

Abstract
Background and purpose
Patients with neuromuscular conditions are at increased risk of suffering perioperative complications related to anaesthesia. There is currently little specific anaesthetic guidance concerning these patients. Here, we present the European Neuromuscular Centre (ENMC) consensus statement on anaesthesia in patients with neuromuscular disorders as formulated during the 259th ENMC Workshop on Anaesthesia in Neuromuscular Disorders.
Methods
International experts in the field of (paediatric) anaesthesia, neurology, and genetics were invited to participate in the ENMC workshop. A literature search was conducted in PubMed and Embase, the main findings of which were disseminated to the participants and presented during the workshop. Depending on specific expertise, participants presented the existing evidence and their expert opinion concerning anaesthetic management in six specific groups of myopathies and neuromuscular junction disorders. The consensus statement was prepared according to the AGREE II (Appraisal of Guidelines for Research & Evaluation) reporting checklist. The level of evidence has been adapted according to the SIGN (Scottish Intercollegiate Guidelines Network) grading system. The final consensus statement was subjected to a modified Delphi process.
Results
A set of general recommendations valid for the anaesthetic management of patients with neuromuscular disorders in general have been formulated. Specific recommendations were formulated for (i) neuromuscular junction disorders, (ii) muscle channelopathies (nondystrophic myotonia and periodic paralysis), (iii) myotonic dystrophy (types 1 and 2), (iv) muscular dystrophies, (v) congenital myopathies and congenital dystrophies, and (vi) mitochondrial and metabolic myopathies.
Conclusions
This ENMC consensus statement summarizes the most important considerations for planning and performing anaesthesia in patients with neuromuscular disorders.
Keywords: anaesthesia, malignant hyperthermia, myopathy, neuromuscular disorders, perioperative care
This consensus statement summarizes the most important recommendations concerning anaesthesia in patients with neuromuscular disorders.
INTRODUCTION
Patients with neuromuscular disorders (NMDs) are at increased risk of peri‐operative complications related to anaesthesia. The reasons for this include associated cardiorespiratory morbidity, altered pharmacodynamics of anaesthetics and neuromuscular blocking agents (NMBAs), impaired temperature and glucose regulation, and specific risks associated with certain underlying genetic defects, or a combination of the above [1, 2, 3].
Whereas in recent years comprehensive standards of care have been formulated for the more common NMDs such as Duchenne muscular dystrophy (DMD) [4] and spinal muscular atrophy (SMA) [5], there is lack of consensus guidance focusing on the anaesthetic management of NMDs throughout the range of neuromuscular conditions. Neurologists are often asked to share the specific considerations regarding their neuromuscular patients requiring surgery, anaesthesia, or procedural sedation. Furthermore, neurologists who provide follow‐up for patients with NMDs are well placed to alert them to any perioperative risks specifically associated with their NMD and the particular precautions to be taken in close liaison with anaesthesiologists [1].
We therefore present the European Neuromuscular Centre (ENMC) consensus statement on anaesthesia in patients with NMDs as discussed and formulated during the 259th ENMC Workshop on Anaesthesia in Neuromuscular Disorders [6]. This consensus statement provides a comprehensive and accessible overview with the aim of guiding neurologists and anaesthesiologists, improving perioperative safety, and optimizing risk assessment and appropriate management of these patients.
METHODS
Our approach to developing this consensus statement was performed according to the AGREE II (Appraisal of Guidelines for Research & Evaluation) reporting checklist [7].
ENMC workshop
For the 259th ENMC Workshop on Anaesthesia in Neuromuscular Disorders, 30 international experts in the field of (paediatric) anaesthesia, neurology, and genetics were selected by the workshop organizers (N.C.V., M.M.J.S., S.R., H.J.) based on their particular expertise in anaesthesia and/or NMDs and geographic balance. The ENMC research board approved the participants list. The virtual workshop consisted of three parts:
Anaesthetic management of various NMDs (11 December 2020);
New developments in the field of malignant hyperthermia (MH) and increasing awareness (28 May 2021); and
Genetic counselling in patients with MH, rhabdomyolysis, and related congenital myopathies (29 May 2021).
The comprehensive workshop report has been published separately [6] but did not allow for the inclusion of the full anaesthesia considerations included in this paper.
In preparation for the ENMC workshop, the organizers conducted a literature search covering the period 1 January 2000 to 14 July 2021 in PubMed and Embase using the methodological framework for scoping reviews [8]. The main findings of this literature search were disseminated to the participants and presented during the first part of the workshop. Furthermore, six couples of workshop participants were invited to present the existing evidence and their expert opinion for anaesthetic management in specific groups of NMDs (see below). These participants provided a written summary of their recommendations, which were merged into a preliminary version of this consensus statement. The presenters were subsequently invited to participate in a virtual meeting to discuss the preliminary consensus statement. After the initial evaluation, a revised version was distributed as a basis for discussion during the second part of the ENMC workshop. Subsequently, the preliminary consensus statement document was modified according to the discussion at the second workshop session and distributed by email. Consensus regarding the consensus statement content was reached after further communication by email. Finally, the consensus statement content was subjected to a modified Delphi process [9]. The whole process description is summarized in Figure 1.
FIGURE 1.
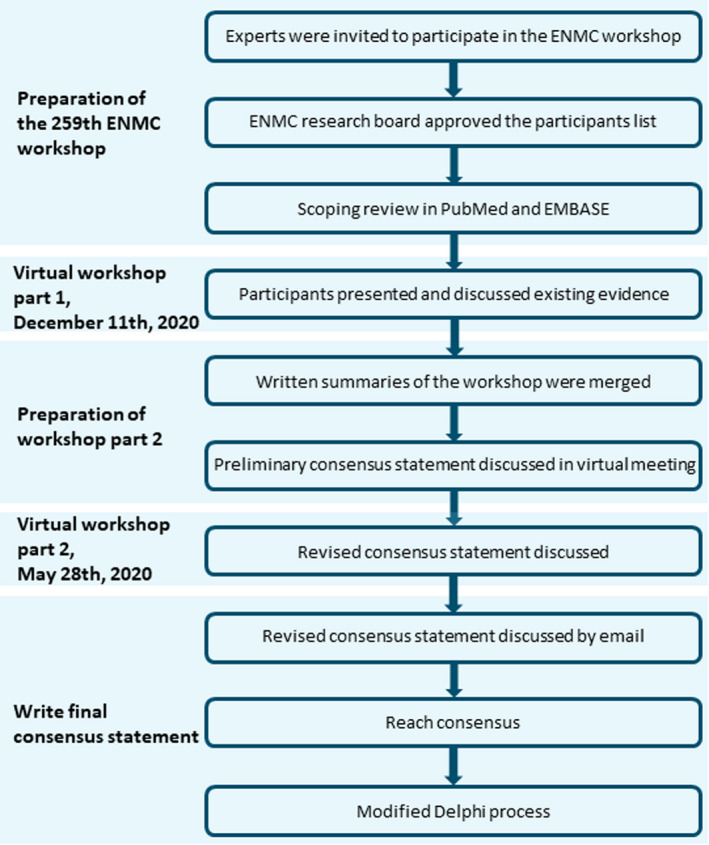
A summary of the process of developing this consensus statement document. ENMC, European Neuromuscular Centre [Colour figure can be viewed at wileyonlinelibrary.com
Modified Delphi process
The consensus statement drafted following the ENMC workshop and subsequent meetings was distributed among its coauthors (n = 22). They were asked to indicate their agreement with each recommendation on a 9‐point scale (score 1: absolutely disagree, score 9: absolutely agree). Given the variable and specific expertise of the workshop participants, for some of them the full content of this consensus statement was not completely within their area of expertise. The participants were asked to abstain from voting if a specific recommendation was not within their area of expertise. The level of agreement for each recommendation is presented as the median score and the percentage of participants who scored ≥7. Consensus was defined as a median score of ≥7 and at least 70% of the respondents scoring the recommendation ≥7.
Focus and approach
This consensus statement entails general recommendations applicable to all patients with NMDs who need anaesthesia or procedural sedation and group‐specific recommendations for:
Neuromuscular junction disorders;
Muscle channelopathies (nondystrophic myotonia and periodic paralysis);
Myotonic dystrophy (types 1 and 2);
Muscular dystrophies;
Congenital myopathies and dystrophies; and
Mitochondrial and metabolic myopathies.
Suggested relevant topics were risks, myopathy‐related treatment, preoperative diagnosis and management, specific recommendation for anaesthesia including nonanaesthetic drugs used in the perioperative phase, and specific recommendations for postoperative care.
We restricted detailed clinical information concerning the six main groups of NMDs to what was essential for our purpose. For complementary disease‐specific anaesthesia considerations, we refer to the following databases:
OrphanAnesthesia (https://www.orphananesthesia.eu/en/); and
Syndromes & Rare Diseases in Pediatrics: Anesthesia (http://tinyurl.com/PED‐RARE).
As we consider the American Society of Anesthesiologists physical status classification [10] too unspecific for this particular patient population, we devised a practical risk assessment tool with a scoring matrix based on the NARCO‐SS‐Risk Assessment Tool [11] and the matrix model developed by Schieren et al. [2].
Level of evidence
The level of evidence for each recommendation has been adapted from the Scottish Intercollegiate Guidelines Network grading system [12] and is indicated in each paragraph:
Level 1+: high‐quality meta‐analyses, systematic reviews of randomized controlled trials (RCTs), or RCTs with very low risk of bias;
Level 1−: meta‐analyses, systematic reviews or RCTs with high risk of bias;
Level 2+: systematic reviews or case–control or cohort studies with low risk of bias;
Level 2−: case–control or cohort studies with high risk of bias, animal studies;
Level 3: case reports, case series; and
Level 4: expert opinion.
RESULTS
Modified Delphi process
A total of 20 participants (90.9%) responded. During the modified Delphi process, consensus was reached for all recommendations.
LEVEL 1: GENERAL RECOMMENDATIONS FOR PATIENTS WITH NMDs
Surgery setting
Surgery and the associated need for sedation and/or anaesthesia pose a burden on the neuromuscular patient. They should only receive anaesthesia or sedation in the setting of a 24‐h high‐care facility [1, 2, 3].
Level of evidence: 4
The median score by the modified Delphi process was 9. Respondents who scored ≥7 totaled 18 of 19 (94.7%); one respondent abstained from voting.
Anaesthesia technique
Surgery or diagnostic procedures should be performed using regional anaesthesia if possible, preferably as a single technique or alternatively in combination with general anaesthesia [1, 2, 3].
Level of evidence: 4
The median score by the modified Delphi process was 8. Respondents who scored ≥7 totaled 18 of 19 (94.7%); one respondent abstained from voting.
Preoperative assessment
A clear understanding of the confirmed or suspected neuromuscular diagnosis, degree of muscle weakness, and current treatment and awareness of associated cardiorespiratory manifestations are paramount for the preoperative assessment. Cardiomyopathy or cardiac conduction defects can be part of certain NMDs [13, 14, 15, 16, 17]. Due to respiratory muscle weakness and spinal deformities, there is an increased risk of restrictive lung disease [16, 18, 19, 20]. Furthermore, chronic hypoxaemia and pulmonary hypertension may lead to cor pulmonale, which must be considered during the preoperative examination [13]. This information can be partly obtained from the history or physical examination, but ancillary assessments may be necessary.
A careful preoperative examination, multisystem evaluation, and clear communication among anaesthesiologists, surgeons, cardiologists, pneumologists, and neurologists are crucial. During the preoperative assessment, specific attention should be paid to craniofacial abnormalities indicating a difficult airway [21]. Depending on the procedure, additional preoperative investigations may include electrocardiography (ECG), chest X‐ray, echocardiography, Holter examination, lung function tests (in the sitting and supine positions), polysomnography, and blood tests including arterial blood gases, haemoglobin, electrolytes, lactate, creatine kinase (CK), and liver and kidney function tests [1, 2, 3, 13, 14, 15, 16, 17, 18, 19, 20, 22].
Some neuromuscular patients are treated with anticholinesterase inhibitors, antiarrhythmic drugs, antisense oligonucleotides, steroids, or other immunosuppressive agents [4, 23]. This needs to be recorded and evaluated during the preoperative assessment.
Level of evidence: 2+
The median score by the modified Delphi process was 9. Respondents who scored ≥7 totaled 19 of 19 (100%); one respondent abstained from voting.
Premedication
Patients with NMDs have an increased sensitivity to sedatives and anaesthetics [1, 2, 3]. Premedication with benzodiazepines may result in central respiratory depression, airway obstruction, and/or worsening of muscle weakness. Premedication should therefore be avoided, and if absolutely required only be used in reduced doses with concurrent pulse oximetry (SpO2) and respiratory rate monitoring [1, 2, 3].
Level of evidence: 4
The median score by the modified Delphi process was 9. Respondents who scored ≥7 totaled 19 of 19 (100%); one respondent abstained from voting.
Preoperative fasting
Patients with NMDs should be scheduled for surgery as the first case of the day, because prolonged fasting may cause hypoglycaemia, in particular in patients with reduced muscle mass [24, 25, 26].
Level of evidence 2+
The median score by the modified Delphi process was 8. Respondents who scored ≥7 totaled 14 of 16 (87.5%); four respondents abstained from voting.
Some NMDs are associated with gastrointestinal dysmotility and dysphagia, and the patient may not have an empty stomach [27]. Gastric ultrasound imaging could be used to evaluate the volume of gastric content before anaesthesia and adapt the induction technique accordingly [28].
Level of evidence 2+
The median score by the modified Delphi process was 8. Respondents who scored ≥7 totaled 12 of 16 (75%); four respondents abstained from voting.
Body temperature management
Anaesthesia causes drop in body temperature due to vasodilation and blunting of thermoregulatory reflexes. This effect is more pronounced in neuromuscular patients because of their reduced muscle mass. Moreover, hypothermia further increases sensitivity to sedatives, anaesthetic agents, and nondepolarizing NMBAs [29]. Preoperative warming reduces the fall in core temperature following induction of anaesthesia in the general population [30] and will be even more beneficial in patients with NMDs.
Level of evidence: 4
The median score by the modified Delphi process was 9. Respondents who scored ≥7 totaled 19 of 19 (100%); one respondent abstained from voting.
On the other hand, increased muscle activity (cramps, myotonia) or excessive external heating can result in hyperthermia with increased muscle tone, cramps, and rhabdomyolysis. Therefore, continuous temperature monitoring is recommended.
Level of evidence: 4
The median score by the modified Delphi process was 9. Respondents who scored ≥7 totaled 18 of 18 (100%); two respondents abstained from voting.
General anaesthesia
Short‐acting opioids, sedatives, and hypnotic agents are preferred to minimize respiratory depression on emergence from anaesthesia [1, 2, 3].
Level of evidence: 4
The median score by the modified Delphi process was 9. Respondents who scored ≥7 totaled 18 of 18 (100%); two respondents abstained from voting.
Neuromuscular blocking agents
Based on their mechanisms of action, NMBAs can be divided into depolarizing NMBAs and nondepolarizing NMBAs [1]. Patients with an NMD are at greatest risk for succinylcholine‐induced hyperkalaemia [31, 32, 33, 34]. Hence, succinylcholine should not be used in patients with a known or suspected NMD.
Level of evidence: 2−
The median score by the modified Delphi process was 9. Respondents who scored ≥7 totaled 19 of 19 (100%); one respondent abstained from voting.
Most neuromuscular patients have an increased sensitivity to nondepolarizing NMBAs because of the reduced muscle mass and strength. A lower initial dose may be sufficient to achieve adequate muscle relaxation. Duration of action is often prolonged, and effects of residual relaxation are more severe than in normal patients [35, 36, 37, 38]. The effect of NMBAs should be measured by quantitative neuromuscular monitoring to prevent residual neuromuscular blockade following emergence from anaesthesia and extubation.
Level of evidence: 2+
The median score by the modified Delphi process was 9. Respondents who scored ≥7 totaled 18 of 18 (100%); two respondents abstained from voting.
When a nondepolarizing NMBA is used, extubation and emergence from anaesthesia should be guided by a train‐of‐four ratio of 1.0. Any residual and potentially clinically relevant muscle relaxation at the end of the intervention should preferably be reversed with sugammadex as a specific pharmacological antagonist of rocuronium and vecuronium [39, 40, 41]. Due to the effectiveness of sugammadex, the latter two nondepolarizing NMBAs should be given preference. When there is residual neuromuscular blockade after use of other NMBAs or sugammadex is unavailable, extubation and emergence from anaesthesia should be postponed until baseline muscular strength has recovered spontaneously. In this scenario, postoperative sedation and ventilatory support are necessary.
Level of evidence: 2−
The median score by the modified Delphi process was 9. Respondents who scored ≥7 totaled 14 of 15 (93.3%); five respondents abstained from voting.
Reversal of muscle relaxation using cholinesterase inhibitors is contraindicated because of side effects in myotonic dystrophies [42, 43] and some congenital myasthenic syndromes. Cholinesterase inhibitors (e.g., neostigmine) are undesirable in other neuromuscular patients because of the unpredictable pharmacodynamics and the increased sensitivity to NMBAs.
Level of evidence: 2−
The median score by the modified Delphi process was 8. Respondents who scored ≥7 totaled 12 of 16 (75%); four respondents abstained from voting.
Volatile anaesthetics
Volatile anaesthetics are only strictly contraindicated in patients with NMDs with variants in the RYR1 gene because of the possibility of MH susceptibility [44, 45, 46, 47, 48, 49, 50], unless the specific variant has been classified as benign for MH according to the ClinGen Variant Curation Expert Panel recommendations for RYR1 pathogenicity classifications in MH susceptibility [51]. Patients with variants in the CACNA1S and/or STAC3 genes should be referred to an MH investigation centre for advice on the risk of MH before exposure to volatile anaesthetics.
Level of evidence: 2+
The median score by the modified Delphi process was 8. Respondents who scored ≥7 totaled 11 of 15 (73.3%); five respondents abstained from voting.
NMDs without these specific genetic backgrounds might be associated with other, rare adverse effects, such as anaesthesia‐induced rhabdomyolysis (AIR) in DMD or Becker muscular dystrophy (BMD) patients [33, 34]. The prolonged effect of NMBAs when used in combination with volatile anaesthetics also has to be taken into account [52].
Level of evidence: 2+
The median score by the modified Delphi process was 9. Respondents who scored ≥7 totaled 18 of 18 (100%); two respondents abstained from voting.
Hence, anaesthesiologists are advised against the prolonged use of volatile anaesthetics in patients with NMDs. Although often recommended as an alternative, total intravenous anaesthesia (TIVA; most frequently based on a propofol, short‐acting benzodiazepines, or dexmedetomidine‐based technique in combination with short‐acting potent opioids) has other risks [53, 54, 55, 56]. This is an important consideration as part of a comprehensive risk–benefit assessment between TIVA and volatile anaesthetic‐based techniques.
Specific indications for use of volatile anaesthetics (which can be administered through a face mask) in patients with an NMD might include situations where awake intravenous access is not tolerated or may be difficult or impossible (e.g., in anxious children).
Level of evidence: 4
The median score by the modified Delphi process was 8. Respondents who scored ≥7 totaled 16 of 17 (94.1%); three respondents abstained from voting.
Postoperative period
After general anaesthesia or sedation, up to 24 h of monitoring (including ECG, SpO2, preferably CO2 monitoring, as well as surveillance for signs of ongoing rhabdomyolysis) may be necessary [1, 2].
Level of evidence: 4
The median score by the modified Delphi process was 8. Respondents who scored ≥7 totaled 16 of 18 (88.9%); two respondents abstained from voting.
Early mobilization and feeding after surgery should be pursued. Respiratory physiotherapy can improve breathing and facilitate coughing.
Level of evidence: 4
The median score by the modified Delphi process was 8. Respondents who scored ≥7 totaled 17 of 18 (94.4%); two respondents abstained from voting.
Medical alert cards, apps, and warnings in electronic patient files
Many patient advocacy organizations provide medical alert cards to patients with NMDs. These cards improve patient safety, supporting individuals and their families in relaying information to emergency services and other medical professionals. Specific and personalized disease information, possible complications, and their specific treatments should be recorded in such formats.
Neurologists and rehabilitation specialists who provide follow‐up for patients with NMDs are in an excellent position to alert their patients regarding perioperative risks and the specific precautions to be taken. Through annual review at follow‐up visits, medical professionals can contribute to the improved use of medical alert cards. Medical alerts should be clearly visible in electronic health records, and recommendations for essential perioperative precautions should be included in correspondence with other health care providers.
Level of evidence: 4
The median score by the modified Delphi process was 9. Respondents who scored ≥7 totaled 19 of 19 (100%); one respondent abstained from voting.
LEVEL 2: GROUP‐SPECIFIC RECOMMENDATIONS
In addition to the general recommendations summarized above, this section gives disease‐specific recommendations concerning the six groups of NMDs.
Neuromuscular junction disorders
Disorders of the neuromuscular junction may be caused by pathogenic antibodies, genetic defects [57], specific drugs, and/or toxins that interfere with the normal signalling between the presynaptic nerve ending and the postsynaptic muscle membrane [58].
Acquired autoimmune disorders of the neuromuscular junction are the most common and include myasthenia gravis [58, 59] and Lambert–Eaton myasthenic syndrome (LEMS). LEMS is frequently associated with an underlying malignancy or another autoimmune process.
Congenital myasthenic syndromes are a phenotypically heterogeneous group of disorders due to a variety of genetic defects resulting in impaired neuromuscular transmission. Muscle weakness typically begins in early childhood, but presentation may be delayed until adolescence or adulthood. Facial muscles, including (extra)ocular and bulbar muscles, are the most consistently affected. Due to muscle weakness, affected infants may have feeding difficulties. Motor development may be delayed. The severity of the myasthenia varies greatly, ranging from an inability to walk to only minor weakness. Weakness may be exacerbated by fever or infection [57, 58]. Specific anaesthesia considerations for patients with neuromuscular junction disorders are summarized in Table 1.
TABLE 1.
Specific anaesthesia recommendations for neuromuscular junction disorders
| Recommendations | SIGN | Median score, modified Delphi process | Respondents who scored ≥7, n | Abstained from voting, n |
|---|---|---|---|---|
| Specific considerations for myasthenia gravis | ||||
|
4 | 9 | 19/19 (100%) | 1 |
|
4 | 9 | 18/19 (94.7%) | 1 |
| Preoperative recommendations | ||||
|
4 | 8 | 15/17 (88.2%) | 3 |
|
4 | 9 | 17/19 (89.5%) | 1 |
|
4 | 8 | 16/17 (94.1%) | 3 |
|
4 | 9 | 19/19 (100%) | 1 |
| Intraoperative recommendations | ||||
| 2− | 9 | 15/15 (100%) | 5 | |
|
2− | 8.5 | 14/14 (100%) | 6 |
| Postoperative recommendations | ||||
|
4 | 9 | 19/19 (100%) | 1 |
|
However, caution is needed, as there are large interindividual differences in bioavailability. |
4 | 9 | 16/16 (100%) | 4 |
|
4 | 9 | 13/13 (100%) | 7 |
Note: For each recommendation, the level of evidence according to the SIGN criteria and the level of agreement are given by the median voting results and the percentage of respondents with a voting result ≥7.
Abbreviations: SIGN, Scottish Intercollegiate Guidelines Network; TIVA, total intravenous anaesthesia.
Muscle channelopathies
Skeletal muscle channelopathies are rare clinically and genetically heterogeneous diseases. They are characterized by altered muscle fibre excitability caused by mutations affecting voltage‐gated ion channels such as chloride channel (ClC‐1), sodium channel (NaV1.4), calcium channel (CaV1.1), or potassium channels (Kir2.1, Kir2.6, Kir3.4). The clinical manifestations vary from an inability to relax after voluntary contraction (myotonia) to transient attacks of generalized or focal flaccid muscle weakness (periodic paralysis). Fluctuation of symptoms are strongly affected by environmental triggers such as exercise, temperature changes, pain, emotional factors, fasting, or alterations in serum potassium concentration [63]. The nondystrophic myotonias include myotonia congenita (due to variants in CLCN1), paramyotonia congenita (due to variants in SCN4A), and sodium channel myotonia (SCN4A) [23]. Periodic paralyses include hyperkalaemic periodic paralysis caused by variants in SCN4A and hypokalaemic periodic paralysis (hypoPP) caused by variants in CACNA1S, SCN4A [23, 64], or rarely RYR1 [65], and Andersen–Tawil syndrome caused by variants in KCNJ2 [66]. Andersen–Tawil syndrome is associated with ECG abnormalities (long Q‐T interval and U‐waves) and predisposition to life‐threatening ventricular arrythmias [14].
Up to 31% of patients with muscle channelopathies report worsening of symptoms and a prolonged recovery time after general anaesthesia [67]. There are reports of myotonic crises in patients with myotonias and prolonged paralysis (hyperkalaemic triggered by succinylcholine or other potassium‐releasing agents in patients with SCN4A variants and hypokalaemic related to hypokalaemia and hyperglycaemia following anaesthesia) [67, 68].
Variants in CACNA1S and, very rarely, RYR1 are associated with hypoPP, and although none of these RYR1 variants has been shown to predispose to MH, the risk is difficult to disprove [65, 67, 69]. A patient with known hypoPP who had a confirmed episode of MH [69] has subsequently been found to harbour a CACNA1S variant pathogenic for hypoPP but also a second variant in CACNA1S and two variants in RYR1 [48] (unpublished data, P.M.H.). Specific anaesthesia considerations for patients with muscle channelopathies are summarized in Table 2.
TABLE 2.
Specific anaesthesia recommendations for muscle channelopathies
| Recommendations | SIGN | Median score, modified Delphi process | Respondents who scored ≥7, n | Abstained from voting, n |
|---|---|---|---|---|
| Preoperative recommendations | ||||
|
2+ | 9 | 18/18 (100%) | 2 |
|
2+ | 9 | 19/19 (100%) | 1 |
|
2+ | 9 | 18/19 (94.7%) | 1 |
| 2+ | 9 | 18/18 (100%) | 2 | |
| Intraoperative recommendations | ||||
|
3 | 9 | 17/17 (100%) | 3 |
|
3 | 9 | 19/19 (100%) | 1 |
|
3 | 9 | 16/16 (100%) | 4 |
|
3 | 9 | 16/16 (100%) | 4 |
|
3 | 9 | 13/13 (100%) | 7 |
|
3 | 9 | 18/18 (100%) | 2 |
Note: For each recommendation, the level of evidence according to the SIGN criteria and the level of agreement are given by the median voting results and the percentage of respondents with a voting result ≥7.
Abbreviations: ECG, electrocardiography; hyperPP, hyperkalaemic periodic paralysis; hypoPP, hypokalaemic periodic paralysis; MH, malignant hyperthermia; SIGN, Scottish Intercollegiate Guidelines Network.
Myotonic dystrophy types 1 and 2
Myotonic dystrophy type 1 (DM1) is a frequent adult muscular dystrophy caused by DNA triplet repeat expansion in the DMPK gene. Larger expansions lead to the infantile or more severe, congenital form resembling a congenital muscular dystrophy, with often profound associated weakness, hypotonia, and bulbar and respiratory involvement. A second form, myotonic dystrophy type 2 (DM2), results from a different repeat expansion in the CNBP gene, showing more proximal weakness [71].
Both disorders have autosomal dominant inheritance and multisystem features, including a myotonic myopathy, cataract, cardiac conduction defects, central and obstructive sleep apnoea, and a range of endocrine abnormalities including diabetes mellitus [71]. Respiratory impairment is more common and more pronounced in DM1.
Much of the available literature does not differentiate the congenital form of DM1 from its other forms, although CTG repeat analysis [72] and grading by presence of proximal weakness [72, 73] argue for a higher risk of complications in the congenital form. Furthermore, myotonia is usually absent in the early life of patients with this form. Specific anaesthesia considerations for patients with DM1 and DM2 are summarized in Table 3.
TABLE 3.
Specific anaesthesia recommendations for myotonic dystrophy types 1 and 2
| Recommendations | SIGN | Median score, modified Delphi process | Respondents who scored ≥7, n | Abstained from voting, n |
|---|---|---|---|---|
| Preoperative recommendations | ||||
|
2− | 9 | 19/19 (100%) | 1 |
|
2− | 9 | 19/19 (100%) | 1 |
| 4 | 7 | 13/17 (76.5%) | 3 | |
| Intraoperative | ||||
| 3 | 9 | 15/15 (100%) | 5 | |
| 3 | 8 | 14/17 (82.4%) | 3 | |
|
3 | 8.5 | 18/18 (100%) | 2 |
| Postoperative | ||||
| 4 | 9 | 17/19 (89.5%) | 1 | |
|
4 | 9 | 16/19 (84.2%) | 1 |
|
4 | 9 | 16/18 (88.9%) | 2 |
| Specific considerations for Myotonic Dystrophy type II (DM2) | ||||
| 2‐ | 9 | 15/15 (100%) | 5 | |
|
2‐ | 9 | 14/14 (100%) | 6 |
Note: For each recommendation, the level of evidence according to the SIGN criteria and the level of agreement are given by the median voting results and the percentage of respondents with a voting result ≥7.
Abbreviations: CK, creatine kinase; CNS, central nervous system; DM1, myotonic dystrophy type 1; DM2, myotonic dystrophy type 2; ECG, electrocardiography; SIGN, Scottish Intercollegiate Guidelines Network; TIVA, total intravenous anaesthesia.
Muscular dystrophies
Several different skeletal muscle disorders, with very different degrees of severity and characterized by various pathophysiological mechanisms, are classified under the definition of muscular dystrophies. These disorders vary widely with regard to age at onset, degree of motor impairment, and associated cardiorespiratory involvement [80]. Classification is based on the underlying gene defect. This paragraph will focus on the most common forms, the dystrophinopathies, limb girdle muscular dystrophies (LGMDs), facioscapulohumeral muscular dystrophy (FSHD), and oculopharyngeal muscular dystrophy (OPMD).
Dystrophinopathy refers to DMD and BMD due to hemizygous X‐linked variants in the DMD gene, encoding dystrophin. Ventilatory support and steroid treatments have improved survival in boys with DMD significantly and have prolonged ambulation, reduced the risk of developing scoliosis, and improved cardiac health [80]. Severe cardiorespiratory involvement is common and needs thorough evaluation during the preoperative assessment [13, 17]. Macroglossia and contracture of neck muscles can cause problems for intubation. Central nervous system (CNS) involvement manifesting as cognitive deficits, speech problems, and psychiatric comorbidities such as anxiety and depression may complicate preoperative counselling [81].
LGMDs are a genetically heterogeneous group of disorders characterized by predominantly proximal muscle weakness and onset after becoming able to walk [80]. Specific forms (e.g., sarcoglycanopathies and alpha‐dystroglycanopathies) may be associated with cardiac involvement, usually dilated cardiomyopathy or, rarely, arrhythmogenic heart disease, which is frequent in POPDC1‐related LGMD R25 [15, 16]. Respiratory failure may be a feature of the most severe forms or of those with spinal deformities, such as the COL6‐related LGMDs. Significant tongue enlargement, which can cause problems for intubation, is occasionally present in FKRP‐related LGMD R9, DMD, and sarcoglycanopathies [16, 21].
FSHD is caused by skeletal muscle misexpression of the DUX4 retrogene and represents the third most common form of muscular dystrophy in adults after DMD and DM1. Phenotypes may be variable and not immediately apparent in presymptomatic carriers [80].
OPMD is another late onset mostly autosomal dominant disorder caused by a trinucleotide repeat expansion in the first exon of the polyadenylate‐binding protein nuclear 1 gene (PABPN1). The main clinical issues are ptosis and dysphagia, although patients may later also develop proximal muscle weakness. Specific anaesthesia considerations for patients with muscular dystrophies are summarized in Table 4.
TABLE 4.
Specific anaesthesia recommendations for muscular dystrophies
| Recommendations | SIGN | Median score, modified Delphi process | Respondents who scored ≥7, n | Abstained from voting, n |
|---|---|---|---|---|
| Preoperative recommendations | ||||
|
2− | 9 | 19/19 (100%) | 1 |
|
2− | 9 | 19/19 (100%) | 1 |
|
2+ | 9 | 19/19 (100%) | 1 |
|
2− | 8.5 | 16/18 (88.9%) | 2 |
| Intraoperative | ||||
|
2+ | 9 | 18/18 (100%) | 2 |
|
2+ | 9 | 19/19 (100%) | 1 |
| 2− | 9 | 17/17 (100%) | 3 | |
|
4 | 9 | 19/19 (100%) | 1 |
| Postoperative | ||||
|
4 | 9 | 19/19 (100%) | 1 |
Note: For each recommendation the level of evidence according to the SIGN criteria and the level of agreement is given by the median voting results and the percentage of respondents with a voting result ≥7.
Abbreviation: AIR, anaesthesia‐induced rhabdomyolysis; BMD, Becker muscular dystrophy; CNS, central nervous system; DMD, Duchenne muscular dystrophy; FSHD, facioscapulohumeral muscular dystrophy; LGMD, limb girdle muscular dystrophy; MH, malignant hyperthermia; OPMD, oculopharyngeal muscular dystrophy; SIGN, Scottish Intercollegiate Guidelines Network.
Congenital myopathies and congenital muscular dystrophies
Congenital myopathies and congenital muscular dystrophies are disorders with early onset muscle weakness. Both groups give rise to proximal, axial, and often facial weakness with slow progression and cardiorespiratory impairment, as well as joint contractures and scoliosis. Despite some overlap, anaesthetic implications are different.
Congenital muscular dystrophies
Congenital muscular dystrophies are classically defined as a group of disorders characterized by onset in the first 6 months of life and muscle biopsy evidence of a muscular dystrophy with histopathological features of de‐ and regeneration [85]. Depending on the underlying disease, cardiorespiratory involvement can be prominent, and some forms may have associated CNS involvement with epilepsy. The genes associated can be classified according to the cellular localization of the encoded protein into the following groups: proteins of the plasma membrane–extracellular matrix interface such as collagen 6 (COL6A 1,2,3), merosin, or laminin α2 (LAMA2), integrin α7 (ITGA7), proteins involved in O‐glycosylation of the dystrophin‐associated glycoprotein (POMT1, POMT2, POMGnT1, LARGE, FKTN, FKRP, ISPD) and other intracellular proteins such as lamin A/C (LMNA), an intermediate filament located at the inner nuclear membrane, and selenoprotein N (SELENON), a protein with redox and calcium level regulation function [80].
It should be noted that the clinical phenotype of congenital DM1 (see paragraph above) is more similar to this group of diseases than to the adult forms of myotonic dystrophy. Specific anaesthesia considerations for patients with congenital muscular dystrophies are summarized in Table 5a.
TABLE 5.
Specific anaesthesia recommendations for congenital muscular dystrophies (a) and congenital myopathies (b)
| Recommendations | SIGN | Median score, modified Delphi process | Respondents who scored ≥7, n | Abstained from voting, n |
|---|---|---|---|---|
| (a) Specific anaesthesia recommendations for congenital muscular dystrophies | ||||
| Preoperative recommendations | ||||
|
3 | 9 | 17/17 (100%) | 3 |
|
3 | 9 | 16/16 (100%) | 4 |
|
3 | 9 | 15/15 (100%) | 5 |
| Intraoperative | ||||
|
4 | 9 | 17/17 (100%) | 3 |
|
4 | 9 | 18/18 (100%) | 2 |
|
4 | 8 | 17/17 (100%) | 3 |
| Postoperative | ||||
| 3 | 9 | 15/15 (100%) | 5 | |
| (b) Specific anaesthesia recommendations for congenital myopathies | ||||
| Preoperative recommendations | ||||
|
3 | 9 | 17/17 (100%) | 3 |
|
3 | 9 | 14/14 (100%) | 6 |
|
3 | 9 | 16/16 (100%) | 4 |
|
3 | 9 | 16/16 (100%) | 4 |
| Intraoperative recommendations | ||||
|
3 | 9 | 15/16 (93.8%) | 4 |
|
2+ | 9 | 19/19 (100%) | 1 |
| 3 | 9 | 17/17 (100%) | 3 | |
Note: For each recommendation, the level of evidence according to the SIGN criteria and the level of agreement are given by the median voting results and the percentage of respondents with a voting result ≥7.
Abbreviations: AIR, anaesthesia‐induced rhabdomyolysis; BMD, Becker muscular dystrophy; DMD, Duchenne muscular dystrophy; MH, malignant hyperthermia; SIGN, Scottish Intercollegiate Guidelines Network; TIVA, total intravenous anaesthesia; XLMTM, X‐linked myotubular myopathy.
Congenital myopathies
Congenital myopathies are a heterogeneous group of early onset inherited disorders. The different subtypes are traditionally based on the predominant histopathological finding; nemaline rods, cores, central nuclei, or fibre type disproportion. There is, however, overlap between these findings, and there is an emerging trend to define the conditions according to which gene is involved. There are >20 genes associated with congenital myopathies. The overall prevalence is estimated at 2–4/100,000 [86, 87, 88], with RYR1‐related myopathies the most common, followed by those due to variants in NEB and ACTA1. TTN‐related myopathies are increasingly diagnosed and represent an important subgroup. The genes causing congenital myopathies predominantly encode proteins implicated in skeletal muscle Ca2+ homeostasis, excitation–contraction coupling, myofilament assembly and interaction, and other mechanisms [88]. Specific anaesthesia considerations for patients with congenital myopathies are summarized in Table 5b.
Mitochondrial and metabolic myopathies
Mitochondrial myopathies
Mitochondrial myopathies are caused by variants in either nuclear or mitochondrial DNA leading to impairment of oxidative phosphorylation or fatty acid metabolism in mitochondria. The accumulation of Acyl‐CoA might result in a secondary carnitine deficiency and impair the citrate cycle, gluconeogenesis, the urea cycle, and fatty‐acid oxidation, resulting in a deficit in energy production in the form of adenosine triphosphate, particularly in skeletal muscle [22, 89]. In addition, patients with a mitochondrial myopathy can also present with multisystemic symptoms. Anaesthetic preparation should therefore always be case‐specific and include a multisystemic assessment. Circumstances causing metabolic disturbance should be avoided in all mitochondrial myopathies whenever possible [22, 25, 89, 90]. Specific anaesthesia considerations for patients with mitochondrial myopathies are summarized in Table 6a.
TABLE 6.
Specific anaesthesia recommendations for mitochondrial myopathies (a), metabolic myopathies (b), and lipid metabolism disorders (c)
| Recommendations | SIGN | Median score, modified Delphi process | Respondents who scored ≥7, n | Abstained from voting, n |
|---|---|---|---|---|
| (a) Specific anaesthesia recommendations for mitochondrial myopathies | ||||
| Preoperative recommendations | ||||
|
4 | 9 | 19/19 (100%) | 1 |
|
4 | 9 | 19/19 (100%) | 1 |
| Intraoperative | ||||
|
3 | 8.5 | 17/18 (94.4%) | 2 |
| 3 | 9 | 16/16 (100%) | 4 | |
| 4 | 9 | 15/15 (100%) | 5 | |
|
4 | 9 | 15/15 (100%) | 5 |
|
4 | 9 | 17/18 (94.4%) | 2 |
|
4 | 9 | 18/18 (100%) | 2 |
| Postoperative | ||||
| 3 | 9 | 18/18 (100%) | 2 | |
|
3 | 9 | 13/13 (100%) | 7 |
| (b) Specific anaesthesia recommendations for muscle glycogen storage diseases | ||||
| Preoperative recommendations | ||||
|
4 | 9 | 19/19 (100%) | 1 |
|
4 | 9 | 19/19 (100%) | 1 |
| Intraoperative recommendations | ||||
| 4 | 9 | 19/19 (100%) | 1 | |
|
4 | 9 | 19/19 (100%) | 1 |
|
4 | 8.5 | 14/14 (100%) | 6 |
|
4 | 8.5 | 17/18 (94.4%) | 2 |
| (c) Specific anaesthesia recommendations for muscle lipid metabolism disorders | ||||
| Preoperative recommendations | ||||
|
4 | 9 | 20/20 (100%) | 0 |
|
4 | 9 | 18/18 (100%) | 2 |
|
4 | 9 | 19/19 (100%) | 1 |
| Intraoperative recommendations | ||||
| 3 | 9 | 16/16 (100%) | 4 | |
Note: For each recommendation, the level of evidence according to the SIGN criteria and the level of agreement are given by the median voting results and the percentage of respondents with a voting result ≥7.
Abbreviations: GSD, glycogen storage disease; ICU, intensive care unit; LCHAD, long‐chain 3‐hydroxyacyl‐CoA dehydrogenase deficiency; MCAD, medium‐chain acyl‐CoA dehydrogenase deficiency; MH, malignant hyperthermia; MTP, Mitochondrial trifunctional protein; SIGN, Scottish Intercollegiate Guidelines Network; TIVA, total intravenous anaesthesia; VLCAD, very‐long‐chain acyl‐CoA dehydrogenase deficiency.
Metabolic myopathies
Metabolic myopathies present with either permanent (fixed) muscle symptoms or episodic abnormalities, such as exercise intolerance, activity‐induced myalgia, muscle contractures, and muscle damage that can progress to rhabdomyolysis. Metabolic myopathies can be classified further as muscle glycogen storage diseases (GSDs; or muscle glycogenosis) and lipid metabolism disorders [22, 91, 92].
Muscle GSDs are a group of inherited disorders that cause glycogen to be improperly stored or utilized in the body. Symptoms and comorbidities are highly diverse; some GSDs only affect skeletal muscle, whereas others feature involvement of other organs (including the heart and liver). This needs special focus, as patients can be severely affected multisystemically [22, 91, 92]. For instance, patients with GSD type II may present as classic Pompe disease with cardiomegaly, juvenile oligosymptomatic cases, or late onset cases with weakness and often severe respiratory involvement. Specific anaesthesia considerations for patients with GSDs are summarized in Table 6b.
Muscle lipid metabolism disorders can also manifest in organs other than skeletal muscle. Abnormalities in enzyme‐processing fats can result in accumulation of fatty acid and its derivatives, including triglycerides, and sterol‐containing metabolites such as cholesterol. This can be harmful to the heart and liver and may result in cardiomyopathy, arrythmias, hepatomegaly, hypoglycaemia, and hyperammonaemia [92]. Specific anaesthesia considerations for patients with muscle lipid metabolism disorders are summarized in Table 6c.
LEVEL 3: RISK PREDICTION MATRIX AND SAFETY OF NONANAESTHETIC DRUGS USED IN THE PERIOPERATIVE PERIOD
Risk prediction matrix
Table 7 illustrates a preoperative assessment matrix designed as a practical tool based on Schieren et al. [2], which can be used to make an inventory of the multisystem features in patients with NMDs. A grey box in the risk prediction matrix indicates that at least a subset of this group of NMDs is associated with this specific condition/complication or disease manifestation. A white box indicates that this NMD is not specifically associated with this specific condition/complication or disease manifestation.
TABLE 7.
A risk prediction matrix to assess the multisystem features of patients with neuromuscular disorders
| Neuromuscular junction disorders | Muscle channelopathies | Myotonic dystrophies | Muscular dystrophies | Congenital muscular dystrophies | Congenital myopathies | Mitochondrial myopathies | Metabolic myopathies | Lipid metabolism disorders | |
|---|---|---|---|---|---|---|---|---|---|
| Neurological signs and muscular impairment | |||||||||
| Ataxia | |||||||||
| Cerebral thrombosis | |||||||||
| Cognitive impairment | |||||||||
| Epilepsy | |||||||||
| Encephalopathy | |||||||||
| Myoclonus | |||||||||
| Strokelike episodes | |||||||||
| Weakness | |||||||||
| Airway | |||||||||
| Aspiration | |||||||||
| Difficult intubation | |||||||||
| Dysphagia | |||||||||
| Macroglossia | |||||||||
| Respiratory | |||||||||
| Respiratory insufficiency | |||||||||
| Cardiac | |||||||||
| Arrythmias | |||||||||
| Cardiomyopathy | |||||||||
| Risk of sudden death | |||||||||
| Other | |||||||||
| Increased bleeding tendency | |||||||||
| Diabetes | |||||||||
| Exercise intolerance | |||||||||
| Hypoglycaemia | |||||||||
| Lactic acidosis | |||||||||
| Rhabdomyolysis | |||||||||
| Spinal deformity | |||||||||
| Malignant hyperthermia susceptibility | |||||||||
| Myotonia | |||||||||
Note: A grey box in the risk prediction matrix indicates that at least a subset of this group of neuromuscular disorders is associated with this specific condition/complication or disease manifestation. A white box indicates that this neuromuscular disorder is not specifically associated with this specific condition/complication or disease manifestation.
After obtaining the relevant information, the perioperative risk can be obtained using the modified NARCO‐SS tool [11]. In this risk assessment tool, a score of 0, 1, or 2 is given for each organ system and/or specific scenario in the NARCO‐SS acronym. A score of 0 indicates no signs or symptoms, a score of 1 indicates mild to moderate symptoms, and a score of 2 indicates moderate to severe symptoms in the specific organ system. For the surgical severity (or “SS” in the acronym), a score of 0 is given for noninvasive or superficial elective procedures. A score of 1 is allocated for minimally invasive, elective procedures of up to 60‐min duration with anticipated moderate blood loss. A score of 2 is given for major surgery/interventions or any type of surgery in an emergency setting.
N: Neurological signs & muscular impairment? [0–1–2]
A: Difficult Airway? [0–1–2]
R: Symptoms of Respiratory disease? [0–1–2]
C: Symptoms of Cardiac disease? [0–1–2]
O: Other comorbidity or metabolic concerns? [0–1–2]
SS: Surgical Severity? [0–1–2]
Result risk assessment based on modified NARCO‐SS
Low risk: total score 0–4 with no individual score > 1;
Moderate risk: total score 5–6 with no individual score > 1;
High risk: total score 7–9 or any individual score = 2;
Seriously reconsider the indication for surgery and anaesthesia when total score > 9.
Unsafe nonanaesthetic drugs
Anaesthetists are often asked by surgeons or other physicians to administer other drugs (e.g., antibiotics or antiepileptics). However, several of these drugs are possibly harmful for patients with NMDs. The following drugs should only be used with caution:
Open questions
Because of the low prevalence of most NMDs, there is a lack of prospective clinical studies with a high level of evidence on the anaesthetic management of patients with NMDs, resulting in several urgent open questions [8]. Observational multicentre studies can be of great value to study the prevalence of perioperative complications in patients with NMDs and the association with certain anaesthetic agents. One of the open questions is the future role of remimazolam, a recently introduced short‐acting benzodiazepine. In patients with NMDs, neither propofol or volatile anaesthetics are ideal, and short‐acting benzodiazepines might be useful in this respect.
Another open question is how to manage a patient suspected of a yet undefined myopathy presenting for a procedure requiring anaesthesia (e.g., surgery, muscle biopsy, magnetic resonance imaging). If it is a diagnostic procedure, it is worth establishing whether lower risk diagnostic options, especially DNA testing, have been exhausted.
Some recommend that volatile anaesthetics be avoided. However, there is no high‐level evidence on this topic, and this remains a matter of debate. Anaesthesiologists should exercise their own professional expertise and judgement to assess the disease‐inherent and circumstantial risks to determine the most appropriate anaesthetic technique.
The following considerations may be helpful:
Infants with congenital myopathies, the group at risk for MH [45, 46, 49, 50], and X‐linked muscular dystrophies due to variants in the dystrophin gene, the group at risk for AIR [33, 34], usually have delayed motor milestones, but cognitive development is usually normal [19, 20, 49, 88].
Infants with hypotonia due to a metabolic disease, the group at risk for propofol infusion syndrome [54, 55, 56], usually present with global developmental delay or other neurological signs, for example, epilepsy and spasticity [22, 91, 92].
Although nonspecific, an increased baseline CK level possibly points to a congenital myopathy or dystrophy. Therefore, these patients might be prone to AIR or MH susceptibility, and TIVA might be the preferred option.
Laboratory evidence of metabolic derangements (e.g., elevated basal lactates) favour a metabolic myopathy, in particular mitochondrial myopathy [22, 92]. Volatile anaesthesia might be the preferred option in such cases, as there is an increased risk of propofol infusion syndrome [54, 55].
Although the balance of risks may be judged to favour a TIVA technique for a particular child, an intravenous induction in an infant can be very challenging or impossible even when using nitrous oxide for sedation and topical local anaesthesia. The use of a short inhalation induction is an option in this situation, subsequently changing to TIVA once the intravenous line has been secured.
Nitrous oxide, barbiturates, benzodiazepines, ketamine, opioids, rocuronium, vecuronium, and local anaesthetics are not linked to MH, AIR, propofol infusion syndrome, or other adverse events specifically linked to certain NMDs [104]. However, one must realize that there are no risk‐free anaesthetic agents, as each agent has its own specific risks, especially in patients with several and severe comorbidities such as neuromuscular patients [105].
CONCLUSIONS
This consensus statement summarizes the most important recommendations concerning anaesthesia in patients with NMDs. Because there have been no RCTs and only few large retrospective studies on this topic, most evidence is based on small case series and expert opinion, highlighting the need for future research. Prospective, observational studies using common databases would be of great value and may provide answers to several unresolved questions. However, carefully curated expert opinion‐based recommendations can still be of value for health care professionals in the field of anaesthesia, NMDs, and genetics. Given the low level of evidence, we expect an update of this consensus statement will be necessary in 10 years or earlier, in the case that new highly relevant evidence outdates the content of this consensus statement.
AUTHOR CONTRIBUTIONS
Luuk van den Bersselaar: Conceptualization (lead); methodology (lead); writing – original draft (lead). Luc Heytens: Conceptualization (lead); methodology (lead); writing – original draft (lead). Helga Cristina de Almeida Silva: Data curation (equal); writing – review and editing (equal). Jens Reimann: Data curation (equal); writing – original draft (equal); writing – review and editing (equal). Giorgio Tasca: Data curation (equal); writing – original draft (equal); writing – review and editing (equal). Oscar Diaz‐Cambronero: Data curation (equal); writing – original draft (equal); writing – review and editing (equal). Nicoline Loekken: Data curation (equal); writing – original draft (equal); writing – review and editing (equal). Anna Hellblom: Data curation (equal); writing – review and editing (equal). Philip M. Hopkins: Data curation (equal); writing – review and editing (equal). Henrik Rueffert: Data curation (equal); writing – review and editing (equal). Börge Bastian: Data curation (equal); writing – review and editing (equal). Juan J Vílchez: Data curation (equal); writing – original draft (equal); writing – review and editing (equal). Robyn Gillies: Data curation (equal); writing – review and editing (equal). Stephan Johannsen: Data curation (equal); writing – review and editing (equal). Francis Veyckemans: Data curation (equal); writing – original draft (equal); writing – review and editing (equal). Tino Muenster: Data curation (equal); writing – review and editing (equal). Andrea Klein: Data curation (equal); writing – original draft (equal); writing – review and editing (equal). Ron Litman: Data curation (equal). H. Jungbluth: Conceptualization (equal); data curation (equal); methodology (equal); writing – review and editing (equal). Sheila Riazi: Conceptualization (equal); data curation (equal); methodology (equal); writing – review and editing (equal). Nicol Voermans: Conceptualization (equal); data curation (equal); methodology (equal); supervision (equal); writing – review and editing (equal). Marc Snoeck: Conceptualization (equal); data curation (equal); methodology (equal); supervision (equal); writing – review and editing (equal).
CONFLICT OF INTEREST
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. The ENMC did not influence the content of this consensus statement.
ACKNOWLEDGMENTS
We are thankful to the ENMC, which financially supported the 259th ENMC workshop. Several authors are members of the Radboud‐NMD, NL‐NMD, EURO‐NMD, and TREAT‐NMD consortium. We would like to dedicate this consensus statement to the late Professor Ron Litman, who participated in and contributed to the first workshop session.
van den Bersselaar LR, Heytens L, Silva HCA, et al. European Neuromuscular Centre consensus statement on anaesthesia in patients with neuromuscular disorders. Eur J Neurol. 2022;29:3486‐3507. doi: 10.1111/ene.15526
Luuk R. van den Bersselaar and Luc Heytens are co‐first authors.
Nicol C. Voermans and Marc M. J. Snoeck are co‐last authors.
DATA AVAILABILITY STATEMENT
The anonymized dataset generated during this study is available from the corresponding author on reasonable request from qualified investigators.
REFERENCES
- 1. van den Bersselaar LR, Snoeck MMJ, Gubbels M, et al. Anaesthesia and neuromuscular disorders: what a neurologist needs to know. Pract Neurol. 2021;21:12‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schieren M, Defosse J, Böhmer A, Wappler F, Gerbershagen MU. Anaesthetic management of patients with myopathies. Eur J Anaesthesiol. 2017;34:641‐649. [DOI] [PubMed] [Google Scholar]
- 3. Katz JA, Murphy GS. Anesthetic consideration for neuromuscular diseases. Curr Opin Anaesthesiol. 2017;30:435‐440. [DOI] [PubMed] [Google Scholar]
- 4. Kinnett K, Rodger S, Vroom E, Furlong P, Aartsma‐Rus A, Bushby K. Imperatives for DUCHENNE MD: a simplified guide to comprehensive care for Duchenne Muscular Dystrophy. PLoS Curr. 2015;7:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mercuri E, Finkel RS, Muntoni F, et al. Diagnosis and management of spinal muscular atrophy: part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018;28:103‐115. [DOI] [PubMed] [Google Scholar]
- 6. Van den Bersselaar LR, Riazi S, Snoeck MMJ, Jungbluth H, Voermans N. 259th ENMC international workshop: Anaesthesia and neuromuscular disorders, December 11th, 2020 and may 28‐29, 2021. Neuromuscul Disord. 2021;32:86‐97. [DOI] [PubMed] [Google Scholar]
- 7. Brouwers MC, Kho ME, Browman GP, et al. AGREE II: advancing guideline development, reporting and evaluation in health care. Cmaj. 2010;182:E839‐E842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van den Bersselaar LR, Gubbels M, Riazi S, et al. Mapping the current evidence on the anesthetic management of adult patients with neuromuscular disorders‐a scoping review. Can J Anaesth. 2022;69:756‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hohmann E, Brand JC, Rossi MJ, Lubowitz JH. Expert opinion is necessary: Delphi panel methodology facilitates a scientific approach to consensus. Art Ther. 2018;34:349‐351. [DOI] [PubMed] [Google Scholar]
- 10. ASA Physical Status Classification System . 2014. https://www.asahq.org/resources/clinical‐information/asa‐physical‐status‐classification‐system. Accessed September 25, 2021.
- 11. Malviya S, Voepel‐Lewis T, Chiravuri SD, et al. Does an objective system‐based approach improve assessment of perioperative risk in children? A preliminary evaluation of the 'NARCO'. Br J Anaesth. 2011;106:352‐358. [DOI] [PubMed] [Google Scholar]
- 12. Scottish Intercollegiate Guidelines Network. SIGN 50 : a guideline developers' handbook. Edinburgh: 2008.
- 13. Cripe LH, Tobias JD. Cardiac considerations in the operative management of the patient with Duchenne or Becker muscular dystrophy. Paediatr Anaesth. 2013;23:777‐784. [DOI] [PubMed] [Google Scholar]
- 14. Mazzanti A, Guz D, Trancuccio A, et al. Natural history and risk stratification in Andersen‐Tawil syndrome type 1. J Am Coll Cardiol. 2020;75:1772‐1784. [DOI] [PubMed] [Google Scholar]
- 15. Schindler RF, Scotton C, Zhang J, et al. POPDC1(S201F) causes muscular dystrophy and arrhythmia by affecting protein trafficking. J Clin Invest. 2016;126:239‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ten Dam L, Frankhuizen WS, Linssen W, et al. Autosomal recessive limb‐girdle and Miyoshi muscular dystrophies in The Netherlands: the clinical and molecular spectrum of 244 patients. Clin Genet. 2019;96:126‐133. [DOI] [PubMed] [Google Scholar]
- 17. Ogiso M, Isogai T, Kato K, Tanaka H, Tejima T, Isozaki E. Electrocardiographic and echocardiographic findings in muscular dystrophy patients with heart failure. Heart Vessels. 2018;33:1576‐1583. [DOI] [PubMed] [Google Scholar]
- 18. Herman GE, Finegold M, Zhao W, de Gouyon B, Metzenberg A. Medical complications in long‐term survivors with X‐linked myotubular myopathy. J Pediatr. 1999;134:206‐214. [DOI] [PubMed] [Google Scholar]
- 19. Yiu EM, Kornberg AJ. Duchenne muscular dystrophy. J Paediatr Child Health. 2015;51:759‐764. [DOI] [PubMed] [Google Scholar]
- 20. Klein A, Lillis S, Munteanu I, et al. Clinical and genetic findings in a large cohort of patients with ryanodine receptor 1 gene‐associated myopathies. Hum Mutat. 2012;33:981‐988. [DOI] [PubMed] [Google Scholar]
- 21. Muenster T, Mueller C, Forst J, Huber H, Schmitt HJ. Anaesthetic management in patients with Duchenne muscular dystrophy undergoing orthopaedic surgery: a review of 232 cases. Eur J Anaesthesiol. 2012;29:489‐494. [DOI] [PubMed] [Google Scholar]
- 22. Cohen BH. Mitochondrial and metabolic myopathies. Continuum (Minneap Minn). 2019;25:1732‐1766. [DOI] [PubMed] [Google Scholar]
- 23. Stunnenberg BC, LoRusso S, Arnold WD, et al. Guidelines on clinical presentation and management of nondystrophic myotonias. Muscle Nerve. 2020;62:430‐444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hayes LH, Yun P, Mohassel P, et al. Hypoglycemia in patients with congenital muscle disease. BMC Pediatr. 2020;20:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Taroni F, Uziel G. Fatty acid mitochondrial beta‐oxidation and hypoglycaemia in children. Curr Opin Neurol. 1996;9:477‐485. [DOI] [PubMed] [Google Scholar]
- 26. Ørngreen MC, Zacho M, Hebert A, Laub M, Vissing J. Patients with severe muscle wasting are prone to develop hypoglycemia during fasting. Neurology. 2003;61:997‐1000. [DOI] [PubMed] [Google Scholar]
- 27. Hilbert JE, Barohn RJ, Clemens PR, et al. High frequency of gastrointestinal manifestations in myotonic dystrophy type 1 and type 2. Neurology. 2017;89:1348‐1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Van de Putte P, Perlas A. Ultrasound assessment of gastric content and volume. Br J Anaesth. 2014;113:12‐22. [DOI] [PubMed] [Google Scholar]
- 29. Heier T, Caldwell JE, Sessler DI, Miller RD. Mild intraoperative hypothermia increases duration of action and spontaneous recovery of vecuronium blockade during nitrous oxide‐isoflurane anesthesia in humans. Anesthesiology. 1991;74:815‐819. [DOI] [PubMed] [Google Scholar]
- 30. Horn EP, Bein B, Böhm R, Steinfath M, Sahili N, Höcker J. The effect of short time periods of pre‐operative warming in the prevention of peri‐operative hypothermia. Anaesthesia. 2012;67:612‐617. [DOI] [PubMed] [Google Scholar]
- 31. Larach MG, Rosenberg H, Gronert GA, Allen GC. Hyperkalemic cardiac arrest during anesthesia in infants and children with occult myopathies. Clin Pediatr (Phila). 1997;36:9‐16. [DOI] [PubMed] [Google Scholar]
- 32. Gronert GA. Cardiac arrest after succinylcholine: mortality greater with rhabdomyolysis than receptor upregulation. Anesthesiology. 2001;94:523‐529. [DOI] [PubMed] [Google Scholar]
- 33. Gurnaney H, Brown A, Litman RS. Malignant hyperthermia and muscular dystrophies. Anesth Analg. 2009;109:1043‐1048. [DOI] [PubMed] [Google Scholar]
- 34. Segura LG, Lorenz JD, Weingarten TN, et al. Anesthesia and Duchenne or Becker muscular dystrophy: review of 117 anesthetic exposures. Paediatr Anaesth. 2013;23:855‐864. [DOI] [PubMed] [Google Scholar]
- 35. Fujimoto M, Terasaki S, Nishi M, Yamamoto T. Response to rocuronium and its determinants in patients with myasthenia gravis: a case‐control study. Eur J Anaesthesiol. 2015;32:672‐680. [DOI] [PubMed] [Google Scholar]
- 36. Caron MJ, Girard F, Girard DC, et al. Cisatracurium pharmacodynamics in patients with oculopharyngeal muscular dystrophy. Anesth Analg. 2005;100:393‐397. [DOI] [PubMed] [Google Scholar]
- 37. Wick S, Muenster T, Schmidt J, Forst J, Schmitt HJ. Onset and duration of rocuronium‐induced neuromuscular blockade in patients with Duchenne muscular dystrophy. Anesthesiology. 2005;102:915‐919. [DOI] [PubMed] [Google Scholar]
- 38. Muenster T, Schmidt J, Wick S, Forst J, Schmitt HJ. Rocuronium 0.3 mg x kg‐1 (ED95) induces a normal peak effect but an altered time course of neuromuscular block in patients with Duchenne's muscular dystrophy. Paediatr Anaesth. 2006;16:840‐845. [DOI] [PubMed] [Google Scholar]
- 39. Wefki Abdelgawwad Shousha AA, Sanfilippo M, Sabba A, Pinchera P. Sugammadex and reversal of neuromuscular block in adult patient with duchenne muscular dystrophy. Case Rep Anesthesiol. 2014;2014:680568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gurunathan U, Kunju SM, Stanton LML. Use of sugammadex in patients with neuromuscular disorders: a systematic review of case reports. BMC Anesthesiol. 2019;19:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mouri H, Jo T, Matsui H, Fushimi K, Yasunaga H. Effect of Sugammadex on postoperative myasthenic crisis in myasthenia gravis patients: propensity score analysis of a Japanese Nationwide database. Anesth Analg. 2020;130:367‐373. [DOI] [PubMed] [Google Scholar]
- 42. Mangla C, Bais K, Yarmush J. Myotonic dystrophy and anesthetic challenges: a case report and review. Case Rep Anesthesiol. 2019;2019:4282305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. White RJ, Bass SP. Myotonic dystrophy and paediatric anaesthesia. Paediatr Anaesth. 2003;13:94‐102. [DOI] [PubMed] [Google Scholar]
- 44. Hopkins PM, Rüffert H, Snoeck MM, et al. European malignant hyperthermia group guidelines for investigation of malignant hyperthermia susceptibility. Br J Anaesth. 2015;115:531‐539. [DOI] [PubMed] [Google Scholar]
- 45. Murayama T, Kurebayashi N, Ogawa H, et al. Genotype‐phenotype correlations of malignant hyperthermia and central Core disease mutations in the central region of the RYR1 channel. Hum Mutat. 2016;37:1231‐1241. [DOI] [PubMed] [Google Scholar]
- 46. Stamm DS, Aylsworth AS, Stajich JM, et al. Native American myopathy: congenital myopathy with cleft palate, skeletal anomalies, and susceptibility to malignant hyperthermia. Am J Med Genet A. 2008;146a:1832‐1841. [DOI] [PubMed] [Google Scholar]
- 47. Monnier N, Krivosic‐Horber R, Payen JF, et al. Presence of two different genetic traits in malignant hyperthermia families: implication for genetic analysis, diagnosis, and incidence of malignant hyperthermia susceptibility. Anesthesiology. 2002;97:1067‐1074. [DOI] [PubMed] [Google Scholar]
- 48. Miller DM, Daly C, Aboelsaod EM, et al. Genetic epidemiology of malignant hyperthermia in the UK. Br J Anaesth. 2018;121:944‐952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dowling JJ, Lillis S, Amburgey K, et al. King‐Denborough syndrome with and without mutations in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul Disord. 2011;21:420‐427. [DOI] [PubMed] [Google Scholar]
- 50. van den Bersselaar LR, Hellblom A, Gashi M, et al. Referral indications for malignant hyperthermia susceptibility diagnostics in patients without adverse anesthetic events in the era of next‐generation sequencing. Anesthesiology. 2022;136:940‐953. [DOI] [PubMed] [Google Scholar]
- 51. Johnston JJ, Dirksen RT, Girard T, et al. Variant curation expert panel recommendations for RYR1 pathogenicity classifications in malignant hyperthermia susceptibility. Genet Med. 2021;23:1288‐1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ye L, Zuo Y, Zhang P, Yang P. Sevoflurane enhances neuromuscular blockade by increasing the sensitivity of skeletal muscle to neuromuscular blockers. Int J Physiol Pathophysiol Pharmacol. 2015;7:172‐177. [PMC free article] [PubMed] [Google Scholar]
- 53. Hemphill S, McMenamin L, Bellamy MC, Hopkins PM. Propofol infusion syndrome: a structured literature review and analysis of published case reports. Br J Anaesth. 2019;122:448‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Savard M, Dupré N, Turgeon AF, Desbiens R, Langevin S, Brunet D. Propofol‐related infusion syndrome heralding a mitochondrial disease: case report. Neurology. 2013;81:770‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vanlander AV, Jorens PG, Smet J, et al. Inborn oxidative phosphorylation defect as risk factor for propofol infusion syndrome. Acta Anaesthesiol Scand. 2012;56:520‐525. [DOI] [PubMed] [Google Scholar]
- 56. Shimizu J, Tabata T, Tsujita Y, et al. Propofol infusion syndrome complicated with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke‐like episodes: a case report. Acute Med Surg. 2020;7:e473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Finsterer J. Congenital myasthenic syndromes. Orphanet J Rare Dis. 2019;14:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ciafaloni E. Myasthenia gravis and congenital myasthenic syndromes. Continuum (Minneap Minn). 2019;25:1767‐1784. [DOI] [PubMed] [Google Scholar]
- 59. Cata JP, Lasala JD, Williams W, Mena GE. Myasthenia gravis and thymoma surgery: a clinical update for the cardiothoracic anesthesiologist. J Cardiothorac Vasc Anesth. 2019;33:2537‐2545. [DOI] [PubMed] [Google Scholar]
- 60. Woodcock T, Barker P, Daniel S, et al. Guidelines for the management of glucocorticoids during the peri‐operative period for patients with adrenal insufficiency: guidelines from the Association of Anaesthetists, the Royal College of Physicians and the Society for Endocrinology UK. Anaesthesia. 2020;75:654‐663. [DOI] [PubMed] [Google Scholar]
- 61. Fujita Y, Moriyama S, Aoki S, et al. Estimation of the success rate of anesthetic management for thymectomy in patients with myasthenia gravis treated without muscle relaxants: a retrospective observational cohort study. J Anesth. 2015;29:794‐797. [DOI] [PubMed] [Google Scholar]
- 62. Della Rocca G, Coccia C, Diana L, et al. Propofol or sevoflurane anesthesia without muscle relaxants allow the early extubation of myasthenic patients. Can J Anaesth. 2003;50:547‐552. [DOI] [PubMed] [Google Scholar]
- 63. Phillips L, Trivedi JR. Skeletal muscle channelopathies. Neurotherapeutics. 2018;15:954‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang XY, Ren BW, Yong ZH, Xu HY, Fu QX, Yao HB. Mutation analysis of CACNA1S and SCN4A in patients with hypokalemic periodic paralysis. Mol Med Rep. 2015;12:6267‐6274. [DOI] [PubMed] [Google Scholar]
- 65. Matthews E, Neuwirth C, Jaffer F, et al. Atypical periodic paralysis and myalgia: a novel RYR1 phenotype. Neurology. 2018;90:e412‐e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vivekanandam V, Munot P, Hanna MG, Matthews E. Skeletal muscle channelopathies. Neurol Clin. 2020;38:481‐491. [DOI] [PubMed] [Google Scholar]
- 67. Raja Rayan DL, Hanna MG. Managing pregnancy and anaesthetics in patients with skeletal muscle channelopathies. Neuromuscul Disord. 2020;30:539‐545. [DOI] [PubMed] [Google Scholar]
- 68. Bandschapp O, Iaizzo PA. Pathophysiologic and anesthetic considerations for patients with myotonia congenita or periodic paralyses. Paediatr Anaesth. 2013;23:824‐833. [DOI] [PubMed] [Google Scholar]
- 69. Marchant CL, Ellis FR, Halsall PJ, Hopkins PM, Robinson RL. Mutation analysis of two patients with hypokalemic periodic paralysis and suspected malignant hyperthermia. Muscle Nerve. 2004;30:114‐117. [DOI] [PubMed] [Google Scholar]
- 70. Rüffert H, Bastian B, Bendixen D, et al. Consensus guidelines on perioperative management of malignant hyperthermia suspected or susceptible patients from the European malignant hyperthermia group. Br J Anaesth. 2021;126:120‐130. [DOI] [PubMed] [Google Scholar]
- 71. Thornton CA. Myotonic dystrophy. Neurol Clin 2014;32:705‐719, viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sinclair JL, Reed PW. Risk factors for perioperative adverse events in children with myotonic dystrophy. Paediatr Anaesth. 2009;19:740‐747. [DOI] [PubMed] [Google Scholar]
- 73. Mathieu J, Allard P, Gobeil G, Girard M, De Braekeleer M, Bégin P. Anesthetic and surgical complications in 219 cases of myotonic dystrophy. Neurology. 1997;49:1646‐1650. [DOI] [PubMed] [Google Scholar]
- 74. Reimann J, Kornblum C. Towards central nervous system involvement in adults with hereditary myopathies. J Neuromuscul Dis. 2020;7:367‐393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Imison AR. Anaesthesia and myotonia‐‐an Australian experience. Anaesth Intensive Care. 2001;29:34‐37. [DOI] [PubMed] [Google Scholar]
- 76. Veyckemans F, Scholtes JL. Myotonic dystrophies type 1 and 2: anesthetic care. Paediatr Anaesth. 2013;23:794‐803. [DOI] [PubMed] [Google Scholar]
- 77. Kim CS, Park JM, Park D, Kim DH, Park JS. Opioid use may be associated with postoperative complications in myotonic dystrophy type 1 with high‐grade muscular impairment. Sci Rep. 2021;11:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kirzinger L, Schmidt A, Kornblum C, Schneider‐Gold C, Kress W, Schoser B. Side effects of anesthesia in DM2 as compared to DM1: a comparative retrospective study. Eur J Neurol. 2010;17:842‐845. [DOI] [PubMed] [Google Scholar]
- 79. Weingarten TN, Hofer RE, Milone M, Sprung J. Anesthesia and myotonic dystrophy type 2: a case series. Can J Anaesth. 2010;57:248‐255. [DOI] [PubMed] [Google Scholar]
- 80. Mercuri E, Bönnemann CG, Muntoni F. Muscular dystrophies. Lancet. 2019;394:2025‐2038. [DOI] [PubMed] [Google Scholar]
- 81. Latimer R, Street N, Conway KC, et al. Secondary conditions among males with Duchenne or Becker muscular dystrophy. J Child Neurol. 2017;32:663‐670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wang S, Peng D. Cardiac involvement in Emery‐Dreifuss muscular dystrophy and related management strategies. Int Heart J. 2019;60:12‐18. [DOI] [PubMed] [Google Scholar]
- 83. Schorling DC, Müller CK, Pechmann A, et al. Coagulation disorders in Duchenne muscular dystrophy? Results of a registry‐based online survey. Acta Myol. 2020;39:2‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Turturro F, Rocca B, Gumina S, et al. Impaired primary hemostasis with normal platelet function in Duchenne muscular dystrophy during highly‐invasive spinal surgery. Neuromuscul Disord. 2005;15:532‐540. [DOI] [PubMed] [Google Scholar]
- 85. Schorling DC, Kirschner J, Bönnemann CG. Congenital muscular dystrophies and myopathies: an overview and update. Neuropediatrics. 2017;48:247‐261. [DOI] [PubMed] [Google Scholar]
- 86. Amburgey K, McNamara N, Bennett LR, McCormick ME, Acsadi G, Dowling JJ. Prevalence of congenital myopathies in a representative pediatric United States population. Ann Neurol. 2011;70:662‐665. [DOI] [PubMed] [Google Scholar]
- 87. Witting N, Werlauff U, Duno M, Vissing J. Phenotypes, genotypes, and prevalence of congenital myopathies older than 5 years in Denmark. Neurol Genet. 2017;3:e140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Jungbluth H, Treves S, Zorzato F, et al. Congenital myopathies: disorders of excitation‐contraction coupling and muscle contraction. Nat Rev Neurol. 2018;14:151‐167. [DOI] [PubMed] [Google Scholar]
- 89. Ahmed ST, Craven L, Russell OM, Turnbull DM, Vincent AE. Diagnosis and treatment of mitochondrial myopathies. Neurotherapeutics. 2018;15:943‐953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mtaweh H, Bayır H, Kochanek PM, Bell MJ. Effect of a single dose of propofol and lack of dextrose administration in a child with mitochondrial disease: a case report. J Child Neurol. 2014;29:Np40‐Np46. [DOI] [PubMed] [Google Scholar]
- 91. Finsterer J. Update review about metabolic myopathies. Life (Basel). 2020;10:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Angelini C, Marozzo R, Pegoraro V, Sacconi S. Diagnostic challenges in metabolic myopathies. Expert Rev Neurother. 2020;20:1287‐1298. [DOI] [PubMed] [Google Scholar]
- 93. Miyamoto Y, Miyashita T, Takaki S, Goto T. Perioperative considerations in adult mitochondrial disease: a case series and a review of 111 cases. Mitochondrion. 2016;26:26‐32. [DOI] [PubMed] [Google Scholar]
- 94. Footitt EJ, Sinha MD, Raiman JA, Dhawan A, Moganasundram S, Champion MP. Mitochondrial disorders and general anaesthesia: a case series and review. Br J Anaesth. 2008;100:436‐441. [DOI] [PubMed] [Google Scholar]
- 95. De Vries MC, Brown DA, Allen ME, et al. Safety of drug use in patients with a primary mitochondrial disease: an international Delphi‐based consensus. J Inherit Metab Dis. 2020;43:800‐818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Conover ZR, Talai A, Klockau KS, Ing RJ, Chatterjee D. Perioperative Management of Children on ketogenic dietary therapies. Anesth Analg. 2020;131:1872‐1882. [DOI] [PubMed] [Google Scholar]
- 97. Stettner GM, Viscomi C, Zeviani M, Wilichowski E, Dutschmann M. Hypoxic and hypercapnic challenges unveil respiratory vulnerability of Surf1 knockout mice, an animal model of Leigh syndrome. Mitochondrion. 2011;11:413‐420. [DOI] [PubMed] [Google Scholar]
- 98. Bollig G, Mohr S, Raeder J. McArdle's disease and anaesthesia: case reports. Review of potential problems and association with malignant hyperthermia. Acta Anaesthesiol Scand. 2005;49:1077‐1083. [DOI] [PubMed] [Google Scholar]
- 99. Hopkins PM, Ellis FR, Halsall PJ. Comparison of in vitro contracture testing with ryanodine, halothane and caffeine in malignant hyperthermia and other neuromuscular disorders. Br J Anaesth. 1993;70:397‐401. [DOI] [PubMed] [Google Scholar]
- 100. Martin JM, Gillingham MB, Harding CO. Use of propofol for short duration procedures in children with long chain 3‐hydroxyacyl‐CoA dehydrogenase (LCHAD) or trifunctional protein (TFP) deficiencies. Mol Genet Metab. 2014;112:139‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Justiz AC, Mayhew JF. Anesthesia in a child with medium‐chain acyl‐CoA dehydrogenase deficiency. Paediatr Anaesth. 2006;16:1293‐1294. [DOI] [PubMed] [Google Scholar]
- 102. Myasthenia Gravis Foundation of America Medical/Scientific Advisory Board . www.Myasthenia.org/cautionary‐drugs. Accessed November 25, 2021.
- 103. Orsucci D, Ienco EC, Siciliano G, Mancuso M. Mitochondrial disorders and drugs: what every physician should know. Drugs Context. 2019;8:212588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Trevisan CP, Accorsi A, Morandi LO, et al. Undiagnosed myopathy before surgery and safe anaesthesia table. Acta Myol. 2013;32:100‐105. [PMC free article] [PubMed] [Google Scholar]
- 105. Hopkins PM. Anaesthesia and the sex‐linked dystrophies: between a rock and a hard place. Br J Anaesth. 2010;104:397‐400. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The anonymized dataset generated during this study is available from the corresponding author on reasonable request from qualified investigators.