

Abstract
Nosocomial infections caused by resistant Gram-positive organisms are on the rise, presumably due to a combination of factors including prolonged hospital exposure, increased use of invasive procedures, and pervasive antibiotic therapy. Although antibiotic stewardship and infection control measures are helpful, newer agents against multidrug-resistant (MDR) Gram-positive bacteria are urgently needed. Here, we describe our efforts that led to the identification of 5-amino-4-quinolone 111 with exceptionally potent Gram-positive activity with minimum inhibitory concentrations (MICs) ≤0.06 μg/mL against numerous clinical isolates. Preliminary mechanism of action and resistance studies demonstrate that the 5-amino-4-quinolones are bacteriostatic, do not select for resistance, and selectively disrupt bacterial membranes. While the precise molecular mechanism has not been elucidated, the lead compound is nontoxic displaying a therapeutic index greater than 500, is devoid of hemolytic activity, and has attractive physicochemical properties (clog P = 3.8, molecular weight (MW) = 441) that warrant further investigation of this promising antibacterial scaffold for the treatment of Gram-positive infections.
Graphical Abstract

INTRODUCTION
Rising antimicrobial resistance (AMR) threatens global public health and, if unabated, may force us to return to a “pre-antibiotic era” when infectious diseases caused nearly one-third of all reported deaths.1 The Gram-positive bacterium methicillin-resistant Staphylococcus aureus (MRSA) is a prototypical multidrug-resistant organism listed by the Centers for Disease Control (CDC) as a top-priority pathogen.2,3 S. aureus is both a commensal microbe found in the nasal mucosa of ~30% of healthy adults4 and a human opportunistic pathogen. Infections with S. aureus typically occur in immunocompromised individuals with underlying medical conditions—such as diabetes,5 acquired immunodeficiency syndrome, or defective neutrophil function6—following disruption of the host’s cutaneous or mucosal barriers. Disruption of these barriers can be caused by injury, surgical procedures,7 medical devices,8 and drug use,9 which can lead to a litany of diseases, including sepsis, severe skin infections, catheter-associated infections, and pneumonia.8 In 2017 alone, severe cases of MRSA led to an estimated 119,000 systemic infections with a mortality rate of 17%.10 While MRSA has historically been recognized for its role in healthcare-associated (HA) infections, community-associated (CA) infections have become more prevalent in the past 25 years, often leading to worse health outcomes.11,12
MRSA was first reported in 1961,13 only 1 year after the introduction of the β-lactamase-resistant penicillin known as methicillin into clinical practice. β-Lactam resistance in MRSA is due to the expression of the altered penicillin-binding protein PBP2a,14,15 which is only weakly inhibited by virtually all β-lactam antibiotics.16 PBP2a is encoded by mecA or similar homologues that are part of a mobile genetic element called the staphylococcal cassette chromosome mec (SCCmec), which can be further classified into 14 types (I–XIV).17,18 SCCmec types I, II, and III are commonly found in healthcare-associated MRSA (HA-MRSA), while SCCmec IV and V are found in both HA-MRSA and community-associated MRSA (CA-MRSA).19 The different SCCmec types contain other genetic elements that confer resistance to other classes of antibacterial agents such as tetracyclines,20 glycopeptides,21 lipopeptides,22 macrolides,23,24 and aminoglycosides.23,25
Despite the growing rise of antimicrobial resistance, there have only been six new first-in-class antibacterial drugs approved in the past 20 years.26–30 Clinicians continue to rely almost exclusively on intravenously administered vancomycin for the treatment of hospitalized patients with serious MRSA infections, while intravenous (i.v.) daptomycin is used for MRSA bacteremia and endocarditis. Linezolid is an attractive oral switch therapy for MRSA infections and is widely used for the treatment of pneumonia and skin and soft tissue infections.31 Resistance to all three agents has been reported.21,22,32 The limited treatment options, inadequate number of antibacterial agents in the drug pipeline, and emerging resistance to standard-of-care treatment options all point to the need for novel therapeutics with unconventional mechanisms of action.
The bacterial membrane has traditionally been overlooked in antibacterial drug research because membrane-targeting agents are generally considered poorly selective.33 However, selectivity can be achieved by binding prokaryotic structural lipids,34 membrane proteins,35 and cell wall components35,36 enabling discrimination from host cell membranes.37 Bacterial membranes represent particularly promising antibacterial targets since they are essential under replicating and nonreplicating conditions as well as in planktonic and biofilm cultures. Moreover, the development of resistance to compounds targeting the bacterial membrane is more difficult than to classical antibiotics directed against proteins that are mutable.37 The naturally occurring cationic antimicrobial peptides (CAMPs) that disrupt bacterial membranes are part of prokaryotes’, eukaryotes’, and plantaes’ innate immune system,38,39 while several classes of Food and Drug Administration (FDA)-approved antibiotics exert their activity through bacterial membrane disruption including the polymyxins,40 bacitracins,41 lipopeptides,42 and select lipoglycopeptides43 (Figure 1). There have also been multiple efforts to produce new membrane-active small molecules reported recently in the literature.44–52 For example, the potent antibacterial activity of the synthetic retinoids CD437 and CD1530 (Figure 1) was recently shown53 to be caused by membrane disruption as the primary mechanism of action. These aforementioned membrane-targeting antibacterial agents are noted for their poor pharmacokinetic (PK) behavior and/or toxicity, which emanates from their amphipathic nature and undesirable physicochemical properties. Herein, we report our investigation of a membrane-disrupting aminoquinoline antibacterial scaffold that led to the identification of a highly potent and bacterial-selective Gram-positive antibacterial agent with attractive physicochemical properties.
Figure 1.

Therapeutic and experimental membrane-disrupting agents. Daptomycin is an FDA-approved antibiotic for the treatment of Gram-positive infections that was initially proposed to insert into the cytoplasmic membrane of the bacteria and permeabilize it via membrane-associated oligomers; however, this mechanism has been recently revised. Daptomycin targets cell wall biosynthesis by forming a tripartite complex with undecaprenyl-coupled intermediates and membrane lipids.54 Guavanin 2 is a cationic antimicrobial peptide (CAMP) that disrupts membranes of bacteria via membrane hyperpolarization. Guavanin 2 structure was taken from PDB 5V1E.39 Polymyxin B1 is part of the polymyxin class of antibiotics and is an FDA-approved antibiotic that disrupts membranes of Gram-negative bacteria. The synthetic retinoids typified by CD437 and CD1530 disrupt bacterial membranes and are active against bacterial persisters. Chemical properties including molecular weight (MW) and the calculated partition or distribution coefficient (clog P or clog D), which is a measure of lipophilicity are included for each compound. Compounds with a molecular weight (MW) greater than 500 generally have poor bioavailability while compounds with a clog P or clog D greater than 5 often have poor solubility, high protein binding, and lower selectivity. Additionally, exceptionally low clog P values (clog P < −3) generally portend poor membrane permeability, absorption, and distribution properties. All of the compounds have undesirable physicochemical properties by these criteria.
RESULTS
We previously reported the identification of the 4-quinolinol derivative DNAC-2 from a high-throughput screening (HTS) campaign with moderate activity (minimum inhibitory concentration (MIC) = 8 μg/mL) against MRSA (Figure 2).55 Intriguingly, DNAC-2 was found to target the membrane of Gram-positive bacteria resulting in partial membrane depolarization while displaying no overt toxicity toward eukaryotic membranes. In addition to DNAC-2, a few other substituted quinolines were identified with the same mechanism of action typified by quinoline 1 (Figure 2) indicating flexibility at the 4-position. We were attracted to the 4-substituted quinoline scaffold based on its promising activity, chemical tractability for analogue synthesis, and prevalence in several approved drugs.56 We initially sought to examine the structure–activity relationships (SAR) of 1 through substitution and replacement of the 4-aryl ring with more polar and nonplanar substituents (Figure 2). In parallel, we wanted to explore modification and substitution to the quinoline heterocycle through the introduction of nitrogen atoms and the introduction of more polar substituents at the 2-, 7-, and 8-positions to decrease the lipophilicity.
Figure 2.

High-throughput screen hits and SAR analysis of the 4-aminoquinoline scaffold.
Chemistry.
The first series of quinoline analogues were synthesized from a common set of quinolinol building blocks 2–10 that were prepared via a modified Conrad–Limpach reaction57,58 between substituted aniline derivatives and 4,4,4-trifluoroacetoacetate in neat polyphosphoric acid (PPA) at 120 °C.59 m-substituted anilines typically formed a mixture of both the 5- and 7-regioisomers that were challenging to separate and led to reduced isolated yields of the desired 7-regioisomers, whereas o-substituted anilines exclusively afforded the 8-regioisomers. The quinolines were evaluated for antibacterial activity and only compounds 2–6 (DNAC-2 is the same as 5) were found to be active (Table 1). Consequently, only these compounds were derivatized by the introduction of a substituent at the 4-position. Quinolinol 2 was converted to the corresponding triflate 2a employing triflic anhydride and reacted with various amines and phenols by nucleophilic aromatic substitution (SNAr)60 to afford 14–21, 23–26, 28–30, 51, 59, 61, 62, and 80–83. This strategy proved less effective for electron-deficient amines as well as quinolinols with electron-donating substituents. In these cases, we utilized a complimentary route by conversion of quinolinols to the corresponding aryl chloride61,62 or aryl bromides63 2b to 6b and 10b followed by Buchwald–Hartwig amination64–66 to provide 11–13, 22, 27, 31–38, 42–50, 52–58, 60, 63–65, and 84 (Scheme 1). Finally, an alternative route from 2c afforded one single amido substitution at the 4-position to afford compound 85 (see Scheme S1 in the Supporting Information (SI)).
Table 1.
Substituted Quinolin-4-ol Analoguesc

|
||||
|---|---|---|---|---|
| cmpd | R1 | R2 | MICa,b (μg/mL) | clog Pb |
| 2 | CF3 | H | 5 | 2.2 |
| 3 | OCF3 | H | 4–8 | 2.4 |
| 4 | H | CF3 | 2–4 | 2.2 |
| 5 | H | OCF3 | 5 | 2.4 |
| 6 | Cl | H | 20 | 2.0 |
| 7 | H | H | 320 | 2.3 |
| 8 | ethyl | H | 640 | 3.1 |
| 9 | OCH3 | H | 640 | 1.3 |
| 10 | SCH3 | H | 640 | 1.9 |
MIC = minimum inhibitory concentration resulting in complete inhibition of observable growth in methicillin-resistant S. aureus (FPR3757).
Oxacillin control used (MIC = 64 μg/mL for FPR3757 cells).
clog P = calculated log of the partition coefficient.
Scheme 1.

4-Aminoquinoline and 4-Oxyquinoline Synthesisa
aConditions: (a) K3PO4, SPhos, Pd2(dba)3·CH2Cl2, tetrahydrofuran (THF), 55 °C; (b) t-BuONa, DPPF, Pd(dppf)Cl2·CH2Cl2, THF, 55 °C; (c) t-BuONa, XantPhos, Pd2(dba)3, dioxane, 80 °C; (d) HCl, EtOH, reflux.
Compounds containing a difluoromethyl C-2 substituent were prepared analogously by Conrad–Limpach reaction between 3-trifluoromethylaniline and 4,4-difluoroacetoacetate to afford quinolinols 67, which was activated by triflic anhydride to 67a and elaborated to 68 and 69 by SNAr substitution with 3,4-dichloroaniline and 3-trifluormethoxyani-line, respectively (Scheme 2). Analogs containing a dimethylaminosulfonyl C-7 substituent could not be synthesized using the usual PPA-mediated procedure and required substantially more thermal energy. Compound 66 was instead prepared by refluxing 3-(dimethylaminosulfonyl)aniline and 4,4,4-trifluoroacetoacetate at 255 °C in diphenyl ether (Scheme 2). Chlorination of 66 with POCl3 yielded 66a that was diversified to 39–41 by Buchwald–Hartwig amination.
Scheme 2.

4-Aminoquinoline Synthesisa
aConditions: (a) dimethyl sulfoxide (DMSO), 80 °C; (b) HCl, EtOH, reflux; (c) Pd2(dba)3, XantPhos, t-BuONa, dioxane, 80 °C.
We synthesized a series of mono-, di-, and trifluorinated analogues of the B-ring in an attempt to replace the lipophilic C-7 trifluoromethyl group. While m-fluoroaniline reacted with 4,4,4-trifluoroacetoacetate in neat polyphosphoric acid (PPA) at 100 °C to furnish 70, the di- and tri-fluoroanilines required heating at 150 °C to effect cyclization to quinolinols 71–73 (Scheme 3). Halogenation of 70–73 to quinolines 70a to 73a followed by Buchwald–Hartwig amination as described previously yielded 74–79.
Scheme 3.

Synthesis of Fluorinated B-Ring Analogues
In an attempt to reduce the lipophilicity of the amino-quinolines, we targeted the synthesis of quinazolines containing an additional nitrogen atom in the A-ring. Synthesis commenced from commercially available 2-amino-4-(trifluoromethyl)benzonitrile that was oxidized from the nitrile to the amide intermediate 86 by treatment with an alkaline solution of hydrogen peroxide (Scheme 4). Treatment of the resulting substituted aniline 86 with 2,2,2-trifluoroacetyl chloride furnished the bis-amide intermediate, which was cyclized to the quinazolone 87 employing potassium hydroxide in ethanol at reflux. Chlorination of quinazolone 87 with thionyl chloride in dimethylformamide (DMF) gave 88 which on SNAr substitution with 3,4-dichloroaniline, 3-trifluoromethoxyaniline, and 2-amino-5-trifluoromethylpyridine afforded the final 4-aminoquinazolines 89–91.
Scheme 4.

Synthesis of 4-Aminoquinazolines
The pyrido[2,3-d]pyrimidine scaffold containing two additional nitrogen atoms was investigated as a quinoline isostere in an attempt to further decrease lipophilicity. The synthesis began by SNAr substitution of 2-chloro-3-cyano-6-trifluoromethylpyridine with ammonia in THF followed by base-promoted hydration of the cyano group to furnish amide 92 (Scheme 5). Subsequent condensation of 92 with ethyl trifluoroacetate and base-catalyzed annulation afforded 93. Pyrido[2,3-d]pyrimidin-4-one derivative 93 was chlorinated with POCl3 and SNAr substitution with 3,4-dichloroaniline yielded 94, while the reaction of 93 with 2-amino-5-trifluoromethylpyridine to provide 95 required the complimentary Buchwald–Hartwig amination.
Scheme 5.

Synthesis of Pyrido[2,3-d]pyrimidines
The cinnoline analogues were the final set of aza-analogues of the quinoline scaffold prepared (Scheme 6). Initial efforts to arrive at the o-acetylaniline intermediate (97) from Grignard alkylation of 2-cyano-5-(trifluoromethyl)aniline repeatedly gave very undesirable yields in our hands (5–19% yields). Instead we employed a base-mediated alkylation of 2-chloro-5-(trifluoromethyl)nitrobenzene with nitroethanone followed by an oxidative Nef reaction from a modified procedure reported by Reid and Reny Runge.67 The reported conditions stated 2 equiv of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) at 0 °C in ethyl acetate were required for the initial alkylation of 2-chloro-5-(trifluoromethyl)nitrobenzene. In our hands, these conditions gave complete conversion to the ortho-DBU adduct of the nitrobenzene starting material which was noted as a trace byproduct in the initial manuscript.67 Optimal base and solvent (see the SI for details) were determined to be sodium hydride and DMSO which gave yields of 96 similar to what was reported in the initial Reid paper.67 The subsequent oxidative Nef reaction of 96 gave the desired acetyl intermediate (97) without modification. In two steps, 97 was reduced to the aniline and subsequently treated with aqueous (aq) sodium nitrite in acetic acid resulting in the diazonium salt which allowed for acid-cyclized annulation of the ketone to give cinnoline (98). The cinnoline (98) was then converted to chloride upon heating with a solution of phosphorus oxychloride and phosphorus pentachloride. The resulting halogenated cinnoline (99) was coupled to 3,4-difluoroaniline and 2-amino-5-trifluoromethylpyridine under Buchwald–Hartwig conditions providing 100 and 101, respectively.
Scheme 6.

Cinnoline Synthesis
We developed an alternate quinoline synthesis to explore the modification of the C-2-position featuring a 2,4-dichloroquinoline intermediate. This was accomplished by intramolecular Claisen-like condensation of ethyl N-acetyl-2-amino-4-trifluoromethylbenzoate (102) mediated by potassium bis(trimethylsilyl)amide (KHMDS) to afford a 4-hydroxyquinoline-(2H)-one (103) intermediate (Scheme 7). Compound 103 was converted to 2,4-dichloroquinoline by refluxing in phosphorus oxychloride followed by SNAr substitution by Boc-protected piperazine to give 104 along with the C-4 regioisomer (not shown). Buchwald–Hartwig coupling of 104 and 2-amino-5-trifluoromethylpyridine followed by trifluoroacetic acid (TFA) deprotection of the Boc group gave the desired analogue 105. The regioisomeric analogues 106 and 108, described in the Supporting Information, were prepared from the corresponding C-4 piperazine intermediate isolated as a side product in the preparation of 104. The C-2 and C-4 morpholino-substituted analogues 107 and 108 were synthesized in an analogous manner.
Scheme 7.

Synthesis of a C-2 Piperazine Analogue
We next conceived of a hybrid scaffold of the initial active quinoline-4-ones and the aryl-substituted 4-aminoquinolines, giving an aryl-substituted 5-aminoquinoline-4-ones scaffold (Scheme 8). The two-step synthesis started from the condensation of 3-bromo-5-trifluoromethylaniline and ethyl 2,2,2-trifluoromethylacetoacetate in neat PPA to give two separable regioisomers 109 and 110. The structure of the regioisomers was assigned by 1H NMR analysis of the debrominated products generated by palladium-catalyzed dehalogenation (not shown). Following optimization of the Buchwald–Hartwig amination conditions, we were able to access the desired C-5 substituted aminoquinoline-4-ones 111–120 from 109. The C-7 fluoro derivative 122 was synthesized analogously from 3-bromo-5-fluoroaniline 121 as described in the Supporting Information.
Scheme 8.
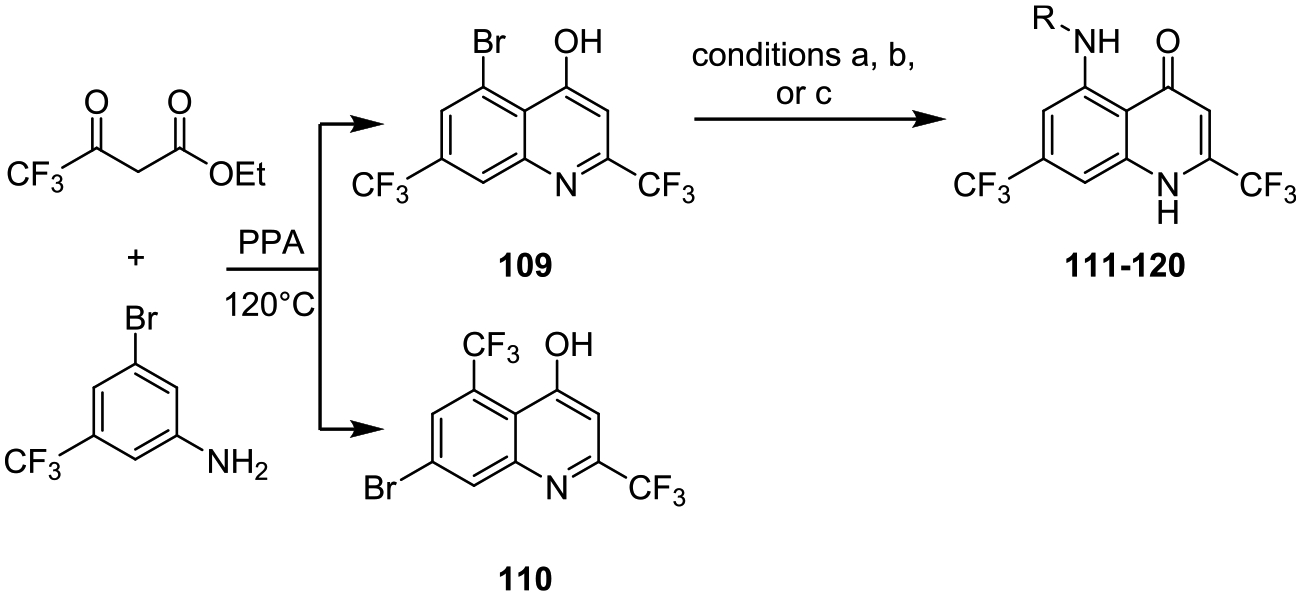
C-5- and C-7-Substituted Quinoline-4-onesa
aConditions: (a) 2-MeBuOH, K3PO4, t-BuBrettPhos Pd G3, 90 °C; (b) dioxane, K3PO4, t-BuBrettPhos, Pd2(dba)3, 100 °C; (c) dioxane, K2CO3, Pd2(dba)3, JohnPhos, dioxane 100 °C.
Microbiology.
The antibacterial activity of compounds was initially determined against a clinical strain of methicillin-resistant S. aureus (FPR3757) in Mueller–Hinton (MH) broth according to the CLSI protocol to determine the minimum inhibitory concentration (MIC) that resulted in complete inhibition of observable growth. Oxacillin was selected as a control for all MIC studies (MIC of 64 μg/mL in FPR3757). The first set of compounds evaluated were analogues at the C-7- and C-8-positions of the initial quinolinol hit DNAC-2 since the other HTS hits identified (data not shown) were substituted at these positions (Table 1). We first explored modification at the C-7-position with a small series of electron-donating and withdrawing substituents. The trifluoromethyl 2 and trifluoromethoxy 3 are the most potent with MICs of 4–8 μg/mL while chloro 6 is slightly weaker with an MIC of 20 μg/mL. However, analogues containing electron-donating groups at C-7 including ethyl 8, methoxy 9, and methylthio 10 are weakly active displaying MICs of 320–640 μg/mL indicating electron-donating substituents at C-7 are poorly tolerated. The impact of electronics is best illustrated with methoxy 9, which is 80–160 less potent that the isosteric trifluoromethoxy 3. The C-8-position was evaluated with trifluoromethyl 4 and trifluoromethoxy 5 containing the optimal C-7 substituents. Both 4 and 5 are equipotent to the corresponding C-7 analogues 2 and 3 indicating some structural tolerance of the quinolinol scaffold.
We next explored the SAR of the C-4 aryl substituent of 4-aminoquinoline 1 whose MIC is 8 μg/mL (Table 2). Our first series of compounds contain a 7-trifluoromethyl rather than the 8-trifluoromethoxy substituent found in 1. The 2′-, 3′-, and 4′-chlorophenyl analogues 15–17 helped to define the steric requirements for activity: 2′-chlorophenyl 15 is inactive while both 3′-chlorophenyl 16 and 4′-chlorophenyl 17 possess MICs of 0.125–0.25 μg/mL, which represents a dramatic 32- to 64-fold increase in potency over 1. Given the enhanced potency of the chloro-substituted analogues, we conducted a halogen scan and evaluated 3′-fluorophenyl 18, 3′-bromophenyl 19, and 3′-iodophenyl 20. The more lipophilic halogens 19 and 20 maintain potent activity with MICs of 0.25 μg/mL while the fluoro analogue has a substantial 16- to 64-fold loss of potency. We also explored a couple of 3′,4′-disubstituted analogues with 3′,4′-fluorophenyl 21 and 3′,4′-dichlorophenyl 22. Both analogues display further improvements in potency relative to the corresponding mono-halogenated analogues and the MIC of 3′,4′-dichlorophenyl 22 decreased to 0.0625 μg/mL, the lowest value among the series of analogues described in Table 2. Additionally, a broader array of substituents were explored at the 3′- and 4′-positions of the aryl ring including 11, 13–14, and 23–30. Analogues containing polar acetyl, cyano, methoxy, hydroxymethyl, methylenedioxy, and morpholino substituents are inactive or weakly active with MICs generally >16 μg/mL. By contrast, analogues containing lipophilic groups including methylthio and trifluoromethyl are potent with MICs of 0.25–0.50 μg/mL, the exception being trifluoromethoxy 27, whose MIC is 8 μg/mL. In an attempt to decrease the lipophilicity, we replaced the phenyl ring of 3′-trifluoromethylphenyl 26 by a pyridine to furnish 5′-(trifluoromethyl)pyridin-2-yl 31, which fortuitously maintains activity providing an identical MIC to 26 while decreasing the calculated log P by 1.2 units to 6.0. While the introduction of an appropriately substituted arylamino group led to a substantial enhancement in activity relative to the simple quinolinols shown in Table 1, this boost in potency came at the expense of substantially increased lipophilicity. This is exemplified by 3′-trifluoromethylphenyl 26 whose 16-fold improvement in potency relative to the parent quinolinol 3 is offset by a 5.1 unit increase in the calculated log P. Attempts to decrease lipophilicity by the introduction of polar substituents onto the aryl ring led to sharp reductions in potency. However, heterocyclic replacement of the phenyl ring by a pyridine in compound 31 is tolerated providing a means to partially address the increased lipophilicity of the N-(arylamino)-quinolines.
Table 2.
Aryl 4-Aminoquinoline Analogues

|
|||||
|---|---|---|---|---|---|
| compound | R1 | R2 | R3 | MIC (μg/mL) | clog P |
| 1 | H | OCF3 | 3-(trifluoromethoxy) phenyl | 8 | 7.9 |
| 11 | CF3 | H | phenyl | 128 | 6.4 |
| 12 | CF3 | H | 2-isopropylphenyl | 320 | 7.8 |
| 13 | CF3 | H | 3-acetylphenyl | >128 | 5.9 |
| 14 | CF3 | H | 3-cyanophenyl | >32 | 5.9 |
| 15 | CF3 | H | 2-chlorophenyl | >16 | 7.1 |
| 16 | CF3 | H | 3-chlorophenyl | 0.25 | 7.1 |
| 17 | CF3 | H | 4-chlorophenyl | 0.12 | 7.1 |
| 18 | CF3 | H | 3-fluorophenyl | 4–16 | 6.5 |
| 19 | CF3 | H | 3-bromophenyl | 0.25 | 7.3 |
| 20 | CF3 | H | 3-iodophenyl | 0.25 | 7.5 |
| 21 | CF3 | H | 3,4-difluorophenyl | 0.125–0.25 | 6.6 |
| 22 | CF3 | H | 3,4-dichloropheyl | 0.0625 | 7.7 |
| 23 | CF3 | H | 3-(hydroxymethyl) phenyl | >16 | 5.3 |
| 24 | CF3 | H | 3-thiomethylphenyl | 0.25–0.5 | 7.0 |
| 25 | CF3 | H | 3-methoxyphenyl | >64 | 6.3 |
| 26 | CF3 | H | 3-(trifluoromethyl) phenyl | 0.25 | 7.3 |
| 27 | CF3 | H | 3-(trifluoromethoxy) phenyl | 8 | 7.4 |
| 28 | CF3 | H | 3,4-(methylenedioxy) phenyl | >4 | 6.2 |
| 29 | CF3 | H | 3-(morpholino) phenyl | >16 | 5.8 |
| 30 | CF3 | H | 4-(morpholino) phenyl | >16 | 5.8 |
| 31 | CF3 | H | 5-(trifluoromethyl) pyridin-2-yl | 0.25 | 6.1 |
The SAR of the N-(arylamino)quinoline scaffold was further probed (see Table 3) at the C-7- and C-8-positions with 7-methoxy, 7-trifluoromethoxy, 7-(dimethylamino)sulfonyl, 8-trifluoromethyl, and 8-trifluoromethoxy substituents with the C-4 aryl moiety selected from representatives of 12–31 including 3′,4′-difluorophenyl 21, 3′,4′-dichlorophenyl 22, 3′-(trifluoromethoxy)phenyl 27, and 5′-(trifluoromethyl)pyridin-2-yl 31. Replacement of the C-7 trifluoromethyl group by a trifluoromethoxy group in 32–34 yielded flat SAR with MICs ranging from 0.5–1 μg/mL. The SAR trend from this limited set of compounds did not parallel the SAR observed with the 7-trifluoromethyl series of compounds (21, 22, 27, and 31) which had MIC ranges from 8 to 0.0625 μg/mL. The isosteric 7-methoxy analogues 35–38 were largely inactive (MICs of ≥32 μg/mL), a result consistent with the quinolinol SAR described in Table 1. The observation that electron-withdrawing substituents at C-7 are favorable prompted exploration of the 7-dimethylaminosulfonyl group with 39–41 since sulfonamides are electron-withdrawing and considerably more polar than a trifluoromethyl group. Unfortunately, this set of compounds was only weakly active with MICs ranging from 8 to >32 μg/mL suggesting optimal quinoline substituents at C-7 should not only be electron-withdrawing but also nonpolar. Analogues bearing trifluoromethyl and trifluoromethoxy substituents at C-8 exhibited remarkably flat SAR with MICs of 0.5–2.0 μg/mL. The SAR trend was inconsistent with the 7-trifluoromethyl substituted analogues, whose MICs varied over 128-fold for the same set of C-4 aryl substituents. Taken together, the SAR from 32–47 demonstrates substitution at C-7 is preferred and optimal substituents at this position should be nonpolar and strongly electron-withdrawing.
Table 3.
Additional Aryl 4-Aminoquinoline Analogues

|
|||||
|---|---|---|---|---|---|
| compound | R1 | R2 | R3 | MIC (μg/mL) | clog P |
| 21 | CF3 | H | 3,4-difluorophenyl | 0.125–0.25 | 6.6 |
| 22 | CF3 | H | 3,4-dichloropheyl | 0.0625 | 7.7 |
| 31 | CF3 | H | 5-(trifluoromethyl)pyridin-2-yl | 0.25 | 6.1 |
| 32 | OCF3 | H | 3,4-dichlorophenyl | 0.5–1 | 8.2 |
| 33 | OCF3 | H | 3-(trifluoromethoxy)phenyl | 1 | 7.9 |
| 34 | OCF3 | H | 5-(trifluoromethyl)pyridin-2-yl | 1 | 6.5 |
| 35 | OMe | H | 3,4-dichlorophenyl | 2 | 6.0 |
| 36 | OMe | H | 3,4-difluorophenyl | >32 | 4.9 |
| 37 | OMe | H | 3-(trifluoromethoxy)phenyl | >32 | 5.7 |
| 38 | OMe | H | 5-(trifluoromethyl)pyridin-2-yl | 32 | 4.3 |
| 39 | S(O)2N(Me)2 | H | 3,4-difluorophenyl | >32 | 5.1 |
| 40 | S(O)2N(Me)2 | H | 3-(trifluoromethoxy)phenyl | 8 | 5.9 |
| 41 | S(O)2N(Me)2 | H | 5-(trifluoromethyl)pyridin-2-yl | 8 | 4.5 |
| 42 | H | OCF3 | 3,4-dichlorophenyl | 0.5 | 8.2 |
| 43 | H | OCF3 | 3-(trifluoromethoxy)phenyl | 1 | 7.8 |
| 44 | H | OCF3 | 5-(trifluoromethyl)pyridin-2-yl | 1 | 6.5 |
| 45 | H | CF3 | 3,4-dichlorophenyl | 0.5–2 | 8.2 |
| 46 | H | CF3 | 3-(trifluoromethoxy)phenyl | 1 | 7.9 |
| 47 | H | CF3 | 5-(trifluoromethyl)pyridin-2-yl | 0.5 | 6.5 |
The promising activity of compound 31 containing a 5′-(trifluoromethyl)pyridin-2-yl-amino moiety appended to C-4 of the quinoline prompted us to explore more diverse heterocyclic substituents at C-4 (Table 4). A primary objective in these analogues was to decrease the overall lipophilicity through the introduction of polar atoms and to reduce the planarity by increasing the sp3 character since lipophilic and planar molecules tend to have poor solubility that adversely impacts drug disposition properties. Replacement of the 5′-(trifluoromethyl)pyridin-3-yl-amino group at C-4 with a closely related 4′-(chloro)pyridin-2-yl-amino group in 48 led to an 8-fold loss of activity while transposition of the pyridine nitrogen by one atom in 2′-(trifluoromethyl)pyridin-5-yl-amino 49 completely abolished activity. These findings foreshadowed our unsuccessful attempts to modify the C-4 substituent. Thus, 50–54 and 56–65 were inactive at the highest concentration evaluated (MIC > 32 μg/mL). Only, aminothiazole 55 demonstrated moderate activity with an MIC of 1–2 μg/mL.
Table 4.
Heterocyclic 4-Substitutions

|
Further structural modifications were focused on reducing the calculated log P by modifications of the quinoline core employing the optimal C-4 substituents: 3′,4′-dichlorophenyl, 3′-(trifluoromethoxy)phenyl and 5′-(trifluoromethyl)pyridin-2-yl from compounds 22, 27, and 31, respectively. The trifluoromethyl groups at the C-2- and C-7-positions contribute significantly to the overall lipophilicity; thus, the next series of analogues explored the replacement of the trifluoromethyl group by difluoromethyl and aryl fluorides, which were predicted to lower the log P by approximately 0.7 units per trifluoromethyl group (Table 5). Replacement of the C-2 trifluoromethyl group of 22 with a difluoromethyl group afforded compound 68, which is 16-fold less potent than 22. Conversely, compound 69 exhibits a 16-fold increase in potency relative to the corresponding trifluoromethyl analogue 27. We cannot reconcile the disparate impact on potency of the difluoromethyl group based on this limited set of analogues, but the difluoromethyl group appears to level the SAR as both 68 and 69 have similar MICs. Replacement of the C-7 trifluoromethyl group by a fluorine was explored with analogues 74–79. Substitution of the C-7 trifluoromethyl group by a 7-fluoro moiety in 74–76 led to uniform 4- to 8-fold reductions in potency relative to the corresponding trifluoromethyl analogues 22, 27, and 31 providing MICs ranging from 0.5 to 2.0 μg/mL. Given the more predictable SAR of the aryl fluoride analogues, we sought to introduce additional fluorine atoms to 76 at the 5-, 6-, and 8-positions with difluorinated analogues 77–78 and trifluorinated analogue 79. While fluorine was poorly tolerated at the 5- and 6-positions, the 6,7-difluoro analogue 78 fully regained the activity of the parent trifluoromethyl analogue 31. Collectively, these results indicate modest attenuation of the log P can be achieved by replacement of the lipophilic trifluoromethyl groups with fluorine atoms while maintaining potent activity.
Table 5.
Fluorine Substitutionsa

|
||||||||
|---|---|---|---|---|---|---|---|---|
| compound | C-5 | C-6 | C-7 | C-8 | C-2 | R | MIC (μg/mL) | clog P |
| 68 | H | H | CF3 | H | CHF2 | 3,4-dichlorophenyl | 1 | 6.3 |
| 69 | H | H | CF3 | H | CHF2 | 3-(trifluoromethoxy)phenyl | 0.5 | 6.6 |
| 74 | H | H | F | H | CF3 | 3,4-dichlorophenyl | 0.5 | 7.0 |
| 75 | H | H | F | H | CF3 | 3-(trifluoromethoxy)phenyl | 2 | 6.7 |
| 76 | H | H | F | H | CF3 | 5-(trifluoromethyl)pyridin-2-yl | 1 | 5.3 |
| 77 | F | F | H | H | CF3 | 5-(trifluoromethyl)pyridin-2-yl | 16–32 | 5.3 |
| 78 | H | F | F | H | CF3 | 5-(trifluoromethyl)pyridin-2-yl | 0.25 | 5.3 |
| 79 | F | F | H | F | CF3 | 5-(trifluoromethyl)pyridin-2-yl | >32 | 5.5 |
C-2 refers to the position of the quinoline scaffold. Similarly, C-5, C-6, C-7, and C-8 refer to the respective positions on the quinoline nucleus.
With extensive coverage of the C-2-, C-4-, C-7-, and C-8-positions of the 4-aminoquinoline, the SAR campaign moved toward heteroatom modifications of the 4-aminoquinoline core. We first studied the importance of the 4-amino group and specifically the importance of an H-bond donor at this position with ether analogues 80–83, N-methyl derivative 84, and amide 85 (Table 6). Compounds 80–85 are inactive with MICs greater than 32 μg/mL indicating an NH moiety is essential for activity and a one atom linker is preferred. We then explored quinazoline analogues 89–91 containing a single aza substitution at the C-3-position, which retains a similar pharmacophore while lowering the calculated log P by 1.5 units. The 3′,4′-dichlorophenyl 89, and 5′-(trifluoromethyl)-pyridin-2-yl 91 quinazoline analogues are 8-fold less active than the parent quinolines 22, 31 while the trifluoromethoxy 90 derivative has an opposite 8-fold increase in potency relative to the parent quinoline 27. The aza substitution thus appears to flatten the SAR as the potency of 89–91 varies only 4-fold from 0.5 to 2.0 μg/mL. The introduction of another nitrogen atom into the quinazoline at the C-8-position led to pyridopyrimidine derivatives 94–95 and an attendant decrease in log P by almost 3 units. Unfortunately, both pyridopyrimidines 94 and 95 have drastically reduced activity with MICs ≥ 32 μg/mL.
Table 6.
Heteroatom Exchanges

|
||||||
|---|---|---|---|---|---|---|
| Compound | X | Y | Z | R | MIC μg/mL | cLogP |
| 80 | CH | O | CH | 3-chlorophenyl | >32 | 6.8 |
| 81 | CH | O | CH | 3-fluorophenyl | >32 | 6.2 |
| 82 | CH | O | CH | 3,4-dichlorophenyl | >32 | 7.4 |
| 83 | CH | O | CH | 3-(trifluoromethoxy)phenyl | >32 | 7.1 |
| 84 | CH | NMe | CH | 4-trifluoromethylphenyl | >32 | 7.4 |
| 85 | CH | N(C=O) | CH | 5-(trifluoromethyl)pyridin-2-yl | >32 | 5.3 |
| 89 | N | NH | CH | 3,4-dichlorophenyl | 0.5 | 6.9 |
| 90 | N | NH | CH | 3-(trifluoromethoxy)phenyl | 1 | 6.6 |
| 91 | N | NH | CH | 5-(trifluoromethyl)pyridin-2-yl | 2 | 5.1 |
| 94 | N | NH | N | 3,4-difluorophenyl | 32 | 4.0 |
| 95 | N | NH | N | 5-(trifluoromethyl)pyridin-2-yl | >32 | 5.8 |

|
||||
|---|---|---|---|---|
| Compound | R | MIC μg/mL | cLogP | |
| 100 | 3,4-difluorophenyl | >32 | 4.8 | |
| 101 | 5-(trifluoromethyl)pyridin-2-yl | >32 | 4.1 |
A few remaining miscellaneous modifications to reduce the log P of the 4-aminoquinoline scaffold are described in Tables 6 and 7. The cinnoline analogues 100 and 101 lacking a C-2 trifluoromethyl group and containing a nitrogen atom at C-2 are unsurprisingly inactive (Table 7). Remarkably, the introduction of a piperazine at C-2 with 105 was reasonably well tolerated yielding an MIC of 0.5–1.0 μg/mL while the morpholine analogue 107 is inactive (Table 7). The piperazine and morpholine constitutional isomers 106 and 108 have modest activity with MICs of 4–8 μg/mL.
Table 7.
C-2 and C-4 Substituted Quinolines

|
||||
|---|---|---|---|---|
| compd | R1 | R2 | MIC (μg/mL) | clog P |
| 105 | piperazinyl | 5-(trifluoromethyl) pyridin-2-yl-amino | 0.5–1 | 5.1 |
| 106 | 5-(trifluoromethyl) pyridin-2-yl-amino | piperazinyl | 4–8 | 5.1 |
| 107 | morpholino | 5-(trifluoromethyl) pyridin-2-yl-amino | >32 | 5.1 |
| 108 | 5-(trifluoromethyl) pyridin-2-yl-amino | morpholino | 8 | 5.1 |
The final series of compounds investigated was a hybrid scaffold of the initial active 4-quinolones (Table 1) and the aryl-substituted 4-aminoquinoline (Tables 2 and 3) to afford an aryl-substituted 5-aminoquinolin-4-one scaffold (Table 8) in an attempt to lower the calculated log P. The 5-aminoquinolin-4-ones 111–122 with the exception of 119 showed good to outstanding antibacterial activity with MICs ranging from <0.0625 to 2 μg/mL while simultaneously decreasing the calculated log P by two and up to five units. The optimal C-4 substituents in the 4-aminoquinoline series yielded extremely potent 5-amino-4-quinolone analogues 111–113 with MICs less than 0.0625 μg/mL and attendant dramatic reductions in lipophilicity. Compound 111 was the first potent derivative synthesized with a calculated log P of less than 4. Given the impressive activity of 111, 112, and 113 we sought to further examine closely related substituents containing polar substituents and/or greater sp3 character including 5-fluoropyridin-2-yl-amino 114, 6-(trifluoromethyl)-pyridazin-3-yl-amino 115, 5-dimethylaminopyridin-2-yl-amino 116, 3-(N,N-dimethylsulfonamide)pyridine-6-yl-amino 117, 4-amino-1H-indazolyl 118, morpholino 119, and 4-(trifluoromethyl)cyclohex-1-yl-amino 120, whose calculated log Ps ranged from 2.0 to 3.9. The SAR exhibited substantially greater flexibility than observed in the 4-aminoquinoline series (Tables 2 and 3) and many of these analogues including 114–118 had respectable MICs ranging from 0.125 to 2 μg/mL (Table 8). Finally, we prepared 122 incorporating a 7-fluoro substituent in place of the trifluoromethyl group of 111 in an attempt to further modulate the lipophilicity. Unfortunately, 122 loses some potency with an MIC of 1 μg/mL which indicates that the 5-position is much more mailable to functional group modification than the 7-position with 114 having 8-fold greater activity than 122 with the same fluoro modification. Collectively, the results from the last series of 5-aminoquinoline-4-one demonstrate high antibacterial activity can be achieved by introduction of appropriate substituents at C-5 of this scaffold and that the C-5-position is somewhat permissive to modification tolerating more polar as well as nonplanar groups.
Table 8.
5- and 7-Substituted Quinolinones

|
||||
|---|---|---|---|---|
| compound | R1 | R2 | MIC (μg/mL) | clog P |
| 111 | CF3 | 5-(trifluoromethyl)pyridin-2-yl-amino | <0.0625 | 3.8 |
| 112 | CF3 | 3,4-dichloroanilino | <0.0625 | 5.2 |
| 113 | CF3 | 3,4-difluoroanilino | <0.0625 | 4.1 |
| 114 | CF3 | 5-(fluoro)pyridin-2-yl-amino | 0.125 | 3.1 |
| 115 | CF3 | 6-(trifluoromethyl)pyridazine-3-yl-amino | 1 | 2.9 |
| 116 | CF3 | 5-dimethylaminopyridin-2-yl-amino | 2 | 3.6 |
| 117 | CF3 | (N,N-dimethyl-6-sulfamoyl) pyridin-2-yl-amino | 2 | 2.1 |
| 118 | CF3 | 1H-indazol-4-yl-amino | 2 | 3.6 |
| 119 | CF3 | morpholino | >32 | 2.1 |
| 120 | CF3 | 4-(trifluoromethyl)cyclohexyl-amino | 0.125 | 3.9 |
| 122 | F | 5-(trifluoromethyl)pyridin-2-yl-amino | 1 | 3.1 |
We selected a few of the most potent compounds from the 4-aminoquinoline (22, 31) and 5-aminoquinolin-4-one (111, 120) series for evaluation against a panel of other MRSA strains and representative Gram-positive and Gram-negative pathogens (Table 9). Compounds 111 and 120 show excellent activity (MIC < 0.125 μg/mL) toward all six S. aureus strains while 31 also displays very good activity with MICs ranging from 0.125 to 0.25 μg/mL. The compounds maintain activity against Staphylococcus epidermidis; however, 31, 111, and 120 still maintain some potency against Enterococcus faecalis and Enterococcus faecium (MIC of 0.125–2 μg/mL for 111 and 120), which contribute heavily, along with MRSA, to healthcare-associated infections.7 The compounds are inactive against the Gram-negative bacilli Escherichia coli, Klebsiella pneumoniae, and Enterococcus cloacae as well as the fungus Candida albicans at the highest concentration (128–256 μg/mL) tested. While the MIC data confirmed that these compounds were at least bacteriostatic, we wanted to test for the bactericidal properties of both the 4-aminoquinoline and 5-amino-4-quinolone scaffolds. The minimum bactericidal concentration (MBC) of 22, 31, 111, and 120 was evaluated against the MRSA clinical strain S. aureus FPR3757 (Table 10). Compounds 22, 31, 111, and 120 possessed MBC values equal to 8 to 32 times of MIC, indicating that these compounds are not potently bactericidal.
Table 9.
Antimicrobial Susceptibility
| MIC (μg/mL) | |||||
|---|---|---|---|---|---|
| species | strain | 22 | 31 | 111 | 120 |
| S. aureus | FPR3757 | 0.0625 | 0.25 | <0.06 | 0.125 |
| MW2 | 0.125 | 0.125 | <0.06 | 0.125 | |
| COL | 0.25 | 0.25 | <0.06 | <0.06 | |
| N315 | 0.25 | 0.25 | <0.06 | 0.125 | |
| NRS71 | 0.25 | 0.25 | <0.06 | 0.0625 | |
| S. epidermidis | NIH04008 | 0.125 | 0.125 | <0.06 | 0.125 |
| NIH04003 | 0.25 | 0.5 | <0.06 | 0.125 | |
| E. faecalis | ATCC700802 | 0.5 | 16 | 2 | 2 |
| DHMC #1 | 0.25 | 1 | 2 | 0.125 | |
| E. faecium | ATCC19579 | 0.125 | 1 | <0.06 | 0.25 |
| DHMC #1 | 0.25 | 4 | 2 | 0.25 | |
| E. coli | DHMC-1 | >128 | >128 | >128 | >128 |
| K. pneumoniae | 7117 | >128 | >128 | >128 | >128 |
| E. cloacae | ND-21 | >128 | >128 | >128 | >128 |
| C. albicans | SC5314 | >256 | >256 | 256 | >256 |
Table 10.
Bactericidal Activity of Lead Compounds in MRSAa
| compound | MIC (μg/mL) | MBC (μg/mL) | fold difference |
|---|---|---|---|
| 22 | 0.0625 | 2 | 32× |
| 31 | 0.25 | 8 | 32× |
| 111 | 0.0625 | 2 | 32× |
| 120 | 0.5 | 4 | 8× |
105 S. aureus FPR3757 cells (USA300 CA-MRSA) used per well. All MICs and MBCs were conducted in triplicate.
Mechanism of Action Studies.
Classical macromolecular synthesis assays were initially performed to provide insight into the putative mechanism of action of the most promising quinolines 22 and 31. Compounds 22 and 31 interfere with all major metabolic activities in the cell as distinguished by macromolecular synthesis assays. More specifically, radiolabeled precursors [3H]-l-isoleucine, [3H]-thymidine, [3H]-uridine, and [3H]-glucosamine were added to a culture of Staphylococcus simulans (OD600 = 0.4) as a surrogate for S. aureus in Mueller–Hinton cation (MHC) adjusted medium at 37 °C along with compounds at 0.5×, 1× and 5× MIC. The control antibiotics ciprofloxacin, rifampicin, vancomycin, and tetracycline were included as inhibitors of DNA, RNA, cell wall, and protein synthesis, respectively. The cells were quenched at various time points with 10% trichloroacetic acid (TCA), filtered, washed and the amount of precursor incorporation was quantified by scintillation counting. Cells were treated with 22 or 31 concentration-dependent inhibition of DNA, RNA, and protein (Figure 3A,B). At 5× MIC, both 22 and 31 completely inhibited all macromolecular processes, a profile that is consistent with disruption of the cellular membrane.55,68–71
Figure 3.

Macromolecular synthesis assays. Percent inhibition of incorporation of radiolabeled precursors [3H]-thymidine (DNA, red), [3H]-uridine (RNA, blue), [3H]-l-isoleucine (protein, green), and [3H]-glucosamine (cell wall, purple) in S. simulans by 22 (3A) and 31 (3B) (at 0.5×, 1× and 5× MIC). Data are expressed as the percentage of inhibition relative to the DMSO-only negative control after 60 min of exposure. The positive control antibiotics ciprofloxacin, rifampicin, tetracycline, and vancomycin were used at 10× MIC. Data represent the mean ± standard error of the mean (SEM) of triplicate experiments.
While macromolecular synthesis assays suggested membrane disruption as a likely mechanism, we wanted to provide additional evidence for bacterial membrane damage. Transmission electron microscopy (TEM) and fluorescence microscopy (FM) allowed us to directly observe the effects of the novel 4-aminoquinolines on 5-amino-4-quinolones on the cell morphology of staphylococci. S. aureus (strain USA300) was treated with 31, 111, and 120. After 10 min exposure, the cells displayed cross-wall-septum formation with reduced splitting compared to the untreated cells in TEM images. Cells treated with 31, 111, and 120 cells also displayed mesosome-like membrane inclusions, membrane “wrinkling,” and bulging of the septum (blue arrows, Figure 4). These cellular defects were not seen in any of the control cells treated with DMSO. Fluorescence microscopy was performed on a S. aureus (strain COL) strain after treatment with either DMSO, 120 and 122 for 30 min followed by staining with FM 4–64 (red membrane stain), bodipy-vancomycin (Van-FL, green stain for cell wall), and Hoescht (blue DNA stain). FM 4–64 staining revealed membrane defects (Figure 5B,C) in the cells treated with 120 and 122 that included large bulges and bulging septum formation. DNA and cell wall staining showed little or no change compared to the DMSO control (Figure 5A). Taken together, these results indicated that 120 and 122 affected the cell membrane causing gross morphological changes in the membrane but did not have a major impact on the cell wall, consistent with the macromolecular synthesis assays.
Figure 4.

Transmission electron microscopy (TEM) imaging of MRSA USA300 treated with 31, 111, 120, DMSO, and daptomycin. Membrane disruption is highlighted by blue arrows by compounds 31 (C), 111 (D), and 120 (E, F) after 1 h of exposure at 1× MIC compared to DMSO (A) and daptomycin (B) controls.
Figure 5.

Fluorescence microscopy (FM) analysis of membrane, cell wall, and DNA in MRSA. FM of COL MRSA strain treated with compound 120 ((B) row) or 122 ((C) row) at 1× MIC and DMSO ((A) row) for 30 min followed by staining with FM-64 (far left column, 0.5 μg/mL), Van-FL (second column, 1 μg/mL), and Hoescht 33342 (third column, 1 μg/mL) for 5 min and washed with 1× phosphate-buffered saline (PBS) before imaging (fourth column, overlay). Scale bars are 2 μm.
We next sought to determine membrane selectivity of representative 4-aminoquinolines (76 and 78) and the most potent 5-aminoquinolin-4-ones (111–113, 120, and 122) by a hemolytic assay employing washed sheep erythrocytes.72 None of the compounds except 78, displayed any hemolysis at 32 μg/mL compared to the positive control Triton X-100 (100% lysis) (Figure 6). The promising 5-aminoquinolin-4-ones tested (111–113) displayed greater than 500-fold selectivity for S. aureus membranes over erythrocyte membranes based on the observed therapeutic index (CC50/MIC). [The CC50 values for these compounds are >32 μg/mL (Figure 6) and the MIC values are <0.0625 μg/mL (Table 8).] Cytotoxicity was also measured by lactate dehydrogenase (LDH) release from cultured Vero cells upon exposure to three 5-amino-quinolinones (111, 114, and 120) at increasing concentrations from 0.47 μM of up to 30 μM. As shown in Figure 7, toxicity was low compared to the positive control (2% sodium dodecyl sulfate (SDS) set at 100%). At the highest concentrations tested (30 μM), the LDH level only approached ~20% of the positive control. These studies suggest that these four compounds are not toxic to mammalian cells at high concentrations.
Figure 6.

Percent hemolysis analysis of sheep erythrocytes. Concentration-dependent hemolysis was measured by monitoring the OD540 of PBS-washed sheep erythrocytes. Complete hemolysis (100%) was confirmed by the treatment of erythrocytes with 2% Triton X-100. Data points represent the mean ± standard deviation (SD) of triplicate experiments.
Figure 7.

Cytotoxicity of compounds 111, 114, and 120. Vero cells grown to confluence were exposed to 5-amino-4-quinolone compounds at various concentrations (0.47–30 μM) and LDH release was monitored as an indicator of cytotoxicity was measured. The positive control is 2% SDS. The amount released is represented as percentage of the positive control.
To identify potential genetic determinants of resistance, we screened the Nebraska Transposon Mutant Library (NTML) of S. aureus JE2 against compound 22 at sub-inhibitory concentrations. Identified among the 51 mutants that displayed reduced survival were six membrane-associated ATPases, five permeases, three lipoproteins, two transcriptional factors, and a conserved hypothetical xenobiotic resistance effector (XRE) Interestingly, one of the identified ATPases, VraF, is the cognate ATPase of the membrane permease VraG which has been implicated in proper sensing of membrane perturbation from antimicrobial peptides.73 Another membrane permease identified was the efflux pump NorB responsible for resistance/efflux of 4-quinolone antibiotics (e.g., ciprofloxacin, norfloxacin, and moxifloxacin).74 These results suggest that our compounds of interest may affect the bacterial membrane and, thus, corroborate our prior assessments.
We next assessed the frequency of resistance (FOR) to these compounds through a multistep resistance selection process by serially passaging S. aureus FPR3757 in duplicate in sub-inhibitory and inhibitory concentrations of compounds 22 and 31. After 65 days of serial passage, no development of resistance was noted against compound 22 but one of the two isolates tested developed 2-fold resistance (0.25–0.5 μg/mL) against compound 31. The genome sequences of these two strains, obtained from two colonies of each isolate, were first evaluated and then compared to those obtained from screening of the transposon library. Sequencing data revealed four single nucleotide variations (SNVs) in intergenic regions and nonsilent SNVs in quorum sensing signal receptor agrC, the clumping factor clfA, and the lysyl transferase gene mprF, which is also involved in daptomycin resistance.75 Notably, the hypothetical XRE protein, identified with the NTML screen, was also identified by the resistant mutant screen. Further work is necessary to understand the independent contribution and biological effects of these mutations as well as those found in uncharacterized intergenic regions to compound.
Pharmacokinetic Analysis of Analogue 111.
With the drastic improvement of the physical properties of our 5-aminoquinolin-4-one scaffolds compared to the initial 4-aminoquinoline hits afforded by our synthetic campaign, we selected compound 111 to assay the pharmacokinetic (PK) properties. Single-dose in vivo PK studies was performed in female CD-1 mice and plasma concentrations of 111 were determined by liquid chromatography–tandem mass spectrometry (LC-MS/MS) at various time points over a 24 h period following both intravenous (i.v.) and oral administration (p.o.) (Figure 8). PK parameters were determined by noncompartmental PK analysis (Table 11). Following i.v. dosing at 0.5 mg/kg, 111 demonstrated a volume of distribution (Vd) of 0.32 L/kg, clearance (CL) of 119 mL/(kg·h), and a half-life (t1/2) of 1.88 h. Single p.o. dosing at 10 mg/kg showed an area under the curve (AUC0–24h) of 43 × 104 h·ng/mL and a maximum concentration (Cmax) of 3100 ng/mL and a bioavailability (F) of 52%.
Figure 8.

Mean plasma concentration versus time curves after single p.o. (10 mg/kg) and i.v. (0.5 mg/kg) administration of compound 111 to mice. Error bars represent standard deviation of the mean (n = 3).
Table 11.
In Vivo Pharmacokinetic Parameters of 111 in Female CD-1 Mice (n = 3, Mean ± SD)
| pharmacokinetic indices | analogue 111 |
|---|---|
| dose i.v., p.o. (mg/kg) | 0.5, 10 |
| AUC0–24h (i.v., h·ng/mL) | (4.20 × 103) ± 308 |
| Vd (i.v., mL/kg) | 322 ± 24 |
| CL (i.v., mL/(kg·h)) | 119 ± 9.2 |
| t1/2 (i.v., h) | 1.88 ± 0.08 |
| AUC0–24h (p.o., h·ng/mL) | (4.33 × 104) ± 543 |
| Cmax (p.o., ng/mL) | (3.10 × 103) ± 780 |
| F (%) | 51.6 ± 5.2 |
Plasma protein binding of 111 along with compounds 114 and 120 was also assayed by rapid equilibrium device (RED) dialysis. The RED device comprises a Teflon base plate, which holds up to 48 disposable dialysis cells. Each dialysis insert is made up of two side-by-side chambers separated by a vertical cylinder of dialysis membrane (molecular weight cut-off (MWCO) 8k), with a high membrane surface area-to-volume ratio.76 Equilibrium is attained at 37 °C in 4 h using this assembly. The protein binding results were validated using oxazepam, which has a known protein binding above 95%.77,78 Simvastatin was chosen as the standard due to its high plasma protein binding. We observed the protein binding of oxazepam to be in the range of the reported values i.e., 88.2%. After validating our method using a standard, the protein binding for the lead compounds (111, 114, and 120) was measured using the same protocol (see Table S1). We observed the protein binding to be 99.7, 99.8, and 99.9% for compounds 111, 114, and 120, respectively.
Plasma protein binding is not usually considered for optimization, but compounds that are protein bound might serve as drug depots, allowing for a slow release and longer duration of action. On the other hand, an extremely high protein binding may reduce the therapeutic efficacy of drugs by reducing free drug levels.79 Thus, analyzing a compound’s protein binding and its effects on the other pharmacokinetic parameters provides a better understanding of the drug distribution and disposition. Despite the extensive protein binding displayed by 111, the favorable oral drug exposure, along with the potent MIC, suggest that 111 warrants further efficacy testing in animal models of MRSA infections. Following i.v. dosing at 0.5 mg/kg total plasma concentrations reached well above the MIC (<0.0625 μg/mL) for about the first 9 h of the study (Figure 8).
To partially assess the effects high plasma protein binding may have on efficacy in animal model studies (as found with the lead compound 111), MIC values were obtained for other potent 4-aminoquinoline and 5-amino-4-quinolone compounds in the presence of serum (Table 12). The MICs in MRSA for each of the 4-aminoquinolines tested were substantially higher (4–8 μg/mL) when the broth was supplemented with serum. The MIC values of lead 111 were substantially affected by the supplementation of both 10% fetal bovine serum (FBS) and 10% mouse serum. Increasing the supplemented FBS to 30% in 111 MIC experiments with MRSA displayed no additional increase in the MIC (4–8 μg/mL). Supplementation of 10% human serum also showed the same reduction of MIC values with compound 111 to 4–8 μg/mL.
Table 12.
Serum Effects on MRSA MIC of Lead 4-Aminoquinoline and 5-Amino-4-quinolone Compounds
| compd | MIC (μg/mL) | MIC + 10% FBS (μg/mL) | MIC + 10% mouse serum (μg/mL) |
|---|---|---|---|
| 22 | 0.0625 | 8 | |
| 31 | 0.25 | 4–8 | |
| 111 | <0.0625 | 4 | 4–8 |
| 120 | 0.125 | 4 |
Finally, aqueous solubility was determined via LC-MS/MS for compounds 111, 114, and 120 at 25 °C in neutral buffered solutions (pH = 7.4). The lead compound 111 had a very poor aqueous solubility of 2.4 ± 0.4 ng/mL, whereas compound 114 was slightly more soluble in aqueous buffer solutions, compared to 111, with a solubility of 1.0 ± 0.0 μg/mL. However, all of these compounds are minimally insoluble in water as per USP guidelines.80 Therefore, future work will need to be done to prepare viable vehicles for i.p. or i.v. dosing in animal models, though oral dosing will still be applicable.
DISCUSSION
The SAR of the quinolinol DNAC-2 (MIC = 8 μg/mL) was systematically explored through the synthesis of more than 100 analogues that examined modification to every position of the scaffold. Strongly electron-withdrawing substituents (CF3 and OCF3) were optimal at the C-2- and C-7-positions, while electron-donating substituents abolished activity. The introduction of a 4-arylamino group at C-4 led to a dramatic increase in potency culminating in 3,4-dichlorophenylamino 22 with an MIC of 0.0625 μg/mL that was offset by a large increase in the calculated log P to 7.7. A great deal of effort was subsequently expended to maintain this outstanding antibacterial activity against Gram-positive organisms while reducing the lipophilicity and planarity of the molecule. Isosteric replacement of the C-4 group with a more polar 5′-(trifluoromethyl)pyridin-2-yl-amino moiety in 31 helped to decrease the calculated log P to 6.1 with an attendant 4-fold loss of potency, but nonconservative changes to introduce more polar or nonplanar heterocycles were not allowed. Examination of replacements for the lipophilic 2- and 7-trifluoromethyl groups revealed 6,7-difluoro substitution of the quinoline in 78 was tolerated while reducing the calculated log P to 5.3; however, further attempts to modulate the lipophilicity through the introduction of nitrogen atoms into the quinoline scaffold indicated potency and lipophilicity could not be separated. A breakthrough in the SAR was observed by the synthesis of a hybrid 5-aminoquinolin-4-one scaffold by combining the quinol-4-one core of DNAC-2 with an N-aryl substituent at C-5, typified by 111 containing a 5′-(trifluoromethyl)pyridin-2-yl-amino group at C-5, whose MIC was less than 0.06 μg/mL with a calculated log P of 3.8.
These novel antibacterial quinoline and quinolone scaffolds we developed are unique even compared to the wealth of bioactive quinoline-containing compounds found throughout drug discovery.81 Historically, quinolines have been found to be privileged scaffolds in many areas of drug development such as anticancer,82 antifungal,83 antimalarial, and antibacterial compounds.56,71 A variety of structures from some of the most prolific quinoline scaffolds in drug discovery are shown in Figure 9, such as the 4-anilinoquinoline and quinazoline anticancer mitogen-activated protein kinase (MAPK) disruptors Gefitinib and experimental inhibitor 123,84 the 2-[alkyl(amino)]quinoline scaffold of antimalarial chloroquine analogues, and antibacterial fluoroquinolones ciprofloxacin. While all of these compounds share a quinoline or 4-quinolone core, they have widely varied targets and mechanisms of action. These novel 5-amino-4-quinolones (111–120, 122) we reported do not share substitution patterns to any of these classic bioactive quinolines and 4-quinolones, most importantly the pharmacophore of these novel 4-quinolones differ greatly from the classic 6-fluoro-(3-carboxy)-4-quinolone scaffolds of the fluoroquinolones which also prevent the growth of Gram-positive bacteria as DNA topoisomerase poisons.
Figure 9.

Quinoline-, quinolone-, and quinazoline-containing drugs and bioactive compounds. Shown are anticancer drug Gefitinib and experimental compound 123,84 antimalarial quinoline drug chloroquine, antifungal quinolinol drug clioquinol,85 antibacterial fluoroquinolone ciprofloxacin, and antiviral quinolone Elvitegravir.
The 4-aminoquinoline and 5-amino-4-quinolone scaffolds represented by 31 and 111 are narrow-spectrum agents with antibacterial activity against strictly Gram-positive organisms, with no activity against Gram-negative bacilli or fungi (Table 9). Staphylococci species including several multidrug-resistant MRSA strains and S. epidermidis are most sensitive with MICs of ≤0.06–0.12 μg/mL, but E. faecalis and E. faecium are susceptible with MICs ranging from 2 to 8 μg/mL. Importantly, the spectrum of activity (Table 9) displayed inhibition of growth in both CA- and HA-MRSA strains that included multiple resistance genes. These lead quinoline and 4-quinolone compounds are bacteriostatic, but show bactericidal properties at 8–32 times their respective MIC (MBC 2–8 μg/mL). The lack of potent bactericidal properties of these compounds is dissimilar to classical membrane disruptors like CAMPS or daptomycin, while the lack of significant resistance seen in the FOR studies of the lead 4-aminoquinolines is similar to classical membrane disruptors.38,86,87
Preliminary mechanism of action studies with the 4-aminoquinoline and 5-aminoquinolines support the notion that these compounds share a similar mechanism of membrane disruption to the initial lead compound DNAC-2.55 In macromolecular synthesis assays (Figure 3), 4-aminoquinolines 22 and 31 inhibited uptake of DNA, RNA, protein precursors to a great extent and to a lesser extent the cell wall precursors which is consistent with disruption of the cellular membrane.55,68–71 TEM (Figure 4) and fluorescence microscopy (Figure 5) imaging studies displayed aberrant cell membrane morphology in cells treated with 4-aminoquinoline and 5-aminoquinolines that was parallel to the membrane disruption produced by daptomycin. While fluorescence microscopy DNA and cell wall staining showed little or no change. Taken together, the novel quinolines and 4-quinolones both affected gross morphological changes in the membrane but did not have a major impact on the cell wall, consistent with the macromolecular synthesis assays. Furthermore, the 4-aminoquinolines and 5-amino-4-quinolones displayed impressive selectivity for S. aureus membranes over erythrocyte membranes by a hemolytic assay (Figure 6) using washed sheep erythrocytes, translating to a therapeutic index of greater than 500 for lead 111.
Single-dose in vivo PK properties of lead 5-aminoquinolin-4-one 111 were favorable. Following oral dosing at 10 mg/kg total plasma concentrations above the MIC (<0.0625 μg/mL) for about the first 9 h of the study (Figure 8). Bioavailability was also promising at 52% indicating that oral dosing should be a viable route of administration. However, the high protein binding of lead 111 coupled with the suppressed bacteriostatic properties of 111 in the presence of serum (Table 12) suggests that higher concentrations may be required in future animal models of infection. Despite the extensive protein binding displayed by 111, the favorable oral drug exposure, along with the potent MIC, suggests that 111 warrants further efficacy testing in animal models of MRSA infections.
CONCLUSIONS
In efforts to develop a novel antibacterial compound that targets Gram-positive drug-resistant bacteria, we successfully identified two novel quinoline-containing antibacterial scaffolds, 4-aminoquinoline, and 5-aminoquinolin-4-ones. An extensive SAR campaign developed around an HTS screen hit 2 drastically improved the physicochemical properties and highlighted common pharmacophores that are active in both the initial 4-aminoquinoline and derived 5-aminoquinolin-4-ones. These quinolines are highly potent, show minimal toxicity at relevant concentrations, do not select for resistance in MRSA, and inhibit the growth of various drug-resistant strains of infectious agents, including MRSA. Initial mechanism of action studies coupled with the lack of resistance in MRSA suggests that bacterial membrane disruption is one of the primary modes of antibacterial activity. Overall, compound 5-amino-4-quinolone 111 is the most promising derivative identified from these studies based on its exceptionally potent activity, excellent therapeutic index, and attractive physicochemical properties including improved solubility, which lends to oral formulation. In this regard, these compounds are distinguished from other membrane-active agents that tend to be large amphipathic molecules. The discovery of 111 is a promising step toward the development novel antibacterial therapeutics to help combat the ever-growing pool of strains of drug-resistant infectious agents.
EXPERIMENTAL SECTION
General Chemistry Materials and Methods.
Chemicals and solvents were purchased from Acros Organics, Alfa Aesar, Sigma-Aldrich, and TCI America and were used as received. An anhydrous solvent dispensing system using two packed columns of neutral alumina was used for drying THF, MeOH, toluene, and CH2Cl2, while two packed columns of molecular sieves were used to dry DMF, and the solvents were dispensed under argon (Ar). EtOAc and hexanes were purchased from Fisher Scientific. Thin-layer chromatography (TLC) analyses were performed on TLC silica gel plates 60F254 from EMD Chemical, Inc. and were visualized with UV light. Purification by flash chromatography was performed using a medium-pressure flash chromatography system equipped with flash column silica cartridges with the indicated solvent system. Preparative reversed-phase high-performance liquid chromatography (HPLC) purification was performed on a Phenomenex Gemini 10 μm C18 250 × 20 mm2 column operating at 21.0 mL/min with detection at 254 nm employing a linear gradient of 5–75% MeCN (solvent B) in water (solvent A) for 13 min and maintaining 75% solvent B for an additional 3 min followed by a linear gradient to 95% MeCN (solvent B) for 5 min (method A). Analytical reversed-phase HPLC was performed on a Waters Symmetry 5 μm C18 4.6 × 20 mm2 column operating at 0.5 mL/min with detection at 250 nm employing a linear gradient from 5 to 95% MeOH in water for 9 min. 1H and 13C spectra were recorded on either 400, 500, or 600 MHz NMR spectrometers. Proton chemical shifts are reported in parts per million (ppm) from an internal standard (IS) of residual chloroform (7.27), methanol (3.31), or dimethyl sulfoxide (2.50); carbon chemical shifts are reported in ppm from an internal standard of residual chloroform (77.0), methanol (49.1), or dimethyl sulfoxide (39.5). Proton chemical data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, dt = doublet of triplets, t = triplet, q = quartet, pentet = pent, m = multiplet, ap = apparent, br = broad, ovlp = overlapping), coupling constant(s), integration. All compounds were determined to be >95% by analytical reversed-phase HPLC (excluding compounds 2, 29, 52, 60, 62, 89, 91, and 118 all of which are >90%) purities for each final compound are given in the Supporting Information.
HPLC Method A.
Preparative reversed-phase HPLC purification was performed on a Phenomenex Gemini 10 μm C18 250 mm × 20 mm column operating at 21.0 mL/min with detection at 254 and 350 nm. A solvent system of A: H2O + 0.1% FA, B: MeCN + 0.1% FA was used with a linear gradient from 5 to 30% B over 5 min which was held at 30% for 5 min followed by a linear gradient to 95% B over an additional 15 min (method A).
HPLC Method B.
Analytical reversed-phase HPLC was performed on a Waters Symmetry 5 μm C18 4.6 × 20 mm2 column operating at 0.5 mL/min with detection at 250 nm employing a linear gradient from 5 to 95% MeOH in water for 9 min (method B).
General Route to 4-Aminoquinolines.
2,7-Bis-(trifluoromethyl)quinolin-4-ol (2).
To a stirring mixture of polyphosphoric acid (1 g/1 mmol aniline) and ethyl 4,4,4-trifluoroacetoacetate (5.44 mL, 37.2 mmol) at 100 °C was added 3-trifluoromethylaniline (4.65 mL, 37.2 mmol) dropwise to the above reaction mixture. The temperature was raised to 120 °C, and the mixture was stirred vigorously for 4 h at which point the reaction was taken off heat and quenched with water once the reaction cooled to 50 °C. The resulting precipitate was filtered and washed with water three to four times. The solid was dissolved in MeOH/CH2Cl2 (1:1) and dried over MgSO4. Flash column chromatography afforded the title compound (7.66 g, 73% yield) as a pale yellow solid: HPLC purity 93.1%, tR = 6.68 min, k′ = 14.53 (method B); 1H NMR (500 MHz, (CD3OD)) δ 8.46 (d, J = 8.4 Hz, 1H), 8.24 (s, 1H), 7.79 (d, J = 8.7 Hz, 1H), 7.03 (s, 1H); high-resolution mass spectrometry (HRMS) (ESI+) m/z calcd for C11H6F6NO+ [M + H]+ 282.0348, found 282.0350 (error 0.61 ppm). We could not obtain useful 13C NMR due to extensive line broadening caused by the 14N nitrogen quadrupole moment, 19F–13C coupling of the trifluoromethyl groups, and prototropic tautomerism in the A quinoline ring despite extensive solvent screening.
4-Bromo-2,7-bis(trifluoromethyl)quinoline (2b).
To a flame-dried round-bottom flask under a nitrogen atmosphere containing toluene (0.5 M) were added 2,7-bis(trifluoromethyl)quinolin-4-one (2), (1.00 g, 2.80 mmol), phosphorus pentoxide (5.6 mmol, 2 equiv), and tetrabutylammonium bromide (4.3 mmol, 1.5 equiv) in one portion. The flask was backfilled with argon and heated to 95 °C for 3.5 h, the top organic layer was decanted, and the lower layer was extracted with refluxing toluene (2 × 30 mL). Toluene layers were combined, diluted with EtOAc, washed with saturated aq NaHCO3 (2 × 30 mL) and water (30 mL), dried over MgSO4, and concentrated under vacuum. Flash column chromatography (SiO2) afforded the product (850 mg, 86% yield) as a white solid: Rf = 0.3 (Hex); HPLC purity 98.3%, tR = 7.58 min, k′ = 16.63 (method B); 1H NMR (500 MHz, CDCl3) δ 8.57 (s, 1H), 8.42 (d, J = 8.8 Hz, 1H), 8.14 (s, 1H), 7.95 (d, J = 8.7 Hz, 1H); could not obtain HRMS via electrospray ionization time-of-flight (ESI TOF) HRMS.
2,7-Bis(trifluoromethyl)-4-[5-(trifluoromethyl)pyridin-2-yl-amino]quinoline (31).
To a flame-dried Schlenk tube under a nitrogen atmosphere were added 4-bromo-2,7-bis(trifluoromethyl)-quinoline (2b, 150 mg, 0.43 mmol), potassium phosphate tribasic (130 mg, 0.6 mmol), 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (26 mg, 0.06 mmol), Pd2(dba)3·CH2Cl2 (22 mg, 0.02 mmol), and 2-amino-5-trifluoromethylpyridine (83.0 mg, 0.51 mmol) at once, and the flask was backfilled with nitrogen. Anhydrous THF (0.15 M, degassed with nitrogen for 5 min) was added, and again degassed the reaction mixture for 5 min. Next, the reaction mixture was heated to 55 °C and stirred for 20 h. Flash column chromatography (SiO2) using a stepwise gradient of EtOAc/Hex (0:1–1:5) afforded the title compound (97 mg, 54% yield) as a yellow powder: Rf = 0.27 (1:5 EtOAc/hexanes); HPLC purity 99.0%, tR = 7.71 min, k′ = 16.94 (method B); 1H NMR (500 MHz, (CD3)2SO) δ 10.3 (s, 1H), 9.04 (s, 1H), 8.81 (d, J = 8.9 Hz, 1H), 8.71 (br. s, 1H), 8.37 (s, 1H), 8.12 (d, J = 8.2 Hz, 1H), 8.02 (d, J = 8.8 Hz, 1H), 7.56 (d, J = 8.8 Hz, 1H); 13C NMR (126 MHz, (CD3)2SO) δ 157.0, 148.6 (q, 2JC–F = 34 Hz), 146.6, 145.9, 144.8 (d, 3JC–F = 3.6 Hz), 135.2, 130.1 (q, 2JC–F = 33.7 Hz), 127.4 (d, 3JC–F = 4.5 Hz), 124.6, 124.1 (q, 1JC–F = 272 Hz), 123.7 (q, 1JC–F = 273 Hz), 122.5, 122.2, 121.5 (q, 1JC–F = 274 Hz), 118.8 (q, 2JC–F = 32.7 Hz), 113.9, 104.3; HRMS (ESI+) m/z calcd for C17H9F9N3+ [M + H]+ 426.0647, found 426.0631 (error 3.76 ppm).
General Route to 4-Amino-7-sulfonamidoquinolines.
N,N-Dimethyl-2-(trifluoromethyl)quinoline-4(1H)-one-7-sulfonamide (66).
To a solution of 3-amino-N,N-dimethyl-benzenesulfonamide (1.00 g, 4.99 mmol) in Ph2O (3 mL) were added ethyl 4,4,4-trifluoro-3-oxo-butanoate (1.09 mL, 7.49 mmol) and HCl (12 M, 208 μL). The mixture was stirred at 265 °C for 2 h. The reaction mixture was quenched by adding NaOH (10%, 50 mL) at 0 °C, then diluted with tert-butyl methyl ether (TBME, 20 mL), and extracted with TMBE (20 mL × 2). The obtained aq layer was acidified by conc. HCl (10 mL), then extracted with EtOAc (20 mL × 3), and the combined organic layers were washed with brine (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by Prep.-HPLC: (column: Phenomenex Luna C18 200 mm × 40 mm × 10 μm; mobile phase: [water (0.1% TFA)–acetonitrile (ACN)]; B%: 30–50%, 10 min) to afford the title compound (400 mg, 25% yield) as a white solid: 1H NMR (400 MHz, (CD3)2SO) δ 8.54 (d, J = 8.8 Hz, 1H), 8.42 (s, 1H), 7.97 (d, J = 8.8 Hz, 1H), 7.22 (br. s, 1H), 2.81 (s, 6H).
4-(3,4-Difluoroanilino)-N,N-dimethyl-2-(trifluoromethyl)-quinoline-7-sulfonamide (39).
To N,N-dimethyl-4-oxo-2-(trifluoromethyl)-1H-quinoline-7-sulfonamide (66, 100 mg, 312 μmol) was added POCl3 (1.45 mL, 15.6 mmol). The mixture was stirred at 120 °C for 1 h. The reaction mixture was quenched by adding H2O (10 mL) at 0 °C and extracted with EtOAc (10 mL × 3). The combined organic layers were washed with brine (10 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to 4-chloro-N,N-dimethyl-2-(trifluoromethyl)quinoline-7-sulfonamide (66a, 100 mg, crude) as a brown oil.
A mixture of 4-chloro-N,N-dimethyl-2-(trifluoromethyl)quinoline-7-sulfonamide (66a, 90.0 mg, 266 μmol), 3,4-difluoroaniline (68.6 mg, 531 μmol), and HCl (12 M, 2.21 μL) in EtOH (2 mL) was degassed and purged with N2 three times, and then the mixture was stirred at 80 °C for 12 h under N2 atmosphere. The reaction mixture was concentrated under reduced pressure to give a residue. The residue was purified by Prep.-HPLC: (column: Kromasil 150 mm × 25 mm × 10 μm; mobile phase: [water (0.04% NH3H2O + 10 mM NH4HCO3)−ACN]; B%: 30–60%, 20 min) gave the title compound (42.2 mg, 36.7% yield, 99.6% purity) as a white solid: 1H NMR (400 MHz, (CD3)2SO) δ 9.84 (s, 1H), 8.71 (d, J = 9.0 Hz, 1H), 8.28 (d, J = 1.5 Hz, 1H), 7.98 (dd, J = 1.8, 8.8 Hz, 1H), 7.65–7.51 (m, 2H), 7.36–7.26 (m, 1H), 7.12 (s, 1H), 2.71 (s, 6H); MS (ESI) [M + H]+ = 432.0; HRMS (ESI) m/z calcd for C18H13F5N3O2S− [M − H]− 430.0654, found 430.0674 (error 4.71 ppm).
General Route to 4-Aminoquinazolines.
2-Amino-4-(trifluoromethyl)benzamide (86).
To a stirred solution of 2-amino-4-(trifluoromethyl)benzonitrile (2.00 g, 10.5 mmol) in DMSO (6.6 mL) at 0 °C were added potassium carbonate (306 mg, 2.22 mmol) and hydrogen peroxide (30% aqueous solution, 2.19 mL, 21.4 mmol). The mixture was allowed to warm up to room temperature and stirred for 2 h. The reaction was then separated between water (100 mL) and EtOAc (100 mL). The organics were separated and washed with brine (50 mL), dried over MgSO4, and concentrated under vacuum. The reaction was taken off heat and concentrated under vacuum onto silica gel. Flash column chromatography afforded the product (3.94 g, 88% yield) as a pale yellow oil. Rf = 0.4 (2:5 EtOAc/Hex); 1H NMR (400 MHz, CD3OD) δ 7.64 (d, J = 8.2 Hz, 1H), 7.01 (s, 1H), 6.79 (dd, J = 8.2, 1.3 Hz, 1H); 13C NMR (101 MHz, CD3OD) δ 173.4, 151.4, 134.9 (q, 3JC–F = 31.9 Hz), 130.7, 125.3 (q, 1JC–F = 271.7 Hz), 118.2, 114.4 (q, 3JC–F = 3.9 Hz), 112.2 (q, 3JC–F = 3.7 Hz).
2,7-Bis(trifluoromethyl)quinazolin-4-one (87).
To a stirred solution of 2-amino-4-(trifluoromethyl)benzamide (86, 525 mg, 2.56 mmol) in THF (9 mL) was added pyridine (5.14 mmol, 0.42 mL), trifluoroacetic anhydride (0.39 mL, 2.80 mol), and 4-dimethylaminopyridine (DMAP) (6.00 mg, 0.05 mmol) at ambient temperature and then stirred at that temperature for 3 h. After completion of the reaction, as indicated by TLC, and the reaction mixture was diluted with water (10 mL) and extracted with CH2Cl2 (3 × 10 mL). The organic extracts were combined, dried with Na2SO4, and evaporated to dryness under reduced pressure to afford the crude 2-(2,2,2-trifluoroacetamido)-4-(trifluoromethyl)benzamide intermediate (0.5 g) as a white powder. The crude trifluoroacetamide was carried on to the next step without further purification: Rf = 0.08 in 1:5 EtOAc/Hex; 1H NMR (500 MHz, (CD3)2SO) δ 13.5 (s, 1H), 8.71 (s, 1H), 8.66 (d, J = 1.82 Hz, 1H), 8.23 (s, 1H), 8.15 (d, J = 8.27 Hz, 1H), 7.73 (dd, J = 8.33, 2.11 Hz, 1H).
To a solution of the crude trifluoroacetamide intermediate in EtOH/H2O (8 mL, 1:1) was added 1 M KOH (5 mL) at room temperature. The reaction mixture was heated at reflux for 1 h. The reaction mixture was then cooled to ambient temperature, diluted with saturated NH4Cl (10 mL) and EtOAc (10 mL), and extracted with EtOAc (2 × 10 mL). The organics extracts were combined, concentrated under vacuum, and dried over MgSO4 to give the title compound (302 mg, 42% yield over two steps) as a white powder: Rf = 0.16 (3:10 EtOAc/Hex); 1H NMR (400 MHz, CD3OD) δ 8.44 (d, J = 8.3 Hz, 1H), 8.11 (s, 1H), 7.91 (dd, J = 8.3, 1.3 Hz, 1H); 13C NMR (151 MHz, CD3OD) δ 162.4, 148.5, 145.9 (q, 2JC–F = 38.8 Hz), 137.4 (q, 2JC–F = 33.1 Hz), 129.1, 126.8, 126.6 (q, 3JC–F = 3.9 Hz), 125.9 (q, 3JC–F = 3.4 Hz), 124.8 (q, 1JC–F = 270.1 Hz), 119.2 (q, 1JC–F = 274.5 Hz); HRMS (ESI−) m/z calcd for C10H3F6N2O− [M − H]− 281.0155, found 281.0156 (error 0.29 ppm).
4-(3,4-Dichlorophenylamino)-2,7-bis(trifluoromethyl)-quinazoline (89).
To a round-bottom flask was dissolved 4-chloro-2,7-bis(trifluoromethyl)quinazoline (88, 35.0 mg, 0.12 mmol) in isopropyl alcohol (IPA) (0.5 mL, 0.24 M) and then added 3,4-dichloroaniline (18.0 mg, 0.12 mmol) in one portion. The reaction was heated to reflux for 17 h, allowed to cool to ambient temperature, and then concentrated under vacuum. Flash column chromatography afforded the product (37 mg, 75% yield) as a white powder: Rf = 0.5 (3:10 EtOAc/Hex); HPLC purity 91.2%, tR = 7.63 min, k′ = 16.72 (method B); 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 8.09 (d, J = 8.7 Hz, 1H), 8.05 (d, J = 2.6 Hz, 1H), 7.89 (dd, J = 8.6, 1.8 Hz, 1H), 7.76 (s, 1H), 7.72 (dd, J = 8.8, 2.6 Hz, 1H), 7.49 (d, J = 8.7 Hz, 1H); 13C NMR (151 MHz, CD3OD) δ 160.3, 154.7 (q, 2JC–F = 35.6 Hz), 150.6, 139.5, 136.5 (q, 2JC–F = 33 Hz), 133.3, 131.4, 128.9, 125.7, 127.1 (q, 3JC–F = 4.0 Hz), 125.2 (q, 3JC–F = 3.0 Hz), 125.1, 124.81 (q, 1JC–F = 273 Hz), 123.0, 120.51 (q, 1JC–F = 276 Hz), 118.0; HRMS (ESI−) m/z calcd for C16H6Cl2F6N3− [M − H]− 423.9848, found 423.9850 (error 0.35 ppm).
General Route to Pyrido[2,3-d]pyrimidines.
2-Amino-6-(trifluoromethyl)nicotinamide (92).
A mixture of 2-chloro-6-(trifluoromethyl)-nicotinonitrile (1.50 g, 7.26 mmol) and NH3 (7 M in THF, 70 mL) was degassed and purged with N2 three times and heated at 80 °C for 12 h under N2 atmosphere. The reaction mixture was concentrated under reduced pressure to give the crude 2-amino-6-(trifluoromethyl)nicotinonitrile (1.3 g, crude) as a white solid.
To a microwave tube were added the crude 2-amino-6-(trifluoromethyl)nicotinonitrile (1.00 g, 5.34 mmol) and aqueous NH3 (21.4 mL, 139 mmol, 25% purity). The sealed tube was stirred at 80 °C for 18 h under microwave. The reaction mixture was concentrated under reduced pressure. Flash column chromatography (SiO2) afforded the title compound (1.05 g, 81% yield, 85% purity) as a pale yellow solid from a EtOAc/petroleum ether (0–7:10) stepwise gradient: 1H NMR (400 MHz, (CD3)2SO) δ 8.15 (br. s, 1H), 8.14–8.12 (d, J = 7.9 Hz, 1H), 7.61 (br. s, 3H), 7.00 (d, J = 7.9 Hz, 1H).
2,7-Bis(trifluoromethyl)pyrido[2,3-d]pyrimidin-4(3H)-one (93).
To a flask charged with MeOH (10 mL) at 0 °C under an N2 atmosphere was added Na (842 mg, 36.7 mmol) portion-wise. The resulting mixture was stirred until Na was consumed completely. 2-Amino-6-(trifluoromethyl)nicotinamide (92, 940 mg, 4.58 mmol) and ethyl 2,2,2-trifluoroacetate (5.21 g, 36.6 mmol, 5.06 mL) were added into the generated NaOMe, and then the mixture was stirred at 70 °C for 12 h. LC-MS showed reactant was consumed completely, and the desired mass was detected. The reaction mixture was concentrated under reduced pressure to give crude 2,7-bis-(trifluoromethyl)pyrido[2,3-d]pyrimidin-4(3H)-one (1.3 g, crude) as a yellow gum: 1H NMR (400 MHz, (CD3)2SO) δ 8.62 (d, J = 7.9 Hz, 1H), 7.74 (d, J = 8.2 Hz, 1H).
N-(3,4-Difluorophenyl)-2,7-bis(trifluoromethyl)pyrido[2,3-d]-pyrimidin-4-amine (94).
To a flask under N2 atmosphere were added 2,7-bis(trifluoromethyl)pyrido[2,3-d]pyrimidin-4(3H)-one (93, 200 mg, 706 μmol) and POCl3 (2.00 mL, 21.5 mmol). The flask was purged with N2 three times, then the mixture was stirred at 110 °C for 1 h under N2 atmosphere. LC-MS showed reactant was consumed completely, and one main peak with desired mass was detected. The reaction mixture was diluted with H2O (30 mL) and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to give the title compound 4-chloro-2,7-bis(trifluoromethyl)pyrido[2,3-d]pyrimidine (93b, 189 mg, crude) as a red solid.
A flask under N2 atmosphere containing 4-chloro-2,7-bis-(trifluoromethyl)pyrido[2,3-d]pyrimidine (93b, 80.0 mg, 265 μmol), 3,4-difluoroaniline (31.9 μL, 318 μmol), and HCl (2.21 μL, 12 M) in EtOH (1 mL) was degassed and purged with N2 three times; then, the mixture was stirred at 80 °C for 12 h under N2 atmosphere. LC-MS showed that the reactant was consumed completely and desired mass was detected. The reaction mixture was concentrated under reduced pressure to give a residue. The residue was purified by Prep.-HPLC (column: Kromasil 150 mm × 25 mm × 10 μm; mobile phase: [water (0.04% NH3H2O + 10 mM NH4HCO3)−ACN]; B%: 50–70%, 10 min) to afford the title compound (47.7 mg, 45% yield, 97.58% purity) as a yellow solid: 1H NMR (400 MHz, (CD3)2SO) δ 10.9 (s, 1H), 9.40 (d, J = 8.4 Hz, 1H), 8.37 (d, J = 8.6 Hz, 1H), 8.08 (ddd, J = 2.6, 7.5, 13.0 Hz, 1H), 7.71–7.64 (m, 1H), 7.61–7.51 (m, 1H); 13C NMR (151 MHz, (CD3)2SO) δ 160.0, 157.1, 155.4 (q, 2JC–F = 35.6 Hz), 152.3 (q, 2JC–F = 34.7 Hz), 148.8 (dd, 1JC–F = 244, 13.2 Hz), 146.5 (dd, 1JC–F = 244, 12.6 Hz), 137.3, 134.8 (dd, 3JC–F = 8.8, 3.1 Hz), 120.83 (q, 1JC–F = 276 Hz), 119.51, 119.48 (q, 1JC–F = 276.4 Hz), 119.1 (dd, 3JC–F = 6.4, 3.3 Hz), 117.5 (d, 2JC–F = 18.0 Hz), 113.1, 111.8 (d, 2JC–F = 21.6 Hz); HRMS (ESI) m/z calcd for C15H5F8N4− [M − H]− 393.0392, found 393.0388 (error 1.11 ppm).
General Route to 4-Aminocinnolines.
2-Nitro-1-(1-nitroethyl)-4-(trifluoromethyl)benzene (96).
To a stirred solution of 4-chloro-3-nitrobenzotrifluoride (1.00 g, 4.43 mmol) and nitroethane (0.66 μL, 8.87 mmol) in DMSO at 10 °C was added sodium hydride (60% on mineral oil, 697 mg, 18.1 mmol). The reaction was stirred for 18 h. The reaction was quenched with the addition of cold aqueous HCl (1 M, 10 mL) and then extracted with EtOAc (3 × 10 mL). Organics were combined, dried over Na2SO4, and concentrated onto silica gel. Column chromatography (SiO2) using a stepwise gradient of EtOAc/hexanes (0:1–1:10 EtOAc/hexanes) afforded the product (1.1 g, 94% yield) as a yellow oil: Rf = 0.34 (1:5 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 7.98 (dd, J = 8.3, 1.9 Hz, 1H), 7.78 (d, J = 8.3 Hz, 1H), 6.29 (q, J = 6.9 Hz, 1H), 2.03 (d, J = 6.9 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 148.6, 134.1, 133.2 (q, 2JC–F = 35 Hz), 130.7 (q, 3JC–F = 3.4 Hz), 129.7, 122.8 (q, 3JC–F = 3.9 Hz), 122.5 (q, 1JC–F = 273 Hz), 80.4, 19.7.
1-[2-Nitro-4-(trifluoromethyl)phenyl]ethan-1-one (97).
To a suspension of the 2-nitro-1-(1-nitroethyl)-4-(trifluoromethyl)benzene (96, 1.69 g, 6.40 mmol) and potassium carbonate (1.36 g, 9.75 mmol) in toluene (2 mL) was added hydrogen peroxide solution (30% solution in water, 4 mL, 35 mmol). The reaction was quenched after 18 h upon addition of aq. HCl (1 M, 10 mL) and then extracted with CH2Cl2 (2 × 20 mL). Organics were combined, dried over Na2SO4, and concentrated onto silica gel for column chromatography. Column chromatography (SiO2) using a stepwise gradient of EtOAc/hexanes (0:1–1:10 EtOAc/hexanes) afforded the product (1.43 g, 69% yield) as a light yellow foam: Rf = 0.11 (1:10 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 7.97 (dd, J = 8.0, 1.7 Hz, 1H), 7.56 (d, J = 7.9 Hz, 1H), 2.57 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 198.6, 141.2, 133.2 (q, 2JC–F = 34 Hz), 131.2 (q, 3JC–F = 3.5 Hz), 128.4, 122.5 (q, 1JC–F = 273 Hz), 122.0 (q, J = 3.8 Hz), 30.3.
7-(Trifluoromethyl)cinnolin-4-ol (98).
To a suspension of 1-[2-nitro-4-(trifluoromethyl)phenyl]ethan-1-one (97, 2.10 g, 8.11 mmol) in deionized water (12 mL) were added iron filings (1.36 g, 24.3 mmol) and ammonium chloride (1.30 g, 24.3 mmol). The reaction was heated at 90 °C for 21 h in a sealed container, after which it was quenched with aqueous NaOH (1 M, 15 mL). The aqueous layer was extracted with EtOAc (3 × 20 mL), organics were combined, dried over Na2SO4, and concentrated onto silica gel. Crude reaction mixture adsorbed to silica gel was filtered over a plug of celite with EtOAc to afford the crude 1-[2-amino-4-(trifluoromethyl)phenyl]ethan-1-one intermediate (1.33 g) as a yellow foam: Rf = 0.20 (1:10 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 8.4 Hz, 1H), 6.89 (s, 1H), 6.84 (dd, J = 8.4, 1.8 Hz, 1H), 6.44 (s, 2H), 2.60 (s, 3H); HRMS (ESI) m/z calcd for C9H9F3NO+ [M + H]+ 204.0630, found 204.0630 (error 0.31 ppm).
To a solution of the crude 1-[2-amino-4-(trifluoromethyl)phenyl]-ethan-1-one intermediate (0.90 g, 3.30 mmol) in concentrated acetic acid (14 mL, 250 mmol) at 0 °C was added a solution of NaNO2 (459 mg, 6.65 mmol) in H2O (9 mL) dropwise over 15 min. The mixture was left to warm to room temperature for 3 h where the reaction was quenched with the addition of saturated aqueous NaHCO3 (10 mL) and extracted with EtOAc (3 × 10 mL). Organics were combined, dried over Na2SO4, and concentrated. Flash column chromatography (SiO2) with a stepwise gradient of EtOAc/hexanes (0:1–2:5) afforded the title compound (480 mg, 68% yield over two steps) as an orange powder: Rf = 0.16 (1:5 EtOAc/hexanes); 1H NMR (400 MHz, CD3OD) δ 8.34 (d, J = 8.6 Hz, 1H), 7.93 (s, 1H), 7.90 (s, 1H), 7.68 (d, J = 8.5 Hz, 1H); 13C NMR (151 MHz, CD3OD) δ 172.6, 142.3, 142.2, 136.4 (q, 2JC–F = 33.1 Hz), 130.3, 128.7, 127.9, 127.4, 125.8, 124.7 (q, 1JC–F = 271.5 Hz), 121.9 (q, 3JC–F = 3.2 Hz), 115.5 (q, 3JC–F = 4.5 Hz); HRMS (ESI) m/z calcd for C9H6F3N2O+ [M + H]+ 215.0427, found 215.0424 (error 1.43 ppm).
4-Chloro-7-(trifluoromethyl)cinnoline (99).
A mixture of 7-(trifluoromethyl)cinnolin-4-ol (98, 97.0 mg, 0.45 mmol), POCl3 (0.43 mL, 4.50 mmol), and PCl5 (94.0 mg, 0.45 mmol) was heated at 90 °C for 1 h. The reaction mixture was poured over cold water (5 mL) and then washed with NaHCO3 (10 mL). The aqueous layer was extracted with CH2Cl2 (3 × 10 mL) and combined organics, dried over Na2SO4, and concentrated onto silica gel. Flash column chromatography (SiO2) with a stepwise gradient of EtOAc/hexanes (0:1–2:5) afforded the title compound (75 mg, 71% yield) as a tan solid: Rf = 0.49 (3:10 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 9.46 (s, 1H), 8.87 (s, 1H), 8.34 (d, J = 8.8 Hz, 1H), 8.02 (d, J = 8.9 Hz, 1H).
N-(3,4-Difluorophenyl)-7-(trifluoromethyl)cinnolin-4-amine (100).
To a flask under an argon atmosphere were added 3,4-difluoroaniline (16.8 mg, 129 μmol), 4-chloro-7-(trifluoromethyl)-cinnoline (99, 25.0 mg, 101 μmol), potassium carbonate (45.0 mg, 320 μmol), Pd2(dba)3 (9.80 mg, 11 μmol), Xantphos (12.0 mg, 22.0 μmol), and degassed dioxane (2 mL). The flask was purged with argon three times, sealed, and then stirred at 100 °C for 3 h. The reaction mixture was then concentrated onto silica gel under reduced pressure, filtered over a pad of Celite, and then reduced under vacuum to give a residue. Flash column chromatography (SiO2) with a stepwise gradient of EtOAc/hexanes (0:1–3:5) afforded the title compound (24 mg, 63% yield, 98.7% purity) as a yellow solid: Rf = 0.33 (1:5 EtOAc/hexanes); 1H NMR (400 MHz, CD3OD) δ 8.75 (s, 1H), 8.54 (d, J = 8.9 Hz, 1H), 8.40 (s, 1H), 7.95 (dd, J = 8.9, 1.8 Hz, 1H), 7.49–7.32 (m, 2H), 7.28–7.25 (m, 1H); 13C NMR (151 MHz, CD3OD) δ 152.1 (dd, 1JC–F = 249, 13.7 Hz), 150.0 (dd, 1JC–F = 247, 12.5 Hz), 147.1, 143.8, 137.0, 134.7 (q, 2JC–F = 32.9 Hz), 132.4, 125.0, 124.8 (q, 1JC–F = 272 Hz), 124.5, 124.0, 121.9 (dd, 3JC–F = 6.2, 3.4 Hz), 119.9, 119.6 (d, 2JC–F = 18.6 Hz), 114.8 (d, 2JC–F = 19.1 Hz); HRMS (ESI) m/z calcd for C15H7F5N3− [M − H]− 324.0566, found 324.0560 (error 1.63 ppm).
General Route to 2-Piperazinyl-4-aminoquinolines.
Methyl 2-Acetamido-4-(trifluoromethyl)benzoate (102).
To a mixture of methyl 2-amino-4-(trifluoromethyl)benzoate (4.40 g, 20 mmol) and TEA (3.35 mL, 24.1 mmol) in THF (40 mL) was added acetyl chloride (1.58 mL, 22.1 mmol) dropwise at −20 °C, and then the mixture was stirred at 18 °C for 12 h under N2 atmosphere. The reaction mixture was diluted with H2O (100 mL) and extracted with EtOAc (30 mL × 3). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to give the title compound (3.94 g, 75% yield) as a pink solid: 1H NMR (400 MHz, CDCl3) δ 11.1 (br. s, 1H), 9.08 (s, 1H), 8.14 (d, J = 8.4 Hz, 1H), 7.32 (dd, J = 1.3, 8.4 Hz, 1H), 3.98 (s, 3H), 2.27 (s, 3H).
4-Hydroxy-7-(trifluoromethyl)quinolin-2(1H)-one (103).
To a stirred solution of methyl 2-acetamido-4-(trifluoromethyl)benzoate (102, 3.94 g, 15.1 mmol) in THF (40 mL) was dropwise added KHMDS (1 M in THF, 45.3 mL) at −78 °C, over 0.5 h; then, the mixture was slowly warmed to 18 °C and stirred for 3.5 h under N2 atmosphere. The reaction mixture was diluted with H2O (100 mL) and extracted with EtOAc (50 mL × 2). The aq. layer was acidified with HCl (4M, 10 mL). The precipitate formed was collected by filtration to give the title compound solid (2.6 g, 11.4 mmol, 75% yield) as a pink solid: 1H NMR (400 MHz, (CD3)2SO) δ 11.7 (br. s, 1H), 11.49 (br. s, 1H), 7.97 (d, J = 8.3 Hz, 1H), 7.58 (s, 1H), 7.43 (dd, J = 1.3, 8.4 Hz, 1H), 5.85 (s, 1H).
tert-Butyl 4-[4-Chloro-7-(trifluoromethyl)quinolin-2-yl]-piperazine-1-carboxylate (104).
To 4-hydroxy-7-(trifluoromethyl)-quinolin-2(1H)-one (103, 2.10 g, 9.16 mmol) was added POCl3 (41.3 g, 269 mmol, 25 mL) in one portion, then the mixture was stirred at 110 °C for 12 h under N2 atmosphere. The reaction mixture was diluted with H2O (400 mL) and extracted with EtOAc (100 mL × 3). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue to give crude 2,4-dichloro-7-(trifluoromethyl)quinoline (103b, 2.8 g, crude) as a red solid.
A mixture of 2,4-dichloro-7-(trifluoromethyl)quinoline (103b, 50.0 mg, 188 μmol), tert-butyl piperazine-1-carboxylate (35.0 mg, 188 μmol), and TEA (26.2 μL, 188 μmol) in DMSO (1 mL) was degassed and purged with N2 three times, then the mixture was stirred at 60 °C for 8 h under N2 atmosphere. The reaction mixture was diluted with H2O (10 mL) and extracted with EtOAc (5 mL × 3). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by Prep.-TLC (SiO2) from a petroleum ether/EtOAc gradient (15:1) to afford the title compound 104 (5 mg, 6% yield) as a yellow solid: 1H NMR (400 MHz, CDCl3) δ 8.30 (s, 1H), 8.19 (d, J = 8.6 Hz, 1H), 7.68 (d, J = 8.6 Hz, 1H), 7.30 (s, 1H), 3.92 (br. s, 4H), 3.73–3.67 (m, 4H), 1.50 (s, 9H).
2-(Piperazin-1-yl)-7-(trifluoromethyl)-4-{[5-(trifluoro-methyl)-pyridin-2-yl]amino}quinoline (105).
To a flask under N2 atmosphere were added tert-butyl-4-[4-chloro-7-(trifluoromethyl)-2-quinolyl]-piperazine-1-carboxylate (104, 100 mg, 240 μmol), 2-amino-5-(trifluoromethyl)pyridine (46.8 mg, 289 μmol), K2CO3 (99.7 mg, 721 μmol), Xantphos (55.7 mg, 96.2 μmol), and Pd2(dba)3 (44.0 mg, 48.1 μmol) in dioxane (2 mL). The solvent was degassed, and the flask was purged with N2 three times. The mixture was then stirred at 100 °C for 6 h under N2 atmosphere. The reaction mixture was concentrated under reduced pressure to remove the solvent. The residue was purified by Prep.-TLC (SiO2) from a petroleum ether/EtOAc gradient (3:1) to afford crude tert-butyl-4-(7-(trifluoromethyl)-4-[(5-(trifluoromethyl)pyridin-2-yl)amino]quinolin-2-yl)-piperazine-1-carboxylate (104b, 70 mg, 51% yield, 95% purity) as a yellow oil: MS (ESI) [M + H]+ = 542.2.
To a stirred solution of tert-butyl-4-(7-(trifluoromethyl)-4-{[5-(trifluoromethyl)-2-pyridyl]amino}-2-quinolyl)piperazine-1-carboxylate (104b, 60.0 mg, 111 μmol) in dichloromethane (DCM) (2 mL) was added TFA (32.8 μL, 443 μmol) at 18 °C, then the mixture was stirred at 18 °C for 12 h under N2 atmosphere. The reaction pH was adjusted to 7 by adding aq NH3·H2O (25%). The reaction mixture was concentrated under reduced pressure to give a residue. The residue was purified by Prep.-HPLC: (column: Kromasil 150 mm × 25 mm × 10 μm; mobile phase: [water (0.04% NH3H2O + 10 mM NH4HCO3)−ACN]; B%: 30–60%, 20 min) to afford the title compound (12.6 mg, 26% yield, 99.9% purity) as a white solid: 1H NMR (400 MHz, (CD3)2SO) δ 9.72 (br. s, 1H), 8.65 (s, 1H), 8.34 (d, J = 8.7 Hz, 1H), 8.22 (s, 1H), 8.04 (dd, J = 2.4, 8.9 Hz, 1H), 7.80 (s, 1H), 7.47 (dd, J = 1.6, 8.7 Hz, 1H), 7.41 (d, J = 8.9 Hz, 1H), 3.65–3.58 (m, 4H), 2.84–2.77 (m, 4H); 13C NMR (151 MHz, (CD3)2SO) δ 158.8, 158.2, 147.7, 145.1 (q, 3JC–F = 4.3 Hz), 143.7, 134.7 (q, 3JC–F = 3.0 Hz), 129.6 (q, 2JC–F = 31.6 Hz), 124.4 (q, 1JC–F = 271 Hz), 124.3 (q, 1JC–F = 272 Hz), 123.8, 123.43 (q, 3JC–F = 4.2 Hz), 118.9, 117.00 (q, 2JC–F = 32.4 Hz), 115.93 (q, 3JC–F = 3.4 Hz), 112.4, 98.8, 45.8, 45.7; MS (ESI) [M+H]+ = 442.1; HRMS (ESI) m/z calcd for C20H16F6N5− [M − H]− 440.1315, found 440.1326 (error 2.32 ppm).
General Route to 5-Aminoquinolin-4-ones.
5-Bromo-2,7-bis(trifluoromethyl)quinolin-4-ol (109).
To a stirring mixture of polyphosphoric acid (19.0 g) at 100 °C was added ethyl 4,4,4-trifluoroacetoacetate (2.38 mL, 15.8 mmol) dropwise. 3-Bromo-5-trifluoromethylaniline (1.98 mL, 15.8 mmol) was added dropwise to the above reaction mixture at 100 °C. The temperature was raised to 120 °C, and the mixture was stirred vigorously for 4 h at that temperature. At 4 h, the reaction was taken off heat and quenched with water once the reaction cooled to 50 °C. The resulting precipitate was filtered and washed with water followed by multiple washes with saturated NaHCO3. The solid was dissolved in MeOH/CH2Cl2 (1:1), dried over MgSO4, and concentrated under vacuum. Flash column chromatography (SiO2) using a stepwise gradient of EtOAc/Hex (0:1–1:5) afforded the title compound (109, 12% yield) as a light yellow powder: Rf = 0.23 (1:5 EtOAc/Hex); and compound (110, 7% yield) as a light yellow powder: Rf = 0.24 (1:5 EtOAc/Hex) as two regioisomers respectively.
Data for 109: HPLC purity 98.5%, tR = 7.17 min, k′ = 15.65 (method B); 1H NMR (500 MHz, (CD3)2SO) δ 9.07 (br. s, 1H), 7.88 (s, 1H), 7.48 (s, 1H), 6.43 (s, 1H); HRMS (ESI) m/z calcd for C11H3BrF6NO− [M − H]− 357.9308, found 357.9309 (error 0.41 ppm).
7-Bromo-2,5-bis(trifluoromethyl)quinolin-4-ol (110).
Data for 110: 1H NMR (500 MHz, (CD3)2SO) δ 8.95 (br. s, 1H), 8.36 (d, J = 1.6 Hz, 1H), 7.96 (s, 1H), 6.99 (s, 1H).
5-[(5-Trifluoromethylpyridin-2-yl)amino]-2,7-bis-(trifluoromethyl)quinolin-4(1H)-one (111).
To a mixture of 5-bromo-2,7-bis(trifluoromethyl) quinolin-4-ol (109, 188 mg, 0.53 mmol), 2-amino-5-(trifluoromethyl)pyridine (120 mg, 0.74 mmol), [2-(2-aminophenyl)phenyl]-methylsulfonyloxy-palladium;dicyclohexyl-[3,6-dimethoxy-2-(2,4,6-triisopropylphenyl)phenyl]phosphane (0.09 mg, 0.1 mmol), K3PO4 (340 mg, 1.6 mmol) were added sequentially under inert atmosphere of nitrogen. Degassed 2-methylbutan-2-ol (0.16 M) was added, then purged with N2 three times, and the mixture was stirred at 100 °C for 12 h under N2 atmosphere. The reaction mixture was concentrated under reduced pressure and purified by Prep.-HPLC (method A) to afford the title compound (21.0 mg, 9% yield); 1H NMR (400 MHz, CD3OD) δ 9.11 (s, 1H), 8.56 (s, 1H), 7.98–7.83 (m, 1H), 7.34 (s, 1H), 6.96 (d, J = 8.6 Hz, 1H), 6.52 (s, 1H); 13C NMR (151 MHz, CD3OD) δ 182.8, 158.3, 146.2 (q, J = 4.44 Hz), 145.1, 143.4, 136.2 (q, J = 32.4 Hz), 135.5 (q, J = 3.55 Hz), 126.6 (q, J = 270 Hz), 124.9 (q, J = 272 Hz), 121.5 (q, J = 275 Hz), 119.8 (q, J = 32.9 Hz), 115.8, 114.3, 109.4 (q, J = 3.53 Hz), 108.1 (q, J = 3.67 Hz), 107.5; MS (ESI) [M + H]+ = 442.0; HRMS (ESI) m/z calcd for C17H7F9N3O− [M − H]− 440.0451, found 440.0472 (error 4.71 ppm).
MIC Assays.
MICs were performed with compounds according to CLSI standards.88 Briefly, bacterial strains were grown in either trypticase soy or Luria–Bertani broth (TSB, LB; Difco) overnight and then diluted to an OD600 of 1 (~109 CFU/mL) in Mueller–Hinton broth (MBH; Difco). Strains were then diluted to 106 CFU/mL in MHB and 100 μL aliquots were applied to a 96-well plate. Amended to each aliquot were 100 μL twofold serial dilutions of compounds. Oxacillin was selected as a control for all MIC studies using FPR3757 cells (MIC of 64 μg/mL) and Gram-positive bacteria. Ceftriaxone was selected as a control for all Gram-negative bacteria. Plates were then incubated stationary at 37 °C overnight for 24 h and MICs were assessed. C. albicans was grown overnight at 30 °C in YPD (yeast extract, peptone; Difco, dextrose; Fischer). Overnight culture is then diluted to an OD600 of 0.1 in 200 μL of Roswell Park Memorial Institute (RPMI) 1640 (Gibco) and 60 μL of this dilution is amended to 5 mL of RPMI 1640 to make the working inoculi. To a 96-well plate, 100 μL aliquots of inoculum were dispensed followed by 100 μL twofold serial dilutions of compounds. Plates were then incubated stationary at 35 °C overnight for 24 h and MICs were assessed.
Determination of MBC Values.
Compound bactericidal activity was assessed according to CLSI standards (same reference as above). Briefly, S. aureus FPR3757 was grown in TSB overnight and then diluted to an OD600 of 1 (~109 CFU/mL) in MHB. The culture was then diluted to 106 CFU/mL in MHB and 100 μL aliquots were applied to a 96-well plate. Amended to each aliquot were 100 μL twofold serial dilutions of compounds beginning. Following stationary incubation at 37 °C for 24 h, dilutions of wells corresponding to 0.5× MIC to 32× MIC were plated on an antibiotic-free plate and incubated overnight at 37 °C. The minimal bactericidal concentration was determined as the lowest compound concentration at which cell viability fell below 99.9% of the initial inoculum.
Nebraska Transposon Mutant Library (NTML) Susceptibility Screening.
Nonessential genetic contributions to compound resistance were assessed through the USA300 JE2 bursa aurelias transposon mutagenized library. MHB was amended with 1× MIC concentration of compound 22, and 200 μL aliquots were then added to wells in enough 96-well plates to cover all 1952 unique mutants. Mutants were inoculated into each well by a 96 solid pin microplate replicator from frozen stock plates. Plates were then incubated overnight stationary at 37 °C. Library hits were deemed to be mutants that grew at half MIC concentrations of compound 22.
Frequency of Resistance (FOR).
Resistance development to compounds 22 and 31 was assessed by a multistep resistance study. Briefly, S. aureus FPR3757 was serially passaged in duplicate in sub-inhibitory concentrations of compounds 22 and 31 for a total of 65 days. Upon completion of passaging, all four strains were genomically sequenced (Dartmouth Genomics and Molecular Biology Shared Resources) and genetic variations were identified and processed by CLC Genomic Workbench (Dartmouth CLC Genomic Workbench Server).
Macromolecular Synthesis Assays.
The effect of compounds on macromolecular synthesis was studied by monitoring the incorporation of 3H- or 14C-labeled precursors (5-[3H] thymidine, [3H] glucosamine hydrochloride, [3H] uridine, and l-[14C] isoleucine) as described previously.89 An overnight culture of S. simulans 22 grown in MHC was diluted 50-fold into fresh medium and cultured at 37 °C to an OD600 of about 0.5. Cultures were aliquoted, diluted to an OD600 of 0.1, and allowed to regrow to an OD600 of 0.4. The respective labeled precursor was then added to each culture (final concentration 1 μCi/mL); the compound was added at 0.5×, 1×, or 2× MIC, while another aliquot was run with 10× MIC of a control antibiotic and one without any antibiotic. Control antibiotics were vancomycin (3.1 μg/mL) to inhibit cell wall synthesis, tetracycline (0.4 μg/mL) to inhibit protein synthesis, ciprofloxacin (0.3 μg/mL) to inhibit DNA synthesis, and rifampicin (0.01 μg/mL) to inhibit RNA synthesis. The incorporation of labeled precursors was monitored for up to 60 min, and representative aliquots were taken at 0, 5, 15, 30, 45, and 60 min. Macromolecules were precipitated with ice-cold TCA (10%) and incubated for at least 30 min on ice before being filtered through glass microfiber filters (Whatman). Filters were washed with 5 mL of TCA (2.5%) containing 10 mm unlabeled metabolite, dried, counted, and the data were expressed as the mean of the counts incorporated from triplicate samples.
Fluorescence Microscopy.
Bacterial strains were incubated overnight in TSB at 37 °C, back-diluted in fresh TSB, and allowed to grow until mid-exponential phase (OD600 ~ 0.5). Each culture was then divided into flasks with antibiotic (compounds 120 or 122 at 1× MIC), DMSO, or TSB alone. Cultures were incubated for 30 min at 37 °C, after which the cells were pelleted, washed in 1× PBS, and mounted on microscope slides with pads of 1.5% agarose in 1× PBS. For staining of membrane, cell wall, or DNA, cells were incubated with FM 4–64 (0.5 μg/mL), BODIPY FL vancomycin (1 μg/mL) or Hoechst 33342 (1 μg/mL) (all from Molecular Probes) for 5 min at 23 °C with shaking and washed with 1× PBS before being imaged. The cells were imaged by super-resolution structured illumination microscopy (SR-SIM) in an ELYRA PS.1 Microscope (Zeiss) using an sCMOS camera (Andor) and a Plan-Apochromat 63×/1.4 oil DIC M27 objective (Zeiss). SR-SIM images were acquired using five phase shifts and five grid rotations for each channel, reconstructed, and analyzed with Zen Black Software (Zeiss). For channel alignment, multicolored beads were imaged to determine the experimental point spread function (PSF), which was used for the alignment of the different channels.
Transmission Electron Microscopy.
Transmission electron microscopy (TEM) was used to analyze membrane disruption in S. aureus FPR3757 after exposure to daptomycin and compounds 22 and 31. Overnight cultures of FPR3757 were diluted 1:100 and allowed to grow at 37 °C with shaking until an OD600 of 1.1. These cultures were then diluted to an OD600 of 0.45 and allowed to grow at 37 °C with shaking for an additional 30 min. To each culture, 1× MIC of compounds, 2 μg/mL daptomycin, or 0.5 μL of DMSO was amended and the cultures were incubated for 1 h at 37 °C with shaking. Cell pellets were then collected at room temperature by centrifugation at 2000 rpm for 10 min, washed once in PBS, and then collected again by centrifugation. The PBS was completely removed and pellets were resuspended in 10× volume of fixative (2% glutaraldehyde/1% paraformaldehyde in 0.1 M sodium cacodylate, pH 7.4) and then left at room temperature for 15 min. The cells were collected by centrifugation and then resuspended in fixative at room temperature for 1 h. The cells are then collected, resuspended in fixative, and then allowed to fix at room temperature for 24 h on a rotator. Pellets are collected, washed three times with 0.1 M sodium cacodylate for 15 min each, and then post-fixed at room temperature with 1% osmium tetroxide in 0.1 M sodium cacodylate for 2 h on a rotator. Fixed cells were then rinsed in 0.1 M sodium cacodylate and then distilled water followed by en bloc staining in 1–2% aqueous uranyl acetate in the dark for 30 min. Stained cells are then rinsed with distilled water and collected at 3000 rpm. The samples are then dehydrated with an ethanol series (30, 50, 70, 95%) for 20 min followed by three times at 100% for 1 h each on a rotator and finally with 100% ethanol overnight on a rotator. The samples are then changed in propylene oxide twice for 30 min each, and the pellet is then immersed in LX112/propylene oxide (1.5:1) several times over 6–8 h and then under vacuum for 24 h. Pellets are then placed in specimen embedding capsules and desiccated for 24 h followed by polymerization by heating to 45 °C for 8 h and then for 40 h at 60 °C. Samples were then sectioned, thin sectioned, stained, and imaged by the Dartmouth College Electron Microscopy Lab.
Red Blood Cell Lysis Assay.
The hemolytic activity of compounds was evaluated as described previously.90 Briefly, 100 μL of 4% sheep erythrocytes (Rockland Immunochemicals, Limerick, PA) was added to 100 μL of twofold serially diluted compounds in PBS, 0.2% DMSO, or 2% Triton X-100 in a 96-well plate and then incubated stationary at 37 °C for 1 h. The plate was then centrifuged at 500g for 5 min, and 50 μL of the supernatant was transferred from each well to a new 96-well plate and measured for absorbance at 540 nm. Percent hemolysis was calculated according to eq 1
| (1) |
LDH Release Assay.
The cellular toxicity of compounds was also evaluated by the release of lactate dehydrogenase from cultured Vero cells exposed to the compound for 24 h. In brief, cultured cells were maintained in RPMI 1640 medium (Sigma, St. Louis, MO) with 10% fetal bovine serum and grown with 5% CO2 in an incubator at 37 °C. When confluent, cells in the flask were released with trypsin, collected, and counted. Various compounds at predetermined concentrations (range of 0.47–30 μM with twofold dilutions) in serum-free media were added to epithelial cells and incubated for 24 h at 37 °C. Triton X-100 at 2% final concentration was used as a positive control. On the following day, 100 μL of the cytotoxicity reagents (TaKaRa LDH cytotoxicity Detection kit Cat# MK401) was added to each well containing CFBE supernatant, and plates were then incubated at room temperature in the dark for 20 min, absorbance read at 490 nm, and LDH release calculated as per manufacturer’s protocol.
Pharmacokinetics Studies.
All animal studies were ethically reviewed and carried out in accordance with the Institutional Animal Care and Use Committee of Hackensack Meridian Health. Six-week-old CD-1 female mice (20–25 g) were used in pharmacokinetic studies. 111 was administered as a single dose by intravenous injection at 0.5 mg/kg using 5% dimethylacetamide (DMA):95% (4% Cremophor EL) vehicle and by oral gavage at 10 mg/kg in 5% DMA:60% poly(ethylene glycol) 300 (PEG 300):35% (5% dextrose in water) vehicle. Aliquots of 20 μL of blood were taken by puncture of the lateral tail vein from each mouse (n = 3 per route and dose) at 1 min, 15 min, 1, 3, 7, and 24 h post-dose following intravenous injection and at 30 min, 1, 3, 5, 7, and 24 h post-dose following oral gavage and captured in CB300 blood collection tubes containing K2EDTA and stored on ice. Plasma was recovered after centrifugation and stored at −80 °C until analyzed by high-pressure liquid chromatography coupled to tandem mass spectrometry.
Pharmacokinetic LC-MS/MS Analytical Methods.
Neat 1 mg/mL DMSO stocks of 111 were serially diluted in 50/50 acetonitrile-water to create standard curves and quality control spiking solutions. Standards and QCs were created by adding 10 μL of spiking solutions to 90 μL of drug-free plasma (CD-1 K2EDTA mouse, Bioreclamation IVT); 10 μL of control, standard, QC, or study sample were added to 100 μL of acetonitrile/methanol 50/50 protein precipitation solvent containing 10 ng/mL of the internal standard Verapamil (Sigma-Aldrich). Extracts were vortexed for 5 min and centrifuged at 4000 rpm for 5 min; 75 μL of supernatant was transferred for HPLC-MS/MS analysis and diluted with 75 μL of Milli-Q deionized water.
LC-MS/MS analysis was performed on a Sciex Applied Biosystems Qtrap 6500+ triple quadrupole mass spectrometer coupled to a Shimadzu Nexera X2 UHPLC system to quantify each drug in plasma. Chromatography was performed on an Agilent SB-C8 (2.1 × 30 mm2; particle size, 3.5 μm) using a reversed-phase gradient. Milli-Q deionized water with 0.1% formic acid was used for the aqueous mobile phase and 0.1% formic acid in acetonitrile for the organic mobile phase. Multiple reaction monitoring of parent/daughter transitions in electrospray positive ionization mode was used to quantify the analytes. The following MRM transitions were used for 111 (441.98/146.10) and Verapamil (455.40/165.00). Sample analysis was accepted if the concentrations of the quality control samples were within 20% of the nominal concentration. Data processing was performed using Analyst software (version 1.6.2; Applied Biosystems Sciex).
Plasma Protein Binding.
The rapid equilibrium dialysis (RED) method was used. Human plasma (Sigma-Aldrich P9523-5ML) was resuspended in sterile water. A stock solution of each active compound in DMSO (5 mg/mL) was prepared and diluted into physiological buffer (Dulbecco’s phosphate-buffered saline (DPBS), pH = 7.4) at a concentration of 100 μg/mL. This compound solution was spiked with human plasma at 10 μg/mL concentrations, and 500 μL was placed in plasma chambers (with the red ring) of the RED device. PBS (500 μL) was placed in the buffer chambers, and the RED device was sealed and allowed to shake in an orbital shaker for 4 h at 37 °C to achieve equilibrium. Equal volumes of plasma were added to the aliquots of the buffer chambers and vice versa to create identical matrices. The compounds were then precipitated using methanol solution containing internal standard (IS) (two times the aqueous phase) and centrifuged; then, the compound concentration in the supernatant was quantified using LC-MS-MS. The calibration curve was established with five-/six-point standards prepared by a serial dilution in the range the unknown sample signal would likely fall and exactly simulating the sample preparation. Starting from a 50 μg/mL organic stock solution of compound, a threefold serial dilution of sample was prepared in PBS. The resulting solutions were 5, 1.67, 0.56, 0.19, 0.06, 0.02 μg/mL, and 0 μg/mL (buffer) concentrations. A solution of methanol containing 1 μg/mL of 111 (for 114 and oxazepam samples) or 114 (for 111 and 120 samples) was prepared. The standard solution (100 μL) was treated with 200 μL of methanol solution, mixed well by vortex, centrifuged (2 min, 15 000g), and the supernatant was analyzed by LC-MS/MS. The standards and samples were quantified using LC-MS/MS, and unknown concentrations were back-calculated from the calibration curve.
The % free and bound drug concentration were calculated using eqs 2 and 3, respectively.
% Free drug concentration
| (2) |
% Bound drug concentration
| (3) |
LC-MS/MS Analysis.
Reversed-phase LC was performed on a Kinetex C18 column (50 mm × 2.1 mm, 2.6 μm; Phenomenex, Torrance, CA) using a Shimadzu UFLC XR instrument. The elution gradient was carried out with a binary solvent system consisting of 0.1% formic acid in H2O (solvent A) and 0.1% formic acid in MeCN (solvent B). A linear gradient profile with the following proportions (v/v) of solvent B was applied (t (min), %B): (0, 5), (0.5, 5), (3, 40), (5, 95), (7, 95), (8, 5) with 2 min for reequilibration to provide a total run time of 10 min. The flow rate was 0.5 mL/min, and the column oven was maintained at 40 °C. The injection volume was 7 μL. The retention times for 111, 114, 120, and oxazepam were 5.50, 5.81, 5.58, and 3.69 min, respectively (see Figure S3). MS/MS analysis was carried out using a triple quadrupole/linear ion trap instrument (AB SCIEX QTRAP 5500). To determine the optimum MRM settings (see Figure S2B), each analyte was infused at a concentration of 10 μM (in 1:1 water/acetonitrile containing 0.2% formic acid) onto the MS by a syringe pump at a flow of 5 μL/min. Samples were analyzed by MS in positive ionization mode by multiple reaction monitoring (MRM) monitoring the 442/424 transition for 111, 392/374 transition for 114, 447/297 transition for 120, and 287/231 transition for oxazepam. The peak areas of 111, 114, 120, and oxazepam were calculated (MultiQuant, version 2.0.2) and normalized to internal standards, and the concentration of samples was determined using the standard curve of 111, 114, 120, and oxazepam (Figure S3C–F, respectively). For the preparation of standard curves, the average area of standard 0 was subtracted from 111, 114, 120, and oxazepam areas of all higher standards prior to area normalization.
Kinetic Aqueous Solubility of 111 and 114.
A stock solution of each active compound in DMSO (5 mg/mL) was prepared and diluted into the physiological buffer (DPBS, pH = 7.4) at a concentration of 100 μg/mL and left for 6 h at room temperature (25 °C). After 6 h, the sample was centrifuged and the supernatant was filtered, and 50 μL of the filtrate was diluted with 50 μL of methanol–water solution containing internal standard (IS). The calibration curve was established with five-point standards prepared by a serial dilution in the range the unknown sample signal would likely fall and exactly simulating the sample preparation. Starting from a 50 μg/mL organic stock solution of compound, a threefold serial dilution of the sample was prepared in PBS. The resulting solutions were 1.67, 0.56, 0.19, 0.06, 0.02, and 0 μg/mL (buffer) concentrations. A solution of 1:1 methanol/water containing 2 μg/mL of 111 (for 114 samples) or 114 (for 111 sample) was prepared. Standard solution (50 μL) was treated with 50 μL of methanol–water solution, mixed well by vortex, centrifuged (2 min, 15 000g), and the supernatant was analyzed by LC-MS/MS. The standards and samples were quantified using LC-MS/MS, and unknown concentrations were back-calculated from the calibration curve.
LC-MS/MS Analysis.
Reversed-phase LC was performed on a Kinetex C18 column (50 mm × 2.1 mm, 2.6 μm; Phenomenex, Torrance, CA) using a Shimadzu UFLC XR instrument. The elution gradient was carried out with a binary solvent system consisting of 0.2% formic acid in H2O (solvent A) and 0.2% formic acid in MeCN (solvent B). A linear gradient profile with the following proportions (v/v) of solvent B was applied (t (min), %B): (0, 5), (0.5, 5), (3, 40), (5, 95), (7, 95), (8, 5) with 2 min for reequilibration to provide a total run time of 10 min. The flow rate was 0.5 mL/min, and the column oven was maintained at 40 °C. The injection volume was 7 μL. The retention times for 111 and 114 were 5.50 and 5.81 min, respectively (Figure S3A). MS/MS analysis was carried out using a triple quadrupole/linear ion trap instrument (AB SCIEX QTRAP 5500). To determine the optimum MRM settings (Figure S3B), each analyte was infused at a concentration of 10 μM (in 1:1 water/acetonitrile containing 0.2% formic acid) onto the MS by a syringe pump at a flow of 5 μL/min. Samples were analyzed by MS in positive ionization mode by Multiple Reaction Monitoring (MRM) the 442/424 transition for 111 and 392/374 transition for 114. The 111 and 114 peak areas were calculated (MultiQuant, version 2.0.2), peak areas were normalized to internal standards, and the concentration of samples was determined using the standard curve of 111 and 11 (Figure S3C,D, respectively). For the preparation of standard curves, the average area of standard 0 was subtracted from 111 and 114 areas of all higher standards prior to area normalization.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by RO1 AI146116 and R21 AI130540. The following reagent S. aureus strain FPR3757 was provided by the Network on Antimicrobial Resistance in S. aureus (NARSA) for distribution by BEI Resources, NIAID, NIH: Nebraska Transposon Mutant Library (NTML) Genetic Toolbox, NR-48850. This study was also funded by a La Caixa Junior Leader Fellowship (LCF/BQ/PI20/11760012) financed by “la Caixa” Foundation (ID 100010434) and by European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no 847648 (P.M.P.), by the European Research Council through grant ERC-2017-CoG-771709 (M.G.P.), and by Project LISBOA-01-0145-FEDER-007660 Microbiologia Molecular, Estrutural e Celular (ITQB-NOVA).
ABBREVIATIONS USED
- AMR
antimicrobial resistance
- CAMPs
cationic antimicrobial peptides
- CA-MRSA
community-associated MRSA
- CDC
Centers for Disease Control
- DMA
dimethylacetamide
- DMF
N,N-dimethylformamide
- DMSO
dimethylsulfoxide
- EtOAc
ethyl acetate
- FM
fluorescence microscopy
- HA-MRSA
healthcare-associated MRSA
- HTS
high-throughput screening
- KHMDS
potassium bis(trimethylsilyl)amide
- MeOH
methanol
- MBC
minimum bactericidal concentration
- MeCN
acetonitrile
- MDR
multidrug-resistant
- MH
Mueller–Hinton
- MIC
minimum inhibitory concentration
- MRSA
methicillin-resistant Staphylococcus aureus
- PPA
polyphosphoric acid
- RED
rapid equilibration dialysis
- SAR
structure–activity relationships
- SCCmec
Staphylococcal cassette chromosome mec
- SNAr
nucleophilic aromatic substitution
- TEM
transmission electron microscopy
- TFAA
trifluoroacetic anhydride
- THF
tetrahydrofuran
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01151.
Synthetic procedures for 2–122, 1H and 13C NMR spectra, and HPLC traces for all final compounds (PDF)
Molecular formula strings (CSV)
Complete contact information is available at: https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01151
The authors declare no competing financial interest.
Contributor Information
John R. Schultz, Department of Medicinal Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States
Stephen K. Costa, Department of Microbiology & Immunology, Geisel School of Medicine at Dartmouth, Hanover, New Hampshire 03755, United States
Gorakhnath R. Jachak, Department of Medicinal Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States
Pooja Hegde, Department of Medicinal Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Matthew Zimmerman, Center for Discovery and Innovation, Hackensack Meridian Health, Nutley, New Jersey 07110, United States.
Yan Pan, Center for Discovery and Innovation, Hackensack Meridian Health, Nutley, New Jersey 07110, United States.
Michaele Josten, Institute for Pharmaceutical Microbiology and Institute for Medical Microbiology, Immunology, and Parasitology, University of Bonn, D-53115 Bonn, Germany.
Chinedu Ejeh, Department of Microbiology & Immunology, Geisel School of Medicine at Dartmouth, Hanover, New Hampshire 03755, United States.
Travis Hammerstad, Department of Medicinal Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Hans Georg Sahl, Institute for Pharmaceutical Microbiology and Institute for Medical Microbiology, Immunology, and Parasitology, University of Bonn, D-53115 Bonn, Germany.
Pedro M. Pereira, Bacterial Cell Biology Laboratory, Instituto de Tecnologia Química e Biológica António Xavier, Universidade Nova de Lisboa, 2781-901 Oeiras, Portugal
Mariana G. Pinho, Bacterial Cell Biology Laboratory, Instituto de Tecnologia Química e Biológica António Xavier, Universidade Nova de Lisboa, 2781-901 Oeiras, Portugal
Véronique Dartois, Center for Discovery and Innovation, Hackensack Meridian Health, Nutley, New Jersey 07110, United States.
Ambrose Cheung, Department of Microbiology & Immunology, Geisel School of Medicine at Dartmouth, Hanover, New Hampshire 03755, United States.
Courtney C. Aldrich, Department of Medicinal Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States
REFERENCES
- (1).NVSS—Mortality—Historical Data. https://www.cdc.gov/nchs/nvss/mortality_historical_data.htm (accessed June 17, 2020).
- (2).CDC. The Biggest Antibiotic-Resistant Threats in the U.S; Centers for Disease Control and Prevention, 2020. https://www.cdc.gov/drugresistance/biggest-threats.html (accessed Jan 28, 2020). [Google Scholar]
- (3).CDC. Antibiotic Resistance Threats in the United States; U.S. Department of Health and Human Services, CDC: Atlanta, GA, 2019. [Google Scholar]
- (4).Wertheim HF; Melles DC; Vos MC; van Leeuwen W; van Belkum A; Verbrugh HA; Nouwen JL The Role of Nasal Carriage in Staphylococcus aureus Infections. Lancet Infect. Dis 2005, 5, 751–762. [DOI] [PubMed] [Google Scholar]
- (5).Dryden M; Baguneid M; Eckmann C; Corman S; Stephens J; Solem C; Li J; Charbonneau C; Baillon-Plot N; Haider S Pathophysiology and Burden of Infection in Patients with Diabetes Mellitus and Peripheral Vascular Disease: Focus on Skin and Soft-Tissue Infections. Clin. Microbiol. Infect 2015, 21, S27–S32. [DOI] [PubMed] [Google Scholar]
- (6).Weinke T; Schiller R; Fehrenbach FJ; Pohle HD Association between Staphylococcus aureus Nasopharyngeal Colonization and Septicemia in Patients Infected with the Human Immunodeficiency Virus. Eur. J. Clin. Microbiol. Infect. Dis 1992, 11, 985–989. [DOI] [PubMed] [Google Scholar]
- (7).Percival SL; Suleman L; Vuotto C; Donelli G Healthcare-Associated Infections, Medical Devices and Biofilms: Risk, Tolerance and Control. J. Med. Microbiol 2015, 64, 323–334. [DOI] [PubMed] [Google Scholar]
- (8).Hidron AI; Edwards JR; Patel J; Horan TC; Sievert DM; Pollock DA; Fridkin SK; National Healthcare Safety Network Team and Participating National Healthcare Safety Network Facilities. Antimicrobial-Resistant Pathogens Associated with Health-care-Associated Infections: Annual Summary of Data Reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect. Control Hosp. Epidemiol 2008, 29, 996–1011. [DOI] [PubMed] [Google Scholar]
- (9).Ronan MV; Herzig SJ Hospitalizations Related to Opioid Abuse/Dependence and Associated Serious Infections from 2002 to 2012. Health Aff. 2016, 35, 832–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kourtis AP; Hatfield K; Baggs J; et al. Vital Signs: Epidemiology and Recent Trends in Methicillin-Resistant and in Methicillin-Susceptible Staphylococcus aureus Bloodstream Infections — United States. Morb. Mortal. Wkly. Rep 2019, 68, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chambers HF; DeLeo FR Waves of Resistance: Staphylococcus aureus in the Antibiotic Era. Nat. Rev. Microbiol 2009, 7, 629–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lakhundi S; Zhang K Methicillin-Resistant Staphylococcus aureus: Molecular Characterization, Evolution, and Epidemiology. Clin. Microbiol. Rev 2018, 31, No. e00020–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Jevons MP “Celbenin” - Resistant Staphylococci. Br. Med. J 1961, 1, 124–125. [Google Scholar]
- (14).Lim D; Strynadka NCJ Structural Basis for the β Lactam Resistance of PBP2a from Methicillin-Resistant Staphylococcus aureus. Nat. Struct. Biol 2002, 9, 870. [DOI] [PubMed] [Google Scholar]
- (15).Hartman BJ; Tomasz A Low-Affinity Penicillin-Binding Protein Associated with β-Lactam Resistance in Staphylococcus aureus. J. Bacteriol 1984, 158, 513–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kosowska-Shick K; McGhee PL; Appelbaum PC Affinity of Ceftaroline and Other β-Lactams for Penicillin-Binding Proteins from Staphylococcus aureus and Streptococcus pneumoniae. Antimicrob. Agents Chemother 2010, 54, 1670–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Shore AC; Coleman DC Staphylococcal Cassette Chromosome Mec: Recent Advances and New Insights. Int. J. Med. Microbiol 2013, 303, 350–359. [DOI] [PubMed] [Google Scholar]
- (18).Urushibara N; Aung MS; Kawaguchiya M; Kobayashi N Novel Staphylococcal Cassette Chromosome Mec (SCCmec) Type XIV (5A) and a Truncated SCCmec Element in SCC Composite Islands Carrying SpeG in ST5 MRSA in Japan. J. Antimicrob. Chemother 2020, 75, 46–50. [DOI] [PubMed] [Google Scholar]
- (19).Bal AM; Coombs GW; Holden MTG; Lindsay JA; Nimmo GR; Tattevin P; Skov RL Genomic Insights into the Emergence and Spread of International Clones of Healthcare-, Community- and Livestock-Associated Meticillin-Resistant Staphylococcus aureus: Blurring of the Traditional Definitions. J. Global Antimicrob. Resist 2016, 6, 95–101. [DOI] [PubMed] [Google Scholar]
- (20).Côrtes MF; Botelho AM; Almeida LG; Souza RC; de Lima Cunha O; Nicolás MF; Vasconcelos AT; Figueiredo AM Community-Acquired Methicillin-Resistant Staphylococcus aureus from ST1 Lineage Harboring a New SCCmec IV Subtype (SCCmec IVm) Containing the TetK Gene. Infect. Drug Resist 2018, 11, 2583–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Sievert DM; Rudrik JT; Patel JB; McDonald LC; Wilkins MJ; Hageman JC Vancomycin-Resistant Staphylococcus aureus in the United States, 2002–2006. Clin. Infect. Dis 2008, 46, 668–674. [DOI] [PubMed] [Google Scholar]
- (22).Mangili A; Bica I; Snydman DR; Hamer DH Daptomycin-Resistant, Methicillin-Resistant Staphylococcus aureus Bacteremia. Clin. Infect. Dis 2005, 40, 1058–1060. [DOI] [PubMed] [Google Scholar]
- (23).Faridi A; Kareshk AT; Fatahi-Bafghi M; Ziasistani M; Ghahraman MRK; Seyyed-Yousefi SZ; Shakeri N; Kalantar-Neyestanaki D Detection of Methicillin-Resistant Staphylococcus aureus (MRSA) in Clinical Samples of Patients with External Ocular Infection. Iran. J. Microbiol 2018, 10, 215–219. [PMC free article] [PubMed] [Google Scholar]
- (24).Mohammadi S; Sekawi Z; Monjezi A; Maleki M-H; Soroush S; Sadeghifard N; Pakzad I; Azizi-Jalilian F; Emaneini M; Asadollahi K; Pourahmad F; Zarrilli R; Taherikalani M Emergence of SCCmec Type III with Variable Antimicrobial Resistance Profiles and Spa Types among Methicillin-Resistant Staphylococcus aureus Isolated from Healthcare- and Community-Acquired Infections in the West of Iran. Int. J. Infect. Dis 2014, 25, 152–158. [DOI] [PubMed] [Google Scholar]
- (25).Mahairas GG; Lyon BR; Skurray RA; Pattee PA Genetic Analysis of Staphylococcus aureus with Tn4001. J. Bacteriol 1989, 171, 3968–3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Butler MS; Blaskovich MA; Cooper MA Antibiotics in the Clinical Pipeline at the End of 2015. J. Antibiot 2017, 70, 3–24. [DOI] [PubMed] [Google Scholar]
- (27).U.S. Food and Drug Administration. Novel Drug Approvals for 2016; FDA, 2020. [Google Scholar]
- (28).U.S. Food and Drug Administration. Novel Drug Approvals for 2017; FDA, 2020. [Google Scholar]
- (29).U.S. Food and Drug Administration. Novel Drug Approvals for 2018; FDA, 2020. [Google Scholar]
- (30).U.S. Food and Drug Administration. New Drug Therapy Approvals 2019; FDA, 2020. [Google Scholar]
- (31).Liu C; Bayer A; Cosgrove SE; Daum RS; Fridkin SK; Gorwitz RJ; Kaplan SL; Karchmer AW; Levine DP; Murray BE; Rybak MJ; Talan DA; Chambers HF Clinical Practice Guidelines by the Infectious Diseases Society of America for the Treatment of Methicillin-Resistant Staphylococcus aureus Infections in Adults and Children. Clin. Infect. Dis 2011, 52, e18–e55. [DOI] [PubMed] [Google Scholar]
- (32).Gu B; Kelesidis T; Tsiodras S; Hindler J; Humphries RM The Emerging Problem of Linezolid-Resistant Staphylococcus. J. Antimicrob. Chemother 2013, 68, 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Silver LL Challenges of Antibacterial Discovery. Clin. Microbiol. Rev 2011, 24, 71–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sohlenkamp C; Geiger O Bacterial Membrane Lipids: Diversity in Structures and Pathways. FEMS Microbiol. Rev 2016, 40, 133–159. [DOI] [PubMed] [Google Scholar]
- (35).Epand RM; Walker C; Epand RF; Magarvey NA Molecular Mechanisms of Membrane Targeting Antibiotics. Biochim. Biophys. Acta, Biomembr 2016, 1858, 980–987. [DOI] [PubMed] [Google Scholar]
- (36).García-Lara J; Weihs F; Ma X; Walker L; Chaudhuri RR; Kasturiarachchi J; Crossley H; Golestanian R; Foster SJ Supramolecular Structure in the Membrane of Staphylococcus sureus. Proc. Natl. Acad. Sci. U.S.A 2015, 112, 15725–15730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hurdle JG; O’Neill AJ; Chopra I; Lee RE Targeting Bacterial Membrane Function: An Underexploited Mechanism for Treating Persistent Infections. Nat. Rev. Microbiol 2011, 9, 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).van’t Hof W; Veerman EC; Helmerhorst EJ; Amerongen AV Antimicrobial Peptides: Properties and Applicability. Biol. Chem 2001, 382, 597–619. [DOI] [PubMed] [Google Scholar]
- (39).Porto WF; Irazazabal L; Alves ESF; Ribeiro SM; Matos CO; Pires ÁS; Fensterseifer ICM; Miranda VJ; Haney EF; Humblot V; Torres MDT; Hancock REW; Liao LM; Ladram A; Lu TK; Fuente-Nunez C; de la Franco OL In Silico Optimization of a Guava Antimicrobial Peptide Enables Combinatorial Exploration for Peptide Design. Nat. Commun 2018, 9, No. 1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ortwine JK; Kaye KS; Li J; Pogue JM Colistin: Understanding and Applying Recent Pharmacokinetic Advances. Pharmacotherapy 2015, 35, 11–16. [DOI] [PubMed] [Google Scholar]
- (41).Katz BE; Fisher AA Bacitracin: A Unique Topical Antibiotic Sensitizer. J. Am. Acad. Dermatol 1987, 17, 1016–1024. [DOI] [PubMed] [Google Scholar]
- (42).Pogliano J; Pogliano N; Silverman JA Daptomycin-Mediated Reorganization of Membrane Architecture Causes Mis-localization of Essential Cell Division Proteins. J. Bacteriol 2012, 194, 4494–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Zhanel GG; Schweizer F; Karlowsky JA Oritavancin: Mechanism of Action. Clin. Infect. Dis 2012, 54, S214–S219. [DOI] [PubMed] [Google Scholar]
- (44).Wang M; Feng X; Gao R; Sang P; Pan X; Wei L; Lu C; Wu C; Cai J Modular Design of Membrane-Active Antibiotics: From Macromolecular Antimicrobials to Small Scorpionlike Peptidomimetics. J. Med. Chem 2021, 64, 9894–9905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Lin S; Koh J-J; Aung TT; Sin WLW; Lim F; Wang L; Lakshminarayanan R; Zhou L; Tan DTH; Cao D; Beuerman RW; Ren L; Liu S Semisynthetic Flavone-Derived Antimicrobials with Therapeutic Potential against Methicillin-Resistant Staphylococcus aureus (MRSA). J. Med. Chem 2017, 60, 6152–6165. [DOI] [PubMed] [Google Scholar]
- (46).Su M; Xia D; Teng P; Nimmagadda A; Zhang C; Odom T; Cao A; Hu Y; Cai J Membrane-Active Hydantoin Derivatives as Antibiotic Agents. J. Med. Chem 2017, 60, 8456–8465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Guo Y; Hou E; Wen T; Yan X; Han M; Bai L-P; Fu X; Liu J; Qin S Development of Membrane-Active Honokiol/Magnolol Amphiphiles as Potent Antibacterial Agents against Methicillin-Resistant Staphylococcus aureus (MRSA). J. Med. Chem 2021, 64, 12903–12916. [DOI] [PubMed] [Google Scholar]
- (48).Zhong R; Li H; Li H; Fang S; Liu J; Chen Y; Liu S; Lin S Development of Amphiphilic Coumarin Derivatives as Membrane-Active Antimicrobial Agents with Potent In Vivo Efficacy against Gram-Positive Pathogenic Bacteria. ACS Infect. Dis 2021, 7, 2864–2875. [DOI] [PubMed] [Google Scholar]
- (49).Hammond K; Lewis H; Faruqui N; Russell C; Hoogenboom BW; Ryadnov MG Helminth Defense Molecules as Design Templates for Membrane Active Antibiotics. ACS Infect. Dis 2019, 5, 1471–1479. [DOI] [PubMed] [Google Scholar]
- (50).Chu W; Yang Y; Cai J; Kong H; Bai M; Fu X; Qin S; Zhang E Synthesis and Bioactivities of New Membrane-Active Agents with Aromatic Linker: High Selectivity and Broad-Spectrum Antibacterial Activity. ACS Infect. Dis 2019, 5, 1535–1545. [DOI] [PubMed] [Google Scholar]
- (51).Niu Y; Wang M; Cao Y; Nimmagadda A; Hu J; Wu Y; Cai J; Ye X-S Rational Design of Dimeric Lysine N-Alkylamides as Potent and Broad-Spectrum Antibacterial Agents. J. Med. Chem 2018, 61, 2865–2874. [DOI] [PubMed] [Google Scholar]
- (52).Rončević T; Vukičević D; Ilić N; Krce L; Gajski G; Tonkić M; Goić-Barišić I; Zoranić L; Sonavane Y; Benincasa M; Juretić D; Maravić A; Tossi A Antibacterial Activity Affected by the Conformational Flexibility in Glycine–Lysine Based α-Helical Antimicrobial Peptides. J. Med. Chem 2018, 61, 2924–2936. [DOI] [PubMed] [Google Scholar]
- (53).Kim W; Zhu W; Hendricks GL; Tyne DV; Steele AD; Keohane CE; Fricke N; Conery AL; Shen S; Pan W; Lee K; Rajamuthiah R; Fuchs BB; Vlahovska PM; Wuest WM; Gilmore MS; Gao H; Ausubel FM; Mylonakis E A New Class of Synthetic Retinoid Antibiotics Effective against Bacterial Persisters. Nature 2018, 556, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Grein F; Müller A; Scherer KM; Liu X; Ludwig KC; Klöckner A; Strach M; Sahl H-G; Kubitscheck U; Schneider T Ca2+-Daptomycin Targets Cell Wall Biosynthesis by Forming a Tripartite Complex with Undecaprenyl-Coupled Intermediates and Membrane Lipids. Nat. Commun 2020, 11, No. 1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Nair DR; Chen J; Monteiro JM; Josten M; Pinho MG; Sahl H-G; Wu J; Cheung A A Quinolinol-Based Small Molecule with Anti-MRSA Activity That Targets Bacterial Membrane and Promotes Fermentative Metabolism. J. Antibiot 2017, 70, 1009–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Kumar P A Review on Quinoline Derivatives as Anti-Methicillin Resistant Staphylococcus aureus (MRSA) Agents. BMC Chem. 2020, 14, No. 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Conrad M; Limpach L Synthesen von Chinolinderivaten Mittelst Acetessigester. Ber. Dtsch. Chem. Ges 1887, 20, 944–948. [Google Scholar]
- (58).Garner R; Suschitzky H Syntheses of Heterocyclic Compounds. Part XI. Quinolines and Pyrones from the Reaction of N-Aminophenyl Heterocycles with β-Ketoesters in Polyphosphoric Acid. J. Chem. Soc. C 1966, 0, 186–189. [DOI] [PubMed] [Google Scholar]
- (59).Reitsema RH The Chemistry of 4-Hydroxyquinolines. Chem. Rev 1948, 43, 43–68. [DOI] [PubMed] [Google Scholar]
- (60).Bunnett JF; Zahler RE Aromatic Nucleophilic Substitution Reactions. Chem. Rev 1951, 49, 273–412. [Google Scholar]
- (61).Borrows ET; Clayton JC; Hems BA; Long AGS 43. The Synthesis of Thyroxine and Related Substances. Part II. The Preparation of Dinitrodiphenyl Ethers. J. Chem. Soc. (Resumed) 1949, S190–S199. [Google Scholar]
- (62).Ashley JN; Harris JO; Dalgliesh CE; Stephenson EFM; Albert A; Magrath D; Baddiley J; Topham A; Reed RA Notes. J. Chem. Soc. (Resumed) 1944, 677–679. [Google Scholar]
- (63).Kato Y; Okada S; Tomimoto K; Mase T A Facile Bromination of Hydroxyheteroarenes. Tetrahedron Lett. 2001, 42, 4849–4851. [Google Scholar]
- (64).Guram AS; Rennels RA; Buchwald SL A Simple Catalytic Method for the Conversion of Aryl Bromides to Arylamines. Angew. Chem., Int. Ed 1995, 34, 1348–1350. [Google Scholar]
- (65).Louie J; Hartwig JF Palladium-Catalyzed Synthesis of Arylamines from Aryl Halides. Mechanistic Studies Lead to Coupling in the Absence of Tin Reagents. Tetrahedron Lett. 1995, 36, 3609–3612. [Google Scholar]
- (66).Dorel R; Grugel CP; Haydl AM The Buchwald–Hartwig Amination After 25 Years. Angew. Chem., Int. Ed 2019, 58, 17118–17129. [DOI] [PubMed] [Google Scholar]
- (67).Reid JG; Reny Runge JH Addition of Nitroalkanes to Ortho-Halo-Nitrobenzenes: A New Synthesis of 4-Chloro-7-(Trifluoromethyl)Quinoline. Tetrahedron Lett. 1990, 31, 1093–1096. [Google Scholar]
- (68).Cotsonas King A; Wu L Macromolecular Synthesis and Membrane Perturbation Assays for Mechanisms of Action Studies of Antimicrobial Agents. Curr. Protoc. Pharmacol 2009, 47, 13A.7.1–13A.7.23. [DOI] [PubMed] [Google Scholar]
- (69).Gerits E; Blommaert E; Lippell A; O’Neill AJ; Weytjens B; De Maeyer D; Fierro AC; Marchal K; Marchand A; Chaltin P; Spincemaille P; De Brucker K; Thevissen K; Cammue BPA; Swings T; Liebens V; Fauvart M; Verstraeten N; Michiels J Elucidation of the Mode of Action of a New Antibacterial Compound Active against Staphylococcus aureus and Pseudomonas aeruginosa. PLoS One 2016, 11, No. e0155139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Hobbs JK; Miller K; O’Neill AJ; Chopra I Consequences of Daptomycin-Mediated Membrane Damage in Staphylococcus aureus. J. Antimicrob. Chemother 2008, 62, 1003–1008. [DOI] [PubMed] [Google Scholar]
- (71).Oliva B; Miller K; Caggiano N; O’Neill AJ; Cuny GD; Hoemann MZ; Hauske JR; Chopra I Biological Properties of Novel Antistaphylococcal Quinoline-Indole Agents. Antimicrob. Agents Chemother 2003, 47, 458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Lee K-H; Oh J-E Design and Synthesis of Novel Antimicrobial Pseudopeptides with Selective Membrane-Perturbation Activity. Bioorg. Med. Chem 2000, 8, 833–839. [DOI] [PubMed] [Google Scholar]
- (73).Falord M; Karimova G; Hiron A; Msadek T GraXSR Proteins Interact with the VraFG ABC Transporter To Form a Five-Component System Required for Cationic Antimicrobial Peptide Sensing and Resistance in Staphylococcus aureus. Antimicrob. Agents Chemother 2012, 56, 1047–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Truong-Bolduc QC; Dunman PM; Strahilevitz J; Projan SJ; Hooper DC MgrA Is a Multiple Regulator of Two New Efflux Pumps in Staphylococcus aureus. J. Bacteriol 2005, 187, 2395–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Bayer AS; Schneider T; Sahl H-G Mechanisms of Daptomycin Resistance in Staphylococcus aureus: Role of the Cell Membrane and Cell Wall. Ann. N. Y. Acad. Sci 2013, 1277, 139–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Waters NJ; Jones R; Williams G; Sohal B Validation of a Rapid Equilibrium Dialysis Approach for the Measurement of Plasma Protein Binding. J. Pharm. Sci 2008, 97, 4586–4595. [DOI] [PubMed] [Google Scholar]
- (77).Lennernäs H; Fager G Pharmacodynamics and Pharmacokinetics of the HMG-CoA Reductase Inhibitors. Clin. Pharmacokinet 1997, 32, 403–425. [DOI] [PubMed] [Google Scholar]
- (78).Davies JE The Pharmacological Basis of Therapeutics. Occup. Environ. Med 2007, 64, No. e2. [Google Scholar]
- (79).Craig WA; Ebert SC Protein Binding and Its Significance in Antibacterial Therapy. Infect. Dis. Clin. North Am 1989, 3, 407–414. [PubMed] [Google Scholar]
- (80).Savjani KT; Gajjar AK; Savjani JK Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012, 2012, No. 195727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Senerovic L; Opsenica D; Moric I; Aleksic I; Spasić M; Vasiljevic B Quinolines and Quinolones as Antibacterial, Antifungal, Anti-Virulence, Antiviral and Anti-Parasitic Agents. In Advances in Microbiology, Infectious Diseases and Public Health; Donelli G, Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, 2020; Vol. 14, pp 37–69. [DOI] [PubMed] [Google Scholar]
- (82).Solomon VR; Lee H Quinoline as a Privileged Scaffold in Cancer Drug Discovery. Curr. Med. Chem 2011, 18, 1488–1508. [DOI] [PubMed] [Google Scholar]
- (83).Pippi B; Machado G. d. R. M.; Bergamo VZ; Alves RJ; Andrade SF; Fuentefria AM Clioquinol Is a Promising Preventive Morphological Switching Compound in the Treatment of Candida Infections Linked to the Use of Intrauterine Devices. J. Med. Microbiol 2018, 67, 1655–1663. [DOI] [PubMed] [Google Scholar]
- (84).Yin K-H; Hsieh Y-H; Sulake RS; Wang S-P; Chao J-I; Chen C Optimization of Gefitinib Analogues with Potent Anticancer Activity. Bioorg. Med. Chem. Lett 2014, 24, 5247–5250. [DOI] [PubMed] [Google Scholar]
- (85).Pippi B; Reginatto P; Machado G. d. R. M.; Bergamo VZ; Lana DFD; Teixeira ML; Franco LL; Alves RJ; Andrade SF; Fuentefria AM Evaluation of 8-Hydroxyquinoline Derivatives as Hits for Antifungal Drug Design. Med. Mycol 2017, 55, 763–773. [DOI] [PubMed] [Google Scholar]
- (86).Silverman JA; Oliver N; Andrew T; Li T Resistance Studies with Daptomycin. Antimicrob. Agents Chemother 2001, 45, 1799–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Taylor SD; Palmer M The Action Mechanism of Daptomycin. Bioorg. Med. Chem 2016, 24, 6253–6268. [DOI] [PubMed] [Google Scholar]
- (88).CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, Approved Guideline, CLSI Document M07-A10, 10th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, 2015. [Google Scholar]
- (89).Schneider T; Kruse T; Wimmer R; Wiedemann I; Sass V; Pag U; Jansen A; Nielsen AK; Mygind PH; Raventós DS; Neve S; Ravn B; Bonvin AMJJ; Maria LD; Andersen AS; Gammelgaard LK; Sahl H-G; Kristensen H-H Plectasin, a Fungal Defensin, Targets the Bacterial Cell Wall Precursor Lipid II. Science 2010, 328, 1168–1172. [DOI] [PubMed] [Google Scholar]
- (90).Nair DR; Monteiro JM; Memmi G; Thanassi J; Pucci M; Schwartzman J; Pinho MG; Cheung AL Characterization of a Novel Small Molecule That Potentiates β-Lactam Activity against Gram-Positive and Gram-Negative Pathogens. Antimicrob. Agents Chemother 2015, 59, 1876–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.