs2CO3 gave full conversion to a range of unisolable products. Testing various conditions (Supporting Information Table S2) indicated that the expected aza‐decalin product 23 is in equilibrium with the corresponding ring‐opened primary amine 24 as a result of the acidic nature of the proton between the ketone carbonyl and nitroarene (Scheme 4). As isolation of this dynamic mixture proved challenging, we opted to acylate the product in situ with chloroacetyl chloride. Performing the reaction in acetone proved key in this transformation, since poorly separable mixtures were formed in other solvents. This procedure finally allowed the isolation of a single product (25 a), albeit in moderate yield (48 %).
Scheme 4.

Nitroaryl transfer/acylation cascade reaction of 16.
To further investigate the scope of this cascade process, we also tested several functionalized bicyclic sulfonamides (26 a–h, Scheme 5). Because of a slightly different approach during the synthesis of these sulfonamides (see Supporting Information), we performed the scope study with the piperidine D ring already closed. A control experiment with the closed parent cascade substrate 26 a showed no difference in yield, suggesting that conjugate addition of the sulfonamide is not rate‐determining in the sequence. The introduction of a fluoro, chloro or methoxy substituent at the 4‐position (26 b,c,e) did not significantly change the outcome of the reaction. Notably, the halide‐substituted substrates 26 b–d were fully converted in 2–3 h (vs. 18 h for 26 a), while methoxy‐substituted 26 e required 72 h to reach full conversion. The strongly electron‐withdrawing trifluoromethyl group at the same position drastically decreased the efficiency (26 d). Substitution at the 3‐position either led to no conversion (26 f) or a range of side products (26 g). Plausibly, delocalization of the lone pairs on the methoxy oxygen atom in 26 f toward the nitro group considerably destabilizes the Meisenheimer intermediate in the Truce–Smiles rearrangement. The reaction with 2‐cyanobenzenesulfonamide 26 h did not show any conversion either.
Scheme 5.
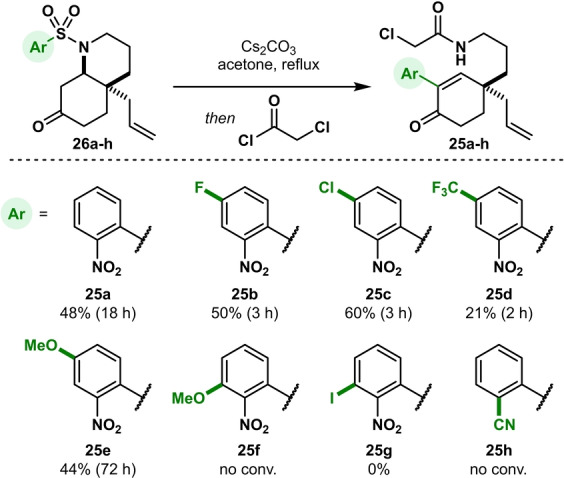
Scope of the cascade reaction.
To continue our efforts toward MIAs (Scheme 6), reduction of the nitro group in 25 a proceeded most efficiently with iron in acetic acid. Advantageously, this reduction/condensation proceeded with concomitant closure of the D ring, affording tetracycle 27 in 62 % yield as a single diastereomer, as corroborated by X‐ray crystallography. [20] Other reducing agents such as zinc or TiCl3 proved less effective. The E ring was closed in 81 % yield by nucleophilic attack of the indole on the chloroacetamide by a Finkelstein halide exchange followed by further activation with AgOTf. Indolenine 14 proved to be prone to degradation, and any further transformations had to be performed immediately afterward. As previously reported by Shao et al., [4e] all attempts to oxidatively cleave the alkene in 14 at this stage proved futile. Instead, we fully reduced indolenine 14 with LiAlH4 to give tertiary amine 28 in 99 % yield. Notably, 28 could be obtained from 27 in 89 % yield over two steps without intermediate purification of 14. Unfortunately, neither conventional ozonolysis nor Lemieux‐Johnson oxidation (both with reductive workup [NaBH4]) provided the desired product 2. Possibly, the basic nitrogen atoms in 28 are prone to oxidation, leading to a range of polar side products. Gratifyingly, conversion of 28 to the corresponding dihydrochloride followed by ozonolysis with reductive workup afforded (−)‐limaspermidine (ent‐ 2) in 59 % yield.
Scheme 6.

Total syntheses of (−)‐limaspermidine (ent‐ 2), (−)‐kopsinine (3), (−)‐kopsinilam (31), and tetrahydrokopsifoline D (33). THF=tetrahydrofuran, LDA=lithium diisopropylamide, DMAP=4‐dimethylaminopyridine, DMF=N,N‐dimethylformamide, HMPA=hexamethylphosphoramide, TFA=trifluoroacetic acid.
Routes towards (−)‐kopsinine and the kopsifoline core were also envisioned from indolenine 14. Carboxylation of similar indolenines using Mander's reagent was previously reported by Andrade in his synthesis of tabersonine. [21] The use of superstoichiometric amounts of base led to formation of a side product with an additional ester moiety at the α‐position of the lactam. Using one equivalent of base did not lead to full conversion of the starting indolenine, but exclusively gave the mono ester in 37 % yield (52 % based on recovered starting material). Boc protection of the indoline nitrogen then gave 29 in quantitative yield. Subsequent ozonolysis with reductive workup followed by Appel reaction cleanly afforded iodide 30. Formation of the bicyclo[2.2.2]octane fragment was efficiently achieved using a SmI2‐mediated diastereoselective free radical cyclization, as previously demonstrated by Boger on a similar substrate.[ 7c , 7e ] Boc deprotection then afforded (−)‐kopsinilam (31). The total synthesis of (−)‐kopsinine (3) was completed by converting 31 to the thiolactam followed by reduction with Raney nickel.[ 6 , 7c ]
Interestingly, when we deprotected iodide 30 and subjected it to slightly basic workup, we observed partial conversion to the bicyclo[1.2.3]octane framework of the kopsifoline alkaloids. We further optimized this transformation by treating the crude deprotected iodide with iPr2NEt. Reduction of the resulting indolenine to indoline 32 with excess NaBH4 surprisingly also reduced the methyl ester to the primary alcohol for a substantial part. To reach the target unnatural alkaloid tetrahydrokopsifoline D (33), [22] we again reduced the γ‐lactam to the analogous pyrrolidine via the thioamide.
In conclusion, we demonstrated the asymmetric total synthesis of three distinct natural product families from a single common intermediate which was prepared via a fully regioselective nitroaryl transfer cascade reaction. Importantly, this cascade approach allows for late‐stage variation by using differently substituted nitroarenesulfonamide precursors.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Acknowledgements
This work was funded by the Dutch Research Council (NWO). We kindly acknowledge Elwin Janssen for NMR measurements, Daniel Preschel for HRMS measurements, and Dr. Jasper Biemolt and Marushka Tirion for experimental work (all VUA). Dr. Christophe M. L. Vande Velde (University of Antwerp) is gratefully acknowledged for advice in solving the crystal structure.
Dedicated to Prof. Dr. Henk Hiemstra on the occasion of his 70th birthday.
B. Horst, D. S. Verdoorn, S. Hennig, G. van der Heijden, E. Ruijter, Angew. Chem. Int. Ed. 2022, 61, e202210592; Angew. Chem. 2022, 134, e202210592.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Woodward R. B., Cava M. P., Ollis W. D., Hunger A., Daeniker H. U., Schenker K., J. Am. Chem. Soc. 1954, 76, 4749–4751. [Google Scholar]
- 2. Medina J. D., Di Genova L., Planta Med. 1979, 37, 165–167. [Google Scholar]
- 3. Ban Y., Honma Y., Ohnuma T., Heterocycles 1976, 5, 47–51. [Google Scholar]
- 4.Racemic syntheses:
- 4a. Overman L. E., Robertson G. M., Robichaud A. J., J. Am. Chem. Soc. 1991, 113, 2598–2610; [Google Scholar]
- 4b. Tan S. H., Banwell M. G., Willis A. C., Reekie T. A., Org. Lett. 2012, 14, 5621–5623; [DOI] [PubMed] [Google Scholar]
- 4c. Guérard K. C., Guérinot A., Bouchard-Aubin C., Ménard M. A., Lepage M., Beaulieu M. A., Canesi S., J. Org. Chem. 2012, 77, 2121–2133; [DOI] [PubMed] [Google Scholar]
- 4d. Jin J., Qiu F. G., Adv. Synth. Catal. 2014, 356, 340–346. Enantioselective syntheses: [Google Scholar]
- 4e. Zhang S.-X., Shen X. L., Li Z. Q., Zou L. W., Wang F. Q., Bin Zhang H., Shao Z. H., J. Org. Chem. 2013, 78, 11444–11449; [DOI] [PubMed] [Google Scholar]
- 4f. Du J. Y., Zeng C., Han X. J., Qu H., Zhao X. H., An X. T., Fan C. A., J. Am. Chem. Soc. 2015, 137, 4267–4273; [DOI] [PubMed] [Google Scholar]
- 4g. Ren W., Wang Q., Zhu J., Angew. Chem. Int. Ed. 2016, 55, 3500–3503; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 3561–3564; [Google Scholar]
- 4h. White K. L., Movassaghi M., J. Am. Chem. Soc. 2016, 138, 11383–11389; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4i. Pritchett B. P., Donckele E. J., Stoltz B. M., Angew. Chem. Int. Ed. 2017, 56, 12624–12627; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12798–12801; [Google Scholar]
- 4j. Martin G., Angyal P., Egyed O., Varga S., Soós T., Org. Lett. 2020, 22, 4675–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Crow W., Michael M., Aust. J. Chem. 1955, 8, 129–135; [Google Scholar]
- 5b. Tan M.-J., Yin C., Tang C.-P., Ke C.-Q., Lin G., Ye Y., Planta Med. 2011, 77, 939–944. [DOI] [PubMed] [Google Scholar]
- 6. Magnus P., Brown P., J. Chem. Soc. Chem. Commun. 1985, 184–186. [Google Scholar]
- 7.
- 7a. Jones S. B., Simmons B., Mastracchio A., MacMillan D. W. C., Nature 2011, 475, 183–188; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Harada S., Sakai T., Takasu K., Yamada K. I., Yamamoto Y., Tomioka K., Chem. Asian J. 2012, 7, 2196–2198; [DOI] [PubMed] [Google Scholar]
- 7c. Xie J., Wolfe A. L., Boger D. L., Org. Lett. 2013, 15, 868–870; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7d. Wu X., Huang J., Guo B., Zhao L., Liu Y., Chen J., Cao W., Adv. Synth. Catal. 2014, 356, 3377–3382; [Google Scholar]
- 7e. Lee K., Boger D. L., Tetrahedron 2015, 71, 3741–3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kam T.-S., Choo Y.-M., Helv. Chim. Acta 2004, 87, 991–998. [Google Scholar]
- 9.For representative total syntheses of the kopsifolines, see:
- 9a. Lee K., Boger D. L., J. Am. Chem. Soc. 2014, 136, 3312–3317; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Myeong S., Avci N. H., Movassaghi M., Org. Lett. 2021, 23, 9118–9122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dewick P. M., Medicinal Natural Products, 3 rd ed., Wiley, Chichester, 2009. [Google Scholar]
- 11. Saya J. M., Ruijter E., Orru R. V. A., Chem. Eur. J. 2019, 25, 8916–8935. [DOI] [PubMed] [Google Scholar]
- 12.See, e.g.:
- 12a. Ueda H., Satoh H., Matsumoto K., Sugimoto K., Fukuyama T., Tokuyama H., Angew. Chem. Int. Ed. 2009, 48, 7600–7603; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 7736–7739; [Google Scholar]
- 12b. Adams G. L., Carroll P. J., A. B. Smith III , J. Am. Chem. Soc. 2012, 134, 4037–4040; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12c. Smith J. M., Moreno J., Boal B. W., Garg N. K., J. Am. Chem. Soc. 2014, 136, 4504–4507; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12d. Moreno J., Picazo E., Morrill L. A., Smith J. M., Garg N. K., J. Am. Chem. Soc. 2016, 138, 1162–1165; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12e. Varga S., Angyal P., Martin G., Egyed O., Holczbauer T., Soós T., Angew. Chem. Int. Ed. 2020, 59, 13547–13551; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 13649–13653. [Google Scholar]
- 13.
- 13a. Kozmin S. A., Iwama T., Huang Y., Rawal V. H., J. Am. Chem. Soc. 2002, 124, 4628–4641; [DOI] [PubMed] [Google Scholar]
- 13b. Banwell M. G., Lupton D. W., Org. Biomol. Chem. 2005, 3, 213–215; [DOI] [PubMed] [Google Scholar]
- 13c. Xu Z., Wang Q., Zhu J., Angew. Chem. Int. Ed. 2013, 52, 3272–3276; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3354–3358; [Google Scholar]
- 13d. Ren W., Wang Q., Zhu J., Angew. Chem. Int. Ed. 2014, 53, 1818–1821; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1849–1852; [Google Scholar]
- 13e. Xu Z., Wang Q., Zhu J., J. Am. Chem. Soc. 2015, 137, 6712–6724; [DOI] [PubMed] [Google Scholar]
- 13f. Dagoneau D., Xu Z., Wang Q., Zhu J., Angew. Chem. Int. Ed. 2016, 55, 760–763; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 770–773; [Google Scholar]
- 13g. Eckermann R., Breunig M., Gaich T., Chem. Eur. J. 2017, 23, 3938–3949. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Saya J. M., Roose T. R., Peek J. J., Weijers B., de Waal T. J. S., Vande Velde C. M. L., Orru R. V. A., Ruijter E., Angew. Chem. Int. Ed. 2018, 57, 15232–15236; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 15452–15456; [Google Scholar]
- 14b. Faltracco M., Ruijter E., Org. Biomol. Chem. 2021, 19, 9641–9644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Graaff C., Bensch L., Boersma S. J., Cioc R. C., Van Lint M. J., Janssen E., Turner N. J., Orru R. V. A., Ruijter E., Angew. Chem. Int. Ed. 2015, 54, 14133–14136; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14339–14342. [Google Scholar]
- 16. Coulibali S., Godou T., Canesi S., Org. Lett. 2016, 18, 4348–4351. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Lawton G., Saxton J. E., Smith A. J., Tetrahedron 1977, 33, 1641–1653; [Google Scholar]
- 17b. Bergès J., García B., Muñiz K., Angew. Chem. Int. Ed. 2018, 57, 15891–15895; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 16118–16122; [Google Scholar]
- 17c. Xu H., Huang H., Zhao C., Song C., Chang J., Org. Lett. 2019, 21, 6457–6460. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Mohr J. T., Behenna D. C., Harned A. M., Stoltz B. M., Angew. Chem. Int. Ed. 2005, 44, 6924–6927; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 7084–7087; [Google Scholar]
- 18b. Liu Y., Liniger M., McFadden R. M., Roizen J. L., Malette J., Reeves C. M., Behenna D. C., Seto M., Kim J., Mohr J. T., Virgil S. C., Stoltz B. M., Beilstein J. Org. Chem. 2014, 10, 2501–2512; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18c. Cusumano A. Q., Stoltz B. M., Goddard W. A., J. Am. Chem. Soc. 2020, 142, 13917–13933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stork G., Danheiser R. L., J. Org. Chem. 1973, 38, 1775–1776. [Google Scholar]
- 20.Deposition Number 2180883 (for 27) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 21. Zhao S., Andrade R. B., J. Am. Chem. Soc. 2013, 135, 13334–13337. [DOI] [PubMed] [Google Scholar]
- 22.Boger named this compound “kopsifoline H”, representing a reduced version of kopsifoline D (see ref. [7e]). However, additional naturally occurring kopsifolines were later isolated from K. fruticose, including kopsifoline H: Long S., Li C., Hu J., Zhao Q., Chen D., Fitoterapia 2018, 129, 145–149.29935259 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.