

Abstract
Recombinant ADAMTS13 is currently undergoing clinical trials as a treatment for hereditary thrombotic thrombocytopenic purpura, a lethal microvascular condition resulting from ADAMTS13 deficiency. Preclinical studies have also demonstrated its efficacy in treating arterial thrombosis and inflammation without causing bleeding, suggesting that recombinant ADAMTS13 may have broad applicability as an antithrombotic agent. Despite this progress, we currently do not understand the mechanisms that regulate ADAMTS13 activity in vivo. ADAMTS13 evades canonical means of protease regulation because it is secreted as an active enzyme and has a long half‐life in circulation, suggesting that it is not inhibited by natural protease inhibitors. Although shear can spatially and temporally activate von Willebrand factor to capture circulating platelets, it is also required for cleavage by ADAMTS13. Therefore, spatial and temporal regulation of ADAMTS13 activity may be required to stabilize von Willebrand factor‐platelet strings at sites of vascular injury. This review outlines potential mechanisms that regulate ADAMTS13 in vivo including shear‐dependency, local inactivation, and biochemical and structural regulation of substrate binding. Recently published structural data of ADAMTS13 is discussed, which may help to generate novel hypotheses for future research.
Keywords: ADAMTS13, antithrombotic, protease regulation, thrombosis, VWF
1. INTRODUCTION
A disintegrin and metalloproteinase with thrombospondin motifs 13 (ADAMTS13) is the primary molecular regulator of von Willebrand factor (VWF) platelet‐binding activity (Figure 1). Dysregulation of the ADAMTS13 and VWF axis can lead to abnormal hemostasis in the form of bleeding or thrombosis. Much of the interest surrounding this interaction has focused on the shear‐dependent availability of VWF required for proteolysis by ADAMTS13, and on the changes in circulating ADAMTS13 levels during acute or chronic illnesses. However, the regulation of ADAMTS13 proteolytic activity during hemostasis remains unclear. Extracellular proteases are often regulated by balancing the rate of activation (or secretion) with the rate of inhibition to limit the spatial and temporal distribution of proteolytic activity in response to a stimulus. ADAMTS13 does not appear to be governed by these canonical mechanisms of protease regulation as it is secreted as an active enzyme and is resistant to natural protease inhibitors found in blood. ADAMTS13 is instead primed to cleave VWF whenever sufficient shear rates impose forces on VWF that expose its scissile bond. This raises the question: how does VWF recruit platelets to the site of blood vessel injury despite the presence of ADAMTS13? It is possible that VWF‐platelet string formation is simply governed by excess stoichiometry of VWF relative to ADAMTS13 1 combined with the mechanosensitive nature of VWF platelet‐binding activity. However, this mechanism would not allow for dynamic changes in VWF platelet‐capturing activity in the early stages of clot development compared to later stages and would leave ADAMTS13 as possibly the only protease in the cardiovascular system without a mechanism of regulation. Therefore, downregulation of ADAMTS13 at sites of vessel injury may contribute to VWF‐platelet string formation and the initial stages of clot formation.
FIGURE 1.
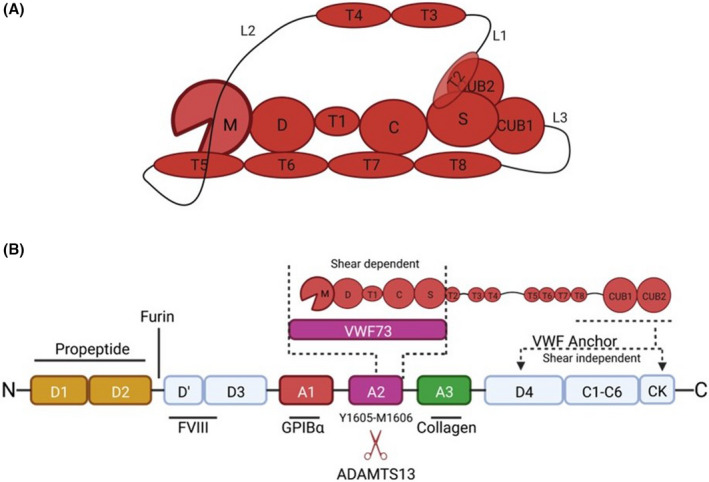
ADAMTS13 binding to VWF and domain organization. A, ADAMTS13 proximal domains include the metalloprotease domains (M), disintegrin‐like domain (D), type‐1 thrombospondin domain (T), cysteine‐rich domain (C), and spacer domain (S). The distal CUB domains are connected to the proximal domains through seven additional type‐1 thrombospondin domains and three linker regions, which provide ADAMTS13 with conformational flexibility. ADAMTS13 adopts a closed conformation in the absence of VWF binding in which the C‐terminal CUB domains bind to the VWF‐binding exosite on the spacer domain. The closed conformation is facilitated by inherent flexibility provided by the linker regions connecting T2‐T3, T4‐T5, and T8‐CUB1. B, ADAMTS13 binds to VWF in both a shear‐independent and a shear‐dependent mechanism. The CUB domains bind to the D4‐CK interval of VWF in a shear‐independent mechanism, which positions MDTCS near the A2 domain. This binding interaction facilitates localization of ADAMTS13 to VWF strings under flow. Upon shear‐activation of the substrate, the spacer, cysteine‐rich, and disintegrin‐like domains bind to the unfolded VWF A2 domain, stabilizing its denatured state and facilitating proteolysis of the Tyr1605‐Met1605 scissile bond by the metalloprotease domain. VWF73, comprising residues Asp1596‐Arg1668 of VWF, is a biochemical tool used to study ADAMTS13 activity in the absence of shear, and only engages the proximal MDTCS domains of ADAMTS13.
1.1. CLINICAL IMPORTANCE OF ADAMTS13 REGULATION
The role of ADAMTS13 in the cardiovascular system is inexorably linked to VWF. The importance of VWF regulation by ADAMTS13 is well‐established for thrombotic thrombocytopenic purpura (TTP) and von Willebrand disease (VWD). However, VWF and ADAMTS13 are now recognized as important contributors to a growing number of diseases and disorders in which vascular inflammation and thrombosis play a role, including cancer, atherosclerosis, sepsis, neurological conditions, and liver disease. 2 , 3 , 4 , 5 , 6 , 7 Therefore, although recombinant ADAMTS13 products continue to make progress in clinical trials for the treatment of hereditary TTP, additional therapeutic applications are also being explored. For example, recombinant ADAMTS13 infusion is beneficial in experimental models of stroke, 8 , 9 myocardial infarction, 10 colitis, 11 skin allograft, 12 thrombotic microangiopathy, 13 heparin‐induced thrombocytopenia, 14 renal disease, 15 and trauma‐induced organ failure. 16 Many of these conditions can be associated with VWF‐dependent inflammation or microvascular thrombosis. Although these studies have not reported increased bleeding risk following recombinant ADAMTS13 administration, this potential adverse effect requires further investigation.
ADAMTS13 is relatively incapable of cleaving VWF unless sufficient shear rates are present to impose enough force on VWF to expose its cryptic scissile bond. 17 This shear‐dependency protects circulating VWF from indiscriminate degradation by ADAMTS13 and may help to explain the low rates of bleeding in experimental animal models following ADAMTS13 infusion. ADAMTS13 therefore exhibits a high degree of specificity for its substrate that distinguishes it from other thrombolytic agents like tissue plasminogen activator (tPA) and its derivatives. The capacity of tPA to catalyze plasmin generation is accelerated in the presence of fibrin, which helps to localize its activity to sites of thrombosis. However, tPA is also capable of catalyzing plasminogen activation on soluble fibrinogen, which can lead to systemic plasmin activity, acquired fibrinogen deficiency, and bleeding. 18 The lack of specificity for tPA limits its therapeutic benefit in the treatment of acute ischemic stroke because of a risk of bleeding. Several studies in experimental animal models have now shown that recombinant ADAMTS13 infusion can improve ischemic stroke recovery in mice receiving low tPA doses, without increasing the risk of bleeding. 19 , 20 , 21 , 22
Therefore, ADAMTS13 exhibits properties that are beneficial to the treatment of thrombosis and inflammation, without causing excessive bleeding. However, there is incomplete evidence for how ADAMTS13 activity is regulated in vivo. Addressing this knowledge gap is needed to understand how recombinant ADAMTS13 therapies can be improved for the treatment of hereditary TTP and other forms of thrombosis. 23 , 24
1.2. SHEAR‐DEPENDENT REGULATION
The antithrombotic activity of ADAMTS13 is partially controlled by the shear‐dependent availability of its substrate, VWF. Under physiological conditions, circulating VWF multimers exist primarily in a globular form. Under conditions of shear stress ranging from approximately 35–70 dyn/cm2, the attractive forces between monomers are overcome by drag and VWF transiently unravels and recompresses, which may allow for ADAMTS13 cleavage in circulation. 25 However, VWF cleavage by ADAMTS13 is more likely to occur at the vessel wall, where VWF is tethered and shear stresses are highest. Thus, ADAMTS13 cleaves VWF during secretion from endothelial cells to establish a hemostatically balanced distribution of multimer lengths, 26 , 27 and at sites of vascular injury where VWF‐platelet strings are adhered to subendothelial collagen. 28
The cleavage site for ADAMTS13 within the VWF A2 domain is cryptic under static conditions and is exposed when shear stresses exceed 10 pN of force across the domain. 25 , 27 A vicinal cysteine pair C1669/C1670 forms a disulfide bond that helps to stabilize the A2 domain, providing a narrow window of shear conditions conducive to unfolding. 28 , 29 This structural stabilization further limits the opportunity for ADAMTS13 to cleave VWF. Mutations in the A2 domain in some patients with type 2A VWD may destabilize the domain and increase its susceptibility to cleavage at lower shear thresholds, increasing the risk for bleeding in these patients. 30 , 31 Once unraveled, exosites in the ADAMTS13 spacer, cysteine‐rich, and disintegrin‐like domains bind to the VWF A2 domain to stabilize its denatured conformation and align the scissile bond to the active site within the metalloprotease domain. 32 Atomic force microscopy experiments have demonstrated that exposure of the scissile bond within the VWF A2 domain requires force thresholds comparable to those required to expose the high affinity platelet‐binding site within the VWF A1 domain. 33 Therefore, shear forces alone may not be sufficient to regulate the balance between VWF platelet‐binding activity and VWF cleavage by ADAMTS13. However, evidence from the literature suggests that ADAMTS13 can be downregulated by agonists generated at sites of vessel injury, which may promote VWF‐mediated platelet recruitment and hemostasis.
1.3. PHYSIOLOGIC REGULATION
ADAMTS13 is primarily expressed in hepatic stellate cells, 34 but is also expressed in endothelial cells, 35 podocytes, 36 astrocytes, and microglial cells. 37 Several studies have shown that ADAMTS13 expression can be downregulated in some cells by inflammatory cytokines, such as interleukin‐6 (IL‐6), IL‐1β, interferon‐γ, IL‐4, and tumor necrosis factor‐α. 36 , 38 , 39 Hepatic stellate cells can activate into a fibroblast‐like cell that deposits extracellular matrix in response to liver injury; however, this phenotype switch does not alter ADAMTS13 mRNA expression. 40 ADAMTS13 mRNA levels are reduced by ~50% in hepatic stellate cells exposed to interferon‐γ, IL‐4, or tumor necrosis factor‐α. 38 Because stellate cells contribute to the majority of ADAMTS13 in circulation, these changes in expression may contribute to reduced ADAMTS13 levels in patients with chronic 41 , 42 , 43 , 44 or acute inflammatory conditions 45 ; however, this remains to be formally investigated. Changes to ADAMTS13 clearance in various disease states has also not been adequately studied. It is known that macrophage scavenger receptor CD163 is capable of binding to fluorescently labeled recombinant ADAMTS13 and mediating its endocytosis in vitro. Whether this, or other clearance mechanisms, are important to regulating ADAMTS13 activity in vivo remains unknown.
ADAMTS13 contains N‐linked, O‐linked, and C‐linked glycans, which together contribute to 20% of its mass. 46 The N‐linked glycosylation and O‐fucosylation are critical for the proper folding and secretion of ADAMTS13. 47 Once secreted into plasma, the removal of N‐glycans does not alter the proteolytic function of ADAMTS13. 47 However, the effects of O‐linked and C‐linked glycosylation on proteolytic function remain unclear. In addition to proper folding and secretion, the glycosylation of ADAMTS13 may be important in immune recognition and clearance. 46 Introduction of N‐glycans at position K608 reduces the binding of autoantibodies to the spacer domain, without altering proteolytic function. 48 This finding suggests that glycosylation may modulate susceptibility to TTP and optimizing glycan structures on recombinant ADAMTS13 products may be useful for developing improved therapeutics to treat immune TTP.
ADAMTS13 is constitutively secreted as an active protease and has an usually long circulating half‐life of several days. ADAMTS13 half‐life following plasma infusion was recently reported between 3.4 and 7.9 days (median 5.4 days), 24 whereas recombinant human ADAMTS13 half‐life was between 2 and 3 days. 23 Because plasma contains high concentrations of natural protease inhibitors known to inhibit members of the ADAMTS protease family, 49 these observations provide indirect evidence that ADAMTS13 is resistant to natural protease inhibitors in circulation. Direct evidence for ADAMTS13 resistance to natural protease inhibitors has been reported as well, 50 demonstrating its resistance to alpha‐2 macroglobulin, the 4 TIMP isoforms, as well as small molecule inhibitors of metalloproteases. These observations have led several groups to hypothesize that ADAMTS13 exists in a latent conformation and becomes activated in the presence of its substrate, VWF. 51 , 52 The biochemical and structural features of ADAMTS13 that contribute to its resistance to inhibition while maintaining activity toward its substrate are currently unknown. Understanding this unique property of ADAMTS13 may reveal novel mechanisms of protease regulation that have not previously been defined.
Although ADAMTS13 is not directly regulated by natural protease inhibitors, its capacity to cleave VWF can be attenuated by other physiological agonists. Platelet factor 4, 53 alpha defensins, 54 IL‐6, 55 thrombospondin‐1, 56 and hemoglobin 57 have all been shown to bind VWF and directly compete with ADAMTS13 docking. Nazy et al. 53 demonstrated that platelet factor 4 binds to the human VWF A2 domain, directly blocking proteolysis by ADAMTS13 in a purified static system. However, Johnston et al. 14 demonstrated that platelet factor 4 did not attenuate the capacity of ADAMTS13 to cleave VWF strings released from endothelial cells in vitro in a flow chamber model, or block the antithrombotic activity of ADAMTS13 in a mouse model of heparin‐induced thrombocytopenia. Similarly, although human neutrophil peptides bind to the VWF A2 domain and block its proteolysis by ADAMTS13 with an IC50 value of 45 μM, this concentration is not likely to be achieved in circulation. 54 Whether PF4 or human neutrophil peptides are present at sufficiently high concentrations locally at sites of vascular injury to exert a transient protection of VWF from ADAMTS13 cleavage remains unknown.
1.4. PROTEOLYTIC MODIFICATION
Proteolytic degradation of ADAMTS13 may contribute to regulating its activity at sites of vascular injury. Degraded forms of ADAMTS13 have been observed in plasma collected from patients with sepsis‐induced disseminated intravascular coagulation and in patients experiencing acute episodes of TTP. 58 , 59 Proteases generated by the coagulation and fibrinolytic system or those released by activated immune cells can be present in high concentrations at sites of vascular injury. Biochemical studies have demonstrated that thrombin, plasmin, factors Xa and XIa, and neutrophil elastase cleave ADAMTS13 in vitro in a time‐ and concentration‐dependent manner. 58 , 60 , 61 , 62 Degradation of ADAMTS13 by these proteases primarily removes the C‐terminal CUB domains from ADAMTS13, leaving the proximal domains MDTCS mostly intact. 61 Plasmin can also cleave VWF, and increasing plasmin generation by infusing recombinant tPA has been shown to partially compensate for ADAMTS13 deficiency in a mouse model of thrombotic microangiopathy. 63 However, endogenous plasmin generation is not capable of downregulating ultra‐large VWF multimers during acute episodes of TTP, suggesting that this pathway does not meaningfully contribute to VWF regulation.
Crawley et al. 60 found that thrombin‐ and plasmin‐mediated cleavage of ADAMTS13 reduces its activity toward purified human VWF under static conditions in vitro. Garland et al. 61 observed that ADAMTS13 degraded by thrombin or factor XIa has an impaired ability to process VWF on the surface of endothelial cells in conditions of flow. These observations are consistent with several in vivo studies showing that the ADAMTS13 CUB domains are essential for regulation of VWF‐platelet string formation in mice. 64 , 65 , 66 For example, unlike other mouse strains like 129/Sv, the C57BL/6 mouse strain express a truncated form of ADAMTS13 lacking the C‐terminal TSP1‐7 to the CUB2 domains because of the insertion of an intracisternal A‐particle retrotransposon into intron 23 of the Adamts13 gene. 67 , 68 As a consequence, C57Bl/6 mice exhibit more rapid occlusion time than 129/Sv in response to ferric chloride injury to mesenteric arterioles and increased stability of VWF‐platelet strings at high shear rates (5000 s−1). 67 De Maeyer et al. 69 found that infusion of an ADAMTS13 variant lacking the CUB domains was 3‐fold less effective than wild‐type ADAMTS13 at cleaving VWF‐platelet strings in a model of ferric chloride injury to the mesenteric vein. These studies suggest that the removal of CUB domains by activated proteases may serve to downregulate ADAMTS13 activity at sites of vascular injury, stabilize VWF‐platelet strings, and promote clot formation.
1.5. ADAMTS13 CONFORMATIONAL DYNAMICS
ADAMTS13 is known to adopt both an open and closed conformation; however, the significance of these conformational changes to its regulation remains poorly understood. The closed conformation occurs as a consequence of CUB1 and CUB2 interactions with the spacer domain. 70 , 71 This interaction is likely facilitated by inherent flexibility within the thrombospondin type‐1 (TSP1) repeats, especially within linker regions following TSP1‐4 and TSP1‐8. 70 , 72 , 73 , 74 Recent crystal structures of MDTCS and CUB1‐2 enabled molecular docking simulations to predict potential points of contact that maintain ADAMTS13 in a closed conformation. These include: (1) spacer domain residues E634 and D635 binding CUB1 residues K1252 and R1272; (2) spacer loop R660‐Y665 binding to CUB2 pocket formed by residues R1326, R1361, E1387, and E1389; (3) spacer residues L591, F592, and L637 with CUB1 residues W1245, L1248, and W1250; (4) spacer domains residue Y664 and CUB2 residue R1326; and (5) a salt bridge between spacer residue R568 with CUB2 residue E1389. 75 Many of these residues overlap with the binding site for the VWF A2 domain on the spacer domain resulting in competition between the CUB domains and biochemical substrates of ADAMTS13 such as VWF73. However, this exosite on the spacer domain site is expected to be unmasked when the ADAMTS13 CUB domains bind VWF strings in circulation or at the vessel wall. Therefore, the contribution of the closed conformation to the regulation of VWF cleavage by ADAMTS13 in vivo remains unclear.
Various groups have investigated the consequence of open and closed conformation on ADAMTS13 activity. Zheng et al. and South et al. used mutagenesis to disrupt the interaction between the spacer and CUB domains and showed ADAMTS13 was 2‐ to 3‐fold faster at cleaving VWF73, consistent with the closed conformation masking exosites on the spacer domain. 76 , 77 Roose et al. 78 showed that antibodies binding to the CUB or spacer domains can arise in patients during acute episodes of TTP and results in increased ADAMTS13 activity. Recently, Schelpe et al. demonstrated that an antibody binding to the spacer domain enhanced the activity of ADAMTS13 through a k cat effect and not a K M effect, suggesting long range allostery is possible between distal exosites of ADAMTS13 and the metalloprotease domain. 79 Acidic pH disrupts the closed conformation of ADAMTS13 and contributes to increased ADAMTS13 activity toward VWF73. 80 , 81 Muia et al. performed an elegant phylogenetic analysis of ADAMTS13 from several vertebrate species and demonstrated that TSP1‐7 and TSP1‐8 are critical to ADAMTS13 allostery. 70 Despite extensive characterization of the sequence determinants of ADAMTS13 conformation, the overall magnitude of effect on proteolytic activity toward VWF73 remains a modest 2‐ to 5‐fold. Currently, there is no compelling evidence to suggest that the reduced activity toward VWF73 caused by the closed conformation is a meaningful mechanism of ADAMTS13 regulation. Whether additional features of ADAMTS13 conformation contribute to its regulation at a greater magnitude remains to be determined.
New insights into the intramolecular contacts that help to maintain a closed conformation are now possible because of the recently released AlphaFOLD2 structure prediction of ADAMTS13. 82 , 83 This model predicts contact points between: (1) TSP1‐8 and the cysteine‐rich domain, (2) CUB2 and the disintegrin‐like domain, (3) TSP1‐7 and the disintegrin‐like domain, and (4) TSP1‐5 and the calcium‐binding loop in the metalloprotease domain (Figure 2). These inter‐domain interactions result in TSP1‐5 to TSP1‐8 aligning with the interface of MDTCS predicted to bind to the shear‐unfolded VWF A2 domain, which could mask substrate access to the metalloprotease domain when ADAMTS13 is folded in its closed conformation. This model provides some evidence that the closed conformation confers global latency to ADAMTS13, which contributes to the ability of ADAMTS13 to circulate as an active protease without exhibiting off‐target proteolysis. However, some predictions from this model are inconsistent with previous mutagenesis and molecular docking simulations, especially the binding site for the CUB domains on the spacer domain and the domain boundaries of the Tsp1 repeats that link the CUB domains to the spacer domain. Therefore, some caution is required when interpreting the AlphaFOLD2 model. Despite these limitations, this structure simulation may help generate new hypotheses to explore the mechanism of ADAMTS13 conformational dynamics, and potential implications for protease regulation.
FIGURE 2.
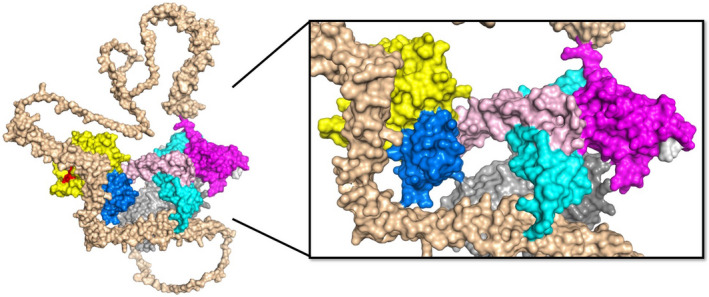
AlphaFOLD2 prediction of full length ADAMTS13 structure. The predicted three‐dimensional structure for full‐length human ADAMTS13 was obtained from AlphaFOLD2 (https://alphafold.ebi.ac.uk/). The domains are indicated as follows: metalloprotease (yellow), disintegrin‐like (blue), TSP1‐1 (rose), cysteine‐rich (cyan), spacer (magenta), TSP1‐2 to TSP1‐8 (wheat), CUB1 (light gray), CUB2 (dark gray). The RRY motif within the spacer domains (white) is primarily associated with autoantibodies in patients with immune TTP and partially occupies the suspected binding site for the CUB domains, but this intramolecular interaction is not represented in this model. Long linker regions extend outward following TSP1‐4 and TSP1‐8, providing flexibility to ADAMTS13 that may facilitate interdomain contacts that promote a closed conformation and confer global latency. This model predicts multiple long‐range interactions between distal TSP1 domains and proximal metalloprotease, disintegrin‐like, and cysteine‐rich domains that may lead to novel hypotheses for the study of ADAMTS13 regulation. (PDB: AF‐Q76LX8‐F1)
1.6. ADAMTS13 RECOGNITION OF VWF
Several groups have provided insights into the domain contacts between ADAMTS13 and VWF that are important for regulating proteolysis (Figure 3). Newly published structural data of ADAMTS13 allows for a reinterpretation of these functional studies, which may help to uncover the structural determinants of ADAMTS13 substrate specificity and regulation. Although these studies highlight key VWF‐binding residues on ADAMTS13, they do not yet offer a complete picture of ADAMTS13 exosite composition. Understanding the key contact points between ADAMTS13 and VWF may enable new variants of ADAMTS13 to be engineered with improved substrate recognition properties and therapeutic potential.
FIGURE 3.
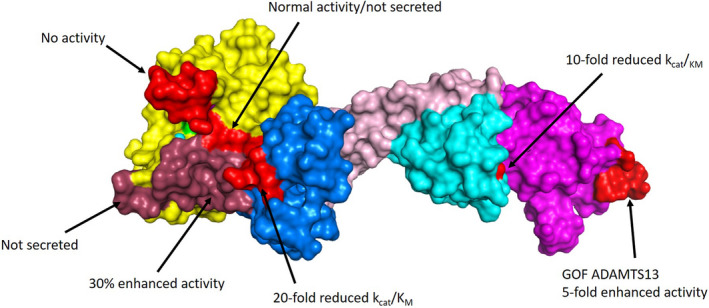
ADAMTS13 exosites. The crystal structure of MDTCS is shown, with domains indicated: metalloprotease (yellow), disintegrin‐like (blue), TSP1 (rose), cysteine‐rich (cyan), and spacer (magenta). The active site zinc (cyan) and catalytic glutamic acid (mutated to glutamine; green) are also indicated within the metalloprotease domain. The structure was obtained using a stabilizing antibody fragment to the metalloprotease domain (not shown). Compared with the AlphaFOLD2 model, this structure is lacking two loops in the cysteine‐rich domain that did not resolve (Asp453‐Tyr468 and I490‐K497). A summary of important mutagenesis work is indicated, with modified residues indicated in light red and dark red for clarity. These studies highlight important contact points for the VWF A2 domain to exosites on ADAMTS13. (PDB: 6qig)
The ADAMTS13 spacer domain is composed of a jelly roll fold of 10 β‐sheets that binds to the C‐terminus of the unfolded VWF A2 domain. 84 , 85 Removal of the spacer domain results in a 25‐fold reduction in the catalytic efficiency (kcat/KM) of VWF73 cleavage. 86 , 87 This binding site is primarily comprised of a hydrophobic cluster surrounded by positively charged arginine residues R636, R660, and R568. 88 These residues are also important for binding to the CUB domains when ADAMTS13 adopts a closed conformation 71 , 81 and are commonly associated with the epitope site of ADAMTS13 autoantibodies in immune TTP. 89 Mutation of these residues results in a 4‐fold increase in the rate of VWF73 cleavage because of disruption of the intramolecular interaction with the CUB domains, and may also exhibit improved antithrombotic activity in vivo. 85 Anti‐spacer domain antibodies that conformationally open ADAMTS13 can also increase the rate of VWF73 cleavage. These studies implicate the spacer domain as a primary exosite for ADAMTS13 recognition of VWF. The cysteine‐rich domain shares structural homology with the disintegrin‐like domain, and is stabilized by 6 disulfide bonds. 84 Deletion of the cysteine‐rich domain results in a 10‐fold reduction in catalytic efficiency for VWF73 cleavage. 90 Residues G471‐V474 forms a hydrophobic pocket that interacts with hydrophobic residues I1642, W1644, I1649, and I1651 on VWF73. 91 , 92 Optimizing the residues within the cysteine‐rich domain exosite may act synergistically with previously identified variants in the spacer domain to facilitate enhanced VWF cleavage, and should be explored in the future.
The disintegrin‐like domain helps present the VWF scissile bond to the active site and forms an extensive interface with the metalloprotease domain. 52 Deletion of the disintegrin‐like domain abolishes detectable proteolytic activity, which suggests that the metalloprotease domain alone does not possess sufficient binding activity to cleave VWF. 86 , 93 Residues P317‐L354 arrange into loops within the disintegrin‐like domain posterior to the active site cleft and are important for positioning VWF within the metalloprotease domain for proteolysis. 52 , 93 Mutations at R349, L350, or V352 reduced the catalytic efficiency of ADAMTS13 by f‐ to 20‐fold toward. 90 Residues L350 and V352 likely interact with A1612 on VWF, and an ionic interaction occurs between residue R349 in the disintegrin domain and D1614 on VWF. 84 , 93
Domain truncation and site‐directed mutagenesis studies assigned important roles to domains of ADAMTS13 that regulate its interaction with VWF. These observations are complemented by a high‐throughput mutagenesis phage display screen that mapped corresponding binding sites on VWF73 that engage with ADAMTS13. For example, mutations at VWF73 residues D1614, I1616, R1618, P1620, and D1622 substantially impaired proteolysis by ADAMTS13. 92 These residues correspond to a disordered loop in the VWF A2 domain that has alternating 180° orientation toward ADAMTS13, suggesting complementary binding sites on the disintegrin‐like domain that have not been completely described. Similarly binding sites on VWF73 were defined for the metalloprotease, cysteine‐rich, and spacer domains that suggest the binding sites for VWF on ADAMTS13 is not completely understood. These mutagenesis data may inform molecular docking simulations or more robust mutagenesis studies to define residues within ADAMTS13 that mediate substrate docking.
The sequential binding of VWF to these exosites on ADAMTS13 both stabilize the A2 domain in its unfolded state and present the scissile bond toward the active site within the metalloprotease domain. The active site of ADAMTS13 is formed by the zinc binding motif (H224, H228, and H234), the catalytic E225, and the S1 and S1′ pockets that flank the active site. 52 , 71 The calcium‐binding loop contributes two of the five residues that comprise the S1 pocket (L185 and V192), and lacks a stabilizing disulfide bridge between residues 184 and 190 found in many other ADAMTS proteases. 52 Therefore, the calcium‐binding loop may have inherent flexibility that contributes to substrate recognition. Mutagenesis of the calcium‐binding loop resulted in a 13‐fold reduction in the catalytic efficiency (kcat/KM), and changes to both kcat and KM, suggesting that it contributes both to the docking of VWF to ADAMTS13 and to substrate turnover. 94 Swapping calcium‐binding residues E184‐R193 with the corresponding region from ADAMTS1 or ADAMTS2 abolished ADAMTS13 activity, whereas point mutations in this region yielded a 2‐ to 10‐fold reduction in catalytic efficiency (k cat/K M). 95 Two additional calcium ions are located in a double calcium‐binding site, which stabilizes the extended disordered loop connecting the metalloprotease and disintegrin‐like domains. 52 The importance of these calcium ions to ADAMTS13 activity or regulation is not currently known. The variable loop (G236‐S263) flanks the catalytic motif across from the calcium‐binding loop. 52 This loop exists as an alpha helix in other ADAMTS protease family members, such as ADAMTS5 and ADAMTS1. However, swapping this loop with alpha helices from other ADAMTS proteases resulted in no change in ADAMTS13 proteolytic activity in vitro. 95
The active site of ADAMTS13 engages the P3‐P3′ residues of VWF (Leu1603‐Val‐Tyr‐Met‐Val‐Thr1609), with an additional requirement for Pro1601. 92 Substrate phage display techniques attempted to define other peptides cleaved by the ADAMTS13 active site in an effort to explore how active site specificity might regulate its activity (Figure 4). However, ADAMTS13 was not able to cleave any of the available 64 million peptides in the library, suggesting other features of VWF are required for substrates to gain access to the active site. Inserting this library into the P3‐P3′ interval of VWF73 resulted in the identification of 1670 cleaved peptide sequences. 51 The resulting substrate recognition motif for ADAMTS13 is dominated by a leucine at the P3 position, followed by large aliphatic residues at the remaining P2‐P3′ positions. Arginine is preferred at the P2 position and is also tolerated at the P1 position, 92 suggesting potential for ionic interactions with surface loops on the metalloprotease domain. These findings suggest that the active site of ADAMTS13 is latent until proximal exosite domains within MDTCS are engaged, permitting substrate access to the active site for subsequent proteolysis. The structural features of ADAMTS13 that govern the latency conversion has been speculated to involve a “gatekeeper triad” in which Asp217 and Asp252 ionic interactions with Arg 193. 52 Previous mutagenesis studies demonstrated that Arg193A exhibits a 4‐fold reduction in kcat/KM compared to wild‐type ADAMTS13, supporting a role for the gatekeeper triad in substrate recognition. 95 However, more work is needed to examine whether the gatekeeper triad contributes to ADAMTS13 latency and to precisely define which of the exosite‐engaging regions of VWF relieve the local latency of the metalloprotease domain.
FIGURE 4.
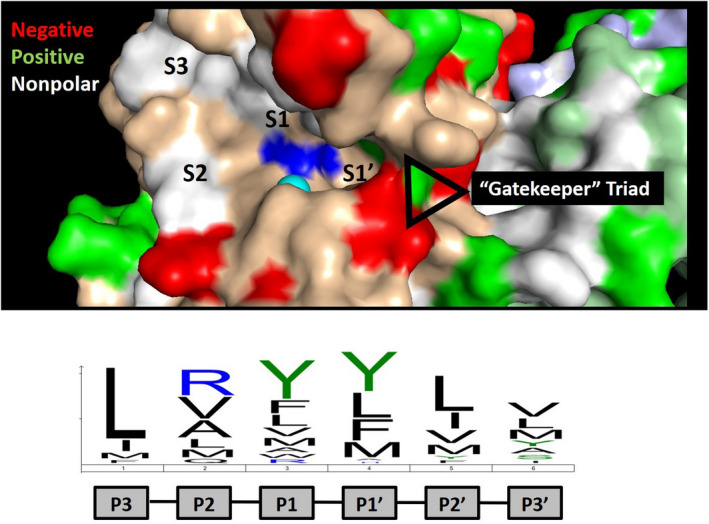
ADAMTS13 active site specificity. The active site of ADAMTS13 is shown with the catalytic zinc (cyan) and glutamic acid (mutated to glutamine; blue) indicated. The active site specificity motif determined by substrate phage display is indicated below, which is dominated by leucine at position P3 and bulky aliphatic amino acids at positions P1 and P1′. 51 These residues are expected to bind to subsites and pockets within the active site, which are indicated. The crystal structure provides evidence of a “gatekeeper” triad at the entrance to the S1′ subsite comprised of Arg193, Asp217, and Asp252 that is predicted to limit access to the active site of ADAMTS13. This ionic interaction is likely overcome by VWF following processive docking to distal exosites, permitting access to the catalytic motif within the active site.
2. CONCLUDING REMARKS
ADAMTS13 circulates as an active protease capable of cleaving its only known substrate, VWF, whenever conditions of shear permit substrate activation. Although this mechanism dictates the conditions under which VWF is cleaved, it is not directly regulating ADAMTS13 protease activity. Autoimmune antibodies and inflammatory cytokines can downregulate systemic ADAMTS13 levels in diseased states, but do not provide spatial and temporal regulation of ADAMTS13 needed to help stabilize VWF‐platelet strings at sites of vessel injury. Localized mediators generated by the coagulation system or released by activated cells may contribute to the stabilization of VWF during hemostasis by attenuating ADAMTS13. Furthermore, some investigators have proposed that ADAMTS13 exists as a latent protease that undergoes zymogen‐to‐protease conversion through an allosteric mechanism when engaged with its substrate. 32 , 51 The contact points for ADAMTS13 onto the unfolded VWF A2 domain have been extensively mapped, 92 and the recently published crystal structure of ADAMTS13 protease domain and proximal exosites may help to uncover the mechanism of its latency conversion. As recombinant ADAMTS13 products continue their journey through clinical trials in the treatment of hereditary TTP and more effective variants of ADAMTS13 are being developed, 73 more work is needed to comprehensively define its role in the cardiovascular system and understand the mechanisms responsible for its regulation.
3. AUTHOR CONTRIBUTIONS
V.D.Y., K.S., and C.A.K. wrote and edited the manuscript.
5. CONFLICT OF INTEREST
All of the authors declare that there are no conflicts of interest.
4. ACKNOWLEDGMENTS
This work was supported by the Canadian Institutes of Health Research PJT‐168987 (C.A.K.) and Hamilton Health Sciences New Investigator Fund (C.A.K.). C.A.K. was the recipient of the McMaster University Department of Medicine Internal Career Award. K.S. was the recipient of the Ontario Graduate Scholarship and the CanVECTOR student scholarship.
DeYoung V, Singh K, Kretz CA. Mechanisms of ADAMTS13 regulation. J Thromb Haemost. 2022;20:2722‐2732. doi: 10.1111/jth.15873
Veronica DeYoung and Kanwal Singh contributed equally to this work.
Manuscript handled by: Karen Vanhoorelbeke
Final decision: Karen Vanhoorelbeke, 06‐Sep‐2022
REFERENCES
- 1. Robertson J, Lillicrap D, Von James PD. Willebrand disease. Pediatr Clin North Am. 2008;55:377‐392, viii‐ix. doi: 10.1016/j.pcl.2008.01.008 [DOI] [PubMed] [Google Scholar]
- 2. Chen X, Cheng X, Zhang S, Wu D. ADAMTS13: an emerging target in stroke therapy. Front Neurol. 2019;10:772. doi: 10.3389/fneur.2019.00772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Favaloro EJ, Henry BM, Lippi G. Increased VWF and decreased ADAMTS‐13 in COVID‐19: creating a milieu for (micro)thrombosis. Semin Thromb Hemost. 2021;47:400‐418. doi: 10.1055/s-0041-1727282 [DOI] [PubMed] [Google Scholar]
- 4. Groeneveld DJ, Poole LG, Luyendyk JP. Targeting von Willebrand factor in liver diseases: a novel therapeutic strategy? J Thromb Haemost. 2021;19:1390‐1408. doi: 10.1111/jth.15312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Katneni UK, Ibla JC, Hunt R, Schiller T, Kimchi‐Sarfaty C. von Willebrand factor/ADAMTS‐13 interactions at birth: implications for thrombosis in the neonatal period. J Thromb Haemost. 2019;17:429‐440. doi: 10.1111/jth.14374 [DOI] [PubMed] [Google Scholar]
- 6. Yang J, Wu Z, Long Q, et al. Insights into immunothrombosis: the interplay among neutrophil extracellular trap, von Willebrand factor, and ADAMTS13. Front Immunol. 2020;11:610696. doi: 10.3389/fimmu.2020.610696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ziliotto N, Bernardi F, Piazza F. Hemostasis components in cerebral amyloid angiopathy and Alzheimer's disease. Neurol Sci. 2021;42:3177‐3188. doi: 10.1007/s10072-021-05327-7 [DOI] [PubMed] [Google Scholar]
- 8. Denorme F, Langhauser F, Desender L, et al. ADAMTS13‐mediated thrombolysis of t‐PA‐resistant occlusions in ischemic stroke in mice. Blood. 2016;127:2337‐2345. doi: 10.1182/blood-2015-08-662650 [DOI] [PubMed] [Google Scholar]
- 9. South K, Denorme F, Salles C II, De Meyer SF, Lane DA. Enhanced activity of an ADAMTS‐13 variant (R568K/F592Y/R660K/Y661F/Y665F) against platelet agglutination in vitro and in a murine model of acute ischemic stroke. J Thromb Haemost. 2018;16:2289‐2299. doi: 10.1111/jth.14275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. de Meyer SF, Savchenko AS, Haas MS, et al. Protective anti‐inflammatory effect of ADAMTS13 on myocardial ischemia/reperfusion injury in mice. Blood. 2012;120:5217‐5223. doi: 10.1182/blood-2012-06-439935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zitomersky NL, Demers M, Martinod K, et al. ADAMTS13 deficiency worsens colitis and exogenous ADAMTS13 administration decreases colitis severity in mice. TH Open. 2017;1:e11‐e23. doi: 10.1055/s-0037-1603927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wong SL, Goverman J, Staudinger C, Wagner DD. Recombinant human ADAMTS13 treatment and anti‐NET strategies enhance skin allograft survival in mice. Am J Transplant. 2020;20:1162‐1169. doi: 10.1111/ajt.15703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Erpenbeck L, Demers M, Zsengellér ZK, et al. ADAMTS13 endopeptidase protects against vascular endothelial growth factor inhibitor‐induced thrombotic microangiopathy. J Am Soc Nephrol. 2016;27:120‐131. doi: 10.1681/ASN.2014121165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Johnston I, Sarkar A, Hayes V, et al. Recognition of PF4‐VWF complexes by heparin‐induced thrombocytopenia antibodies contributes to thrombus propagation. Blood. 2020;135:1270‐1280. doi: 10.1182/blood.2018881607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou S, Guo J, Zhao L, et al. ADAMTS13 inhibits oxidative stress and ameliorates progressive chronic kidney disease following ischaemia/reperfusion injury. Acta Physiol (Oxf). 2021;231:e13586. doi: 10.1111/apha.13586 [DOI] [PubMed] [Google Scholar]
- 16. Kleinveld DJB, Simons DDG, Dekimpe C, et al. Plasma and rhADAMTS13 reduce trauma‐induced organ failure by restoring the ADAMTS13‐VWF axis. Blood Adv. 2021;5:3478‐3491. doi: 10.1182/bloodadvances.2021004404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang X, Halvorsen K, Zhang CZ, Wong WP, Springer TA. Mechanoenzymatic cleavage of the ultralarge vascular protein von Willebrand factor. Science. 2009;324:1330‐1334. doi: 10.1126/science.1170905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barra ME, Feske SK, Sylvester KW, et al. Fibrinogen concentrate for the treatment of thrombolysis‐associated hemorrhage in adult ischemic stroke patients. Clin Appl Thromb Hemost. 2020;26:1076029620951867. doi: 10.1177/1076029620951867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cai P, Luo H, Xu H, et al. Recombinant ADAMTS 13 attenuates brain injury after intracerebral hemorrhage. Stroke. 2015;46:2647‐2653. doi: 10.1161/STROKEAHA.115.009526 [DOI] [PubMed] [Google Scholar]
- 20. Fan M, Xu H, Wang L, et al. Tissue plasminogen activator neurotoxicity is neutralized by recombinant ADAMTS 13. Sci Rep. 2016;6:25971. doi: 10.1038/srep25971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang L, Fan W, Cai P, et al. Recombinant ADAMTS13 reduces tissue plasminogen activator‐induced hemorrhage after stroke in mice. Ann Neurol. 2013;73:189‐198. doi: 10.1002/ana.23762 [DOI] [PubMed] [Google Scholar]
- 22. Xu H, Cao Y, Yang X, et al. ADAMTS13 controls vascular remodeling by modifying VWF reactivity during stroke recovery. Blood. 2017;130:11‐22. doi: 10.1182/blood-2016-10-747089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scully M, Knöbl P, Kentouche K, et al. Recombinant ADAMTS‐13: first‐in‐human pharmacokinetics and safety in congenital thrombotic thrombocytopenic purpura. Blood. 2017;130:2055‐2063. doi: 10.1182/blood-2017-06-788026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Taylor A, Vendramin C, Oosterholt S, Della Pasqua O, Scully M. Pharmacokinetics of plasma infusion in congenital thrombotic thrombocytopenic purpura. J Thromb Haemost. 2019;17:88‐98. doi: 10.1111/jth.14345 [DOI] [PubMed] [Google Scholar]
- 25. Springer TA. Biology and physics of von Willebrand factor concatamers. J Thromb Haemost. 2011;9(Suppl 1):130‐143. doi: 10.1111/j.1538-7836.2011.04320.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Batlle J, Lopez‐Fernandez MF, Lopez‐Borrasca A, et al. Proteolytic degradation of von Willebrand factor after DDAVP administration in normal individuals. Blood. 1987;70:173‐176. [PubMed] [Google Scholar]
- 27. Zimmerman TS, Dent JA, Ruggeri ZM, Nannini LH. Subunit composition of plasma von Willebrand factor. Cleavage is present in normal individuals, increased in IIA and IIB von Willebrand disease, but minimal in variants with aberrant structure of individual oligomers (types IIC, IID, and IIE). J Clin Invest. 1986;77:947‐951. doi: 10.1172/JCI112394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gogia S, Neelamegham S. Role of fluid shear stress in regulating VWF structure, function and related blood disorders. Biorheology. 2015;52:319‐335. doi: 10.3233/BIR-15061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang Q, Zhou YF, Zhang CZ, Zhang X, Lu C, Springer TA. Structural specializations of A2, a force‐sensing domain in the ultralarge vascular protein von Willebrand factor. Proc Natl Acad Sci U S A. 2009;106:9226‐9231. doi: 10.1073/pnas.0903679106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lynch CJ, Cawte AD, Millar CM, Rueda D, Lane DA. A common mechanism by which type 2A von Willebrand disease mutations enhance ADAMTS13 proteolysis revealed with a von Willebrand factor A2 domain FRET construct. PLoS One. 2017;12:e0188405. doi: 10.1371/journal.pone.0188405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lynch CJ, Lane DA, Luken BM. Control of VWF A2 domain stability and ADAMTS13 access to the scissile bond of full‐length VWF. Blood. 2014;123:2585‐2592. doi: 10.1182/blood-2013-11-538173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Crawley JT, de Groot R, Xiang Y, Luken BM, Lane DA. Unraveling the scissile bond: how ADAMTS13 recognizes and cleaves von Willebrand factor. Blood. 2011;118:3212‐3221. doi: 10.1182/blood-2011-02-306597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim J, Zhang CZ, Zhang X, Springer TA. A mechanically stabilized receptor‐ligand flex‐bond important in the vasculature. Nature. 2010;466:992‐995. doi: 10.1038/nature09295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou W, Inada M, Lee TP, et al. ADAMTS13 is expressed in hepatic stellate cells. Lab Invest. 2005;85:780‐788. doi: 10.1038/labinvest.3700275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Turner N, Nolasco L, Tao Z, Dong JF, Moake J. Human endothelial cells synthesize and release ADAMTS‐13. J Thromb Haemost. 2006;4:1396‐1404. doi: 10.1111/j.1538-7836.2006.01959.x [DOI] [PubMed] [Google Scholar]
- 36. Shen L, Lu G, Dong N, Ma Z, Ruan C. Simvastatin increases ADAMTS13 expression in podocytes. Thromb Res. 2013;132:94‐99. doi: 10.1016/j.thromres.2013.05.024 [DOI] [PubMed] [Google Scholar]
- 37. Tauchi R, Imagama S, Ohgomori T, et al. ADAMTS‐13 is produced by glial cells and upregulated after spinal cord injury. Neurosci Lett. 2012;517:1‐6. doi: 10.1016/j.neulet.2012.03.002 [DOI] [PubMed] [Google Scholar]
- 38. Cao WJ, Niiya M, Zheng XW, Shang DZ, Zheng XL. Inflammatory cytokines inhibit ADAMTS13 synthesis in hepatic stellate cells and endothelial cells. J Thromb Haemost. 2008;6:1233‐1235. doi: 10.1111/j.1538-7836.2008.02989.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Frentzou GA, Bradford C, Harkness KA, Haddock G, Woodroofe MN, Cross AK. IL‐1beta down‐regulates ADAMTS‐13 mRNA expression in cells of the central nervous system. J Mol Neurosci. 2012;46:343‐351. doi: 10.1007/s12031-011-9591-6 [DOI] [PubMed] [Google Scholar]
- 40. Velasco M, Saavedra T, Sepulveda C, Suarez M. Prolonged treatment with acyclovir in recurrent genital herpes. Clinical, virological, and immunological response. Rev Med Chil. 1991;119:876‐880. [PubMed] [Google Scholar]
- 41. Langholm LL, Rønnow SR, Sand JMB, et al. Increased von Willebrand factor processing in COPD, reflecting lung epithelium damage, is associated with emphysema, exacerbations and elevated mortality risk. Int J Chron Obstruct Pulmon Dis. 2020;15:543‐552. doi: 10.2147/COPD.S235673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moller C, Schutte AE, Smith W, Botha‐Le Roux S. Von Willebrand factor, its cleaving protease (ADAMTS13), and inflammation in young adults: the African‐PREDICT study. Cytokine. 2020;136:155265. doi: 10.1016/j.cyto.2020.155265 [DOI] [PubMed] [Google Scholar]
- 43. Taniguchi S, Hashiguchi T, Ono T, et al. Association between reduced ADAMTS13 and diabetic nephropathy. Thromb Res. 2010;125:e310‐e316. doi: 10.1016/j.thromres.2010.02.013 [DOI] [PubMed] [Google Scholar]
- 44. Zeng M, Chen Q, Liang W, He W, Zheng H, Huang C. Predictive value of ADAMTS‐13 on concealed chronic renal failure in COPD patients. Int J Chron Obstruct Pulmon Dis. 2017;12:3495‐3501. doi: 10.2147/COPD.S151983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Singh K, Kwong AC, Madarati H, et al. Characterization of ADAMTS13 and von Willebrand factor levels in septic and non‐septic ICU patients. PLoS One. 2021;16:e0247017. doi: 10.1371/journal.pone.0247017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Verbij FC, Stokhuijzen E, Kaijen PHP, van Alphen F, Meijer AB, Voorberg J. Identification of glycans on plasma‐derived ADAMTS13. Blood. 2016;128:e51‐e58. doi: 10.1182/blood-2016-06-720912 [DOI] [PubMed] [Google Scholar]
- 47. Zhou W, Tsai HM. N‐Glycans of ADAMTS13 modulate its secretion and von Willebrand factor cleaving activity. Blood. 2009;113:929‐935. doi: 10.1182/blood-2008-07-167775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Velásquez Pereira LC, Roose E, Graça NAG, et al. Immunogenic hotspots in the spacer domain of ADAMTS13 in immune‐mediated thrombotic thrombocytopenic purpura. J Thromb Haemost. 2021;19:478‐488. doi: 10.1111/jth.15170 [DOI] [PubMed] [Google Scholar]
- 49. Rose KWJ, Taye N, Karoulias SZ, Hubmacher D. Regulation of ADAMTS Proteases. Front Mol Biosci. 2021;8:701959. doi: 10.3389/fmolb.2021.701959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Guo C, Tsigkou A, Lee MH. ADAMTS13 and 15 are not regulated by the full length and N‐terminal domain forms of TIMP‐1, −2, −3 and −4. Biomed Rep. 2016;4:73‐78. doi: 10.3892/br.2015.535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kretz CA, Tomberg K, Van Esbroeck A, Yee A, Ginsburg D. High throughput protease profiling comprehensively defines active site specificity for thrombin and ADAMTS13. Sci Rep. 2018;8:2788. doi: 10.1038/s41598-018-21021-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Petri A, Kim HJ, Xu Y, et al. Crystal structure and substrate‐induced activation of ADAMTS13. Nat Commun. 2019;10:3781. doi: 10.1038/s41467-019-11474-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nazy I, Elliott TD, Arnold DM. Platelet factor 4 inhibits ADAMTS13 activity and regulates the multimeric distribution of von Willebrand factor. Br J Haematol. 2020;190:594‐598. doi: 10.1111/bjh.16553 [DOI] [PubMed] [Google Scholar]
- 54. Pillai VG, Bao J, Zander CB, et al. Human neutrophil peptides inhibit cleavage of von Willebrand factor by ADAMTS13: a potential link of inflammation to TTP. Blood. 2016;128:110‐119. doi: 10.1182/blood-2015-12-688747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bernardo A, Ball C, Nolasco L, Moake JF, Dong JF. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell‐derived ultralarge von Willebrand factor multimers under flow. Blood. 2004;104:100‐106. doi: 10.1182/blood-2004-01-0107 [DOI] [PubMed] [Google Scholar]
- 56. Novelli EM, Kato GJ, Hildesheim ME, et al. Thrombospondin‐1 inhibits ADAMTS13 activity in sickle cell disease. Haematologica. 2013;98:e132‐e134. doi: 10.3324/haematol.2013.092635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhou Z, Han H, Cruz M, López J, Dong JF, Guchhait P. Haemoglobin blocks von Willebrand factor proteolysis by ADAMTS‐13: a mechanism associated with sickle cell disease. Thromb Haemost. 2009;101:1070‐1077. [PubMed] [Google Scholar]
- 58. Feys HB, Vandeputte N, Palla R, et al. Inactivation of ADAMTS13 by plasmin as a potential cause of thrombotic thrombocytopenic purpura. J Thromb Haemost. 2010;8:2053‐2062. doi: 10.1111/j.1538-7836.2010.03942.x [DOI] [PubMed] [Google Scholar]
- 59. Ono T, Mimuro J, Madoiwa S, et al. Severe secondary deficiency of von Willebrand factor‐cleaving protease (ADAMTS13) in patients with sepsis‐induced disseminated intravascular coagulation: its correlation with development of renal failure. Blood. 2006;107:528‐534. doi: 10.1182/blood-2005-03-1087 [DOI] [PubMed] [Google Scholar]
- 60. Crawley JT, Lam JK, Rance JB, Mollica LR, O'Donnell JS, Lane DA. Proteolytic inactivation of ADAMTS13 by thrombin and plasmin. Blood. 2005;105:1085‐1093. doi: 10.1182/blood-2004-03-1101 [DOI] [PubMed] [Google Scholar]
- 61. Garland KS, Reitsma SE, Shirai T, et al. Removal of the C‐Terminal domains of ADAMTS13 by activated coagulation factor XI induces platelet adhesion on endothelial cells under flow conditions. Front Med (Lausanne). 2017;4:232. doi: 10.3389/fmed.2017.00232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lam JK, Chion CK, Zanardelli S, Lane DA, Crawley JT. Further characterization of ADAMTS‐13 inactivation by thrombin. J Thromb Haemost. 2007;5:1010‐1018. doi: 10.1111/j.1538-7836.2007.02514.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tersteeg C, de Maat S, de Meyer SF, et al. Plasmin cleavage of von Willebrand factor as an emergency bypass for ADAMTS13 deficiency in thrombotic microangiopathy. Circulation. 2014;129:1320‐1331. doi: 10.1161/CIRCULATIONAHA.113.006727 [DOI] [PubMed] [Google Scholar]
- 64. Feys HB, Anderson PJ, Vanhoorelbeke K, Majerus EM, Sadler JE. Multi‐step binding of ADAMTS‐13 to von Willebrand factor. J Thromb Haemost. 2009;7:2088‐2095. doi: 10.1111/j.1538-7836.2009.03620.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zanardelli S, Chion ACK, Groot E, et al. A novel binding site for ADAMTS13 constitutively exposed on the surface of globular VWF. Blood. 2009;114:2819‐2828. doi: 10.1182/blood-2009-05-224915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang P, Pan W, Rux AH, Sachais BS, Zheng XL. The cooperative activity between the carboxyl‐terminal TSP1 repeats and the CUB domains of ADAMTS13 is crucial for recognition of von Willebrand factor under flow. Blood. 2007;110:1887‐1894. doi: 10.1182/blood-2007-04-083329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Banno F, Chauhan AK, Kokame K, et al. The distal carboxyl‐terminal domains of ADAMTS13 are required for regulation of in vivo thrombus formation. Blood. 2009;113:5323‐5329. doi: 10.1182/blood-2008-07-169359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhou W, Bouhassira EE, Tsai HM. An IAP retrotransposon in the mouse ADAMTS13 gene creates ADAMTS13 variant proteins that are less effective in cleaving von Willebrand factor multimers. Blood. 2007;110:886‐893. doi: 10.1182/blood-2007-01-070953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. de Maeyer B, de Meyer SF, Feys HB, et al. The distal carboxyterminal domains of murine ADAMTS13 influence proteolysis of platelet‐decorated VWF strings in vivo. J Thromb Haemost. 2010;8:2305‐2312. doi: 10.1111/j.1538-7836.2010.04008.x [DOI] [PubMed] [Google Scholar]
- 70. Muia J, Zhu J, Greco SC, et al. Phylogenetic and functional analysis of ADAMTS13 identifies highly conserved domains essential for allosteric regulation. Blood. 2019;133:1899‐1908. doi: 10.1182/blood-2018-11-886275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. South K, Luken BM, Crawley JTB, et al. Conformational activation of ADAMTS13. Proc Natl Acad Sci USA. 2014;111:18578‐18583. doi: 10.1073/pnas.1411979112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hubbard AR, Heath AB, Kremer Hovinga JA, Subcommittee on von Willebrand, F . Establishment of the WHO 1st International Standard ADAMTS13, plasma (12/252): communication from the SSC of the ISTH. J Thromb Haemost. 2015;13:1151‐1153. doi: 10.1111/jth.12881 [DOI] [PubMed] [Google Scholar]
- 73. South K, Saleh O, Lemarchand E, et al. Robust thrombolytic and anti‐inflammatory action of a constitutively active ADAMTS13 variant in murine stroke models. Blood. 2022;139:1575‐1587. doi: 10.1182/blood.2021012787 [DOI] [PubMed] [Google Scholar]
- 74. Zhu J, Muia J, Gupta G, et al. Exploring the "minimal" structure of a functional ADAMTS13 by mutagenesis and small‐angle X‐ray scattering. Blood. 2019;133:1909‐1918. doi: 10.1182/blood-2018-11-886309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kim HJ, Xu Y, Petri A, Vanhoorelbeke K, Crawley JTB, Emsley J. Crystal structure of ADAMTS13 CUB domains reveals their role in global latency. Sci Adv. 2021;7:1‐12. doi: 10.1126/sciadv.abg4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. South K, Freitas MO, Lane DA. Conformational quiescence of ADAMTS‐13 prevents proteolytic promiscuity. J Thromb Haemost. 2016;14:2011‐2022. doi: 10.1111/jth.13445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zheng X, Nishio K, Majerus EM, Sadler JE. Cleavage of von Willebrand factor requires the spacer domain of the metalloprotease ADAMTS13. J Biol Chem. 2003;278:30136‐30141. doi: 10.1074/jbc.M305331200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Roose E, Vidarsson G, Kangro K, et al. Anti‐ADAMTS13 autoantibodies against cryptic epitopes in immune‐mediated thrombotic thrombocytopenic purpura. Thromb Haemost. 2018;118:1729‐1742. doi: 10.1055/s-0038-1669459 [DOI] [PubMed] [Google Scholar]
- 79. Schelpe AS, Petri A, Roose E, et al. Antibodies that conformationally activate ADAMTS13 allosterically enhance metalloprotease domain function. Blood Adv. 2020;4:1072‐1080. doi: 10.1182/bloodadvances.2019001375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. di Stasio E, Lancellotti S, Peyvandi F, Palla R, Mannucci PM, de Cristofaro R. Mechanistic studies on ADAMTS13 catalysis. Biophys J. 2008;95:2450‐2461. doi: 10.1529/biophysj.108.131532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Muia J, Zhu J, Gupta G, et al. Allosteric activation of ADAMTS13 by von Willebrand factor. Proc Natl Acad Sci USA. 2014;111:18584‐18589. doi: 10.1073/pnas.1413282112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583‐589. doi: 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Varadi M, Anyango S, Deshpande M, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein‐sequence space with high‐accuracy models. Nucleic Acids Res. 2022;50:D439‐D444. doi: 10.1093/nar/gkab1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Akiyama M, Takeda S, Kokame K, Takagi J, Miyata T. Crystal structures of the noncatalytic domains of ADAMTS13 reveal multiple discontinuous exosites for von Willebrand factor. Proc Natl Acad Sci USA. 2009;106:19274‐19279. doi: 10.1073/pnas.0909755106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jian C, Xiao J, Gong L, et al. Gain‐of‐function ADAMTS13 variants that are resistant to autoantibodies against ADAMTS13 in patients with acquired thrombotic thrombocytopenic purpura. Blood. 2012;119:3836‐3843. doi: 10.1182/blood-2011-12-399501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ai J, Smith P, Wang S, Zhang P, Zheng XL. The proximal carboxyl‐terminal domains of ADAMTS13 determine substrate specificity and are all required for cleavage of von Willebrand factor. J Biol Chem. 2005;280:29428‐29434. doi: 10.1074/jbc.M505513200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Gao W, Anderson PJ, Majerus EM, Tuley EA, Sadler JE. Exosite interactions contribute to tension‐induced cleavage of von Willebrand factor by the antithrombotic ADAMTS13 metalloprotease. Proc Natl Acad Sci USA. 2006;103:19099‐19104. doi: 10.1073/pnas.0607264104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Fang X, Lin J, Fang Y, Wu J. Prediction of spacer‐alpha6 complex: a novel insight into binding of ADAMTS13 with A2 domain of von Willebrand factor under forces. Sci Rep. 2018;8:5791. doi: 10.1038/s41598-018-24212-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Jin SY, Skipwith CG, Zheng XL. Amino acid residues Arg(659), Arg(660), and Tyr(661) in the spacer domain of ADAMTS13 are critical for cleavage of von Willebrand factor. Blood. 2010;115:2300‐2310. doi: 10.1182/blood-2009-07-235101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gao W, Anderson PJ, Sadler JE. Extensive contacts between ADAMTS13 exosites and von Willebrand factor domain A2 contribute to substrate specificity. Blood. 2008;112:1713‐1719. doi: 10.1182/blood-2008-04-148759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. de Groot R, Lane DA, Crawley JT. The role of the ADAMTS13 cysteine‐rich domain in VWF binding and proteolysis. Blood. 2015;125:1968‐1975. doi: 10.1182/blood-2014-08-594556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kretz CA, Dai M, Soylemez O, et al. Massively parallel enzyme kinetics reveals the substrate recognition landscape of the metalloprotease ADAMTS13. Proc Natl Acad Sci USA. 2015;112:9328‐9333. doi: 10.1073/pnas.1511328112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. de Groot R, Bardhan A, Ramroop N, Lane DA, Crawley JT. Essential role of the disintegrin‐like domain in ADAMTS13 function. Blood. 2009;113:5609‐5616. doi: 10.1182/blood-2008-11-187914 [DOI] [PubMed] [Google Scholar]
- 94. Gardner MD, Chion CKNK, de Groot R, Shah A, Crawley JTB, Lane DA. A functional calcium‐binding site in the metalloprotease domain of ADAMTS13. Blood. 2009;113:1149‐1157. doi: 10.1182/blood-2008-03-144683 [DOI] [PubMed] [Google Scholar]
- 95. de Groot R, Lane DA, Crawley JT. The ADAMTS13 metalloprotease domain: roles of subsites in enzyme activity and specificity. Blood. 2010;116:3064‐3072. doi: 10.1182/blood-2009-12-258780 [DOI] [PMC free article] [PubMed] [Google Scholar]