

Abstract
The incorporation of the gem‐difluoromethylene (CF2) group into organic frameworks is highly sought due to the influence of this unit on the physicochemical and pharmacological properties of molecules. Herein we report an operationally simple, mild, and switchable protocol to access various gem‐difluoro compounds that employs chlorodifloroacetic anhydride (CDFAA) as a low‐cost and versatile fluoroalkylating reagent. Detailed mechanistic studies revealed that electron‐transfer photocatalysis triggers mesolytic cleavage of a C−Cl bond generating a gem‐difluoroalkyl radical. In the presence of alkene, this radical species acts as a unique intermediate that, under solvent‐controlled reaction conditions, delivers a wide range of gem‐difluorinated γ‐lactams, γ‐lactones, and promotes oxy‐perfluoroalkylation. These protocols are flow‐ and batch‐scalable, possess excellent chemo‐ and regioselectivity, and can be used for the late‐stage diversification of complex molecules.
Keywords: Anhydrides, gem-Difluoro Compounds, Late-Stage Functionalization, Photoredox, Radical Mechanisms
Electron‐transfer photocatalysis triggers selective fragmentation of a C−Cl bond of chlorodifloroacetic anhydride (CDFAA) generating a gem‐difluoroalkyl radical intermediate. In the presence of olefin and under solvent‐controlled reaction conditions, a notable synthetic divergence is achieved to access a wide array of gem‐difluoro compounds.

Introduction
The evolution of classical organic synthesis over the last 40 years has resulted in the formation of significant new fields, opening doors to previously elusive chemical transformations. Carbon‐hydrogen bond activation, [1] cross‐coupling reactions, [2] organo‐, [3] and enzyme‐catalysis [4] are a few examples that currently represent the fundamental principles of reactivity in modern chemistry. Furthermore, non‐classical photo‐[ 5 , 6 , 7 , 8 , 9 , 10 ] and electrochemical [11] activation modes of small molecules have revitalized the field of single‐electron transfer (SET) transformations. The way chemists approach synthetic organic chemistry is also constantly evolving. Unconventional, yet valuable concepts have been introduced into the field, including but not limited to late‐stage functionalization, [12] atom economy,[ 13 , 14 ] bifunctional reagents, [15] divergent syntheses,[ 16 , 17 , 18 , 19 , 20 ] and multicomponent reactions. [21] Indeed, these strategies allow scientists to readily access a broader chemical space for achieving synthetic endeavors. One example is the application of switchable divergent protocols in catalysis as they involve the switch of one of the reaction parameters such as photocatalyst, light source, temperature, etc. to diversify the outcome of the transformation (Scheme 1A). Although significant progress has been made in this regard, it is still underexplored and most reported strategies pertain to two products only, whereas divergence between three and more structures from the same starting material comes with inherent difficulties. [22]
Scheme 1.
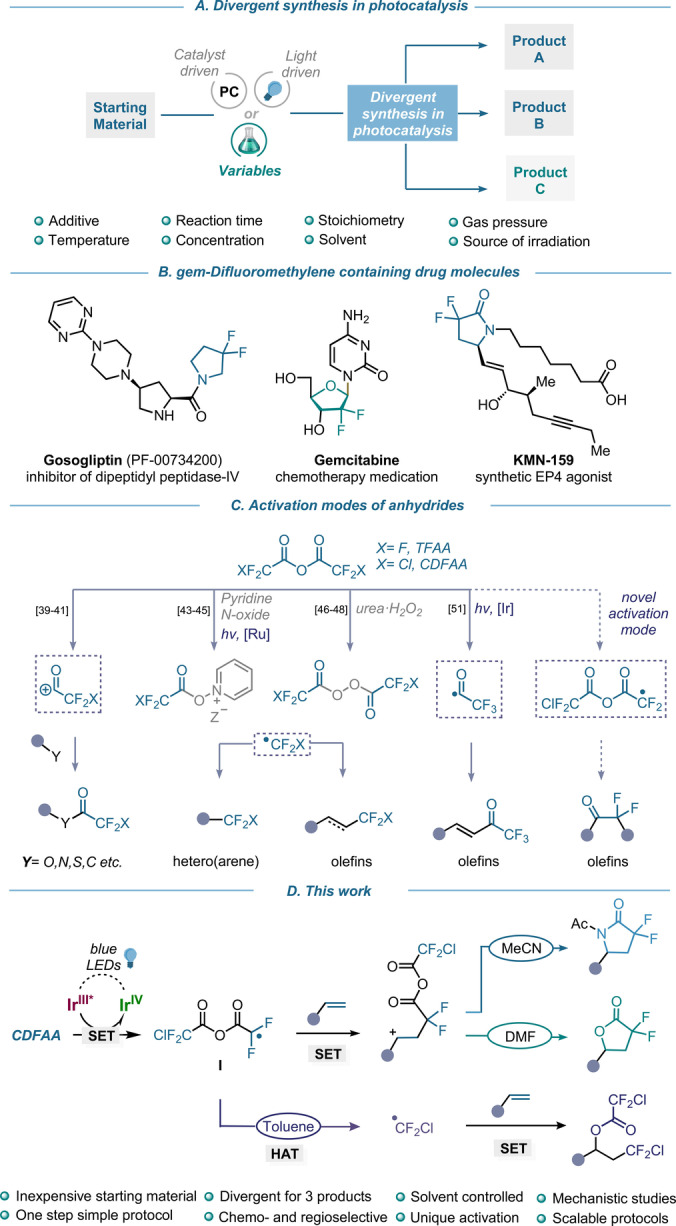
Existing and new reaction modes of halogenated anhydrides. A) Divergent synthesis in photocatalysis. B) Notable examples of gem‐difluoromethylene (CF2) containing biorelevant structures. C) Established and new activation modes of anhydrides. D) Photoredox activation of CDFAA for the switchable synthesis of gem‐difluoro cyclic and acyclic compounds.
Another powerful platform for increasing molecular complexity is the use of functional group transfer reagents (FGTRs).[ 23 , 24 ] This strategy is based on the development of organic and inorganic reagents, which have the capacity to enforce the transfer of the desired functional group (FG) to a molecule in a straightforward fashion. The field of fluorine chemistry is one of the privileged examples,[ 25 , 26 ] which has advanced from the discovery of teflon and freon [27] to an extremely fertile bench of novel functionalities, reagents, and reactivities, such as fluoroalkylation, [28] sulfur‐based fluorination, [29] acyl fluorides chemistry, [30] etc. This is alluring by the fact that more than 20 % of drugs now contain fluorine atoms or fluorinated chains and that the development of the medicinal chemistry of fluorine has grown exponentially for already almost 20 years.[ 31 , 32 ] In particular, the difluoromethylene group (CF2) has recently received great attention because of its relevance in bioisostere design, [33] and to modulate physicochemical and pharmacological properties of a molecule. [34] It is also present in many biologically active molecules, namely dipeptidyl peptidase IV inhibitor Gosogliptin, [35] chemotherapy medication Gemcitabine, [36] while the difluoro analog of γ‐lactam KMN‐159 is five‐folds more active than its nonfluorinated analog (Scheme 1B). [37]
Over the past few decades, various electrophilic, nucleophilic, radical, and cross‐coupling methods have been used to introduce the difluoromethyl moiety. [38] Among many developed reagents, perfluorocarboxylic anhydrides, such as trifluoroacetic anhydride (TFAA) and chlorodifluoroacetic anhydride (CDFAA), have been employed as efficient protecting groups as well as electrophilic acylation reagents of nitrogen, oxygen, sulfur, and carbon centers in organic synthesis (Scheme 1C).[ 39 , 40 , 41 ] These well‐known reagents are now opening up new facets of their reactivity, allowing the preparation of fluoroalkyl radical species in a straightforward fashion. [42] The groups of Stephenson[ 43 , 44 , 45 ] and Sodeoka[ 46 , 47 , 48 ] utilized stoichiometric amounts of oxidants to access CF2X (X=F, Cl) reactive species under photoredox and copper catalysis or metal free conditions, respectively (Scheme 1C). These methodologies have found wide applications for the direct functionalization of (hetero)arenes and olefins, including complex molecules and organic functional materials.[ 49 , 50 ] Recently our group has explored a powerful divergent photocatalytic approach that occurs via a remarkable C−O bond fragmentation of trifluoroacetic anhydride, to accomplish either radical trifluoroacetylation or trifluoromethylation of olefins (Scheme 1C). [51] While a great effort in the activation of perfluorocarboxylic anhydrides for introducing fluorine‐containing functional groups (CF2X (X=F, Cl), CF3C(O)) has been previously reported, similar procedures for generating the corresponding, yet chemically distinct, gem‐difluoromethylene moiety remain elusive. Immediate activation of CDFAA to originate gem‐difluoroalkyl radical intermediates is expected to be an atom‐ and step‐economic strategy to synthesize diverse fluorinated molecules from fundamental feedstock. In this context, the synthesis of gem‐difluoro‐γ‐lactams and lactones has been a topic of interest for many researchers while demonstrating their preparation via transition‐metal catalyzed radical cascade cyclization reactions,[ 52 , 53 , 54 ] oxidative intramolecular cyclizations, [55] and photocatalytic hydroxydifluoroacetylation of alkenes. [56] Although these methods provide routes to access fluorinated ring systems, the incorporation of the gem‐difluoromethylene group or a surrogate moiety via a multi‐step synthetic sequence is often required prior the cyclization. [57] Hence, the perspective of synthetic efficiency, divergence, and atom economy to introduce CF2 group into organic frameworks remains under investigation. Herein, we report a visible light‐mediated strategy for the direct unlock of chlorodifluoroacetic anhydride, a low‐cost, well‐known, and readily available reagent, to access the difluoromethylene anhydride radical I which can participate in Giese‐type addition to olefins (Scheme 1D). [58] The reactivity of subsequent carbocation obtained following photocatalytic oxidation is further determined by the solvent, detailed mechanistic studies of this effect have shown to occur by three distinct pathways, enabling a divergent approach to the important gem‐difluoro scaffolds such as α,α‐difluoro‐γ‐lactams, γ‐lactones, as well as chlorodifluoroalkanes with excellent level of chemo‐ and regioselectivity. These methodologies are easily scalable in batch and in flow, while the corresponding products are versatile, high‐value fluorine‐containing building blocks for organic synthesis. This catalytic manifold promoted by visible light was also successfully applied for the activation of diverse halogenated anhydrides.
Results and Discussion
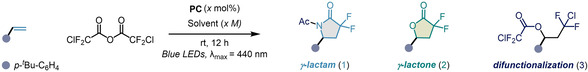
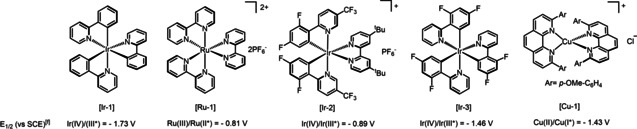
Our research group is interested in perfluoroalkylation reactions,[ 59 , 60 ] especially driven by perfluorocarboxylic anhydrides. [51] We therefore set out to prove the feasibility of CDFAA as an inherent source of gem‐difluoroalkyl radical intermediates for organic synthesis. We suspected, that subjecting this reagent to a single electron transfer (SET) process, an irreversible and selective reductive C−Cl bond fragmentation may occur, resulting in the formation of intermediate I bearing radical and nucleophilic reactive sites (Scheme 1D). The latter species would be incorporated into an alkene via sequential deployment of the reactive centers. It must be emphasized, that reaction rates of the active sites of I is sufficiently distinct towards the target unsaturated system and can permit post‐derivatization of intermediates by solvent molecules, resulting in varied product outcome with complete chemo‐ and regioselectivity. By rational reaction design, we envisioned that in the presence of acetonitrile the sequence will proceed via a multicomponent Ritter type reaction in a single chemical operation to produce gem‐difluoro‐γ‐lactams, while other polar coordinating solvents could accelerate an intramolecular nucleophilic cyclization to access gem‐difluoro‐γ‐lactones. Based on this hypothesis, we commenced our investigations by evaluating the reduction potential of CDFAA. Cyclic voltammetry measurements revealed a cathodic wave at a negatively shifted potential of −0.83 V (vs. SCE, see Supporting Information Figure 6.5, page S24). Accordingly, the use of a photocatalyst (PC), such as tris‐(2‐phenylpyridine)iridium (III) (fac‐Ir(ppy)3), was found to be optimal due to its fairly low reduction potential of the excited state of E 0(IrIV/IrIII*)=−1.73 V (vs. SCE). The reaction between 4‐tert‐butylstyrene and CDFAA was selected as the prototypical system. A thorough evaluation of reaction conditions, including solvents, concentrations, and photocatalysts has been carried out, and key observations are summarized in Table 1 (for details, see Supporting Information, pages S5–S13). The reaction mixture consisting of alkene, CDFAA (2 equiv), photocatalyst [Ir‐I] (1 mol %), and acetonitrile (0.1 M) under blue‐light irradiation in a high‐intensity visible light photoreactor after 12 h provided the desired gem‐difluoro‐γ‐lactam 1 in 56 % yield (Table 1, entry 1). Subsequent modulation of the alkene concentration in solvent (0.03 M) significantly improved the product formation up to 92 % (entry 2). Screening of several photocatalysts showed that [Ru‐1] and [Ir‐2] were incompatible with our reaction system, while PCs with slightly lower E 1/2 (M+/M*) values ([Ir‐3] and [Cu‐1]) than CDFAA showed encouraging results (entry 5–6).[ 61 , 62 ] Higher catalyst loadings and the use of water as an additive both lowered the efficiency, whilst pleasingly 2‐bromo‐2,2‐difluoroacetic anhydride was also demonstrated to be a competent reagent for the transformation (Table 1, entry 7–9). Having found optimal conditions for the synthesis of 1, we next assessed the effect of other solvents on the reaction outcome. The use of dichloromethane (DCM) resulted in the formation of gem‐difluoro‐γ‐lactone 2 under standard reaction conditions (Table 1, entry 10).
Table 1.
Reaction development and optimization.
|
| ||||||
|---|---|---|---|---|---|---|
|
Entry |
PC (1 mol%) |
Solvent |
Concentration [M] |
1 (%)[a] |
2 (%)[a] |
3 (%)[a] |
|
1 |
[Ir‐1] |
MeCN |
0.10 |
56 |
5 |
– |
|
2 |
[Ir‐1] |
MeCN |
0.03 |
92 (90)[e] |
2 |
– |
|
3 |
[Ru‐1] |
MeCN |
0.03 |
4 |
– |
– |
|
4 |
[Ir‐2] |
MeCN |
0.03 |
18 |
– |
– |
|
5 |
[Ir‐3] |
MeCN |
0.03 |
88 |
– |
– |
|
6 |
[Cu‐1] |
MeCN |
0.03 |
80 |
4 |
– |
|
7 |
[Ir‐1] |
MeCN[b] |
0.10 |
32 |
13 |
– |
|
8 [c] |
[Ir‐1] |
MeCN |
0.03 |
64 |
10 |
– |
|
9 |
[Ir‐1][d] |
MeCN |
0.03 |
83 |
6 |
– |
|
10 |
[Ir‐1] |
DCM |
0.03 |
– |
30 |
53 |
|
11 |
[Ir‐1] |
DMF |
0.03 |
– |
80 |
3 |
|
12 |
[Ir‐1] |
DMF |
0.17 |
– |
86 (81)[e] |
3 |
|
13 |
[Ir‐1] |
DMA |
0.03 |
– |
6 |
– |
|
14 |
[Ir‐1] |
CHCl3 |
0.03 |
– |
56 |
28 |
|
15 |
[Ir‐1] |
Dry Et2O |
0.03 |
– |
9 |
75 |
|
16 |
[Ir‐1] |
Toluene |
0.03 |
– |
16 |
76 |
|
17 |
[Ir‐1] |
Toluene |
0.17 |
– |
14 |
81 (75)[e] |
|
18 |
[Ir‐1] |
THF : CHCl3 (1 : 1) |
0.03 |
– |
44 |
14 |
|
| ||||||
[a] General conditions: 4‐tert‐butylstyrene (0.5 mmol, 1 equiv), catalyst (1.0 mol %), and CDFAA (2 equiv), solvent (x M), 350 W blue LEDs, rt, 12 h. Yields of 1, 2, and 3 were determined by GC‐MS against an internal standard of n‐decane. [b] H2O (1 equiv) was added. [c] Reaction was performed with 2‐bromo‐2,2‐difluoroacetic anhydride. [d] 2.5 mol % of fac‐Ir(ppy)3. [e] Yields in parentheses represent isolated yields. [f] E 1/2 values were taken from refs. [5, 61, 62].
Meanwhile, thorough analysis of the latter experiment revealed the intriguing formation of product 3, resulting from oxy‐fluoroalkylation of 4‐tert‐butylstyrene (Table 1, entry 10). After a detailed examination of the effect of solvent and concentration, a complete synthetic discrepancy between products 2 and 3 was reached. When carrying out the reaction in N,N‐dimethylformamide (DMF) (0.17 M), gem‐difluoro‐γ‐lactone 2 was formed in 86 %, while switching the solvent to toluene (0.17 M), chlorodifluoroalkane 3 was isolated in excellent chemical yield (Table 1, entry 11–18). The results demonstrate, that entire chemo‐ and regioselectivity between all three types of fluorinated scaffolds was achieved by simply controlling the reactivity of radical intermediate I with a solvent under photoredox catalysis.
With the optimal conditions established, the modularity and scope of the reaction with respect to a wide array of readily available olefin building blocks bearing common functionalities were investigated. As shown in Scheme 2, up to 90 % isolated yield of exclusively gem‐difluoro‐γ‐lactams or gem‐difluoro‐γ‐lactones were obtained by performing the reaction in either MeCN or in DMF, respectively. Electron neutral (4) and electron‐donating (1–3, 5–14, 37–41, 44, 52) aryl olefins substituted at the ortho‐, meta‐, and para‐positions were well tolerated. Of note, halogen atoms, located at different parts of a molecule scaffold, remained untouched, confirming the robustness and mildness of this methodology and enabling further post derivatization (14–21, 26–28, 30–31, 43, 48–51). Adducts containing polycyclic aromatics such as naphthalene (9, 42), fluorene (10), and annulene (46) were generated with good yields in these reactions. Additional functional groups were also examined to obtain the corresponding lactams and lactones with fluorinated ether (21), amide (22, 53), ester (23), and ether (39, 40) moeties. β‐Substituted styrenes with phenyl (24), methyl (25, 52), and oxazolidine (53) underwent the desired transformation successfully, giving the corresponding cyclic adducts in decent isolated yields and diastereoselectivities (d.r. >20 : 1). The structures of lactams 1 and 24 were unambigously demonstrated by single‐crystal X‐ray crystallography. [63] Disubstituted olefins with remote functional groups like trifluoromethoxy (26), a labile iodine atom (27), and other multi‐substituted aromatics with methoxy, methyl, and halogen groups were also smoothly converted to their corresponding products with excellent chemoselectivities (26–32, 46–51). The formation of spirocycles is an intriguing transformation since it opens the possibility of discovering novel fluorinated derivatives with promising biological properties. [64] Using this photoredox methodology, we successfully synthesized six‐ (33) and seven‐membered (34, 46) spirolactams and lactones. A terminal alkene was found to be more reactive than internal one (46), whereas an alkyne group remains completely unmarked (45), displaying good site selectivity. We also demonstrated that the reaction is applicable to other nitrile solvents affording the corresponding lactam derivatives (35, 36). However, the scope was limited to aryl substituted olefins, most likely due to the inefficiency of the photocatalyst to oxidize a non‐benzylic radical species. Encouraged by the broad functional group tolerance, we next aimed to illustrate the use of this protocol for the late‐stage functionalization of complex molecules, such as natural products and drug derivatives (Scheme 3). This methodology was equally successful in the formation of gem‐difluoro‐γ‐lactams for the derivatives of 6‐acetyl‐1,1,2,4,4,7‐hexamethyltetralin (AHTN) (55) and L‐Menthol (56). Tocopherol, a structurally complex bioactive precursor, also reacted smoothly to yield fluorinated lactone (57), showcasing the practical utility for late‐stage diversification. Camphorsulfonyl imitate (58) underwent the lactone reaction showing endurance to sulfonamides. Similarly, the reaction of a diacetone fructose derivative in either acetonitrile or dimethylformamide afforded lactam (59) and lactone (60) in 72 % and 74 % yield, respectively. Even polycyclic derivatives of estrone, which is poorly soluble in many organic solvents, yielded the desired products (61, 62) with high divergence. The reaction of L‐Camphanic acid derivative also illustrated the potential of this methodology. When a variety of solvents were applied, the result diverged into gem‐difluoro‐γ‐lactam in MeCN (63), gem‐difluoro‐γ‐lactone in DMF (64) and chlorodifluoroacetate derivative in toluene (65). To be noted, the synthesis of these complex fluorinated scaffolds is remarkably arduous with existing synthetic methodologies.
Scheme 2.

Reaction scope. A) Scope of γ‐lactams. B) Scope of γ‐lactones. [a] General conditions I: olefin (1 equiv), fac‐Ir(ppy)3 (1 mol%), CDFAA (2 equiv), MeCN (0.03 M), blue LEDs, rt, 12 h, isolated yields. [b] X‐Ray figures were created with the CYLview 2.0 Software, Legault, C. Y., Sherbrooke University 2020 (www.cylview.com). Hydrogen atoms were omitted for clarity. [c] Solvent EtCN (0.03 M). [d] Solvent iPrCN (0.03 M). [e] General conditions II: olefin (1 equiv), fac‐Ir(ppy)3 (1 mol%), CDFAA (2 equiv), DMF (0.17 M), blue LEDs, rt, 12 h, isolated yields. [f] Reaction conducted for 24 h.
Scheme 3.

Late‐stage functionalization of complex molecules. [a] General conditions I. [b] General conditions II. [c] General conditions III.
To further display the value of these operationally facile protocols, the process was applied to gram‐scale synthesis (10 mmol, 1.95 g) in batch, and after 24 hours under standard reaction conditions the corresponding products 1 and 2 were formed without significant decrease in isolated yields (Scheme 4A). To reduce the reaction time and to enhance chemical efficiency, we additionally demonstrated the scalability of this manifold in continuous flow using an automatic syringe pump and a high intensity blue LED photoreactor equipped with a 100 mL tube of an internal diameter 0.1016 cm (see Supporting Information, page S41). Having this technology in hand, we carried out the lactonization reaction on 20 mmol scale with a flow rate of 0.3 mL min−1 and a residence time (t R) of 2.74 h, affording product 2 in 70 %. The resulting molecules 1 and 2 can be further manipulated in expedient manner to provide added‐value cyclic and acyclic building blocks for organic synthesis including gem‐difluoro pyrrolidin‐2‐one (66), 3,3‐difluoro‐pyrrolidine (67), 2,2‐difluoro‐1,4‐hydroxy amides (68, 69), and 2,2‐difluoro‐1,4‐diols (70, 71) (Scheme 4B).
Scheme 4.
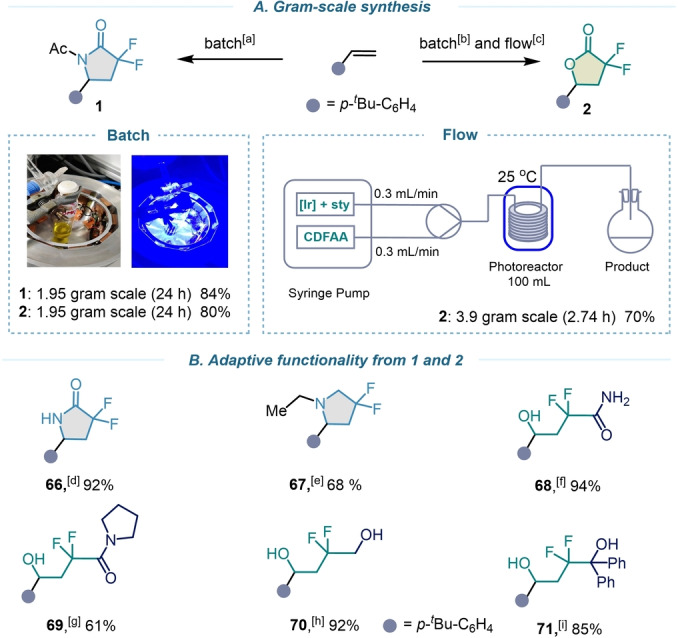
A) Gram‐scale synthesis. [a,b] 4‐tert‐Butylstyrene (10 mmol), fac‐Ir(ppy)3 (0.1 mmol), CDFAA (20 mmol), MeCN (300 mL) or DMF (60 mL), blue LEDs, rt, 24 h, isolated yield. [c] 4‐tert‐Butylstyrene (20 mmol), fac‐Ir(ppy)3 (0.2 mmol), CDFAA (40 mmol), DMF (60 mL), flow rate=0.3 mL min−1, τ R=2.74 h, isolated yields. B) Adaptive functionality from 1 and 2. [d] 1 (0.5 mmol), pentylamine (0.6 mmol), THF (1 mL), rt, 16 h. [e] 1 (0.5 mmol), LiAlH4 (2.5 mmol), THF (1 mL), rt, 2 h. [f] 2 (0.5 mmol), 1 mL 30 % NH4OH, rt, 12 h. [g] 2 (0.5 mmol), Et3N (0.55 mmol), pyrrolidine (0.55 mmol), 1 mL MeOH/THF (1 : 4), rt, 12 h. [h] 1 (0.5 mmol), LiAlH4 (2.5 mmol), THF (1 mL), rt, 4 h. [i] 2 (0.5 mmol), PhLi (2.5 mmol), Et2O (1.5 mL), 0 °C to rt, 6 h.
By a combination of experimental, spectroscopic, [65] and DFT [66] calculations, we next turned our attention to shed light on the key steps of this highly switchable divergent protocol that unequally argue in favor of a radical mechanism. Preliminary examination revealed that in the presence of a radical scavenger such as (2,2,6,6‐tetramethylpiperidin‐1‐yl)oxyl (TEMPO) (see Supporting Information, page S45), the formation of the desired product was completely suppressed. No product formation was observed at elevated temperature (90 °C), in the absence of a photocatalyst nor visible‐light. Carrying out the reaction in the presence of air under established conditions marginally affected the product yield (Scheme 5E). We proposed that the process begins by excitation of the photocatalyst under blue light irradiation. Experimentally, this hypothesis is fully consistent with our UV/Vis spectroscopic analysis that shows the absorption feature of fac‐Ir(ppy)3 at 440 nm to generate IrIII* (Scheme 5B, see Supporting Information, page S29). Owing to its low reduction potential E 0(IrIV/IrIII*)=−1.73 V (vs. SCE), initiation of the catalytic cycle may arise by a single electron photoreduction of CDFAA, triggering the reagent's intermolecular deactivation process. Upon SET reduction, the ensuing fragmentation process could afford either the corresponding acyl radical or the gem‐difluoroalkyl radical I via C−O bond or C−Cl bond fragmentation, respectively. However, DFT calculations showed highly exergonic, exclusive, and irreversible C−Cl bond cleavage with the immediate release of radical species I and chloride ion expulsion in both solvent models (ΔG MeCN=−71.6 kcal mol−1; ΔG DMF=−71.5 kcal mol−1) (Scheme 5A). Attempts to optimize the structure of the distinct radical anion intermediate of CDFAA were unsuccessful, consistently leading to the breaking of C−Cl bond. This indicates a particularly high instability for this species and the presence of a labile C−Cl bond. Cyclic voltammetry data provided an onset potential of −0.83 V (vs. SCE) for the irreversible reduction of CDFAA. Moreover, Stern–Volmer quenching studies indicated a highly efficient quenching of the excited T1‐state of IrIII by the reagent (K=2×109 M−1 s−1) (Scheme 5B, see Supporting Information, page S21). Upon addition of species I to the olefin, and the subsequent single electron oxidation of II (E 0=0.34 V) by the PC (E 0 (IrIV/IrIII)=0.77 V vs. SCE), the stabilized carbocation III is formed, and IrIII is regenerated, closing the catalytic cycle. The radical nature of species II in initial addition step to the olefin was also indicated by Hammett studies (Scheme 5C, see Supporting Information, page S34) showing a slightly negative reaction parameter in both solvents (ρ=−0.52 in MeCN and ρ=−0.42 in DMF). To gain additional insights into the process, we coupled the photochemical depletion of the excited state IrIII by CDFAA with cyclic voltammetry analysis (Scheme 5B, see Supporting Information, page S28). Irradiation of the mixture of PC and the reagent for 6 min at 440 nm resulted in the complete depletion of the IrIII/IrIV couple in cyclic voltammetry, and a novel species oxidized at 0.55 V vs. Ag/AgNO3 was detected, showing irreversible oxidation of fac‐Ir(ppy)3. Addition of the olefin to this reaction mixture caused nondestructive effect on IrIII/IrIV cycle, due to the oxidation of formed benzylic radical II to III and regeneration of photocatalyst. When carbocation III is stabilized by direct interaction with nucleophilic solvents such as MeCN or DMF, two distinct intermediates can be generated. In the case of DMF, iminium species IV is formed, while coordination of acetonitrile to III, affords intermediate VII. To uphold theoretical values for these exergonic steps, a series of experiments in a mixture of solvents was performed, which demonstrated that even at low concentration of DMF, the formation of lactone is highly dominated (Scheme 5C). The experimental equilibrium constants (K DMF/K MeCN=57 (experimental)) are in excellent agreement with the calculated values (K DMF/K MeCN=59 (calculated)) (see Supporting Information, pages S38–S40). In the case of DMF, intramolecular cyclization of IV followed by deacylation leads to the formation of γ‐lactones. On the other hand, intermediate VIII is unstable and undergoes a rearrangement to afford gem‐difluoro‐γ‐lactams. Since the latter example represents a three‐component intramolecular Ritter type reaction, [67] we set out to investigate the origin of oxygen in the N‐acetate moiety. Our experiments conclude that the only source of oxygen would be CDFAA, as the competition experiments using H2O18 showed no incorporation of 18O in the product, supporting our proposed mechanism. Experiments carried out with both β‐d2 labelled styrene and d3 ‐MeCN confirmed the origin of chemo‐ and regioselectivities (74, 75) (Scheme 5E). Capitalizing on these observations, we then evaluated a series of commercially available halogenated anhydrides (−1.14 to −0.85 V vs. SCE (see Supporting Information, page S27)) under established reaction conditions, and we were delighted to observe the formation of the corresponding lactones (72, 2, 73) in good chemical yields (Scheme 5D).
Scheme 5.

Mechanistic studies towards formation of 1 and 2. A) Proposed mechanism (for details, see Supporting Information, pages S50–S51). B) Spectroscopic studies. C) Competition experiments. D) Screening of different anhydrides. E) Control experiments. General reaction conditions: 4‐tert‐butylstyrene (1 equiv), fac‐Ir(ppy)3 (1 mol%), CDFAA (2 equiv) in MeCN or DMF, 12 h, GC yields. [a] For structure I‐14, see Supporting Information, page S51. [b] Extension coefficients for CDFAA are multiplied by 10 times for representation reasons. [c] Reaction was performed under air. [d] Acetonitrile‐d 3 was used as solvent. [e] Ethenyl‐2,2‐d 2‐benzene (1 equiv) was used as olefin. [f] H2O18 (2 equiv, 97 atom % 18O) was added in the reaction mixture after the irradiation under argon and stirred for 30 min. Incorporation of 18O was not observed either in GC‐MS or in NMR. Relative Gibbs free‐energies (ΔG) were calculated at 298 K at the CPCM(Solvent)/M06‐2X/GD3/def2‐TZVP//M06‐2X/GD3/def2‐TZVP level of theory.
Finally, a series of experiments were carried out to verify the plausible mechanism for the oxy‐perfluoroalkylation of olefins that takes place in toluene and supposingly proceeds via generation of chlorodifluoromethyl radicals (XIV) (Scheme 6A). Our theoretical investigations in both gas phase and solvent system were not in line with the reductive mesolytic C−O bond fragmentation mechanism, which would be a direct pathway to access ⋅CF2Cl upon decarbonylation (see Supporting Information, pages S52–S53). However, ensuing experimental determination of chloride concentration utilizing Mohr's approach [68] corroborated our early hypothesis for the generation of I (see Supporting Information, pages S45–S46). Furthermore, various control experiments and computational studies support a hydrogen atom abstraction process (HAT) by I from the solvent molecule. This is also evident from the fact that product 3 was only produced in solvents capable of forming stabilized radicals upon HAT (Scheme 6B). Detailed examination of the reaction mixture by GC‐MS analysis displayed the presence of XVII, which is likely formed by recombination of ⋅CF2Cl and ⋅Bn radicals (Scheme 6B, Supporting Information page S43), and further validates the HAT hypothesis. [69] The resulting benzylic radical X provokes the evolution of ⋅CF2Cl through successive reactions. Upon addition of the ⋅CF2Cl to the olefin, alkyl radical XV is formed and further undergoes oxidation (E 0=0.25 V) by IrIV to generate the benzyl cation intermediate XVI. The attack of another molecule of CDFAA on XVI followed by deacylation with Cl− results in product 3 (for details, see Supporting Information, page S52). Due to the competitive reaction of intermediate I to form either 2 or 3, we expected a high sensitivity of the reaction outcome dependent on the collision probability with the substrate. Indeed, during the competition experiment in a mixture of solvents (toluene‐DMF), we observed a significant inhibition of the HAT process when lowering the concentration of toluene in the reaction (Scheme 6C). This finding is in good agreement with the calculated energy values for reaction of I with toluene vs olefin. Lastly, we solely observed oxy‐perfluoroalkylation reaction in toluene using electronically dissimilar styrenes (3, 76, 77) exposing high synthetic divergence of CDFAA.
Scheme 6.

Mechanistic studies towards formation of 3. A) Proposed mechanism for oxy‐perfluoroalkylation of olefins. B) Key experiments. C) Competition experiments. D) Oxy‐perfluoroalkylation scope. General conditions: 4‐tert‐butylstyrene (1 equiv), fac‐Ir(ppy)3 (1 mol%), CDFAA (2 equiv), toluene (0.17 M), blue LEDs, rt, 12 h, GC yields. [a] For structure I‐17, see Supporting Information, page S52. [b] Reaction was performed under air. [c] See Supporting Information for details, page S47. Relative Gibbs free‐energies (ΔG) were calculated at 298 K at the CPCM(Solvent)/M06‐2X/GD3/def2‐TZVP//M06‐2X/GD3/def2‐TZVP level of theory.
Conclusion
In summary, we have developed a highly divergent synthesis of gem‐difluorinated compounds, including γ‐lactams, γ‐lactones, and chlorodifluoromethyl acetate derivatives from the same set of readily available starting materials. CDFAA was activated in a unique fashion with the help of visible light‐mediated photoredox catalysis, generating an intermediate capable of exhibiting remarkable orthogonal evolutions by a simple choice of solvents. This methodology showcases high levels of chemo‐ and regioselectivity with exceptional substrate scope and functional group tolerance, and can also be extended to late‐stage functionalization of biorelevant molecules. Furthermore, mechanistic studies strongly suggest the presence of reactive radical species. We anticipate that our easy‐to‐operate photocatalytic concept for the activation of halogenated anhydrides will be a valuable avenue in the preparation of fluorinated molecules with direct relevance to the synthetic chemist in drug discovery and development as well as across many other fields.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Acknowledgements
D.K. acknowledges the Swiss National Science Foundation (SNSF, PCEFP2_186964) for financial support of this research. Dr. Nils Trapp (ETH Zürich) is acknowledged for obtaining all X‐ray crystallographic data. D.K. and R.G. thank Prof. Christian Bochet and Dr. Albert Ruggi for their fruitful discussions during the work. Dr. Krzysztof Piech is acknowledged for his support with NMR measurements. Anne Schuwey and synthesis lab. of the University of Fribourg is acknowledged for preparation of olefin molecules. We also acknowledge Manvendra Singh, Markus Andrey, and Olivier Graber for giving insights related to instrument handling and maintenance, and Edmée‐Constance Lyon for insightful comments and suggestions on the manuscript. Generous and continued support by the University of Fribourg is acknowledged. The computational study presented in this work was carried out on facilities provided by Digital Research Alliance of Canada (https://alliancecan.ca/en) and Compute Canada (www.computecanada.ca). Open Access funding provided by Université de Fribourg.
R. Giri, I. Mosiagin, I. Franzoni, N. Y. Nötel, S. Patra, D. Katayev, Angew. Chem. Int. Ed. 2022, 61, e202209143; Angew. Chem. 2022, 134, e202209143.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv‐2022‐8rq77‐v2).
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Rogge T., Kaplaneris N., Chatani N., Kim J., Chang S., Punji B., Schafer L. L., Musaev D. G., Wencel-Delord J., Roberts C. A., Sarpong R., Wilson Z. E., Brimble M. A., Johansson M. J., Ackermann L., Nat. Rev. Methods Primers 2021, 1, 43. [Google Scholar]
- 2. Campeau L. C., Hazari N., Organometallics 2019, 38, 3–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. List B., Chem. Rev. 2007, 107, 5413–5415. [Google Scholar]
- 4. Winkler C. K., Schrittwieser J. H., Kroutil W., ACS Cent. Sci. 2021, 7, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shaw M. H., Twilton J., MacMillan D. W. C., J. Org. Chem. 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Prier C. K., Rankic D. A., MacMillan D. W. C., Chem. Rev. 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang X., Wang D., ACS Appl. Energy Mater. 2018, 1, 6657–6693. [Google Scholar]
- 8. Candish L., Collins K. D., Cook G. C., Douglas J. J., Gómez-Suárez A., Jolit A., Keess S., Chem. Rev. 2022, 122, 2907–2980. [DOI] [PubMed] [Google Scholar]
- 9. Marzo L., Pagire S. K., Reiser O., König B., Angew. Chem. Int. Ed. 2018, 57, 10034–10072; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10188–10228. [Google Scholar]
- 10. Romero N. A., Nicewicz D. A., Chem. Rev. 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
- 11. Horn E. J., Rosen B. R., Baran P. S., ACS Cent. Sci. 2016, 2, 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guillemard L., Kaplaneris N., Ackermann L., Johansson M. J., Nat. Chem. Rev. 2021, 5, 522–545. [DOI] [PubMed] [Google Scholar]
- 13. Trost B. M., Science 1991, 254, 1471–1477. [DOI] [PubMed] [Google Scholar]
- 14. Newhouse T., Baran P. S., Hoffmann R. W., Chem. Soc. Rev. 2009, 38, 3010–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang H. M., Bellotti P., Ma J., Dalton T., Glorius F., Nat. Rev. Chem. 2021, 5, 301–321. [DOI] [PubMed] [Google Scholar]
- 16. Zhan G., Du W., Chen Y. C., Chem. Soc. Rev. 2017, 46, 1675–1692. [DOI] [PubMed] [Google Scholar]
- 17. China H., Kumar R., Kikushima K., Dohi T., Molecules 2020, 25, 6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sakakibara Y., Murakami K., ACS Catal. 2022, 12, 1857–1878. [Google Scholar]
- 19. Meng J. P., Wang W. W., Chen Y. L., Bera S., Wu J., Org. Chem. Front. 2020, 7, 267–272. [Google Scholar]
- 20. Maitland J. A. P., Leitch J. A., Yamazaki K., Christensen K. E., Cassar D. J., Hamlin T. A., Dixon D. J., Angew. Chem. Int. Ed. 2021, 60, 24116–24123; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 24318–24325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. John S. E., Gulati S., Shankaraiah N., Org. Chem. Front. 2021, 8, 4237–4287. [Google Scholar]
- 22. Li J., Luo Y., Cheo H. W., Lan Y., Wu J., Chem 2019, 5, 192–203. [Google Scholar]
- 23. Rössler S. L., Jelier B. J., Magnier E., Dagousset G., Carreira E. M., Togni A., Angew. Chem. Int. Ed. 2020, 59, 9264–9280; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 9350–9366. [Google Scholar]
- 24. Patra S., Mosiagin I., Giri R., Katayev D., Synthesis 2022, 54, 3432–3472. [Google Scholar]
- 25. Gouverneur V., Seppelt K., Chem. Rev. 2015, 115, 563–565. [DOI] [PubMed] [Google Scholar]
- 26. Liang T., Neumann C. N., Ritter T., Angew. Chem. Int. Ed. 2013, 52, 8214–8264; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 8372–8423. [Google Scholar]
- 27. Peer K., Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley-VCH, Weinheim, 2013. [Google Scholar]
- 28. Charpentier J., Früh N., Togni A., Chem. Rev. 2015, 115, 650–682. [DOI] [PubMed] [Google Scholar]
- 29. Ni C., Hu M., Hu J., Chem. Rev. 2015, 115, 765–825. [DOI] [PubMed] [Google Scholar]
- 30. Ogiwara Y., Sakai N., Angew. Chem. Int. Ed. 2020, 59, 574–594; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 584–605. [Google Scholar]
- 31. Inoue M., Sumii Y., Shibata N., ACS Omega 2020, 5, 10633–10640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Purser S., Moore P. R., Swallow S., Gouverneur V., Chem. Soc. Rev. 2008, 37, 320–330. [DOI] [PubMed] [Google Scholar]
- 33. Meanwell N. A., J. Med. Chem. 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]
- 34. Holovach S., Melnykov K. P., Skreminskiy A., Herasymchuk M., Tavlui O., Aloshyn D., Borysko P., Rozhenko A. B., V. Ryabukhin S., Volochnyuk D. M., Grygorenko O. O., Chem. Eur. J. 2022, 28, e202200331. [DOI] [PubMed] [Google Scholar]
- 35. Sharma R., Sun H., Piotrowski D. W., Ryder T. F., Doran S. D., Dai H., Prakash C., Drug Metab. Dispos. 2012, 40, 2143–2161. [DOI] [PubMed] [Google Scholar]
- 36. Li X., Hou Y., Zhao J., Li J., Wang S., Fang J., Chem. Sci. 2020, 11, 3215–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barrett S. D., Holt M. C., Kramer J. B., Germain B., Ho C. S., Ciske F. L., Kornilov A., Colombo J. M., Uzieblo A., O'Malley J. P., Owen T. A., Stein A. J., Morano M. I., J. Med. Chem. 2019, 62, 4731–4741. [DOI] [PubMed] [Google Scholar]
- 38.
- 38a. Booth H. S., Bixby E. M., Ind. Eng. Chem. 1932, 24, 637–641; [Google Scholar]
- 38b. Ni C., Hu J., Synthesis 2014, 46, 842–863; [Google Scholar]
- 38c. Feng Z., Xiao Y. L., Zhang X., Acc. Chem. Res. 2018, 51, 2264–2278; [DOI] [PubMed] [Google Scholar]
- 38d. Gao F., Li B., Wang Y., Chen Q., Li Y., Wang K., Yan W., Org. Chem. Front. 2021, 8, 2799–2819; [Google Scholar]
- 38e. Sap J. B. I., Meyer C. F., Straathof N. J. W., Iwumene N., Am Ende C. W., Trabanco A. A., Gouverneur V., Chem. Soc. Rev. 2021, 50, 8214–8247. [DOI] [PubMed] [Google Scholar]
- 39. Sakakibara S., Inukai N., Bull. Chem. Soc. Jpn. 1964, 37, 1231–1232. [Google Scholar]
- 40. Arlow S. I., Hartwig J. F., Angew. Chem. Int. Ed. 2016, 55, 4567–4572; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4643–4648. [Google Scholar]
- 41. Tedder J. M., Chem. Rev. 1955, 55, 787–827. [Google Scholar]
- 42. Wu W., You Y., Weng Z., Chin. Chem. Lett. 2022, 33, 4517–4530. [Google Scholar]
- 43. Beatty J. W., Douglas J. J., Cole K. P., Stephenson C. R. J., Nat. Commun. 2015, 6, 7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Beatty J. W., Douglas J. J., Miller R., McAtee R. C., Cole K. P., Stephenson C. R. J., Chem 2016, 1, 456–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McAtee R. C., Beatty J. W., McAtee C. C., Stephenson C. R. J., Org. Lett. 2018, 20, 3491–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kawamura S., Sodeoka M., Angew. Chem. Int. Ed. 2016, 55, 8740–8743; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 8882–8885. [Google Scholar]
- 47. Kawamura S., Henderson C. J., Aoki Y., Sekine D., Kobayashi S., Sodeoka M., Chem. Commun. 2018, 54, 11276–11279. [DOI] [PubMed] [Google Scholar]
- 48. Valverde E., Kawamura S., Sekine D., Sodeoka M., Chem. Sci. 2018, 9, 7115–7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lewis S. E., Wilhelmy B. E., Leibfarth F. A., Chem. Sci. 2019, 10, 6270–6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lewis S. E., Wilhelmy B. E., Leibfarth F. A., Polym. Chem. 2020, 11, 4914–4919. [Google Scholar]
- 51. Zhang K., Rombach D., Nötel N. Y., Jeschke G., Katayev D., Angew. Chem. Int. Ed. 2021, 60, 22487–22495; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 22661–22669. [Google Scholar]
- 52. Nagashima H., Isono Y., Iwamatsu S., R N., J. Org. Chem. 2001, 66, 315–319. [DOI] [PubMed] [Google Scholar]
- 53. Wang X., Lei J., Li G., Meng J., Li C., Li J., Sun K., Org. Biomol. Chem. 2020, 18, 9762–9774. [DOI] [PubMed] [Google Scholar]
- 54. Zhang C., Liu Y., J. Heterocycl. Chem. 2022, 59, 809–819. [Google Scholar]
- 55. Surmont R., Verniest G., Thuring J. W., Holte P. T., Deroose F., De Kimpe N., Org. Biomol. Chem. 2010, 8, 4514–4517. [DOI] [PubMed] [Google Scholar]
- 56. Luo X., Zhang B., Xi C., Green Chem. 2021, 23, 2324–2328. [Google Scholar]
- 57. Nguyen Q. P. B., Kim B. M., Song M. S., Yoon K. H., Chai K. Y., Bull. Korean Chem. Soc. 2014, 35, 313–315. [Google Scholar]
- 58.
- 58a. Giese B., Angew. Chem. Int. Ed. Engl. 1983, 22, 753–764; [Google Scholar]; Angew. Chem. 1983, 95, 771–782; [Google Scholar]
- 58b. Giese B., Angew. Chem. Int. Ed. Engl. 1985, 24, 553–565; [Google Scholar]; Angew. Chem. 1985, 97, 555–567; [Google Scholar]
- 58c. Gant Kanegusuku A. L., Roizen J. L., Angew. Chem. Int. Ed. 2021, 60, 21116–21149; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 21286–21319. [Google Scholar]
- 59. Katayev D., Kajita H., Togni A., Chem. Eur. J. 2017, 23, 8353–8357. [DOI] [PubMed] [Google Scholar]
- 60. Calvo R., Comas-Vives A., Togni A., Katayev D., Angew. Chem. Int. Ed. 2019, 58, 1447–1452; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1461–1466. [Google Scholar]
- 61. Koike T., Akita M., Inorg. Chem. Front. 2014, 1, 562–576. [Google Scholar]
- 62. Pirtsch M., Paria S., Matsuno T., Isobe H., Reiser O., Chem. Eur. J. 2012, 18, 7336–7340. [DOI] [PubMed] [Google Scholar]
- 63.Deposition numbers 2048568 (1) and 2050590 (24) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 64. Hiesinger K., Dar'In D., Proschak E., Krasavin M., J. Med. Chem. 2021, 64, 150–183. [DOI] [PubMed] [Google Scholar]
- 65. Buzzetti L., Crisenza G. E. M., Melchiorre P., Angew. Chem. Int. Ed. 2019, 58, 3730–3747; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3768–3786. [Google Scholar]
- 66. Zhao Y., Truhlar D. G., Acc. Chem. Res. 2008, 41, 157–167. [DOI] [PubMed] [Google Scholar]
- 67. Bolsakova J., Jirgensons A., Chem. Heterocycl. Compd. 2017, 53, 1167–1177. [Google Scholar]
- 68. Sheen R. T., Kahler H. L., Ind. Eng. Chem. Anal. Ed. 1938, 10, 628–629. [Google Scholar]
- 69. Zargari N., Winter P., Liang Y., Lee J. H., Cooksy A., Houk K. N., Jung K. W., J. Org. Chem. 2016, 81, 9820–9825. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.