

Abstract
Background
Filaminopathies A are rare disorders affecting the brain, intestine, or skeleton, characterized by dominant X‐linked filamin A (FLNA) gene mutations. Macrothrombocytopenia with functionally defective platelets is frequent. We have described a filaminopathy A male patient, exhibiting a C‐terminal frame‐shift FLNa mutation (Berrou et al., Arterioscler Thromb Vasc Biol. 2017;37:1087–1097). Contrasting with female patients, this male patient exhibited gain of platelet functions, including increased platelet aggregation, integrin αIIbβ3 activation, and secretion at low agonist concentration, raising the issue of thrombosis risk.
Objectives
Our goal is to assess the thrombotic potential of the patient FLNa mutation in an in vivo model.
Methods
We have established a mutant FlnA knock‐in mouse model.
Results
The mutant FlnA mouse platelets phenocopied patient platelets, showing normal platelet count, lower expression level of mutant FlnA, and gain of platelet functions: increased platelet aggregation, secretion, and αIIbβ3 activation, as well as increased spreading and clot retraction. Surprisingly, mutant FlnA mice exhibited a normal bleeding time, but with increased re‐bleeding (77%) compared to wild type (WT) FlnA mice (27%), reflecting hemostatic plug instability. Again, in an in vivo thrombosis model, the occlusion time was not altered by the FlnA mutation, but arteriolar embolies were increased (7‐fold more frequent in mutant FlnA mice versus WT mice), confirming thrombus instability.
Conclusions
This study shows that the FlnA mutation found in the male patient induced gain of platelet functions in vitro, but thrombus instability in vivo. Implications for the role of FLNa in physiology of thrombus formation are discussed.
Keywords: filaminopathy, gain‐of‐function mutation, hemostasis, platelets, thrombosis
Essentials.
Filaminopathies A are multi‐organ and platelet ailments due to X‐linked filamin A gene mutations.
We assessed the thrombosis ability of a patient gain‐of‐function filamin A (FLNa) mutation in a mouse model.
Mouse platelets showed gain of platelet functions, but paradoxical thrombus instability in vivo.
Thrombus instability suggests FLNa is involved in the strength of platelet–platelet interaction.
1. INTRODUCTION
Filamin(s) (FLN), a family of three members (FLNa, FLNb, and FLNc), are essential components of the cell cytoskeleton by stabilizing the actin network. FLN(s) are large dimeric proteins composed of N‐terminal actin‐binding domains followed by 24 immunoglobulin (Ig)‐like repeats interrupted by two hinge regions and a C‐terminal region that ensures the dimerization. 1 , 2 FLNa and FLNb are ubiquitously expressed whereas FLNc is restricted to smooth and striated muscles. 3 , 4 FLNa, which is the most abundant isoform, is encoded by the FLNA gene located on chromosome Xq28, and is predominantly expressed in platelets. Today more than 90 FLNa partners have been described. 5 Most of them bind to Ig repeats 16–23 of FLNa. These interactions are essential for a large variety of cellular functions such as migration, adhesion, and spreading. Deletion of FLNa in mouse platelets, as shown with FlnA loxP GATA1‐Cre mice, induces larger platelets, macrothrombocytopenia, and increased tail‐bleeding. 6 In platelets, FLNa interacts with glycoprotein (GP)Ibα through the Ig repeat 17 and ensures the mechanical stabilization of the plasma membrane under high shear. 7 The GPIbα–FLNa interaction is also essential for the trafficking of GPIb to the plasma membrane. 8 Another essential partner of FLNa in platelets is the integrin αIIbβ3. FLNa by its Ig repeat 21 binds to β3 tail (amino acids 747–755) and regulates the activation of αIIbβ3 integrin. 9 It has been proposed that constitutively bound FLNa to β3 in resting platelets maintains the integrin in an inactive state, possibly by clasping αIIb and β3 tails together, 10 but definitive experimental evidence is still lacking. Likewise, it has been proposed that the activation of αIIbβ3 integrin requires the dissociation of FLNa from β3 allowing talin binding, but hard evidence is missing as well. Recently, our group showed clearly that the FLNa–β3 interaction exerts a negative regulation on talin–β3 interaction after activation, using a megakaryocytic cell line (HEL). 11
The tyrosine kinase Syk involved in the GPVI signaling is another partner of FLNa (Ig repeat 5) highlighted in Flna −/− mice. 6 This interaction, essential in platelet activation under arterial shear, allows Syk recruitment to GPVI.
Recently, new partners have been described in platelets. The adaptor protein PACSIN2, which interacts with FLNa Ig repeat 20, regulates membrane tubulation in megakaryocytes and platelets. 12 STIM1, which regulates store‐operated calcium entry (SOCE) and Ca2+ influx, interacts with FLNa. 13 Finally, the Rho GTPase, RhoA, which binds constitutively to FLNa via the C‐terminal Ig repeat 24, has recently been reported to be dependent on αIIbβ3. This interaction modulates proplatelet formation in megakaryocytes 14 and outside‐in signaling in platelets. 11 How the interactions of FLNa with its partners integrate cellular signaling and cellular functions remains an open question. Investigating FLNA gene variants is of great help. Today, FLNA mutations are associated with a wide spectrum of 10 distinct genetic syndromes called filaminopathies A affecting numerous organs including the brain, skeleton, heart, and skin. 15 Periventricular nodular heterotopia (PVNH), the most frequent syndrome, is characterized by a defect of neuron migration. 16 , 17 Most of PVNH, observed in heterozygous females exhibiting loss‐of‐function mutations, ablate FLNa expression. 17 , 18 , 19 In males, filaminopathy A is generally associated with perinatal lethality; only rare males survivie. 20 , 21 The PVNH phenotype can be associated with Ehlers–Danlos syndrome, with skeletal dysplasia, congenital intestinal pseudo‐obstruction, and familial cardiac valvular dystrophy. 22 , 23 , 24 PVNH is often associated with macrothrombocytopenia and bleeding tendency. 17 We have previously shown in female patients with PVNH that have low platelet count characterized by giant and normal platelets was associated with a reduced FLNa level whereas normal platelet count was associated with a normal FLNa content. 14 , 19 In PVNH, depending on the level of FLNa, platelet functions were often impaired, causing bleeding tendency. 19 , 25 Moreover, we have shown that the absence of FLNa protein in megakaryocytes leads to their inability to produce platelets. 14 More recently, we have studied the rare case of a male filaminopathy A patient with a novel mutant FLNa with a 100 amino acid–long C‐terminal extension. 20 Surprisingly, platelet functions were significantly enhanced, contrary to functional inhibition in heterozygous female patients. This was the first observation in humans of the role of FLNa in the downregulation of αIIbβ3 activation, a key feature in platelet physiology and pathology. However, a number of questions remain unanswered. The aim of this study was to establish an “in vivo” mutated FLNa knock‐in mouse model and to address the question of the potential role of this FLNa mutation in thrombosis.
2. MATERIAL AND METHODS
2.1. Reagents
Fibrillar collagen (equine type I) and adenosine diphosphate (ADP) were obtained from Kordia. Monoclonal antibody directed against GPVI was generously provided by Dr. M. Jandrot‐Perrus (Paris, France). Apyrase grade VII, bovine thrombin, rhodamine 6G, and prostaglandin E1 were from Sigma‐Aldrich. Fibrinogen was obtained from HYPHEN BioMed SAS. Convulxin was purchased from Cryopep. D‐Phe‐Pro‐Arg chlormethylketone dihydrochloride (PPACK) was from Calbiochem‐VWR. Adenosine triphosphate (ATP) determination kit, Alexa Fluor 488‐labeled phalloidin, and Alexa Fluor secondary antibodies were from Molecular Probes. The rat monoclonal antibody against the activated form of mouse αIIbβ3 (JON/A‐Phycoerythrin PE), fluorescein isothiocyanate (FITC)‐labeled rat anti‐mouse GPVI monoclonal antibody (mAb; JAQ1), FITC‐labeled rat anti‐mouse CD62P (P‐selectin) mAb (Wug.E9), and FITC‐labeled rat anti‐mouse GPIb amAb (Xia.B2) were from emfret ANALYTICS. FITC‐labeled rat anti‐mouse integrin αIIb chain (CD41) mAb (MWReg30) was purchased from BD Biosciences. A rabbit polyclonal antibody was raised against the extended C‐terminal FLNa sequence of a patient by GenScript, from a mix of two synthetic FLNa peptides (APPSSPQPRPRPPWC and DGGPVHNHPLVLFSSQEEC) coupled to the carrier protein keyhole limpet hemocyanin, followed by affinity purification of the IgG fraction. The mAb specific for N‐terminal FLNa and the polyclonal antibody specific for phosphorylated linker for activation of T cells (LAT) were obtained from Millipore. The polyclonal antibodies against phosphorylated Syk and protein kinases C (PKCs) substrates were from Cell Signaling Technology. Peroxidase‐conjugated AffiniPure secondary antibodies were obtained from Jackson ImmunoResearch Laboratories, Inc.
2.2. Mouse strains
The mutant FlnA knock‐in mouse strain was generated by genOway. A construct was built (Figure 1A) by polymerase chain reaction (PCR), consisting of mouse FlnA exon 45 added to a polyadenylation site, and flanked with 3′ and 5′ loxP sites, followed by a neomycin cassette and a second mouse exon 45 where the STOP codon and the mouse 300 bp long 3′ downstream untranslated sequence were swapped for the mutated STOP (ct deletion, see Figure S1A in supporting information) followed by the human 300 bp 3′ untranslated sequence of the patient human FLNA gene. The human 3′ sequence was used instead of the mouse 3′ sequence because of the very poor homology between the two sequences, in particular at the peptide sequence level (see Figure S1). The construct was intended to be recombined after Cre expression with deletion of the wild‐type (WT) exon 45, leaving only the mutant human sequence‐extended exon 45 (Figure 1A). This cassette was recombined at the site of the WT exon 45 of the chromosome X‐linked mouse FlnA gene by CRISPR‐Cas9 technology on mouse embryonic stem cells. After injection into blastocysts, chimeric animals were selected for germ‐line transmission. Absence of leakage of recombinant FlnA was checked by real‐time PCR. Flna loxP females were then selected and crossed with PF4‐Cre male mice, to generate mice expressing platelet FlnA with the extended human C‐terminal sequence. Only hemizygous males were studied further.
FIGURE 1.
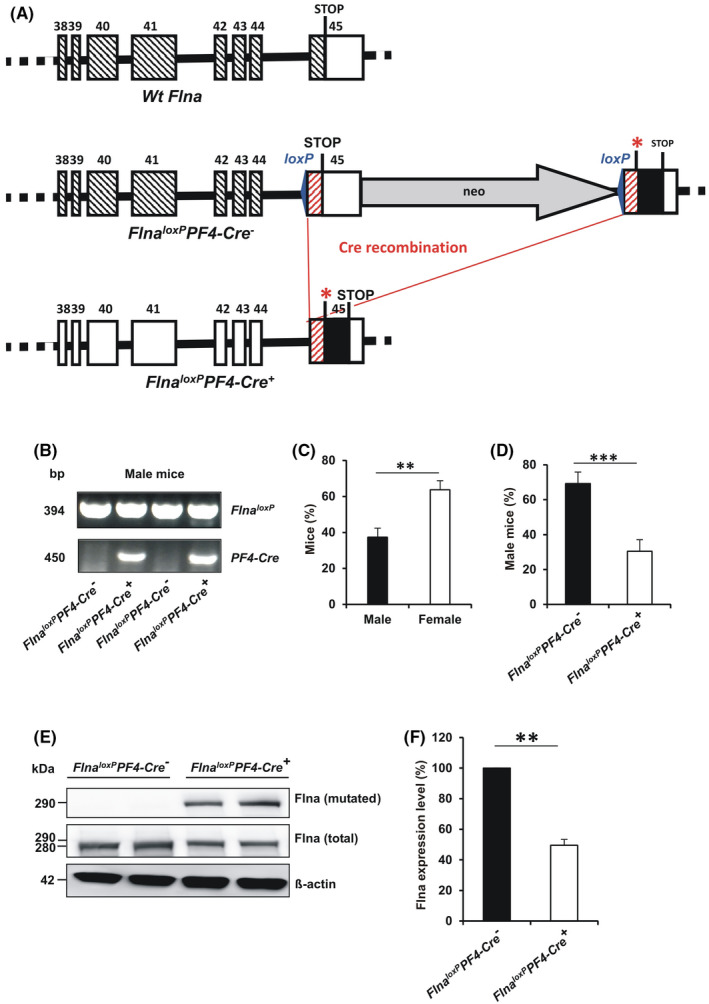
Schematic representation of the mutant humanized FlnA cloning strategy and FlnA expression in platelets of male mice. AThe region of the mouse Flna gene targeted for homologous recombination is shown. Flna WT includes exons from #38 to 45 (hashed boxes). Exon 45 shows the relative position of the stop codon and the non‐coding 3′ sequence of the exon is drawn as a white box. Flna loxP shows the FLNA gene after recombination of the exon38‐‐45 region with a construct including mouse exon 45 with an upstream lox P site (blue arrowhead), and followed by the neomycin (“neo”) negative selection gene and a second exon 45 harboring the downstream loxP site (blue arrowhead), the human stop mutation (red *) followed by the 3′ 300 bp human sequence (black box), which includes an in‐frame second stop codon, shown on the drawing. Flna loxP PF4‐Cre + shows the same exon 38–45 region after expression of the Cre recombinase in the platelet–megakaryocyte lineage: deletion of the loxP–loxP fragment (shown by the red lines and the “Cre recombination” mention), leads to swapping of the wild‐type exon 45 for the mutated one. B, PF4‐Cre + males were crossed with Flna LoxP females. PF4‐Cre genomic insertion in Flna loxP male offsprings was investigated by genomic polymerase chain reaction and agarose gel electrophoresis. C, Percentage of male over female mice during breeding. D, In a population of 200 mice, distribution of males and females and of Flna loxP PF4‐Cre + versus Flna loxP PF4‐Cre − males. E, F, FlnA expression in Flna loxP PF4‐Cre + and Flna loxP PF4‐Cre − platelets as assessed by western blotting. The graph represents the means ± standard error of the mean of four independent Flna loxP PF4‐Cre + male mice and four independent Flna loxP PF4‐Cre − male mice, **p < .01; ***p < .001 (unpaired Student t‐test)
All experimental procedures were carried out in accordance with the European legislation concerning the use of laboratory animals and approved by the ethical committee CEEA26 under the number Apafis #13732–2 018 092 708 484 303.
2.3. Isolation of mouse platelets
Mice were anesthetized by intraperitoneal injection of xylazin (10 mg/kg) and ketamine (100 mg/kg), then whole blood was collected by cardiac puncture into 80 μM PPACK and 10% ACD/C buffer (124 mM sodium citrate, 130 mM citric acid, 110 mM dextrose, pH 6.5). Isolated platelets were obtained as previously described. 26
2.4. Tail bleeding time
Bleeding time of anesthetized mice (10 weeks old) was determined by cutting off the tip of the tail (3 mm of distal mouse tail) and immediate immersion of the tail into 37°C isotonic saline (0.9% NaCl). Bleeding time was defined as the first cessation of bleeding for at least 60 s, to ensure that bleeding did not start again, otherwise it was considered rebleeding.
2.5. Hematologic analysis
Blood counts were determined with an automatic cell counter (Scil Vet abc Plus, Horiba Medical).
2.6. Flow cytometry
Washed platelets (3 × 108/ml) were stimulated with a range of activators (convulxin, ADP, or thrombin). After incubation for 10 min without stirring, platelets were diluted at 108/ml and incubated with the appropriate fluorophore‐conjugated antibodies for 20 min at room temperature. The samples were then directly analyzed with an Accuri C6+ flow cytometer (BD Biosciences).
2.7. Platelet aggregation
Platelet aggregation was monitored by measuring light transmission through the stirred suspension of washed platelets (3 × 108/ml) at 37°C using a Chronolog aggregometer. Platelet aggregation was triggered by adding fibrillar collagen, convulxin, ADP, or thrombin. Representative traces for aggregation were obtained from at least three independent experiments.
2.8. Measurement of platelet dense granule secretion
Dense granule secretion was evaluated by measuring ATP release with the ATP determination kit at the end of aggregation as previously described. 26 Briefly, platelet aggregation was stopped by adding ice‐cold ethylenediaminetetraacetic acid (EDTA; 16 mM) before centrifugation (12 000 g; 1 min). Supernatants were then incubated with recombinant firefly luciferase (0.25 mg/ml) and its substrate, D‐luciferin (0.5 mM), according to the manufacturer's instructions. Light emission was assessed with a luminometer (Fluoroskan Ascent FL; Thermo LabSystems). Dense granule secretion was expressed as the amount of ATP released (in picomoles) from 3 × 108 platelets.
2.9. Western blotting
Washed platelets (300 μl; 3 × 108/ml) were activated for 5 min at 37°C by various concentrations of Cvx (0 to 400 pM) in unstirred conditions and in the presence of apyrase (5 U/ml) and indomethacin (5 μM). Then, platelets were lysed in denaturing buffer (50 mM Tris, 100 mM NaCl, 50 mM NaF, 5 mM EDTA, 40 mM β‐glycerophosphate, 100 μM phenylarsine oxide, 1% sodium dodecyl sulfate [SDS], 5 μg/ml leupeptin, 10 μg/ml aprotinin, pH 7.4). Proteins were separated by SDS‐polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Membranes were incubated with primary antibodies followed by horseradish‐peroxidase‐labeled secondary antibodies. Immunoreactive bands were visualized with enhanced chemiluminescence detection reagents (Perbio Science) using a G:BOX Chemi XT16 Image System and quantified using Gene Tools version 4.03.05.0 (Syngene).
2.10. Spreading
Platelet spreading was performed on fibrinogen matrix (100 μg/ml). Platelets were incubated at 37°C for 30 min, fixed with 4% paraformaldehyde, permeabilized, and stained by incubation for 30 min with Alexa Fluor488–labeled phalloidin (0.3 μM). Then platelets were visualized under an epifluorescence microscope (Nikon, Eclipse 600). Cell surfaces were analyzed using Fiji software (rsb.info.nih.gov/ij).
2.11. Clot retraction
Clot retraction studies were performed at 37°C in aggregometer tubes. First, a 6% (w/v) polyacrylamide cushion was polymerized at the bottom of the tube to avoid clot adherence to the glass bottom. Then, washed platelets (300 μl at 3 × 108 platelets/ml) was added to fibrinogen 500 μg/ml, thrombin 2 U/ml, and Ca2+ 2 mM in the tubes. Clot retraction was monitored for 1 h.
2.12. In vitro thrombus formation under flow conditions
Thrombus formation was evaluated in a whole blood perfusion assay on a fibrillar collagen matrix (50 μg/ml) under arterial shear conditions (shear rate of 1500 s−1). Thrombus formation was quantified by assessment of the mean fluorescence intensity (MFI) of adherent thrombi.
2.13. Ferric chloride‐induced thrombosis model
Ferric chloride (FeCl3) injury was induced in 4‐ to 5‐week‐old mice, as previously described. 26 To facilitate visualization of thrombus formation, platelets from anesthetized mice were fluorescently labeled in vivo by injection of rhodamine 6G (3.3 mg/kg) into the retro‐orbital plexus. After topical deposition on the mesenteric vessels of an FeCl3 solution (10%), thrombus growth was monitored in real time with an inverted epifluorescent microscope (×10; Nikon Eclipse TE2000‐U). During the observation period, the number of full detachments of thrombi from the vessel wall at the injury site was recorded and counted as embolization events.
2.14. Statistical analysis
Statistical significance was evaluated with unpaired Student's t‐tests or one‐way analysis of variance followed by a post hoc test, as indicated in the figure legends. All analyses were performed with GraphPad Prism software (version 6, GraphPad Inc.).
3. RESULTS
3.1. Decreased expression level of mutated FlnA in platelets
Because the patient we reported previously is a male, thus hemizygous for the mutation of the X‐linked FLNA gene, the effect of the FlnA mutant was investigated on male mice expressing PF4‐Cre or not (Figure 1B). Only mutated FlnA was detected in Flna loxP PF4‐Cre + platelets whereas only WT FlnA was detected in Flna loxP PF4‐Cre − platelets. Note that the population of male mice was significantly lower than that of female mice (male mice: 37.4% ± 5.4% vs. female mice: 63.7% ± 5.4%; p = .003; Figure 1C) and that this difference was the result of a lower number of Flna loxP PF4‐Cre + male mice (30.6% ± 6.6%; p = 000.1) compared to Flna loxP PF4‐Cre − male mice (Figure 1D). During this study, we never noticed dead bodies of newborn mice suggesting strongly embryonic lethality. Western blotting using an antibody specific for the extended C‐terminal tail of the patient FLNa showed first that mutated FlnA was only detected in platelets of Flna loxP PF4Cre + mice (Figure 1E). In contrast, western blotting using an anti‐whole FLNa antibody showed that both Flna loxP PF4‐Cre + and Flna loxP PF4‐Cre − mice expressed FlnA in platelets but at a lower level in Flna loxP PF4‐Cre + platelets (49.5% ± 3.9%) compared to Flna loxP PF4‐Cre − platelets (100%; Figure 1E, F). In contrast, the expression level of FlnB was similar in Flna loxP PF4‐Cre + platelets compared to Flna loxP PF4‐Cre − platelets (Figure S2 in supporting information).
Moreover, analysis of the hematologic parameters showed a normal platelet count (Flna loxP PF4‐Cre −: 844 ± 32 × 109/L vs. Flna loxP PF4‐Cre +: 880 ± 44 × 109/L; n = 6) and normal platelet volume (Flna loxP PF4‐Cre −: 5.55 ± 0.07 fL vs. Flna loxP PF4‐Cre +: 5.50 ± 0.07 fL; n = 6; Table 1). Moreover, no difference was observed in the expression level of the major receptors (αIIbβ3, GPVI, and GPIbα) analyzed by flow cytometry and of granule content (von Willebrand factor) measured by western blotting (Figure S3 in supporting information).
TABLE 1.
Hematological parameters
| Flna loxP PF4‐Cre − | Flna loxP PF4‐Cre + | |
|---|---|---|
| Platelets (×109/L) | 844 ± 32 | 880 ± 44 |
| WBC (×109/L) | 8.1 ± 1.0 | 9.4 ± 0.4 |
| RBC (×1012/L) | 7.7 ± 0.8 | 9.4 ± 0.6 |
| Hematocrit (%) | 49.1 ± 1.5 | 47.2 ± 1.1 |
| MPV (fL) | 5.55 ± 0.07 | 5.50 ± 0.07 |
Abbreviations: MPV, mean platelet volume; RBC, red blood cells; WBC, white blood cells.
Altogether, these results show that this mouse platelet model with an extended human sequence at the FlnA C‐terminal tail specifically expressed in platelets reproduces the results obtained with the patient's platelets 20 with a normal platelet count and a lower level of mutated FLNa (patient: 30%, mice: 50%).
3.2. Unstable hemostasis in mutated FlnA mice
We next investigated the role of mutated FlnA in hemostasis by comparing tail bleeding times in Flna loxP PF4‐Cre − and Flna loxP PF4‐Cre + mice in vivo. Similar bleeding times were observed in Flna loxP PF4‐Cre − and Flna loxP PF4‐Cre + mice (Flna loxP PF4‐Cre −: 51.9 ± 6.3 s vs. Flna loxP PF4‐Cre +: 54.4 ± 6.7 s; Figure 2A). In contrast, rebleeding was observed in a large percentage of Flna loxP PF4‐Cre + mice (77%) compared to Flna loxP PF4‐Cre − mice (27%; Figure 2B) consistent with an instability of the thrombus in Flna loxP PF4‐Cre + mice.
FIGURE 2.
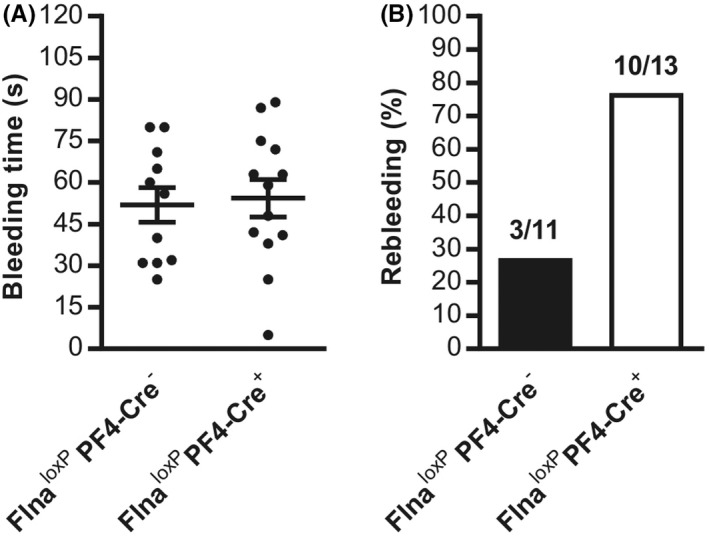
Mouse tail bleeding time. A, Tail bleeding was assessed in Flna loxP PF4‐Cre + (mutated FlnA) and Flna loxP PF4‐Cre − mice (wild‐type FlnA). B, Quantification of rebleedings is expressed as mean ± standard error of the mean of 11 Flna loxP PF4‐Cre − male mice and 13 Flna loxP PF4‐Cre + male mice
3.3. Mutated FlnA increases platelet aggregation induced by thrombin and GPVI agonists
The next step was to investigate the impact of the FlnA mutation on platelet function in vitro. First, platelet aggregation and dense granule secretion measured by the quantification of the ATP release was higher in Flna loxP PF4‐Cre + platelets compared to Flna loxP PF4‐Cre − platelets in conditions of low concentration of thrombin (40 mU/ml; aggregation: 77.1% ± 2.6% for Flna loxP PF4‐Cre + platelets vs. 63.1% ± 3.3% for Flna loxP PF4‐Cre − platelets, p = .007; secretion: 20.1 ± 0.5 pmoles for Flna loxP PF4‐Cre + platelets vs. 14.4 ± 0.2 pmoles for Flna loxP PF4‐Cre − platelets, p < .001 n = 5; Figure 3A–C). In contrast, in conditions of unstirred platelets after thrombin activation, no difference between Flna loxP PF4‐Cre − platelets and Flna loxP PF4‐Cre + platelets was observed for αIIbβ3 integrin activation and α granule secretion, measured by flow cytometry with a rat mAb JON/A(PE) specific for the activated conformation of the mouse integrin and a FITC‐labeled rat anti‐mouse CD62P mAb for P‐selectin expression (Figure 3D,E) suggesting that outside‐in signaling and/or shear were essential for the enhanced effect observed in Flna loxP PF4‐Cre + platelets after thrombin activation. Finally, aggregation and αIIbβ3 activation induced by another agonist of G protein‐coupled receptor such as ADP (5 to 20 μM) were normal (Figure S4 in supporting information). These results show that mutated FlnA may affect platelet activation induced by G‐coupled receptors.
FIGURE 3.
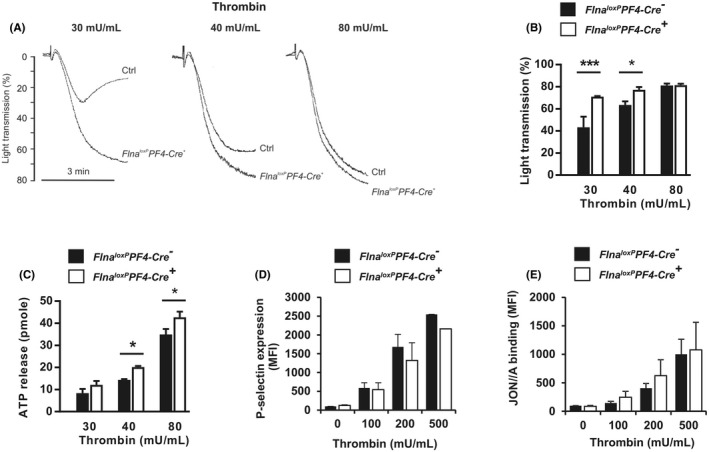
Aggregation, secretion, and αIIbβ3 activation induced by thrombin. A, B, Aggregation of washed Flna loxP PF4‐Cre − (Ctrl) and Flna loxP PF4‐Cre + platelets was induced by thrombin (30, 40, and 80 mU/ml). C, Dense granule secretion measured after 3 min of platelet aggregation was quantified by measuring adenosine triphosphate release. D, E, Quantification of P‐selectin exposure and αIIbβ3 activation in conditions of unstirred platelets was assessed by flow cytometry using an anti‐P‐selectin antibody and the JON/A monoclonal antibody specific for the activated form of αIIbβ3. The graph represents the means ± standard error of the mean of three independent determinations. *p < .05, ***p < .001 (one‐way analysis of variance with Sidak's post hoc test for multiple comparisons)
Then the role of mutated FlnA was investigated in platelet activation induced by GPVI, the collagen receptor. First, in Flna loxP PF4‐Cre + platelets, aggregation induced by different concentrations of agonists (Cvx: 100 to 400 pM; Figure 4A,B) and collagen (2 to 4 μg/ml; Figure 4F,G) was significantly increased but only with agonists at low concentration (Cvx: 100 pM; p = .02 and collagen 2 and 3 μg/ml; p = .002 p = .006, respectively). In parallel, ATP secretion from dense granules in conditions of platelet aggregation was also significantly increased with Cvx (100 pM) reaching 18.8 pmoles for Flna loxP PF4‐Cre + platelets versus 3.0 pmoles for Flna loxP PF4‐Cre − platelets (p < .0001) and collagen (2 μg/ml: Flna loxP PF4‐Cre +: 10.4 ± 0.2 pmoles vs. Flna loxP PF4‐Cre −: 9.2 ± 0.3 pmoles, p = .02; 3 μg/ml: Flna loxP PF4‐Cre +: 11.5 ± 0.3 pmoles vs. Flna loxP PF4‐Cre −: 9.5 ± 0.3 pmoles, p = .002; Figure 4C‐H). Note the plateau of ATP secretion was obtained at 200 pM Cvx for both Flna loxP PF4‐Cre − and Flna loxP PF4‐Cre + platelets (Figure 4C). Furthermore, the activation of αIIbβ3 measured with mAb JON/A in flow cytometry showed a significant increase of JON/A binding (p = .02) in Flna loxP PF4‐Cre + platelets at Cvx 400 pM (Flna loxP PF4‐Cre +: 2157.5 ± 49.8 MFI vs. Flna loxP PF4‐Cre −: 1519.1 ± 199.7 MFI; Figure 4D). Finally, the quantification of P‐selectin exposure induced by various Cvx concentrations (0 to 400 pM) in unstirred conditions confirmed the gain of function of mutated FlnA (Figure 4E). Note that no activation of αIIbβ3 was shown when platelets were activated by collagen in contrast to Cvx. The unstirring conditions of flow cytometry are not compatible with fibrillar collagen activation.
FIGURE 4.
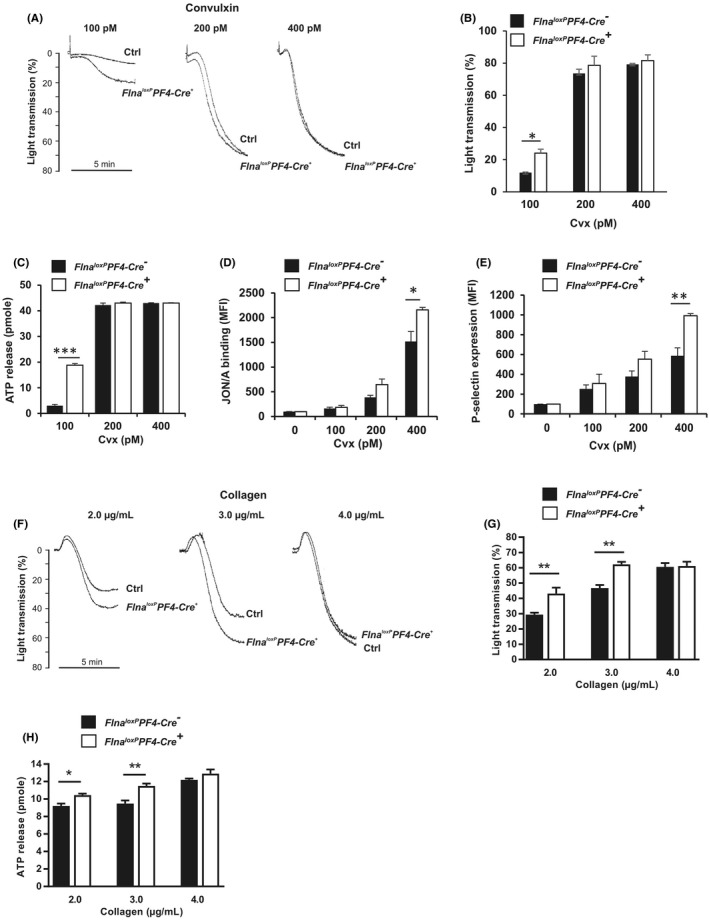
Aggregation, secretion, and αIIbβ3 activation induced by convulxin or collagen. A, B, F, G, Platelet aggregations of washed platelets were initiated by various doses of Cvx (100 to 400 pM) and collagen (2 to 4 μg/ml). C, H, Dense granule secretion measured after 3 min of platelet aggregation was quantified by assessing adenosine triphosphate release. D, E, Quantification of P‐selectin exposure and αIIbβ3 activation in conditions of unstirred platelets was assessed by flow cytometry using an anti‐P‐selectin antibody and the JON/A monoclonal antibody specific for the activated form of αIIbβ3. The graph represents the means ± standard error of the mean of four independent determinations. *p < .05, **p < .01, ***p < .001 (one‐way analysis of variance with Sidak's post hoc test for multiple comparisons)
We then assessed GPVI signaling induced by various concentrations of Cvx (0 to 400 pM) in unstirred conditions and in the presence of apyrase and indomethacin. The phosphorylation of two proteins (Syk and LAT) essential in GPVI signaling was tested by western blotting. Indeed, the phosphorylation of Syk was shown to be dependent on its interaction with FlnA. 6 In Flna loxP PF4‐Cre + platelets, Syk phosphorylation was significantly decreased after activation by 200 pM (p = .007) and 400 pM Cvx (p < .0001; Figure 5A), as well as LAT phosphorylation after activation by 400 pM Cvx (p < .001; Figure 5B). These decreases in Syk and LAT phosphorylation reproduce the results obtained with the patient's platelets, and are probably the consequence of the low platelet level of FlnA (50% and 30%, respectively).
FIGURE 5.
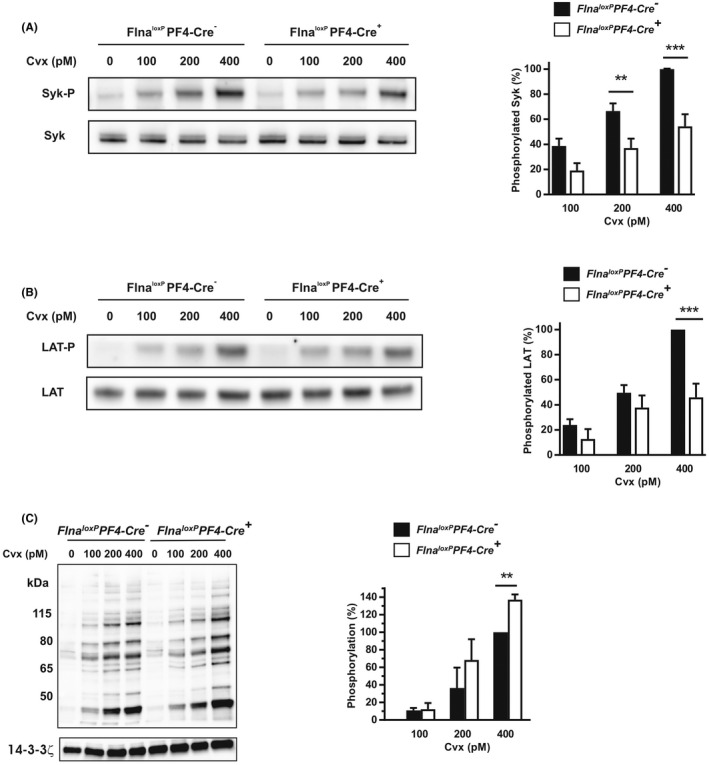
Cell signaling induced by convulxin. Western blotting of phosphorylated Syk (A), LAT (B), and PKC substrates (C) of platelets activated for 5 min by various doses of Cvx (100 to 400 pM) in the presence of apyrase (5 U/ml) and indomethacin (5 μM). Phosphorylation levels were normalized in relation to the expression level of total Syk (A), total LAT (B) of 14–3‐3ζ (C), and then compared with each other to the phosphorylation of Flna loxP PF4‐Cre − (Ctrl) at 400 pM Cvx which was set to 100%. The graphs represent the means of phosphorylation ± standard error of the mean of at least three independent determinations. **p < .01, ***p < .001 (one‐way analysis of variance with Sidak's post hoc test for multiple comparisons)
Finally, we decided to test the activation of PKCs, previously shown to be involved in secretion, 27 , 28 by measuring phosphorylated PKC substrates induced by Cvx (0 to 400 pM) in unstirred conditions (Figure 5C). The intensity of phosphorylated bands was significantly increased by 37% (p = .004) at 400 pM Cvx in Flna loxP PF4‐Cre + platelets compared to Flna loxP PF4‐Cre − platelets indicating that mutated FlnA increased the PKC activity. The results indicate that mutated FlnA does not affect proximal GPVI signaling but increases PKC activities required for platelet secretion.
3.4. Mutated FlnA induces increased outside‐in signaling
We next addressed the question of the impact of mutated FlnA on outside‐in signaling. First, platelet spreading on fibrinogen matrix was examined. Platelet area of Flna loxP PF4‐Cre + mice was slightly but significantly increased (127% ± 12%; p = .03) compared to platelet area of Flna loxP PF4‐Cre − mice (100%; Figure 6A). Then, we examined the role of mutated FlnA on clot retraction (Figure 6B‐C). Clot retraction of Flna loxP PF4‐Cre + platelets was significantly accelerated comparatively to Flna loxP PF4‐Cre − platelets, reaching 40.3 ± 4.1% (p = .04) versus 25.7% ± 3.9% for Flna loxP PF4‐Cre− platelets after 10 min, consistent with mutated FlnA upregulating clot retraction and outside‐in signaling in the absence of shear.
FIGURE 6.
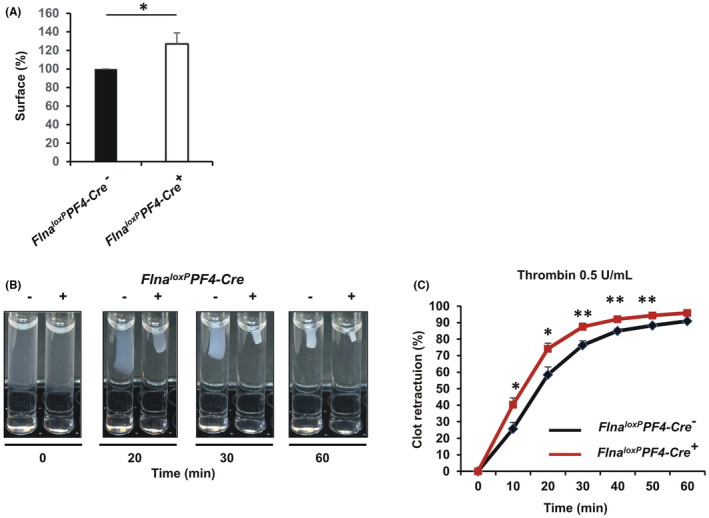
Outside‐in signaling of washed platelets. A, Platelet spreading was performed on a fibrinogen matrix (100 μg/ml). Platelets were incubated at 37°C for 30 min and then stained with Alexa Fluor488‐labeled phalloidin (0.3 μM). Platelets were visualized under an epifluorescence microscope (Nikon, Eclipse 600). Cell surfaces were analyzed using the Fiji software. The graph represents the means ± standard error of the mean (SEM) of four independent determinations. *p < .05, (unpaired Student t‐test). B, Clot retraction of washed platelets was initiated by adding fibrinogen (500 μg/ml) and thrombin (0.5 U/ml). Photographs were taken at different times and (C) the area of clot was measured using the Fiji software. The graph represents the means ± SEM of four independent determinations. *p < .05, **p < .01 (unpaired Student t‐test)
3.5. Mutated FlnA induces thrombus instability in vivo
We next investigated the effect of mutated FlnA by assessing thrombus formation in vitro and in vivo. In vitro, thrombus formation was assessed on a collagen matrix at arterial shear rate (1500s−1). Thrombus size was similar in Flna loxP PF4‐Cre + mice and in Flna loxP PF4‐Cre − mice (Figure 7A). Then in vivo thrombosis induced by ferric chloride on exposed mesenteric vessels was assessed. The initial thrombus was formed much more rapidly in venules of Flna loxP PF4‐Cre + mice (5.4 ± 1.8 min; p = .0013) than in venules of Flna loxP PF4‐Cre − mice (14.6 ± 1.2 min; Figure 7B). In contrast, no difference was found in arterioles. Surprisingly, if the occlusion times were similar between Flna loxP PF4‐Cre + and Flna loxP PF4‐Cre − mice in arterioles and in venules (Figure 7C), thrombus instability was observed in arterioles of all Flna loxP PF4‐Cre + mice (100%) compared to only 14% of Flna loxP PF4‐Cre − mice (Figure 7D). These results indicate that mutated FlnA affects in vivo thrombus stability but not thrombus size and occlusion.
FIGURE 7.
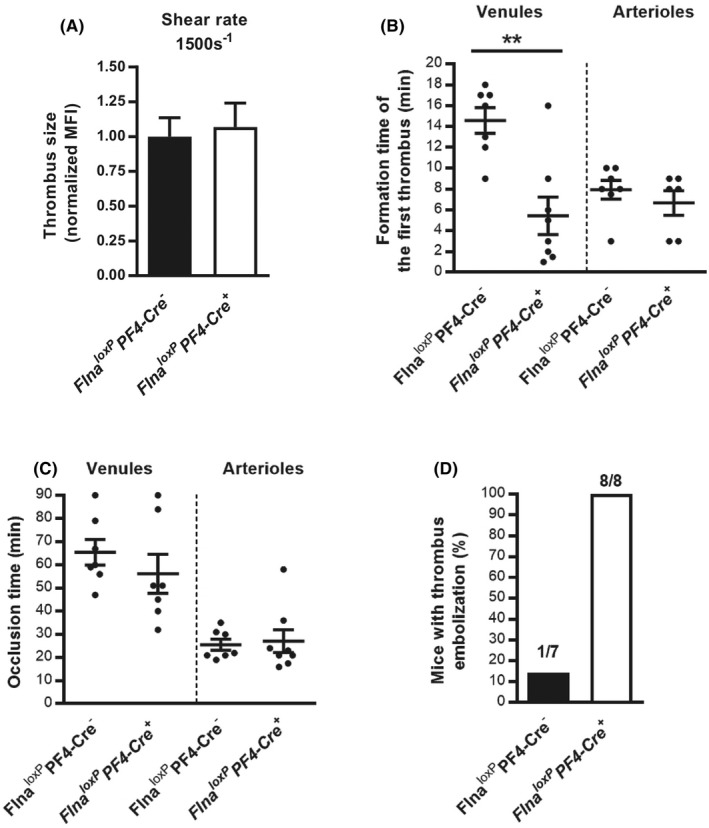
In vivo thrombus instability of Flna loxP PF4‐Cre + mice. The impact of the FlnA mutation on thrombus formation was investigated in vitro. A, A whole‐blood perfusion assay was carried out on collagen matrix (50 μg/ml) at an arterial shear rate (1500 s−1). Thrombus formation was evaluated by fluorescence microscopy and the thrombus size was quantified by measurement of the mean fluorescence intensity (MFI) of thrombi. B‐D, In vivo thrombosis was assessed in exposed mesenteric vessels (venules and arterioles) after FeCl3‐induced injury. Thrombus formation of fluorescently labeled platelets was monitored by intravital videomicroscopy. The graphs represent (B) the time point of the formation of the first thrombus of 30 μm, (C) the occlusion time defined as the stopping blood flow for at least 1 min, and (D) the number of emboli shedding from thrombi in venules throughout the procedure. The graph represents the means ± standard error of the mean of eight Flna loxP PF4‐Cre + and seven Flna loxP PF4‐Cre − mice. Statistical difference between Flna loxP PF4‐Cre − mice and Flna loxP PF4‐Cre + mice was evaluated by the unpaired Student t‐test (**p < .01)
4. DISCUSSION
In this study, we have shown that the expression in platelets of FlnA mutated in the C‐terminal tail leads in vitro to a gain‐of‐function counteracted by thrombus instability in vivo. The gain‐of‐functions was translated by an increased platelet aggregation induced by low concentrations of agonists, increased spreading, and clot retraction. In contrast, if the time of the first thrombus formation in vivo was shorter in Flna loxP PF4‐Cre + mice, unexpectedly, thrombosis in vivo was characterized by emboli formation in arterioles.
We found that the expression level of mutant FlnA (49% of total FlnA) in mice reproduces the expression level of mutant FLNa (30%) previously described in a male patient with the same mutation and exhibiting a PVNH and a congenital intestinal pseudo‐obstruction. 20 Moreover, normal platelet count and normal platelet volume, both described in the patient 20 and in mice, indicate that this mouse platelet model with the extended sequence in the C‐terminal tail present in platelets reproduces the results obtained with the male patient's platelets. 20
Platelet aggregation and αIIbβ3 activation induced by low concentrations of thrombin, collagen, and Cvx were upregulated in Flna loxP PF4‐Cre + platelets. In the patient, a gain of platelet aggregation was also observed with low concentrations of Cvx (aggregation and αIIbβ3 activation induced by thrombin and collagen as well as αIIbβ3 activation induced by Cvx were not performed in the patient). 20 Again, and as found in the patient, the first steps of GPVI signaling (Syk and LAT phosphorylations) were not at the origin of increased platelet GPVI signaling. The phosphorylation of Syk and LAT was decreased and probably the result of a lower level of FlnA (49%) and consequently lower interaction of mutated FlnA with Syk required for GPVI signaling. 6 In contrast, in the advanced steps of GPVI signaling, PKC activities measured by the phosphorylation of PKC substrates were upregulated by mutated FlnA. This upregulation could explain the increased Flna loxP PF4‐Cre + platelet secretion. A possible mechanistic explanation could be that, because the interaction of FLNa with PKCs and notably with PKCα, may be direct, 5 , 29 and because PKCs are mostly cytosolic, the FLNa/PKCα interaction increases PKC translocation to the plasma membrane, upon activation. Further studies will be required to explore this hypothesis.
Surprisingly, in contrast to the patient, platelet aggregation and αIIbβ3 activation induced by ADP were not upregulated in mice. If the platelet cascade is genetically well conserved between humans and mice, some differences have been reported. 30 For example, purinergic receptor signaling regulated by the ADP receptor P2Y12 is clearly higher in mice compared to humans. 30 , 31 This difference could be the cause of a difference observed in ADP‐induced platelet signaling between FLNa mutated human versus mouse platelets.
Whereas FlnA full ablation in mice resulted in a diminution in the level of GPIb‐IX‐V complex, 6 a level of 49% of mutated FlnA, as seen in our model, did not affect the expression level of GPIbα. This is most likely due to the partial diminution of FlnA, but we cannot rule out a possible gain‐of‐function effect of the mutation on GPIb‐IX‐V trafficking.
Finally, spreading on fibrinogen and clot retraction were also upregulated in Flna loxP PF4‐Cre + mice. These upregulations could be the result of the FlnA mutation per se and/or of the different levels of FlnA in Flna loxP PF4‐Cre − mice (100%) versus Flna loxP PF4‐Cre + mice (49%). The only way to answer this question is to design a new mouse model expressing the same level of FlnA in both Flna loxP PF4‐Cre − and Flna loxP PF4‐Cre + mice.
In vivo results are highly contrasted with in vitro results. A rebleeding was observed in a large percentage of Flna loxP PF4‐Cre + mice (77%) compared to Flna loxP PF4‐Cre − mice (27%) indicating thrombus instability in mutated FlnA mice. Moreover, the in vivo thrombosis model induced by ferric chloride clearly showed the presence of embolies in arterioles (not in venules) of all Flna loxP PF4‐Cre + mice (100%) compared to only 14% of Flna loxP PF4‐Cre − mice. The apparent discrepancy between a gain‐of‐function in vitro and thrombus instability in vivo is interesting. A defect of platelet force generation, cytoskeletal reorganization, or fibrin formation are indeed hypotheses to study and require a full study in another paper. αIIbβ3 activation is regulated by talin, kindlin‐3, and FLNa interactions with β3 integrin where talin and kindlin‐3 synergistically coactivate αIIbβ3 32 , 33 , 34 : talin modulating αIIbβ3 affinity and kindlin‐3 modulating clustering and avidity. 33 In our thrombosis model, a defect in αIIbβ3 clustering required for cytoskeleton reorganization could be the cause of the thrombus instability. Non‐exclusive with the first hypothesis, cytoskeleton remodeling per se in response to mechanical forces could be affected by mutated FLNa. FLNa, described as an actin‐crosslinking protein, is known to interact with 90 binding partners. 35 In addition to the dimerization domain, 36 the Ig repeats 24 and 23 of FLNa interact with different partners involved in cytoskeleton remodeling, notably the Rho GTPases (Ral1, Cdc42, Rac, and Rho) 37 , 38 and the GTPase FilGAP, specific for Rac. Dissociation occurs when FLNa–actin networks are subjected to mechanical shear strain. 35 In our model, we do not know the incidence of the extended (100 amino acids) C‐terminal region of mutated FLNa on the dimerization and on the interaction partners with Ig repeat 24 and 23 domains and with the extended C‐terminal region. Further experiments (proteomic analyses) will be required to identify new partners.
Finally, does the thrombus instability in knock‐in mice correspond to clinical conditions for the patient? For obvious ethical reasons, the patient was not explored with in vivo tests. However, he underwent several surgery interventions, but for which the patient was unaware of any hemostatic outcomes. However, one must note that first, there may have been some bleeding, but not mentioned to the patient, if not dramatic; second, human surgery and mouse tail bleeding time may not be compared, because they are conducted in different conditions (different anesthetics, challenge of the wound [mouse] or not [surgery], use of local procoagulation during surgery, and so on).
In conclusion, this study shows evidence for thrombus instability of mutated FlnA and raises a possible cardiovascular risk in the patient with this FLNA mutation.
AUTHOR CONTRIBUTIONS
F.A., A.K., L.L., J.S., C.S., and C.R., performed experiments; F.A., A.K., and M.B. designed experiments; F.A., A.K., H.R., J.‐P.R., and M.B wrote the manuscript.
FUNDING INFORMATION
This work was supported by INSERM and by a grant from Fondation pour la Recherche Médicale (LPC20170637458).
CONFLICTS OF INTERESTS
The authors declare no competing financial interests.
Supporting information
Figure S1
{kind=link}
Figure S2
{kind=link}
Figure S3
{kind=link}
Figure S4
{kind=link}
Adam F, Kauskot A, Lamrani L, et al. A gain‐of‐function filamin A mutation in mouse platelets induces thrombus instability. J Thromb Haemost. 2022;20:2666‐2678. doi: 10.1111/jth.15864
Manuscript handled by: Katsue Suzuki‐Inoue
Final decision: Andreas Greinacher, 22 August 2022
REFERENCES
- 1. Stossel TP, Condeelis J, Cooley L, et al. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol. 2001;2(2):138‐145. [DOI] [PubMed] [Google Scholar]
- 2. Nakamura F, Osborn TM, Hartemink CA, Hartwig JH, Stossel TP. Structural basis of filamin a functions. J Cell Biol. 2007;179(5):1011‐1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goetsch SC, Martin CM, Embree LJ, Garry DJ. Myogenic progenitor cells express filamin C in developing and regenerating skeletal muscle. Stem Cells Dev. 2005;14(2):181‐187. [DOI] [PubMed] [Google Scholar]
- 4. Vorgerd M, van der Ven PF, Bruchertseifer V, et al. A mutation in the dimerization domain of filamin C causes a novel type of autosomal dominant myofibrillar myopathy. Am J Hum Genet. 2005;77(2):297‐304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nakamura F, Stossel TP, Hartwig JH. The filamins: organizers of cell structure and function. Cell Adh Migr. 2011;5(2):160‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Falet H, Pollitt AY, Begonja AJ, et al. A novel interaction between FlnA and Syk regulates platelet ITAM‐mediated receptor signaling and function. J Exp Med. 2010;207(9):1967‐1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nakamura F, Pudas R, Heikkinen O, et al. The structure of the GPIb‐filamin a complex. Blood. 2006;107(5):1925‐1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feng S, Lu X, Kroll MH. Filamin A binding stabilizes nascent glycoprotein Ibalpha trafficking and thereby enhances its surface expression. J Biol Chem. 2005;280(8):6709‐6715. [DOI] [PubMed] [Google Scholar]
- 9. Kiema T, Lad Y, Jiang P, et al. The molecular basis of filamin binding to integrins and competition with Talin. Mol Cell. 2006;21(3):337‐347. [DOI] [PubMed] [Google Scholar]
- 10. Liu J, Das M, Yang J, et al. Structural mechanism of integrin inactivation by filamin. Nat Struct Mol Biol. 2015;22(5):383‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lamrani L, Adam F, Soukaseum C, et al. New insights into regulation of αIIbβ3 integrin signaling by filamin A Res Pract Thromb Haemost . 2022;(in press). [DOI] [PMC free article] [PubMed]
- 12. Begonja AJ, Pluthero FG, Suphamungmee W, et al. FlnA binding to PACSIN2 F‐BAR domain regulates membrane tubulation in megakaryocytes and platelets. Blood. 2015;126(1):80‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lopez JJ, Albarran L, Jardin I, et al. Filamin A modulates store‐operated ca(2+) entry by regulating STIM1 (stromal interaction molecule 1)‐Orai1 Association in Human Platelets. Arterioscler Thromb Vasc Biol. 2018;38(2):386‐397. [DOI] [PubMed] [Google Scholar]
- 14. Donada A, Balayn N, Sliwa D, et al. Disrupted filamin A/alphaIIbbeta3 interaction induces macrothrombocytopenia by increasing RhoA activity. Blood. 2019;133(16):1778‐1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wade EM, Halliday BJ, Jenkins ZA, O'Neill AC, Robertson SP. The X‐linked filaminopathies: synergistic insights from clinical and molecular analysis. Hum Mutat. 2020;41(5):865‐883. [DOI] [PubMed] [Google Scholar]
- 16. Lange M, Kasper B, Bohring A, et al. 47 patients with FLNA associated periventricular nodular heterotopia. Orphanet J Rare Dis. 2015;10:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Parrini E, Ramazzotti A, Dobyns WB, et al. Periventricular heterotopia: phenotypic heterogeneity and correlation with filamin A mutations. Brain. 2006;129(Pt 7):1892‐1906. [DOI] [PubMed] [Google Scholar]
- 18. Sheen VL, Dixon PH, Fox JW, et al. Mutations in the X‐linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Hum Mol Genet. 2001;10(17):1775‐1783. [DOI] [PubMed] [Google Scholar]
- 19. Berrou E, Adam F, Lebret M, et al. Heterogeneity of platelet functional alterations in patients with filamin A mutations. Arterioscler Thromb Vasc Biol. 2013;33(1):e11‐e18. [DOI] [PubMed] [Google Scholar]
- 20. Berrou E, Adam F, Lebret M, et al. Gain‐of‐function mutation in filamin A potentiates platelet integrin alphaIIbbeta3 activation. Arterioscler Thromb Vasc Biol. 2017;37(6):1087‐1097. [DOI] [PubMed] [Google Scholar]
- 21. Kasper BS, Kurzbuch K, Chang BS, et al. Paternal inheritance of classic X‐linked bilateral periventricular nodular heterotopia. Am J Med Genet A. 2013;161A(6):1323‐1328. [DOI] [PubMed] [Google Scholar]
- 22. Sole G, Coupry I, Rooryck C, et al. Bilateral periventricular nodular heterotopia in France: frequency of mutations in FLNA, phenotypic heterogeneity and spectrum of mutations. J Neurol Neurosurg Psychiatry. 2009;80(12):1394‐1398. [DOI] [PubMed] [Google Scholar]
- 23. Sheen VL, Walsh CA. Periventricular heterotopia: new insights into Ehlers‐Danlos syndrome. Clin Med Res. 2005;3(4):229‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oegema R, Hulst JM, Theuns‐Valks SD, et al. Novel no‐stop FLNA mutation causes multi‐organ involvement in males. Am J Med Genet A. 2013;161A(9):2376‐2384. [DOI] [PubMed] [Google Scholar]
- 25. Rosa JP, Raslova H, Bryckaert M. Filamin A: key actor in platelet biology. Blood. 2019;134(16):1279‐1288. [DOI] [PubMed] [Google Scholar]
- 26. Adam F, Kauskot A, Nurden P, et al. Platelet JNK1 is involved in secretion and thrombus formation. Blood. 2010;115(20):4083‐4092. [DOI] [PubMed] [Google Scholar]
- 27. Gilio K, Harper MT, Cosemans JM, et al. Functional divergence of platelet protein kinase C (PKC) isoforms in thrombus formation on collagen. J Biol Chem. 2010;285(30):23410‐23419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Konopatskaya O, Gilio K, Harper MT, et al. PKCalpha regulates platelet granule secretion and thrombus formation in mice. J Clin Invest. 2009;119(2):399‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tigges U, Koch B, Wissing J, Jockusch BM, Ziegler WH. The F‐Actin cross‐linking and focal adhesion protein filamin A is a ligand and in vivo substrate for protein kinase C alpha. J Biol Chem. 2003;278(26):23561‐23569. [DOI] [PubMed] [Google Scholar]
- 30. Balkenhol J, Kaltdorf KV, Mammadova‐Bach E, et al. Comparison of the central human and mouse platelet signaling cascade by systems biological analysis. BMC Genomics. 2020;21(1):897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zeiler M, Moser M, Mann M. Copy number analysis of the murine platelet proteome spanning the complete abundance range. Mol Cell Proteomics. 2014;13(12):3435‐3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Harburger DS, Bouaouina M, Calderwood DA. Kindlin‐1 and ‐2 directly bind the C‐terminal region of beta integrin cytoplasmic tails and exert integrin‐specific activation effects. J Biol Chem. 2009;284(17):11485‐11497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ye F, Petrich BG, Anekal P, et al. The mechanism of kindlin‐mediated activation of integrin alphaIIbbeta3. Curr Biol. 2013;23(22):2288‐2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu Z, Chen X, Zhi H, et al. Direct interaction of kindlin‐3 with integrin alphaIIbbeta3 in platelets is required for supporting arterial thrombosis in mice. Arterioscler Thromb Vasc Biol. 2014;34(9):1961‐1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ehrlicher AJ, Nakamura F, Hartwig JH, Weitz DA, Stossel TP. Mechanical strain in Actin networks regulates FilGAP and integrin binding to filamin A. Nature. 2011;478(7368):260‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sim DW, Lee YS, Kim JH, Seo MD, Lee BJ, Won HS. HP0902 from helicobacter pylori is a thermostable, dimeric protein belonging to an all‐beta topology of the cupin superfamily. BMB Rep. 2009;42(6):387‐392. [DOI] [PubMed] [Google Scholar]
- 37. Ohta Y, Suzuki N, Nakamura S, Hartwig JH, Stossel TP. The small GTPase RalA targets filamin to induce filopodia. Proc Natl Acad Sci USA. 1999;96(5):2122‐2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Del Valle‐Perez B, Martinez VG, Lacasa‐Salavert C, et al. Filamin B plays a key role in vascular endothelial growth factor‐induced endothelial cell motility through its interaction with Rac‐1 and Vav‐2. J Biol Chem. 2010;285(14):10748‐10760. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4