

Abstract
Background
In clinical oncology, systemic 5‐fluorouracil (5‐FU) and its oral pro‐drugs are used to treat a broad group of solid tumours. Patients with dihydropyrimidine dehydrogenase (DPD) enzyme deficiency are at elevated risk of toxicity if treated with standard doses of 5‐FU. DPYD genotyping and measurements of plasma uracil concentration (DPD phenotyping) can be applied as tests for DPD deficiency. In April 2020, the European Medicines Agency recommended pre‐treatment DPD testing to reduce the risk of 5‐FU‐related toxicity.
Objectives
The objective of this study is to present the current evidence for DPD testing in routine oncological practice.
Methods
Two systematic literature searches were performed following the PRISMA guidelines. We identified studies examining the possible benefit of DPYD genotyping or DPD phenotyping on the toxicity risk.
Findings
Nine and 12 studies met the criteria for using DPYD genotyping and DPD phenotyping, respectively.
Conclusions
The evidence supporting either DPYD genotyping or DPD phenotyping as pre‐treatment tests to reduce 5‐FU toxicity is poor. Further evidence is still needed to fully understand and guide clinicians to dose by DPD activity.
Keywords: acute, cancer chemotherapy, gene expression/regulation, pharmacokinetics, safety evaluation, SNPs, toxicity
1. INTRODUCTION
In this review, we describe the underlying clinical evidence for pre‐treatment use of DPYD genotyping and uracil measurements [U] prior to the systemic use of 5‐fluorouracil (5‐FU) capecitabine and tegafur (S‐1). The latter two are oral prodrugs that biotransform to 5‐fluorouracil (5‐FU). 1 These 5‐FU‐based fluoropyrimidines are in this review referred to as FP.
We aim to provide a short, focused overview of the use of DPYD genotyping and DPD phenotyping with relevance to clinical practice in oncology.
FPs are the cornerstone in the chemotherapeutic treatment of several solid tumours, including gastrointestinal and breast cancer. 2 FPs are antimetabolites that mimic pyrimidines and induces cytotoxic effects. 2 The FPs are approved for both monotherapy and in combination with other drugs—both cytotoxics and targeted agents. 3 It is estimated that approximately 600 000 patients are treated with systemic FP each year in Europe. 1
Systemic FP‐associated toxicities (FP‐TOX) include nausea, diarrhoea, vomiting, mucositis, neutropenia, and palmar‐plantar erythrodysesthesia (PPE). Severe toxicity (grades 3–5) is seen in 20–30% of treated patients. FP‐TOX can be fatal. 4 , 5
The dihydropyrimidine dehydrogenase (DPD) enzyme is the primary enzyme responsible for eliminating 5‐FU. 4 , 6 , 7 The DPD enzyme is encoded by the DPYD gene. 8 Since the 1980s, it has been known that patients with reduced uracil metabolism might be at increased risk of severe toxicity when exposed to standard doses of FP. 9 Several methods have been developed to assess the FP‐TOX risk in patients before systemic FP therapy. Ideally, this assessment should reduce the risk of severe FP‐TOX through pre‐treatment dose reduction. 10
In the spring of 2020, the European Medicines Agency (EMA) recommended preemptive testing for DPD deficiency in patients with an indication for treatment with systemic 5‐FU, capecitabine or tegafur. It was stated that patients with partial DPD deficiency should receive a lower dose of FP, and patients with complete DPD deficiency should avoid FP completely. 1 The United States Food and Drug Administration (FDA) has not issued recommendations along the lines of routine pre‐treatment testing for DPD deficiency.
EMA recommends that DPD testing is carried out by measuring the levels of uracil [U] (phenotype test) or DPYD genotyping. Both the DPYD genotype and phenotype tests have strengths and weaknesses. 11 Other more advanced methods exist, such as measuring the DPD activity in peripheral mononuclear cells. Despite being considered a more accurate method to determine the DPD enzyme activity, this method is too complex to implement in routine practice. 11
Several single nucleotide polymorphisms (SNPs) in the DPYD gene have been demonstrated to cause a clinically relevant decrease in the DPD enzyme activity. 12 , 13 A DPYD genotype test is cheap and reliable, and the results do not change over time. 12 , 14 Still, the test only examines the gene for a limited number of known variants and cannot identify rare variants that may hamper the DPD activity.
Measurement of the endogenous metabolite, uracil [U], which is metabolized by DPD, is the most frequently used phenotypic approach. The level may vary depending on preanalytical conditions because [U] concentration is affected by food intake and the circadian rhythm. Furthermore, the [U] concentration is unstable after sampling, and blood must be centrifuged immediately after drawing the sample. 15 , 16 , 17 The [U] concentration is also affected by kidney function with higher values observed in patients with end stage renal desease. 18 A recent study reported significant between‐centre differences in the [U], underlining that measurement of uracil is sensitive to preanalytical conditions. 19
We performed a systematic literature search on the use of DPYD genotyping and DPD phenotyping, focusing on the risk of FP‐TOX in cancer patients.
2. METHODS
We performed two literature searches (genotype and phenotype) following the Preferred Reporting Items for Systematic Reviews and META‐analyses (PRISMA) guideline. 20 PubMed (Medline) and EMBASE (Exerpta Medica, Elsevier; Ovid) were used for both searches. The search was carried out by a consultant and professor in clinical pharmacology [PD] and a junior researcher (medical doctor) [NHP].
The first author [NHP], [PSE] and a clinical pharmacologist [MRH], all medical doctors, screened the articles by title and abstract. All conflicts were solved by consensus involving [PD]. For detailed information regarding the search terms used, see Appendix A and Figure 1.
FIGURE 1.
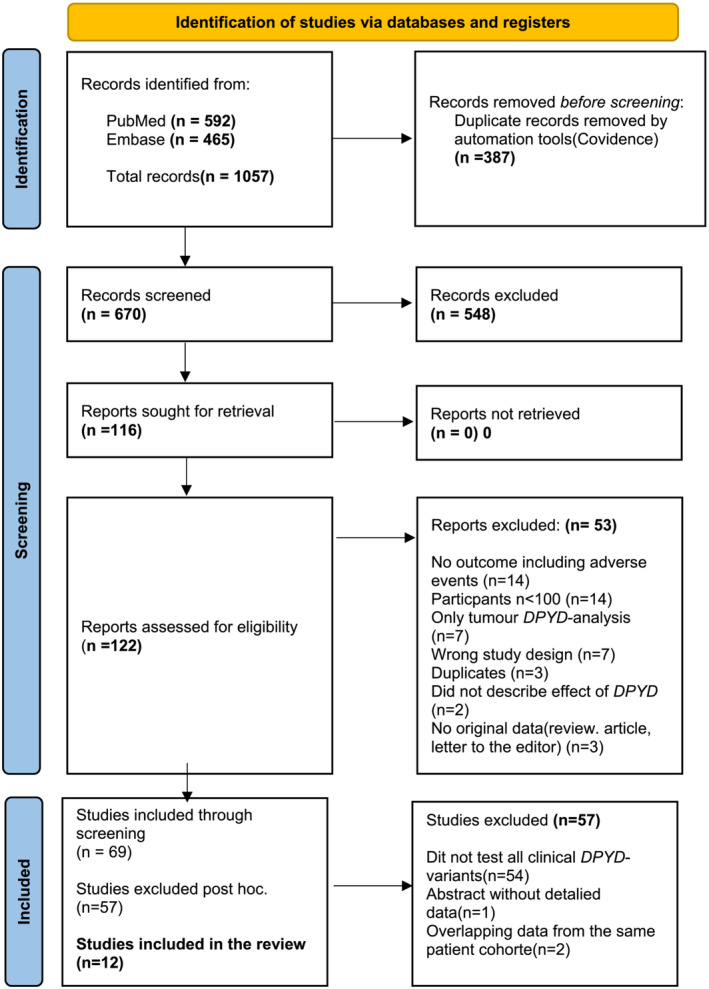
Genotype (DPYD‐genotype test). From Page et al. 20
2.1. DPYD genotype
The literature search was performed on 10 June 2021. The inclusion criteria were as follows: human clinical trials; study participants n > 100; measurement of the participants' DPYD genotype; treatment with systemic 5‐FU, Capecitabin or Tegafur (teysuno); data regarding adverse reactions after treatment; and cancer treatment.
2.2. DPD phenotype
The search was performed on 10 June 2021. The inclusion criteria were as follows: human clinical trials; study participants n > 100, measurement of the participants' DPD phenotype in plasma (uracil and/or dihydrouracil); treatment with systemic 5‐FU; Capecitabine or Tegafur (teysuno); data regarding adverse reactions after treatment; and cancer treatment.
3. RESULTS
Figures 1 and 2 show the flow charts for the two independent searches.
FIGURE 2.
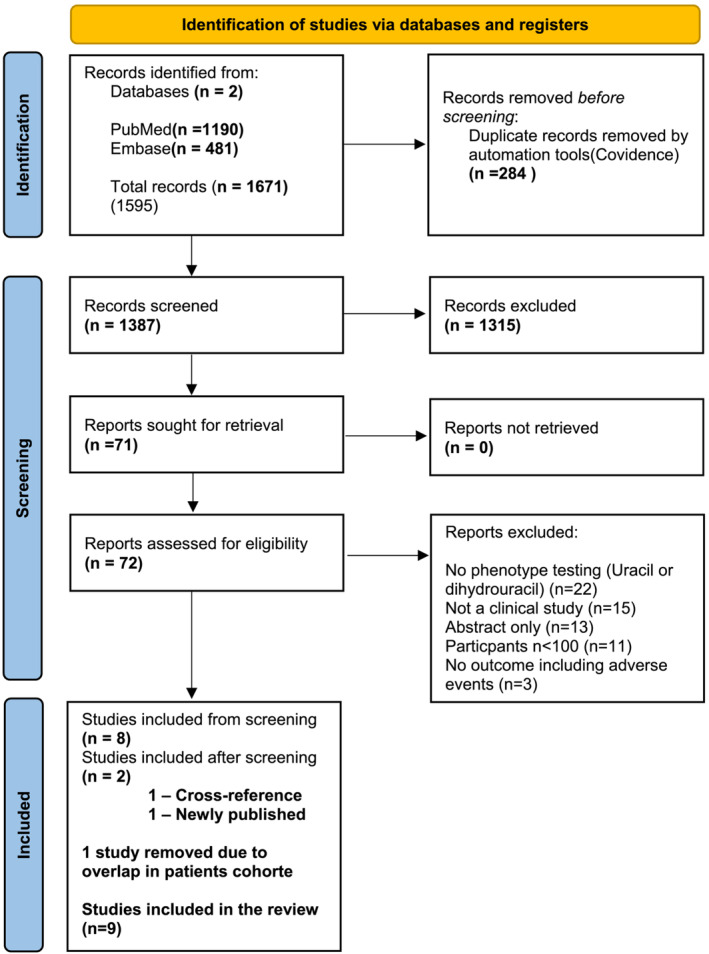
Phenotype (uracil + dihydrouracil). Page et al. 20
3.1. DPYD genotype
The total number of potentially relevant records was 1057. The automatic duplication tool in Covidence® (Covidence systematic review software, Veritas Health Innovation, Melbourne, Australia, https://www.convidence.org) removed 387 duplicate records. After screening the abstracts, we excluded another 554 records because they did not hold relevant or original data. After screening 122 full‐text records, we found that 69 met the inclusion criteria.
The 69 included studies varied widely in design and the specific DPYD variants examined. A larger part of the studies did not examine DPYD variants corresponding to a clinically relevant decrease in DPD activity. Furthermore, some studies examined only the rare variant rs3918290 (DPYD*2A).
Consequently, we decided to adjust the inclusion criteria and only include studies that examined the four most used variants corresponding to a clinically relevant decrease in DPD activity. 12 , 14 Table 1 shows these DPYD variants. Abstracts‐only records were also excluded at this stage. (For data regarding the excluded records, see Appendix A, Table A1.)
TABLE 1.
Overview of clinically relevant DPYD variants
| rs number | DPYD variant | Nucleotide change | Dose recommendation |
|---|---|---|---|
| rs3918290 | DPYD *2A | c.1905 + 1G > A/IVS14 + 1G > A |
50% dose reduction of 5‐FU based treatment a A gradual dose escalation is recommended in patients tolerating the 50% dose well. |
| rs67376798 | D949V | c.2846A > T | |
| rs55886062 | DPYD*13 | c.1679 T > G | |
| rs56038477 or rs75018182 | “HapB3” | c.1236G > A | |
| c.1129‐5923C > G | |||
| c.483 + 18G > A |
Patients heterozygous for one of the listed DPYD‐variants. [Correction added on 26 September 2022, after first online publication: Minor formatting/typographical changes have been made to Table 1.]
After applying the adjusted inclusion criteria, the number of included studies was reduced to 14. As two selected studies used data from the same cohort, 21 , 22 we decided to include only the original paper that focused on DPYD genotyping. 21 Another pair of studies by Meulendijks et al. 16 and Amstutz 22 used data from the same clinical study (NCT00838370). Thus, we decided to include only the paper with the most data regarding DPYD genotyping. 22
The 12 included studies reported data on 10 696 genotyped cancer patients treated with FP. None of the studies examined the effect of DPYD genotyping in a randomized controlled setting; in fact, no randomized controlled trial of this intervention has been published.
Table 2 shows the included studies with the number of DPYD variants found in each study. The design varied across the included studies. Nine retrospective studies examined pre‐treatment DPYD genotyping. Three of these 13 , 23 , 24 examined dosing strategies based on guidelines that have since been outdated and are no longer in use. For detailed data on the included DPYD genotype studies, see Table 3.
TABLE 2.
Overview of included studies
| DPD phenotype | |||||||
|---|---|---|---|---|---|---|---|
| First author, year published (PMID) | n, measurements of [U] and/or [UH2] | DPYD*2A | DPYD*13 | D949V | HapB3 | Homozygous or compound for DPYD variants | Total DPYD variants |
| Ciccolini 2006 (17038885) | 140 | 0 | Not tested | Not tested | Not tested | 0 | 0 |
| Boisdron‐Celle 2007 (17064846) | 252 | 2 | Not tested | 7 | Not tested | 1 | 10 |
| Cai 2017 (28278081) | 244 | Not tested | Not tested | Not tested | Not tested | 0 | 0 |
| Meulendijks 2017 (28427087) | 550 | Excluded | 3 | 6 | 22 | 0 | 31 |
| Etienne‐Grimaldi 2017 a (28481884) | 205 | 3 b | 1 b | 3 b | 4 b | 0 | 0 |
| Boisdron‐celle‐2017 (28395758) | 1,116 | 12 | 2 | 11 | Not Tested | 1 | 26 |
| Launay 2017 (29682445) | 221 | Not Tested | Not Tested | Not Tested | Not Tested | 0 | 0 |
| Capitain 2020 a (32973417) | 472 | 37 | 3 | 42 | 8 | 2 | 92 |
| With 2022 (35397172) | 955 | Excluded | Excluded | Excluded | Excluded | Excluded | Excluded |
| Total DPD phenotype | 4,155 | 54 | 9 | 69 | 34 | 4 | 159 |
| DPYD genotype | |||||||
|---|---|---|---|---|---|---|---|
| First author, year published (PMID) | n, tested for DPYD variants | DPYD*2A | DPYD*13 | D949V | HapB3 | Homozygous or compound for DPYD variants | Total DPYD variants |
| Ruzzo 2017 (29065426) | 508 | 3 | 0 | 6 | 10 | 0 | 19 |
| Etienne‐Grimaldi 2017 a (28481884) | 243 | 3 | 1 | 3 | 4 | 0 | 11 |
| Froehlich 2015 (24923815) | 500 | 4 | 2 | 3 | 24 | 0 | 33 |
| Deenen 2011 (21498394) | 568 | 7 | 1 | 8 | 28 | c | 44 |
| Shakeel 2021 (33410339) | 582 | 8 | 1 | 12 | 15 | 2 | 38 |
| Wigle 2021 (33620159) | 1435 | 9 | 1 | 19 | 41 | 0 | 70 |
| Boige 2016 (26794347) | 1545 | 11 | 4 | 21 | 53 | 0 | 89 |
| Lunenburg 2018 (30361102) | 894 | 13 | 1 | 10 | 31 | 1 | 56 |
| Henricks 2018 (30348537) | 1103 | 16 | 1 | 17 | 51 | 4 | 85 |
| Capitain 2020 a (32973417) | 472 | 37 | 3 | 42 | 8 | 5 | 95 |
| DelRe 2019 (30723313) | 1254 | 61 | 0 | 24 | 0 | 0 | 85 |
| Meulendijks 2016 (26804235) | 1592 | Excluded | 3 | 19 | 57 | 0 | 79 |
| Total DPYD genotype | 10,696 | 172 | 18 | 184 | 322 | 12 | 704 |
Abbreviations: DPD, dihydropyrimidine dehydrogenase; DPYD, gene encoding dihydropyrimidine dehydrogenase; [U], plasma uracil concentration; [UH2], plasma dihydrouracil concentration.
Studies included in both screenings.
Of 243 tested.
Patients homozygous for HapB3 was found. Number not specifed in article. Homozygous patients was pooled with patients heterzygous for HapB3
[Correction added on 26 September 2022, after first online publication: Minor formatting/typographical changes have been made to Table 2.]
TABLE 3.
Detailed data from included DPYD genotype studies
| First author, year published (PMID) | Study design | Pre‐treatment dose reduction based on DPYD genotype (yes/no) | n with DPYD genotype data | n compound heterzygous or homozygous for DPYD variants | Toxcity (grade ≥ 3) in DPYD wild‐type patients | Toxcity (grade ≥ 3) in DPYD variant carriers (pre‐treatment dose reductions based on DPYD variants) | Toxcity (grade ≥ 3) in DPYD variant carriers without pre‐treatment dose reductions (control group) | Toxicity (grade ≥ 3) in patients homozygous or compound heterozygous for DPYD variants (control group) |
|---|---|---|---|---|---|---|---|---|
| Henricks 2018 (30348537) | Prospective | Yes (DPYD variant carriers received an initial dose reduction of 25% (D949V and HapB3) or 50% (DPYD*2A and DPYD*13)) | 1103 | 4 patients were found. All 4 Excluded before treatment | n = 1018/8% | n = 85/20% | No | 4 patients were found. Excluded. |
| Wigle 2021 (33620159) | Retrospective | Yes. 50% (HapB3 25–50%) | 1435 | No patients were found. | n = 1347/21.1% | n = 47/13% | n = 41/24%. Retrospective samples. Full dose | No patients found |
| Lunenburg 2018 (30361102) | Prospective/Retrospective analysis | Yes/no. 50 or 75% dose reduction | 894 | 2 patients received normal doses. (no tox data reported) | n = 7761/13.6% | N = 22/22,7 | n = 34/23,5% | Pooled with heterozygous. n = 34/23,5% |
| Etienne‐Grimaldi 2017 (28481884) | Retrospective | No dose reduction | 243 | No dose intervention | n = 239/11.8% | No dose intervention | n = 11/45.5% | No patients found |
| Meulendijks 2016 (26804235) | Retrospective | No dose reduction | 1592 | No dose intervention | n = 1429/10% | No dose intervention | n = 79/14% (11/79) | No patients found |
| Froehlich 2015 (24923815) | Retrospective | No dose reduction | 500 | No dose intervention | All patients including DPYD‐variant carriers 14% | No dose intervention | n = 33/15.3% | No patients found |
| Shakeel 2021 (33410339) | Retrospective | No dose reduction | 582 | No dose intervention | n = 543/18.1% | No dose intervention | n = 39/36.5% | 2 patients. HapB3/HabB3 + *2A/HapB3 |
| Capitain 2020 (32973417) | Retrospective | No dose reduction | 472 | No dose intervention | n = 377/48% | No dose intervention | n = 90/89% | n = 5/100% (4 dead of tox. Last 1 grade 4) |
| Ruzzo 2017 (29065426) | Retrospective | No dose reduction | 508 | No dose intervention | ALL PATIENTS INCLUDING VARIANTS n = 508/38.2% | No dose intervention | DPYD *2A (HR 15.34, 4.72–49.89) D494V (HR 3.02, 1.12–8.16) HapB3 (HR 0.99 [0.37–2.6]) No DPYD*13 found | No patients found |
| Boige 2016 (26794347) | Retrospective | No dose reduction | 1545 | No dose intervention | ALL PATIENTS INCLUDING VARIANTSn = 1545/49.5% | No dose intervention | D949V (OR 6.3 [2–27]) DP YD*2A (OR 2.2[0.6–10]) DPYD*13 (OR 0.7 [0.079–6.2]) HapB3 (OR 1 [0.55–1.8]) | No patients found |
| DelRe 2019 (30723313) | Retrospective | No dose reduction | 1254 | No dose intervention | Comparison of two cohorts. Cohort 1(tox) (n = 982) grade 3 neutropenia 4.7% /Cohort 2 n = 272/0% tox | No dose intervention | *2A(Febrile neutropenia: OR 4.2, p < 0.5) D949V (diarrhoea: OR 2.78, p < 0.05) No DPYD*13 or HapB3 found | No patients found |
| Deenen 2011 (21498394) | Retrospective | No dose reduction | 568 | No dose intervention | ALL PATIENTS INCLUDING VARIANTS/n = 568/85% * Any (non)hematologic toxicity | No dose intervention | Any (non)hematologic toxicity: DPYD*13/n = 1/0% HabB3 n = 28 / 93% DPYD*2A n = 7/100% D949V n = 8/88% Total = 40/44/91% | Some for HapB3. Unknown number. Pooled with HapB3 heterzyous |
Abbreviations: DPYD, gene encoding dihydropyrimidine dehydrogenase; toxcity grading, Common Terminology Criteria for Adverse Events (CTCAE); [U], plasma uracil concentration; [UH2], plasma dihydrouracil concentration.
3.2. DPD phenotype
The total number of potentially relevant records was 1671. The automatic duplication tool in Covidence® removed 284 duplicate records. After screening the abstracts, we excluded 1315 records because they did not meet the inclusion criteria. After screening 71 full‐text records, we found eight records that met the criteria.
We identified one relevant study through a cross‐reference search (25). Two of the selected studies by Meulendijks et al. 16 and Meulendijks et al. 25 used data from the same clinical study (NCT00838370). Meulendijks et al. 16 focused on pre‐treatment [U], whereas the other study included only the [U] as a secondary parameter. Therefore, we chose to exclude the second study. 25
While writing this manuscript, a large clinical study was published 19 describing the uracil concentrations in patients enrolled in the study by Henricks et al. 13 We decided to include this study in this review.
Table 2 shows the nine studies on DPD phenotyping included in this review. In total, 4155 patients were tested with [U] and/or dihydrouracil [UH2] in the included studies.
3.3. Dihydrouracil (UH2)/uracil(U) ratio or uracil concentration
The DPD enzyme converts uracil [U] to dihydrouracil [UH2]. Therefore, the uracil concentration reflects the DPD enzyme activity, with high values indicating low DPD activity. Some of the included studies also or exclusively used the ratio between [U] and [UH2] to measure DPD activity. A low [UH2]/[U] ratio or a high [U]/[UH2] ratio would indicate low DPD activity.
Currently, EMA 1 only lists and gives threshold values for [U] as a recommended phenotyping method. The EMA states that [U] values of ≥16 ng/ml are indicative of partial DPD deficiency and ≥150 ng/ml of complete DPD deficiency, respectively.
The study by Ciccolini et al. 26 was the only one focusing on the [U]/[UH2] ratio, whereas two other studies 16 , 19 focused only on [U] measurements. The last six studies reported data on the [UH2]/[U] ratio including 15 , 27 , 28 that also examined [U]. For further details regarding the design and results of the included phenotype studies, see Table 4.
TABLE 4.
Detailed data from included DPD phenotype studies
| First author, year published (PMID) | Study design | Pre‐treatment dose reduction based on phenotype | n with phenotype data | Data using the [U]/[UH2] or [UH2]/[U] ratio | Data using plasma uracail concentration [U] |
|---|---|---|---|---|---|
| Ciccolini 2006 (17038885) | Retrospective | No | 140 | U/UH2 ratio: 80 patients with grade ≥ 3 toxicity were compared to 60 patients with no toxicity. The mean [U]/[UH2] ratio in the reference population was 1.4 (±0.6) compared to 3.8 in the toxicity group. 57 of 80(71%) of the patients in the toxicity group had a [U]/[UH2] ratio above 2 (cut‐off value) | |
| Boisdron‐Celle 2007 (17064846) | Retrospective | No | 252 | [UH2]/[U] ratio: A significant correlation was found between the [UH2]/[U] ratio and treatment toxicity. The mean [UH2]/[U] ratio decreased with toxicity grade: grade 0, 8 ± 2.5; grade I, 6.5 ± 3.2; grade II, 6.7 ± 1.7; grade III, 5 ± 3.1; grade IV, 3.7 ± 2.7. | The mean [U] increased with toxicity grade: grade 0, 14 ± 6.7 μg/L; grade I, 14 ± 2.6 μg/:L; grade II, 17.5 ± 4.8 μg/L; grade III, 25 ± 11 μg/L; grade IV, 24.4 ± 10 μg/L |
| Cai 2017 (28278081) | Retrospective | No | 244 | [UH2]/[U] ratio: Only subgroup analysis was reported in the study. UH2/U comparison between different CSN‐38 1.5 h and CSN‐38 49 h subsets with or without adverse effects: The [UH2]/[U] ratio in patients with toxcity were statistically lower than those in patients without toxcity in CSN‐38 1.5 h > 50.24 ng/ml (B) p < 0.001 for bone marrow hypocellular, p < 0.001 for diarrhoea and p < 0.001 for oral mucositis and CSN‐38 49 h > 15.25 ng/ml subgroups (C) p = 0.005 for bone marrow hypocellular, p = 0.001 for diarrhoea, and p = 0.002 for oral mucositis. | |
| Meulendijks 2017 (28427087) | Retrospective | No | 550 | Odds ratio (OR) for global severe toxcity was reported for different [U] intervals: [U] < 13 n = 500 OR = 1 (reference)|[U] 13–13. 8 n = 16 OR1.2(0.24–6.14)|[U] = 13.9–16 n = 17 OR 8.2(2.55–26.1)|[U] > 16 n = 17 OR 5.3 (1.53–18.7) | |
| Etienne‐Grimaldi 2017(28 481 884) | Retrospective | No | 205 | [UH2]/[U] ratio: In patients with validated phenotypic data, distribution of [UH2]/[U] was not different according to toxicity. No specific data is reported | Relative risk (RR) for grade 3 and 4 toxicity was reported. RR for Grade 3–4 toxicity. [U] > 16 n = 18 , RR 1.47(0.48–4.45)|RR for Grade 4 toxicity. [U] > 16 n = 18 RR 20.56(1.96–215.8) |
| Boisdron‐Celle 2017 (28395758) | Prospective with control arm. | Patented treatment algorithm. A Multiparametric approach | 1116 | [UH2]/[U] ratio: A Multiparametric approach was used including genotyping and UH2/U ratio. Risk of ≥ grade 3 toxicity was compared between the two arms in the study. Arm with intervention (Multiparametric approach) n = 718 / 10.8% ( n = 78 )|Arm without MAP n = 398 /17.6% ( n = 70 ). The percentage of death was reduced from 2.5/1000 in arm B to 0 in arm with intevtion | |
| Launay 2017 (29682445) | Prospective | [UH2]/[U] ratio| > 4 Standard dose|3–4 Alert for reduced activity, without systematic dose reduction |2–3 20% dose reduction |1–2 30% dose reduction |0.5–1 50% dose reduction| < 0.5 5‐FU precluded | 221 | [UH2]/[U] ratio: Extensive metabolizers (normal) [UH2]/[U] ratio > 4. 13% of 200 patients ( n = 26 ) suffered severe toxicity. Poor metabolizers [UH2]/[U] ratio < 4 < 18 ( n = 2 ) 11% suffered severe toxcity. In the poor metabolizers revived a 20–50% reduced dose of 5‐FU compared to extensive metabolizers. Mean dose of 5‐FU was 19% lower in the poor metabolizers group | |
| Capitain 2020 (32973417) | Retrospective | No | 472 | [UH2]/[U] ratio: Incidence of overall grade 3 ≥ toxicity was compared across different [UH2]/[U] ratio intervals|[UH2]/[U] ≥ 6 n = 267 /25.1% ( n = 67 )| [UH2]/[U] ≥ 2 ‐ < 6 n = 180 /96.7% ( n = 174 )|[UH2]/[U] < 2 n = 25 /100% ( n = 25 ) | Incidence of overall grade ≥ 3 toxicity was compared across different [U] intervals|[U] < 16 n = 280 /39% ( n = 108 )| [U] ≥ 16 ‐ < 150 n = 178 /80.9% ( n = 144 )|[U] > 150 n = 14 /100% ( n = 14 ) |
| With 2022 (35397172) | Retrospective | No | 955 | DPYD variants carriers were excluded as they had received prereceivedemtive dose reductions based. The median pretreatment [U] was 10.10 ng/ml in patients without grade ≥ 3 overall severe toxicity compared to 10.35 ng/ml in the patients with severe toxicity (P = 0.73) |
Abbreviations: Toxcity grading, Common Terminology Criteria for Adverse Events (CTCAE); DPYD, gene encoding dihydropyrimidine dehydrogenase; [U], plasma uracil concentration; [UH2], plasma dihydrouracil concentration.
[Correction added on 26 September 2022, after first online publication: Author entries in Table 4 (rows 2, 5) have been amended.]
4. DISCUSSION
Below, we discuss the most important papers substantiating the evidence for DPD testing in an everyday clinical setting.
4.1. Genotype
4.1.1. Studies with no pre‐treatment dose reduction based on DPYD genotype
Most of the included papers included toxicity analysis in patients with DPYD variants receiving regular doses of FP. In total, 12 studies of 8328 patients met our inclusion criteria. Of the tested patients, 561 had one of the four clinically relevant DPYD variants.
Patients carrying DPYD variants are at a higher risk of grade ≥3 FP‐TOX when treated with a standard FP dose than wild‐type patients (Table 3). Not all studies reported detailed data regarding toxicity in patients treated with FP. Furthermore, the prevalence of severe toxicity (grade ≥3 toxicity) varied substantially between the studies ranging from 10% to 49% in wild‐type patients. In patients with DPYD variants, the grade ≥3 toxicity prevalence varied from 14% to 89%. This wide range of results indicates that the studies are heterogeneous concerning population and treatment regimens. Therefore, performing a pooled analysis of data is not relevant.
In several studies, the exact prevalence of overall grade ≥3 toxicity was not reported but only data on specific adverse events like thrombocytopenia or febrile neutropenia. Some studies reported no actual number of patients with toxicity; others reported only odds ratios or risk ratios for selected adverse reactions.
Shakeel et al. 29 retrospectively examined patients treated with FP drugs, and genetic data were available. Clinical data regarding toxicity were scored retrospectively by use of electronic health records. The authors found that the incidence of grade ≥3 toxicity was 37% in DPYD variant carriers (n = 39) compared to 18% in wild‐type patients (n = 543) (OR: 2.6, 1.2–5.9).
Meulendijks et al. 30 examined the FP‐TOX rate in 1592 patients retrospectively genotyped for DPYD variants. Patients with the DPYD*2A genotype were excluded beforehand (n = 18). The frequency of grade ≥3 toxicity in patients with DPYD variants was 14% (n = 79) compared to 10% in the overall population.
4.1.2. Studies using pre‐treatment dose reductions based on DPYD genotype
Three studies that met our inclusion criteria examined the effect of pre‐treatment DPYD genotyping and a relevant dose reduction of FP. One of the studies examined patients treated as part of chemoradiation therapy (CRT), and the two remaining studies examined the use of systemic FP alone.
Lunenburg et al. 23 studied patients that received FP as part of CRT. Data were collected from medical records of 828 patients, of which some patients received upfront genotyping and dose reduction, whereas others were genotyped retrospectively. In patients with pre‐treatment genotype data, dose reduction was performed according to the current guidelines (25–50% dose reduction). The incidence of grade ≥3 toxicity (CTC‐AE) was compared among patients with and without pre‐treatment genotyping. Of the 771 wild‐type patients that received standard doses, 14% (105/771) were reported to have grade ≥3 global toxicities. In patients treated with a standard dose despite a DPYD variant, the toxicity rate was 21% (8/34). Of the 22 patients that received reduced FP doses based on their DPYD genotype, the toxicity rate was 22% (5/22). The authors conclude that DPYD variant carriers have an increased risk of toxicity when treated with standard FP as part of CRT. The study failed to show that DPYD variant carriers who received reduced doses of FP had toxicity rates comparable to wild‐type patients. Even though the DPYD variant carriers that received reduced doses had toxicity rates equal to those that received regular doses, the authors advise that FP dose reduction should be applied to DPYD variant carriers.
Henricks et al. 13 conducted a multi‐centre study in 13 Dutch hospitals and included 1018 patients planned for systemic 5‐FU treatment. All patients were genotyped prior to treatment, and patients heterozygous for DPYD variants received an initial dose reduction of 25% (rs67376798 [D949V] and rs56038477/rs75018182[HapB3]) or 50% (DPYD*2A and rs55886062 [DPYD*13]). The authors concluded that pre‐treatment DPYD genotyping was feasible and that toxicity was still more prevalent in patients carrying DPYD variants despite dose reduction but was less prevalent compared to patients from a historical control group. Overall, grade ≥3 toxicity was recorded in 39% of the DPYD variant carriers (n = 85) compared to 23% (n = 1018) among wild‐type patients. Henricks et al. 13 suggested that rather than a 25% initial dose reduction, a 50% dose reduction might be more appropriate in patients with the D949V and HapB3 DPYD variants.
A recent study by Wigle et al. 31 genotyped patients and adjusted the dose before treatment with FP. The DPYD variant HapB3 was included after the study had been initiated. After this variant was included, the recommended dose reduction for patients carrying this specific variant was 25–50%. For the other three clinically relevant variants, a 50% reduction of the FP dose was recommended. Data regarding adverse reactions were available for 1435 genotyped patients. The incidence of overall grade ≥3 (CTC‐AE) toxicity was 21.1% in the wild‐type cohort (n = 1347) compared to 13% in the cohort with DPYD variant that received pre‐treatment dose reductions (n = 47). Post‐hoc genotyping revealed that 41 of the patients were carriers of the HapB3 variant. They had all received standard starting doses. This group of HapB3 carriers had an overall frequency of grade ≥3 toxicity of 24%. The authors conclude that the data support future efforts to study and implement pre‐treatment DPYD genotyping in North America.
4.2. Phenotype
4.2.1. Studies with no pre‐treatment dose reduction based on phenotype
Most of the included studies investigating DPD phenotyping were retrospective studies including less than 250 participants. For details, see Table 4.
Meulendijks et al. 16 examined the toxicity in 550 cancer patients where blood samples had been collected before FP treatment. Patients with the DPYD*2A genotype were excluded beforehand as part of another clinical study (n = 18). All patients received standard doses of FP. The results showed that the [U] concentration was superior to the [UH2]/[U] ratio as a predictor of overall grade ≥3 (CTC‐AE), with high [U] concentrations (>16 ng/ml) strongly associated with severe global toxicity (odds ratio 5.3 [CI 1.5–18.7]).
De With et al. 19 examined the pretreatment [U] of patients enrolled in the study by Henricks et al. 13 and found that the median pretreatment [U] was comparable in patients with grade 3 ≥ toxicity (n = 218) and without toxicity (n = 737) (10.35 ng/ml vs. 10.10 ng/ml, p = 0.73). Patients with DPYD variants (n = 82) were excluded from the analyses because they received pretreatment dose reductions of FP. The authors conclude that there is no association between pretreatment [U] and FP‐related toxicity. Furthermore, significant between‐centre differences in pretreatment [U] were identified, underlining that the measurements of [U] are sensitive to pre‐analytical errors and may be affected by circadian rhythm and food intake. This conclusion can be doubted as it is not substantiated by the data due to the exclusion of DPYD variant carriers.
4.2.2. Studies using pre‐treatment dose reductions based on phenotype
Launay et al. 32 included 218 patients treated with FP after upfront DPD phenotyping by using the [UH2]/[U] ratio. Table 4 shows the dose recommendations used in the study based on the [UH2]/[U] ratio. Twenty patients received a 20% to 30% FP dose reduction. The rate of severe toxicity between patients receiving FP standard doses and those reduced was comparable (13% vs. 11%). The authors conclude that upfront DPD phenotyping based on the [UH2]/[U] ratio may reduce toxicity significantly.
Boisdron‐Celle et al. 33 examined using a multiparametric approach in a non‐randomized multi‐centre cohort study with two treatment arms. Patients in arm A (n = 718) received an upfront assessment of their DPD activity using a multiparametric approach, including DPYD genotyping, [U] and [UH2] measurements. The results from the genotype and phenotype were used to calculate a dose recommendation using a commercially available algorithm (ODPM Tox™). The specific dose recommendations and cut‐off values used were not available in the published material. Patients in arm B (n = 398) were treated with a standard dose of FP. In total, 1116 patients were included in the study. In arm A (n = 718), the incidence of ≥3 toxicity was 10.8% compared to 17.6% in arm B (n = 398). The study was stopped prematurely after external experts' decision in conformity with the protocol due to a toxicity‐related death in arm B. The authors conclude that pretreatment detection of DPD deficiency is cost‐effective and can decrease the incidence of early severe life‐threatening toxic events.
4.3. Synthesis of the evidence
4.3.1. DPYD genotype
Several studies have shown that pre‐treatment DPYD genotyping may benefit patient safety, with two studies demonstrating DPYD variants guided dose reductions giving a comparable incidence of toxicity. 13 , 31 No randomized clinical trials (RCT) have tested this intervention, and an RCT would now be considered unethical given the evidence supporting the test. As for now, the recommended starting dose reduction for patients with the four variants is 50% for the first dose of FP. A gradual dose escalation is recommended in patients who can tolerate the first dose. 14 In the clinical study by Henricks et al., only 13% (n = 11) of DPYD variant carriers received dose escalations. In five of the patients that received dose escalations, the higher dose was not well tolerated. 13
The main four DPYD variants discussed herein have primarily been examined in a Western European population. The examined variants are rare in other populations, such as the Japanese; thus, the benefit of implementing testing must be assessed specifically for different populations. The benefit will most likely be associated with the frequency of the variants in question. 34 However, it is possible that another contingent of variants with clinical relevance is present in non‐Caucasian populations but studies supporting this are lacking. This difference in prevalence is a weakness of DPYD testing, as evidence of benefit demonstrated in one part of the world might not be valid elsewhere. When new variants are identified, it is necessary to validate them in clinical trials before they can be added to treatment algorithms. In the future, whole DPYD gene sequencing may be feasible on a large scale. This kind of new data will most certainly disclose new variants, but it will still be necessary to determine the clinical relevance of each such new variant. The number of variants is not infinite (outside of the discrete occurrence of genuinely new mutations). This effectively means that the identification of new variants with clinical significance will eventually dry out. The benefit of DPYD genotyping is that it is fast and reliable with a low risk of preanalytical variations. The result is categorical and quickly operational in the daily clinic. It is a strength of this approach that the genotype never changes. However, the genotypes correlate poorly with phenotype and clinical outcomes and will not detect patients with FP vulnerability caused by gene variants not included in the test.
4.3.2. DPD phenotype
Several clinical studies have examined the use of [U] and [UH2] measurements. The use of [U] measurement is the preferred DPD phenotyping approach. Although several studies of this method have been published, prospective trials are still lacking. In one of the most notable studies, which included a control group, the authors did not disclose details on the employed treatment algorithm. This, unfortunately, limits the potential for extrapolation to a clinical utility. 33 The evidence supporting the current threshold values of [U] proposed by EMA is sparse. There is a clear need for proper validation in adequately powered prospective clinical trials.
Measurements of the endogenous substances [U] and [UH2], i.e., phenotyping, could be superior to genotyping. Indirect measurements of the DPD enzyme activity identify patients with rare DPYD variants but theoretically also capture other possible causes of outlying DPD activity. The DPD phenotype approach results in a point‐estimate assessment of the patient's DPD activity before treatment. Possible changes in the DPD activity over time could also be considered during prolonged treatment with FP. However, as highlighted by de With et al., 19 [U] is prone to preanalytical conditions that can impact [U] profoundly. They reported that concentrations of [U] varied widely between different laboratories, underlining the need for strict standardizations before implementation. Moreover, as the phenotype is based on a numerical value, it is worthless without an adequate and robust clinical cut‐off value to assign individual patients to the relevant dosing categories. Inherently, the confidence in such categorizing will be less in patients closer to the defined cut‐off.
4.3.3. Phenotype and DPYD genotype correlation
Three studies determined DPD phenotype and the four clinically relevant DPYD variants. 19 , 27 , 28 However, the correlation between [U] in wild‐type patients and DPYD variants carriers is still uncertain. De With et al. 19 found that the median level of [U] differed between patients with DPYD variants and wild‐type patients (wild type: 10.1 ng/ml, HapB3: 12.2, D949V: 14.6, DPYD*2A: 16.8, DPYD*13: 40.1 ng/ml). In contrast, Etienne‐Grimaldi et al. 28 reported that only DPYD variant D949V was associated with elevated [U]. The study by Capitain et al. 27 did not report data regarding [U] across the different DPYD variants. The authors claimed that combining DPYD genotyping and DPD phenotyping is superior to using only one test method.
4.4. Current guidelines, recommendations and clinical practice
France was the first country to require mandatory DPD testing for all patients back in December 2018 when The French National Authority for Health (The Haute Autorité de santé) recommended testing for DPD deficiency by measurement of [U]. 35
EMA recommends DPD testing using either DPYD genotyping or [U] measurements prior to treatment with FP. EMA recommends a dose reduction of FP in patients with partial DPD deficiency and avoiding FP in patients with complete DPD deficiency, respectively. However, EMA does not provide any recommendations with regards to dosing cut‐off levels. 1
The Dutch Pharmacogenetics Working Group (DPWG) and Clinical Pharmacogenetics Implementation Consortium (CPIC) recommend using the DPYD variants listed in Table 1. 12 , 14 Both groups recommended an FP starting dose of 50% of FP in patients carrying any of the specified DPYD variants. Supplementary DPD phenotyping is recommended, if possible, in patients categorized as compound heterozygous. It is recommended that FP treatment be avoided in patients that are homozygous for DPYD variants due to the risk of severe and life‐threatening FP‐TOX.
The European Society for Medical Oncology (ESMO) also recommends a 50% dose reduction in patients with DPYD variants. Regarding [U] measurements, ESMO recommends that patients with [U] above 16 ng/ml should be treated with a 50% FP dose. If [U] is higher than >150 ng/ml, FP is not advised. 36
National clinical guidelines vary across countries in Europe, with some recommending both screening methods simultaneously while others only use one of the methods. 35 , 37 , 38 In the United Kingdom, the National Health Service in 2020 recommended screening for DPD deficiency using DPYD genotyping. 39 In Table 5, the pros and cons of DPYD genoyping and DPD phenotyping are listed.
TABLE 5.
Pros and cons of using different DPD testing methods
| DPYD genotype | DPD phenotype (uracil concentration) |
|---|---|
|
|
|
|
4.5. Improving the level of evidence
As is evident from our review, the level of evidence to support a clinically meaningful efficacy of preemptive phenotype/genotype testing is insufficient. While ongoing trials are likely to clarify some of the underlying issues (Clinical Trials Gov: NCT04194957), the true effect size of either or combined interventions requires a double‐blinded, randomized controlled study with predefined levels of efficacy. This is a high bar, but if we are to convince patients, physicians and other healthcare party interests of the value of this intervention and justify the associated allocation of resources, such is a must. We suggest a design as illustrated in Figure 3.
FIGURE 3.
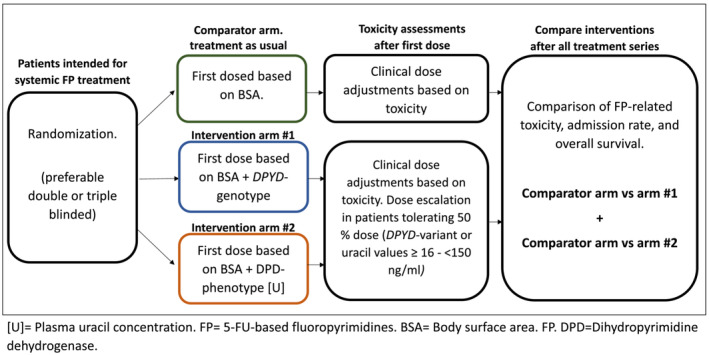
Suggested study design to conclusively determine the efficacy and safety of DPYD‐genotyping and DPD‐phenotyping [U].
5. CONCLUSION
In summary, the present evidence supporting either genotype or phenotype as a pre‐treatment test to prevent severe adverse reactions through personalized dose adjustments of FP is inadequate at this stage. The concept appears promising, but more work is needed to substantiate the effectiveness. Current guidelines and the regulatory recommendations by EMA are, accordingly, inadequately supported. Prospective data have weakly supported pre‐treatment genotyping. The widely used cut‐off value for [U]‐based phenotyping appears insufficiently substantiated. There is still an unmet need for prospective evidence connecting pre‐treatment testing, dose adjustments and clinical outcomes.
CONFLICT OF INTEREST
NHP, FV, SEA, TKB, ME, PPl, PSE, and PD declare no conflict of interest. MRH has received grants from Pfizer, paid to his employer outside the submitted work; has received speaking fees from Novartis outside the submitted work; owns stocks in Novo Nordisk and is employed by Novo Nordisk from 1 February 2022.
ACKNOWLEDGEMENT
The authors have nothing to acknowledge.
APPENDIX A.
Search terms
DPD‐phenotype
For the DPD‐phenotype part of the literature search, the following terms were used:
| (DPYD OR DPD OR [dihydropyrimidine dehydrogenase]) |
| AND |
| (phenotypic OR Phenotype OR phenotyping OR uracil OR dihydrouracil) |
| AND |
| (ONCOLOGY OR CANCER) |
| AND |
| (TREATMENT OR THERAPY) |
| AND |
| (5‐FU OR fluoropyrimidines OR Fluorouracil OR capecitabine OR Teysuno OR tegafur OR S‐1) |
The search was performed on the 16th of September 2020 and updated on the 10th of June 2021.
The inclusion criteria for the studies were as follows:
| Human clinical trials, |
| Study participants n > 100 |
| Measurement of the participant's DPD‐phenotype in plasma (uracil and/or dihydrouracil) Treatment with systemic treatment with 5‐FU, Capecitabine or Tegafur (teysuno), |
| Data regarding adverse events after treatment, |
| Cancer treatment |
DPYD‐genotype
For the DPYD‐genotyping part of the study, the following terms were used:
| (DPYD OR DPD OR [dihydropyrimidine dehydrogenase]) |
| AND |
| (GENOTYPE OR GENE OR GENOTYPING) |
| AND |
| (ONCOLOGY OR CANCER) |
| AND |
| (TREATMENT OR THERAPY) |
| AND |
| (5‐FU OR fluoropyrimidines OR Fluorouracil OR capecitabine OR Teysuno OR tegafur OR S‐1) |
The inclusion criteria for the DPYD‐studies were as follows:
| Human clinical trials |
| Study participants n > 100 |
| Measurement of the participants' DPYD‐genotype |
| Treatment with systemic treatment with 5‐FU, Capcitabin or Tegafur (teysuno), |
| Data regarding adverse events after treatment |
| Cancer treatment |
TABLE A1.
List of excluded DPYD‐genotype studies.
| First author, year published (PMID if available) | n (total) | DPYD*2A | DPYD*13 | D949V | HapB3 | Homo/compound | Total patients with clinical DPYD‐variants | Test all 4 variants |
|---|---|---|---|---|---|---|---|---|
| Amstutz 2015 (26804235) Excluded due to data overlap with Froehlich et al. (24923815) | 514 | 7 | 2 | 3 | 23 | 2 | 37 | yes |
| Lee 2020 (Abstract only) (Asia‐Pacific Journal of Clinical Oncology / 2020;16(SUPPL 8):172 Netherlands Blackwell Publishing Ltd 2020 /) | 176 | Tested no data | Tested no data | Tested no data | Tested no data | 0 | 0 | yes |
| Meulendijks 2017(28427087) (Included in the phenotype part of the review) | 550 | Tested before. Excluded | 3 | 6 | 22 | 0 | 31 | No |
| Thomas 2011(22045187) | 131 | 0 | Not tested | Not tested | Not tested | 0 | 0 | No |
| Deng 2020 (33280607) | 104 | 0 | Not tested | Not tested | Not tested | 0 | 0 | No |
| Etienne‐Grimaldi 2010 (20078613) | 117 | 0 | Not tested | Not tested | Not tested | 0 | 0 | No |
| Kit 2020 (Abstract only) (Journal of Clinical Oncology / 2020;38(4 Supplement): Netherlands American Society of Clinical Oncology 2020) | 104 | 0 | 0 | 0 | Not tested | 0 | 0 | No |
| Joerger 2015 (25677447) | 140 | 1 | Not tested | 3 | Not tested | 0 | 4 | No |
| Rosmarin 2015(24647007) | 1.046 | 1 | 1 | 2 | Not tested | 0 | 4 | No |
| Amstutz 2009 (19530960) | 111 | 1 | 1 | Not tested | Not tested | 0 | 2 | No |
| Borro 2017 (27738344) | 107 | 1 | Not tested | Not tested | Not tested | 0 | 1 | No |
| Onesti 2017 (27845948) | 126 | 1 | Not tested | Not tested | Not tested | 0 | 1 | No |
| Roberto 2017 (27864592) | 142 | 1 | Not tested | Not tested | Not tested | 0 | 1 | No |
| Largillier 2006 (17000685) | 105 | 1 | Not tested | Not tested | Not tested | 0 | 1 | No |
| Ribelles 2008 (18473752) | 136 | 1 | Not tested | Not tested | Not tested | 0 | 1 | No |
| Sulzyc‐Bielicka 2008 (18443386) | 252 | 1 | Not tested | Not tested | Not tested | 0 | 1 | No |
| Puerta‐García 2020 (33035787) | 194 | 1 | 0 | 0 | Not tested | 0 | 1 | No |
| Vivaldi 2021 (33462346) | 104 | 1 | 0 | 0 | Not tested | 0 | 1 | No |
| Boisdron‐Celle 2007 (17064846) | 252 | 2 | Not tested | 7 | Not tested | 1 | 10 | No |
| Milano 2016 (27454530) | 243 | 2 | Not tested | 2 | Not tested | 0 | 4 | No |
| Kristensen 2010 (20819423) | 122 | 2 | Not tested | 1 | Not tested | 0 | 3 | No |
| Boige 20103 (20385995) | 346 | 2 | Not tested | Not tested | Not tested | 0 | 2 | No |
| AlvaradoFernandez 2021 (Abstract only) (European Journal of Hospital Pharmacy 2021;28(SUPPL 1):A132 Netherlands BMJ Publishing Group 202) | 171 | 3 | Not tested | 3 | Not tested | 0 | 6 | No |
| Chi 2020 (Abstract only) (DOI: 10.1200/JCO.2020.38.15_suppl.e16138 Journal of Clinical Oncology 38, no. 15_suppl) | 226 | 3 | Not tested | 1 | Not tested | 0 | 4 | No |
| Loganayagam 2013 (23736036) | 430 | 4 | 1 | 5 | Not tested | 0 | 10 | No |
| Magnani 2013 (23585145) | 180 | 4 | Not tested | Not tested | Not tested | 0 | 4 | No |
| Burnett 2020 (Abstract only) (doi.org/10.1016/j.annonc.2020.04.268) | 506 | 5 | Not tested | 5 | Not tested | 0 | 10 | No |
| Cremolini 2018 (29487697) | 443 | 5 | 0 | 5 | Not tested | 0 | 10 | No |
| Kleibl 2009 (19473056) | 124 | 5 | Not tested | Not tested | 4 | 0 | 9 | No |
| Gross 2008 (19104657) | 181 | 5 | Not tested | 1 | (1) | 0 | 7 | No |
| Botticelli 2017 (28296649) | 642 | 6 | Not tested | Not tested | Not tested | 0 | 6 | No |
| Nahid 2018 (29134491) | 161 | 8 | Not tested | Not tested | Not tested | 0 | 8 | No |
| Morel 2006 (17121937) | 487 | 9 | 1 | 10 | Not tested | 1 | 21 | No |
| Schwab 2008 (18299612) | 683 | 13 | Not tested | 5 | 1 | 0 | 13 | No |
| Deligonul 2021 (33877893) | 100 | 15 | Not tested | Not tested | Not tested | 0 | 15 | No |
| Deenen 2016 (26573078) | 2.038 | 22 | Not tested | Not tested | Not tested | Not tested | 22 | No |
| Jolivet 2021 (33274825) | 2.617 | 24 | Not tested | Not tested | Not tested | 1 | 25 | No |
| Lee 2014 (25381393) | 2.886 | 27 | 4 | 32 | Not tested | 0 | 63 | No |
| Iachetta 2019 (30858516) | 1.827 | 31 | 2 (668) | 5 (668) | Not tested | 0 | 38 | No |
| Henricks 2019 (30485432) | 1.646 | 40 | Not tested | Not tested | Not tested | 0 | 40 | No |
| Toffoli 2015 (26099996) | 603 | 11 (603) | 1 (595) | 5 (588) | Not tested | 1 | 17 | No |
| Madi 2018 (30114658) | 2.183 | 23 (2105) | Not tested | 30 (2116) | Not tested | 0 | 53 | No |
| Graham 2020 (Abstract only) (Journal of Clinical Oncology / 2020;38(4 Supplement): Netherlands American Society of Clinical Oncology 2020 /) | 201 | No Specific data | No Specific data | No Specific data | No Specific data | 0 | 0 | No |
| Kanai 2020 (Abstract only) (Annals of Oncology / 2020;31(Supplement 6):S1359 Netherlands Elsevier Ltd 2020 /) | 3135 | No Specific data | No Specific data | No Specific data | No Specific data | 0 | 0 | No |
| Lee 2016 (26658227) | 1.953 | Not tested | Not tested | Not tested | 78 | 0 | 78 | No |
| Meulendijks 2017 (27995989) | 185 | Not tested | Not tested | 3 | 5 | 0 | 8 | No |
| Pare 2010 (20653680) | 234 | Not tested | Not tested | Not tested | Not tested | 0 | 0 | No |
| Zhang 2012 (22490566) | 362 | Not tested | Not tested | Not tested | Not tested | 0 | 0 | No |
| Yokoi 2020 (32619063) | 301 | Not tested | Not tested | Not tested | Not tested | 0 | 0 | No |
| Murphy 2020 (Abstract only) (Journal of Clinical Oncology / 2020;38(15): Netherlands American Society of Clinical Oncology 2020 /) | 742 | Not tested | Not tested | Not tested | Not tested | 0 | 0 | No |
| Personeni 2020 (Abstract only) (Tumori / 2020 ;106(2 SUPPL):181‐182 Netherlands SAGE Publications Ltd 2020 /) | 179 | Not tested | Not tested | Not tested | Not tested | 0 | 0 | No |
| Cortejoso 2014 (25137161) | 157 | Not tested | Not tested | Not tested | Not tested | 0 | 0 | No |
| Celik 2002 (12025228) | 200 | Not tested | Not tested | Not tested | Not tested | 0 | 0 | No |
| Fidlerova 2010 (19649633) | 113 | Not tested | Not tested | Not tested | Not tested | 0 | 0 | No |
| Pellicer 2017 (28347776) | 301 | Not tested | Not tested | Not tested | Not tested | 0 | 0 | No |
| Zhao 2016 (26846104) | 249 | Not tested | Not tested | Not tested | Not tested | 0 | 0 | No |
| Varma 2019 (31653159) | 234 | Not tested | Not tested | Not tested | Not tested | 0 | 0 | No |
[Correction added on 26 September 2022, after first online publication: PMID numbers have been added to Table A1.]
Paulsen NH, Vojdeman F, Andersen SE, et al. DPYD genotyping and dihydropyrimidine dehydrogenase (DPD) phenotyping in clinical oncology. A clinically focused minireview. Basic Clin Pharmacol Toxicol. 2022;131(5):325‐346. doi: 10.1111/bcpt.13782
Funding information Danish Cancer Society, Grant/Award Number: R231‐A14057; The Region of Southern Denmark, Grant/Award Number: A197
REFERENCES
- 1. EMA . 5‐Fluorouracil (i.v.), capecitabine and tegafur containing products: Pre‐treatment testing to identify DPD‐deficient patients at increased risk of severe toxicity|European Medicines Agency [Internet]. [cited 2022 Mar 1]. Available from: https://www.ema.europa.eu/en/medicines/dhpc/5-fluorouracil-iv-capecitabine-tegafur-containing-products-pre-treatment-testing-identify-dpd#documents-sectio
- 2. Vodenkova S, Buchler T, Cervena K, Veskrnova V, Vodicka P, Vymetalkova V. 5‐fluorouracil and other fluoropyrimidines in colorectal cancer: past, present and future. Pharmacol Ther. 2020;206:107447. doi: 10.1016/j.pharmthera.2019.107447 [DOI] [PubMed] [Google Scholar]
- 3. van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27(8):1386‐1422. doi: 10.1093/annonc/mdw235 [DOI] [PubMed] [Google Scholar]
- 4. Hoff PM, Ansari R, Batist G, et al. Comparison of oral capecitabine versus intravenous fluorouracil plus Leucovorin as first‐line treatment in 605 patients with metastatic colorectal cancer: results of a randomized phase III study. JCO. 2001;19(8):2282‐2292. doi: 10.1200/JCO.2001.19.8.2282 [DOI] [PubMed] [Google Scholar]
- 5. Twelves C, Wong A, Nowacki MP, et al. Capecitabine as adjuvant treatment for stage III colon cancer. N Engl J Med. 2005;9. [DOI] [PubMed] [Google Scholar]
- 6. Traut TW, Loechel S. Pyrimidine catabolism: individual characterization of the three sequential enzymes with a new assay. Biochemistry. 1984;23(11):2533‐2539. doi: 10.1021/bi00306a033 [DOI] [PubMed] [Google Scholar]
- 7. Sommadossi JP, Gewirtz DA, Diasio RB, Aubert C, Cano JP, Goldman ID. Rapid catabolism of 5‐fluorouracil in freshly isolated rat hepatocytes as analyzed by high performance liquid chromatography. J Biol Chem. 1982;257(14):8171‐8176. doi: 10.1016/S0021-9258(18)34313-8 [DOI] [PubMed] [Google Scholar]
- 8. Yokota H, Fernandez‐Salguero P, Furuya H, et al. cDNA cloning and chromosome mapping of human dihydropyrimidine dehydrogenase, an enzyme associated with 5‐fluorouracil toxicity and congenital thymine uraciluria. J Biol Chem. 1994;269(37):23192‐23196. doi: 10.1016/S0021-9258(17)31638-1 [DOI] [PubMed] [Google Scholar]
- 9. Tuchman M, Stoeckeler JS, Kiang DT, O'Dea RF, Ramnaraine ML, Mirkin BL. Familial pyrimidinemia and pyrimidinuria associated with severe fluorouracil toxicity. N Engl J Med. 1985;313(4):245‐249. doi: 10.1056/NEJM198507253130407 [DOI] [PubMed] [Google Scholar]
- 10. Hodroj m.fl . 2021. Issues and limitations of available biomarkers for.pdf. [DOI] [PMC free article] [PubMed]
- 11. Hodroj K, Barthelemy D, Lega JC, et al. Issues and limitations of available biomarkers for fluoropyrimidine‐based chemotherapy toxicity, a narrative review of the literature. ESMO Open. 2021;6(3):100125. doi: 10.1016/j.esmoop.2021.100125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lunenburg CATC, van der Wouden CH, Nijenhuis M, et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene–drug interaction of DPYD and fluoropyrimidines. Eur J Hum Genet. 2020;28(4):508‐517. doi: 10.1038/s41431-019-0540-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Henricks LM, Lunenburg CATC, de Man FM, et al. DPYD genotype‐guided dose individualisation of fluoropyrimidine therapy in patients with cancer: a prospective safety analysis. Lancet Oncol. 2018;19(11):1459‐1467. doi: 10.1016/S1470-2045(18)30686-7 [DOI] [PubMed] [Google Scholar]
- 14. Amstutz U, Henricks LM, Offer SM, et al. Clinical pharmacogenetics implementation consortium (CPIC) guideline for Dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin Pharmacol Ther. 2018;103(2):210‐216. doi: 10.1002/cpt.911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boisdron‐Celle M, Remaud G, Traore S, et al. 5‐fluorouracil‐related severe toxicity: a comparison of different methods for the pretherapeutic detection of dihydropyrimidine dehydrogenase deficiency. Cancer Lett. 2007;249(2):271‐282. doi: 10.1016/j.canlet.2006.09.006 [DOI] [PubMed] [Google Scholar]
- 16. Meulendijks D, Henricks LM, Jacobs BAW, et al. Pretreatment serum uracil concentration as a predictor of severe and fatal fluoropyrimidine‐associated toxicity. Br J Cancer. 2017;116(11):1415‐1424. doi: 10.1038/bjc.2017.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. PubMed . Development and validation of a rapid and sensitive UPLC‐MS/MS method for determination of uracil and dihydrouracil in human plasma. [cited 2022 Mar 3]. Available from: https://pubmed.ncbi.nlm.nih.gov/27179185/ [DOI] [PubMed]
- 18. Gaible C, Narjoz C, Loriot MA, Roueff S, Pallet N. Pretherapeutic screening for Dihydropyrimidine deshydrogenase deficiency in measuring uracilemia in dialysis patients leads to a high rate of falsely positive results. Cancer Chemother Pharmacol. 2021;88(6):1049‐1053. doi: 10.1007/s00280-021-04354-7 [DOI] [PubMed] [Google Scholar]
- 19. de With M, Knikman J, de Man FM, et al. Dihydropyrimidine dehydrogenase phenotyping using pretreatment uracil: a note of caution based on a large prospective clinical study. Clin Pharma Ther. 2022;112(1):62‐68. doi: 10.1002/cpt.2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Page MJ, Moher D, Bossuyt PM, et al. PRISMA 2020 explanation and elaboration: updated guidance and exemplars for reporting systematic reviews. BMJ. 2021;372:n160. doi: 10.1136/bmj.n160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Froehlich TK, Amstutz U, Aebi S, Joerger M, Largiadèr CR. Clinical importance of risk variants in the dihydropyrimidine dehydrogenase gene for the prediction of early‐onset fluoropyrimidine toxicity: DPYD and fluoropyrimidine toxicity. Int J Cancer. 2014;136(3):730‐739. doi: 10.1002/ijc.29025 [DOI] [PubMed] [Google Scholar]
- 22. Amstutz U, Offer SM, Sistonen J, Joerger M, Diasio RB, Largiadèr CR. Polymorphisms in MIR27A associated with early‐onset toxicity in fluoropyrimidine‐based chemotherapy. Clin Cancer Res. 2015;21(9):2038‐2044. doi: 10.1158/1078-0432.CCR-14-2817 [DOI] [PubMed] [Google Scholar]
- 23. Lunenburg CATC, Henricks LM, Dreussi E, et al. Standard fluoropyrimidine dosages in chemoradiation therapy result in an increased risk of severe toxicity in DPYD variant allele carriers. Eur J Cancer. 2018;104:210‐218. doi: 10.1016/j.ejca.2018.07.138 [DOI] [PubMed] [Google Scholar]
- 24. Boige V, Vincent M, Alexandre P, et al. DPYD genotyping to predict adverse events following treatment With fluorouracil‐based adjuvant chemotherapy in patients With stage III colon cancer: a secondary analysis of the PETACC‐8 randomized clinical trial. JAMA Oncol. 2016;2(5):655‐662. doi: 10.1001/jamaoncol.2015.5392 [DOI] [PubMed] [Google Scholar]
- 25. Meulendijks D, van Hasselt JGC, Huitema ADR, et al. Renal function, body surface area, and age are associated with risk of early‐onset fluoropyrimidine‐associated toxicity in patients treated with capecitabine‐based anticancer regimens in daily clinical care. Eur J Cancer. 2016;54:120‐130. doi: 10.1016/j.ejca.2015.10.013 [DOI] [PubMed] [Google Scholar]
- 26. Ciccolini J, Mercier C, Evrard A, et al. A rapid and inexpensive method for anticipating severe toxicity to fluorouracil and fluorouracil‐based chemotherapy. Ther Drug Monit. 2006;28(5):678‐685. doi: 10.1097/01.ftd.0000245771.82720.c7 [DOI] [PubMed] [Google Scholar]
- 27. Capitain O, Seegers V, Metges JP, et al. Comparison of 4 screening methods for detecting fluoropyrimidine toxicity risk: identification of the most effective, cost‐efficient method to save lives. Dose‐Response. 2020;18(3):155932582095136. doi: 10.1177/1559325820951367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Etienne‐Grimaldi MC, Boyer JC, Beroud C, et al. New advances in DPYD genotype and risk of severe toxicity under capecitabine. PLoS ONE. 2017;12(5):e0175998. doi: 10.1371/journal.pone.0175998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shakeel F, Fang F, Kwon JW, et al. Patients carrying DPYD variant alleles have increased risk of severe toxicity and related treatment modifications during fluoropyrimidine chemotherapy. Pharmacogenomics. 2021;22(3):145‐155. doi: 10.2217/pgs-2020-0154 [DOI] [PubMed] [Google Scholar]
- 30. Meulendijks D, Henricks LM, Amstutz U, et al. Rs895819 in MIR27A improves the predictive value of DPYD variants to identify patients at risk of severe fluoropyrimidine‐associated toxicity: MIR27A variants and fluoropyrimidine‐associated toxicity. Int J Cancer. 2016;138(11):2752‐2761. doi: 10.1002/ijc.30014 [DOI] [PubMed] [Google Scholar]
- 31. Wigle TJ, Povitz BL, Medwid S, et al. Impact of pretreatment dihydropyrimidine dehydrogenase genotype‐guided fluoropyrimidine dosing on chemotherapy associated adverse events. Clin Transl Sci. 2021;14(4):1338‐1348. doi: 10.1111/cts.12981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Launay M, Ciccolini J, Fournel C, et al. Upfront DPD deficiency detection to secure 5‐FU Administration: part 2‐ application to head‐and‐neck cancer patients. CCAND. 2018;4(2):122‐128. doi: 10.2174/2212697X04666170817123425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boisdron‐Celle M, Capitain O, Faroux R, et al. Prevention of 5‐fluorouracil‐induced early severe toxicity by pre‐therapeutic dihydropyrimidine dehydrogenase deficiency screening: assessment of a multiparametric approach. Semin Oncol. 2017;44(1):13‐23. doi: 10.1053/j.seminoncol.2017.02.008 [DOI] [PubMed] [Google Scholar]
- 34. White C, Scott RJ, Paul C, Ziolkowski A, Mossman D, Ackland S. Ethnic diversity of DPD activity and the DPYD gene: review of the literature. PGPM. 2021;14:1603‐1617. doi: 10.2147/PGPM.S337147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Haute Autorité de Santé . Screening for dihydropyrimidine dehydrogenase deficiency to decrease the risk of severe toxicities related to fluoropyrimidines (5‐fluorouracil or capecitabine) ‐ INAHTA Brief [Internet]. Haute Autorité de Santé. [cited 2022 Aug 2]. Available from: https://www.has-sante.fr/jcms/c_2891090/en/screening-for-dihydropyrimidine-dehydrogenase-deficiency-to-decrease-the-risk-of-severe-toxicities-related-to-fluoropyrimidines-5-fluorouracil-or-capecitabine-inahta-brief
- 36. Argilés G, Tabernero J, Labianca R, et al. Localised colon cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow‐up. Ann Oncol. 2020;31(10):1291‐1305. doi: 10.1016/j.annonc.2020.06.022 [DOI] [PubMed] [Google Scholar]
- 37. Hamzic S, Aebi S, Joerger M, et al. Fluoropyrimidine chemotherapy: recommendations for DPYD genotyping and therapeutic drug monitoring of the Swiss Group of Pharmacogenomics and Personalised Therapy. 2020. Swiss Med Wkly [Internet] Nov 24 [cited 2022 May 12]. Available from: https://doi.emh.ch/smw.2020.20375 [DOI] [PubMed]
- 38. Casneuf V, Borbath I, van den Eynde M, et al. Joint Belgian recommendation on screening for DPD‐deficiency in patients treated with 5‐FU, capecitabine (and tegafur). Acta Clin Belg. 2022;77(2):346‐352. doi: 10.1080/17843286.2020.1870855 [DOI] [PubMed] [Google Scholar]
- 39. NHS England . Clinical Commissioning Urgent Policy Statement: pharmacogenomic testing for DPYD polymorphisms with fluoropyrimidine therapies [Internet]. [cited 2022 Aug 2]. Available from: https://www.england.nhs.uk/publication/clinical-commissioning-urgent-policy-statement-pharmacogenomic-testing-for-dpyd-polymorphisms-with-fluoropyrimidine-therapies/