

Summary
Mutant protein kinase A catalytic subunit (PKAc) drives adrenal Cushing’s syndrome, though its signaling interactions remain unclear. This protocol details steps to use live-cell proximity labeling to identify subcellular compartments and proteins closely associated with variants of PKAc in human adrenal cells. We include instructions for clonal cell line generation, live biotin labeling of proximal proteins, isolation of biotinylated proteins, and sample processing for proteomic analysis using the biotin ligase miniTurbo with wild-type and mutant PKAc.1,2
For complete details on the use and execution of this protocol, please refer to Omar et al. (2022).3
Subject areas: Cell culture, Signal Transduction, Molecular/Chemical Probes, Protein Biochemistry, Proteomics, Mass Spectrometry
Graphical abstract
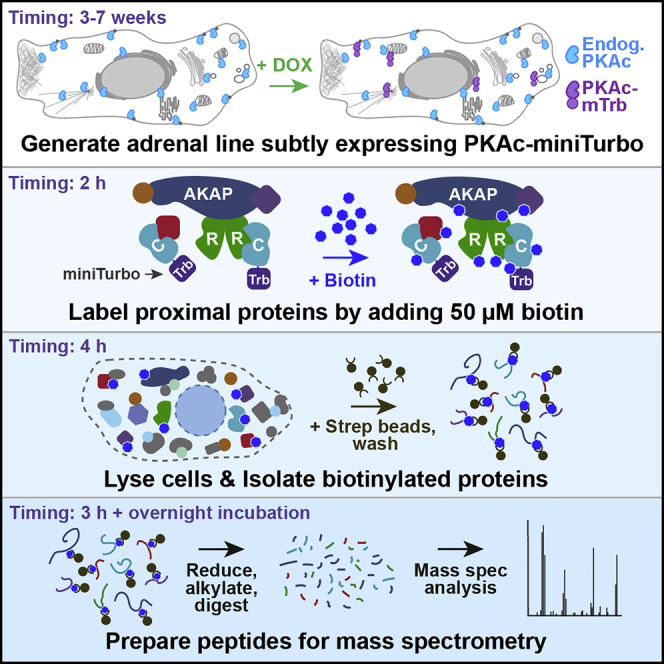
Highlights
-
•
Improves on co-immunoprecipitation by detecting transient and weak interactions
-
•
Describes lentiviral cell line generation and bait protein expression optimization
-
•
Details labeling and isolating proteins proximal to PKA catalytic subunit in live cells
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Mutant protein kinase A catalytic subunit (PKAc) drives adrenal Cushing’s syndrome, though its signaling interactions remain unclear. This protocol details steps to use live-cell proximity labeling to identify subcellular compartments and proteins closely associated with variants of PKAc in human adrenal cells. We include instructions for clonal cell line generation, live biotin labeling of proximal proteins, isolation of biotinylated proteins, and sample processing for proteomic analysis using the biotin ligase miniTurbo with wild-type and mutant PKAc.
Before you begin
This protocol describes the specific steps to label proteins proximal to a miniTurbo-tagged protein kinase A catalytic subunit (PKAc-miniTurbo) in NCI-H295R adrenal cells.3 Slight variations of this protocol have also been used for labeling proteins associated with AKAP18 variants in HEK cells and PKAc fusion proteins in AML12 hepatocytes.2
Preparation of Tris-HCl buffers
Timing: Prepare in advance. Allow ∼1 h
-
1.Prepare 1 M stock Tris-HCl solutions from Tris-HCl powder.
-
a.Weigh and dissolve the appropriate amount of Tris-HCl powder into ∼90% final volume of ddH20.
-
b.Bring solutions to the appropriate pH with NaOH or HCl.
-
c.Finalize the volume with additional ddH20.
-
d.Sterile filter through a 0.2 μm membrane.
-
a.
-
2.
Use these stocks to make specific dilutions needed throughout the protocol.
Cell culture conditions
-
3.
Grow HEK293T cells used for generation of lentiviral particles at standard mammalian conditions (37°C and 5% CO2) using Dulbecco’s modified eagle medium + 10% fetal bovine serum.
-
4.
Grow NCI-H295R cells at 37°C and 5% CO2 in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12) with 2.5% NuSerum I and 1% insulin, transferrin, selenium plus (ITS+) supplement.
-
5.
Allow cells one or two passages after thaw to grow at normal rates. Healthy HEK293T cells will double every 22 h or so. NCI-H295R cells grow somewhat slower with a doubling time near two days.
Lentiviral production
-
6.Clone lentiviral tet-responsive vectors containing PKAc-miniTurbo variants.
-
a.Use Addgene plasmid 80921 or similar. This vector contains resistance to blasticidin, which is superior to puromycin for selection of NCI-H295R cells. Our cloning conditions were optimal when using NEB Stable or Invitrogen Stbl3 competent bacteria and 30°C growth conditions.
-
a.
-
7.Produce lentivirus in HEK293T cells using pMD2.G (Addgene 12259) and psPAX2 (Addgene 12260). Our protocol uses viral supernatant from a 10 cm dish. The steps to viral production are as follows:
-
a.Transfect HEK293T cells at ∼60% confluence using Mirus Trans-IT LT and following manufacturer recommendations. Our protocol uses 2 μg pMD2.G, 6 μg psPAX2, and 6 μg of the viral vector.
-
b.After 6–16 h, change media to remove transfection reagents.
-
c.Harvest media 24 h after media change, and again at 48 h if desired.
-
d.Spin viral media at 500 × g for 5 min and take supernatant.
-
e.Use acutely or aliquot and store frozen at -80°C.
-
a.
Note: For viral production from HEK293T cells, we use destination cell growth medium (H295R) for media change after transfection. This helps to avoid introducing FBS and variation in media percentage at the time of infection. Alternatively, viral particles can be concentrated and resuspended in PBS with 25 mM HEPES.
Biotin stock solution preparation
-
8.
Measure and dissolve biotin in DMSO to 100 mM.
-
9.
Aliquot and store at -20°C.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| V5-Tag; 1:800 dilution | Thermo Fisher |
R96025 RRID: AB_2556564 |
| Alexa Fluor™ 488, donkey anti-mouse; 1:1000 dilution | Thermo Fisher | A21202 RRID: AB_141607 |
| Purified Mouse Anti-PKA[C]; 1:2000 dilution | BD Biosciences | 610981 RRID: AB_398293 |
| Pierce™ High Sensitivity NeutrAvidin™-HRP; 1:4000 dilution | Thermo Fisher | 31030 |
| Bacterial and virus strains | ||
| Stable competent cells | NEB | C3040I |
| One Shot™ Stbl3™ Competent Cells | Invitrogen | C737303 |
| Chemicals, peptides, and recombinant proteins | ||
| Polybrene | Santa Cruz | sc-134220 |
| Blasticidin S HCl | Invitrogen | 46-1120 |
| DMEM: F-12 medium | ATCC | 30-2006 |
| DPBS (PBS) | Thermo Fisher | 14190144 |
| 0.25% Trypsin-EDTA, phenol red | Thermo Fisher | 25200056 |
| Paraformaldehyde, 16% solution, EM grade | Electron Microscopy Sciences | 15710 |
| Albumin, bovine (BSA) | VWR | 0332 |
| Triton™ X-100 | Sigma | T9284 |
| Doxycycline hyclate | Sigma | D9891 |
| Biotin | Sigma | B4501 |
| DMSO | Sigma | D2438 |
| NanoLink® magnetic streptavidin beads | Tri-link Biotechnologies, Vector Labs | M-1002-020 |
| Urea | Fisher Scientific | BP169-10 |
| Tris HCl powder | Fisher Scientific | H5123 |
| TCEP-HCl | Goldbio | TCEP25 |
| 2-Chloroacetamide (CAM) | Sigma | C0267 |
| Triethylammonium bicarbonate buffer, 1 M, pH 8.5 | Sigma | T7408 |
| Endoproteinase LysC | NEB | P8109S |
| Pierce™ Trypsin protease, MS-grade | Thermo Fisher | 90057 |
| Sodium fluoride | Sigma | S7920 |
| Tergitol (NP-40) | Sigma | NP-40 |
| Sodium deoxycholate | Sigma | D6750 |
| Sodium dodecyl sulfate | Fisher | BP166-500 |
| AEBSF | Sigma | A8456 |
| Benzamidine hydrochloride hydrate | Sigma | B6506 |
| Leupeptin | Sigma | L2884 |
| Pepstatin A | MP Biomedicals | 195368 |
| Β-Glycerophosphate | Sigma | G6251 |
| Cellstar Easystrainers 40 μm | VWR | 89508-342 |
| DAPI (1 mg/mL); 1:5000 dilution | Life Technologies | 62248 |
| TBST (10×) | Cell Signaling Technologies | 9997S |
| Sodium hydroxide | Fisher | S318-500 |
| Hydrochloric acid | Fisher | A144S-500 |
| 0.2 μm sterile filter | Fisher | 09-741-04 |
| GemCell Fetal Bovine Serum | Gemini | 100-500 |
| DMEM | Life Technologies | 11965-118 |
| NuSerum I | VWR | 47743-700 |
| Corning ITS+ | Fisher | 354352 |
| Mirus Trans-IT LT | Mirus Bio | MIR 2305 |
| HEPES sodium salt | Sigma | H7006-100 |
| Sodium chloride | Fisher | S671-3 |
| EDTA | Invitrogen | 15576-028 |
| Acetic acid, glacial | Fisher Scientific | 0714 |
| Formic acid | Sigma | F4636 |
| Critical commercial assays | ||
| Pierce™ BCA Protein Assay Kit | Thermo Fisher | PI23227 |
| Deposited data | ||
| Proximity biotinylation mass spectrometry proteomics | MassIVE (deposited) | MSV000088654 |
| Experimental models: Cell lines | ||
| HEK293T cell line | GE Lifesciences | HCL4517 |
| NCI-H295R cell line | ATCC | CRL-2128 |
| Recombinant DNA | ||
| pCW57-MCS1-P2A-MCS2 (Blast) | Adam Karpf; http://n2t.net/addgene:80921 | 80921; RRID: Addgene_80921 |
| pMD2.G | Didier Trono; http://n2t.net/addgene:12259 | 12259; RRID: Addgene_12259 |
| psPAX2 | Didier Trono; http://n2t.net/addgene:12260 | 12260; RRID: Addgene_12260 |
| Software and algorithms | ||
| ImageJ (FIJI) | https://imagej.net/downloads | N/A |
| Other | ||
| DynaMag™-2 Magnet | Thermo Fisher | 12321D |
| Thermo Scientific™ Low Protein Binding Collection Tubes, 1.5 mL | Thermo Fisher | 90410 |
| Eppendorf ThermoMixer® F1.5 | Eppendorf | 5384000020 |
Materials and equipment
RIPA Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl buffer pH 7.5 (1 M) | 20 mM | 0.5 mL |
| Sodium chloride (2.5 M) | 130 mM | 1.3 mL |
| EDTA (500 mM) | 2 mM | 0.1 mL |
| Sodium fluoride (500 mM) | 20 mM | 1 mL |
| NP-40 (25% v/v) | 1% v/v | 1 mL |
| Sodium deoxycholate (10% w/v) | 0.5% v/v | 1.25 mL |
| Sodium dodecyl sulfate (20% w/v) | 0.1% v/v | 0.125 mL |
| 4-benzenesulfonyl fluoride hydrochloride (AEBSF) (200 mM) | 1% v/v | 0.25 mL |
| Benzamidine (1 M) | 0.1% v/v | 0.025 mL |
| Leupeptin/pepstatin (2 mg/mL) | 0.1% v/v | 0.025 mL |
| Β-glycerophosphate (1 M) | 1% v/v | 0.25 mL |
| ddH2O | N/A | 19.175 mL |
| Total | N/A | 25 mL |
Note on storage conditions: Make fresh and use same day. Keep on ice or at 4°C.
Step-by-step method details
Clonal cell line generation
Proximity labeling to identify physiologically associated proteins requires normal (similar to endogenous) localization of the bait protein. To avoid localization artifacts due to strong or variable overexpression, we used clonal NCI-H295R cell lines with doxycycline-inducible PKAc-miniTurbo variants. This strategy allows for titration to achieve uniform, subtle expression levels of the exogenous kinase (∼20% of endogenous PKAc) and matched expression of variants across experimental conditions (Figure 1).
-
1.H295R lentiviral transduction.
-
a.Plate approximately 300,000 cells in 6-well plate 2 days before applying virus. Plan for 70% confluence at time of transduction.
-
b.Plan for different viral amount conditions: No virus, low, mid, and high. Transduction media for a 6-well plate well totals 1 mL. It is composed of 1) harvested lentiviral supernatant from HEK293T viral production cells, 2) 5 μg/mL polybrene, and 3) fresh H295R media to bring volume up to 1 mL. For our experiments and titers, low = 10–50 μL virus-containing supernatant, medium = 50–150 μL, and high = 250–450 μL. Combine these in 1.5 mL tubes in a sterile biosafety cabinet just prior to transduction. An example setup is shown in Table 1.
-
c.Aspirate old media and apply the viral transduction mixture.
-
d.Incubate 6–8 h in cell culture incubator.
-
e.Add 1.5 mL fresh media (do not aspirate viral media) and continue incubating overnight (∼12–16 h).
-
a.
-
2.Administer selection agent.
-
a.After at least 24 h (and up to 48), change media and add selection agent (blasticidin, 5 μg/mL final concentration).
-
b.Monitor cells for several days, changing media every 2–3 days to keep antibiotic fresh. There should be increasing death in the no virus control well. The low, medium, and high infections should demonstrate a dosage-dependent survival curve. Control cells should be completely dead within a week. Troubleshooting problem 1 and problem 2.
-
c.Expand well of best condition to 10-cm plate and continue selection.
-
d.This is your population cell line. Grow enough of this cell line to freeze down multiple tubes for a backup cache.
-
a.
Note: When deciding which conditions to expand, consider the viral impact on the cell genome. A linear increase in cell viability from low to high would suggest no over-saturation in the high condition. A plateau between medium and high conditions, however, would suggest the high condition contains excess copies of virus per cell. This may increase the genomic disruption as more copies of viral DNA are being integrated into the chromosomes.
-
3.Isolating clones.Note: There are two main options for clonal isolation: FAC sorting and dilution plate colony picking. If the viral vector includes a fluorescent protein, FACS is ideal to separate cells based on predicted expression level. Even without a fluorescent marker, FACS combined with single cell plating is a good way to isolate clones. If FACS is not possible, diluting to very low cell density and picking colonies after growth is a viable option. This is detailed here.
-
a.Wash a confluent plate of cells with PBS.
-
b.Aspirate and add 1 mL 0.25% trypsin-EDTA.
-
c.Place in incubator for ∼5 min (monitor cell rounding on microscope).
-
d.Thoroughly dissociate cells by pipetting up and down with 9 mL cell media using a 10-mL pipet.
 CRITICAL: Greater percentage of single cells vs. multiple cells equals greater percentage of true clones upon picking colonies.
CRITICAL: Greater percentage of single cells vs. multiple cells equals greater percentage of true clones upon picking colonies. -
e.Dilute 20 μL into 2 mL media.
-
f.Filter out large multi-cell aggregates by passing through a 40-μm cell strainer.
-
g.From the strained cells, plate 100 μL and 200 μL in 10-cm dish with 10 mL media. This will yield 1:10,000 and 1:5,000 dilution colony plates, respectively. Due to dilutions and straining out aggregate cells, approximate cell numbers plated should be somewhat less than 500 and 1000 per plate, respectively. Factors such as failure to attach or grow will reduce these numbers to a manageable number of cell colonies well-spaced for discrete picking and transfer.
-
h.Change 5 mL media every 4–5 days. Grow until most colonies are between 0.5 and 2 mm in diameter.
-
a.
-
4.Transferring colonies.
-
a.Once the clonal colonies have grown to appropriate size, they must be isolated.
-
b.In a fresh 24-well plate, add 60 μL 0.25% trypsin-EDTA in each well and keep in hood.
-
c.Also, have a new, empty 24-well plate adjacent.
-
d.Replace media in either the low or high-dilution colony plate with 15 mL serum-free media.
-
e.Place colony plate on a tissue culture or dissection microscope and use a low magnification objective (2.5× – 4×) to see multiple colonies at once.Note: Reduce traffic in the area and minimize air flow in area around microscope to decrease chance of contamination. While we have not experienced contamination in the 24-well plate, fungal contaminants have occurred in retained 10 cm colony plates after transferring colonies outside a laminar flow hood.
-
f.While observing through the eye pieces, use a 1,000 μL pipette with tip to manually scrape and aspirate each desired colony. Only take up the smallest volume possible, ∼20–100 μL.
-
g.Expel cells into the 0.25% trypsin-EDTA of the 24-well plate well.
-
h.Continue picking in this manner. After 5–10 min of trypsin incubation, pipette cells up and down 10–20× with a 200 μL pipette.
-
i.Add 1 mL H295R cell culture media (complete with NuSerum and ITS+), pipet to mix, and move 500 μL to the corresponding well in adjacent clean 24-well plate. This duplicate plate will be tested when screening clones.
-
j.Once all colonies are picked, add extra 1 mL of complete media to each well in order to dilute trypsin.
-
k.Change media the next day.Note: Depending on your expected success rate, you will need multiple 24-well plates of clones to ensure success. We generally screen 48–96 colonies per condition.
-
a.
-
5.Screening clones.
-
a.Once cells have grown sufficiently to evaluate heterogeneity, they can be screened via cell staining.
-
b.For each duplicate 24-well clone plate, replace media with fresh media containing 1 μg/mL doxycycline.
-
c.Grow cells in doxycycline-containing media for 48 h.
-
d.Aspirate media and add 4% paraformaldehyde in 1× PBS. A volume of 250–300 μL should be sufficient to cover cells.
-
e.Incubate covered at room temperature (RT, ∼25°C) for 15 min.
-
f.Wash 3 × 5 min in 500 μL 1× PBS.
-
g.Block with 3% BSA and 0.3% Triton X-100 in PBS for 30 min at RT (∼25°C).
-
h.Replace solution with Invitrogen anti-V5 tag antibody at a dilution of 1:800 in blocking solution.
-
i.Incubate at 4°C overnight (∼12–16 h).Note: If reducing the total amount of needed antibody is desired, plates can be stained sequentially, reusing the diluted antibody for each plate. In this case, fixed and washed plates can be stored, sealed with parafilm around the edges to avoid dehydration, for up to three weeks at 4°C.
-
j.Next day, wash 3 × 5 min with 1× PBS.
-
k.Add Alexa Fluor® 488-conjugated donkey anti-mouse secondary antibody at 1:1000 and DAPI at 1:5000 in blocking solution.
-
l.Protect from light and incubate at RT (∼25°C) for 1 h.
-
m.Wash 1–2× with 1× PBS.
-
n.Cells can now be imaged using a fluorescent microscope.
-
o.Ideal clones would have 100% of cells expressing construct at close to equal levels (with variance during the cell cycle). See “expected outcomes” and Figure 5. Troubleshooting problem 3.
-
a.
-
6.Doxycycline induction optimization.
-
a.Grow up selected clones and maintain a passage plate/freeze down tubes for backup.
-
b.Split cells into a 12-well plate to test doxycycline response. Four wells per clone, including 0, 250, 500, and 1,000 ng/mL doxycycline conditions.
-
c.Next day, add appropriate doxycycline and incubate for 48 h in tissue culture (TC) incubator.
-
d.After incubation, scrape cells and perform western blot analysis with antibody to PKAc. Typical dilution factor for BD PKAc antibody is 1:2000. Troubleshooting problem 4.
- e.
-
f.Once ideal clones and doxycycline induction parameters are established, plate H295R cells in 10-cm dishes and allow to grow until 60–70% confluent in preparation for live cell labeling.
-
a.
Figure 1.

Optimizing expression levels of exogenous miniTurbo fusion proteins in NCI-H295R cells
(A) Immunoblot of PKAc expression after 48 h of doxycycline induction. Top bands represent exogenous miniTurbo-tagged WT PKAc (lane 1), PKAc mutant 1 (lane 2), and PKAc mutant 2 (lane 3). Bottom bands represent native PKAc.
(B) Quantitation of immunoblot in A. Percent expression calculated according to Figure 6. Data are represented as mean ± SEM. No significant differences across conditions by 1-way ANOVA. n ≥ 3. Adapted from Omar et al.3
Table 1.
Example viral transduction conditions
| No virus | Low | Medium | High | |
|---|---|---|---|---|
| Virus media | 0 μL | 50 μL | 150 μL | 450 μL |
| Polybrene (1 mg/mL) | 5 μL | 5 μL | 5 μL | 5 μL |
| Fresh H295R media | 995 μL | 945 μL | 845 μL | 545 μL |
Figure 5.

Evaluating clonal PKAc-miniTurbo cell lines by immunofluorescent staining
(A and B) Immunofluorescence imaging of a mixed cell line demonstrating variable expression (A) and true clonal cell line demonstrating consistent and ubiquitous expression (B) of PKAc-miniTurbo (green). DAPI stain (blue) indicates presence of cells. Area of inset denoted by dashed box. Inset was brightened for clarity. Scale bars: large images, 100 μm; insets, 20 μm.
Figure 6.

Quantification of PKAc-miniTurbo expression levels in clonal line
(A) Inverted immunoblot demonstrating expression of PKAc-miniTurbo in lysate samples from four biological replicates. Color-coded rectangular selections indicate region of measured integrated density for each band (red and green) and their corresponding background measurements (magenta and blue).
(B) Equation to calculate percent overexpression of exogenous miniTurbo-tagged proteins.
Live-cell labeling and sample preparation
Once 10-cm plates are 60–70% confluent, you can induce expression of the bait protein and biotinylate. Consider appropriate controls when setting up experiment. For instance, “no biotin” and “no doxycycline” conditions are two slightly different methods to control for non-specific binding in the pulldown. In addition, further validation of labeling efficiency and specificity are recommended as detailed in Hung et al.4 An example of the impact of increased doxycycline concentration on PKAc-miniTurbo expression and the resulting biotin labeling efficiency is shown in Figure 2A.
-
7.Biotinylate proximal proteins.
-
a.Add biotin (100 mM stock in DMSO) to cell media at 50 μM. Gently rock plate to mix.
-
b.Incubate in TC incubator for 1 h at 37°C. This time can be increased or decreased to address different labeling objectives.
-
a.
Note: Results from an experiment testing how increased biotin (Figure 2B) and labeling time (Figure 2C) affect biotinylation extent are shown in Figures 2B and 2C. While increased labeling will yield more peptides detected by mass spectrometry, thereby decreasing false negatives, high amounts of labeling will likely increase false positives. The parameters used in this protocol aim to strike a balance between these two undesirable results. If increased biotinylation is needed, longer labeling time is favorable to higher doxycycline (increased PKAc-miniTurbo expression) as the latter will likely result in mislocalized bait protein.
-
8.Sample preparation.
-
a.Wash cells in 10 mL RT (∼25°C) 1× PBS twice for 1 min each to remove excess biotin.
-
b.Aspirate thoroughly.
-
c.Lyse and scrape cells in 750 μL RIPA buffer containing protease and phosphatase inhibitors.
-
d.Move to a 1.5-mL tube on ice.
-
e.Pipette up and down several times.
-
f.Let lyse for 5 min on ice.
-
g.Centrifuge at 15,000 × g for 8 min in cold room or refrigerated centrifuge set to 4°C.
-
h.Move supernatant to a clean 1.5-mL tube on ice.
-
i.Determine sample protein concentration (e.g., using Pierce™ BCA).
-
a.
-
9.
Run 15–25 μg of experimental and any control samples on western blot to test for biotin labeling (Figure 3). Neutravidin™-HRP is used overnight (∼12–16 h) at 4°C. The typical dilution is 1:4000 in azide-free 5% BSA TBST. Troubleshooting problem 5.
-
10.
Proceed to pulldown or freeze down samples in liquid nitrogen and store in -80°C freezer.
Figure 2.

Testing labeling variables
(A) Immunoblot showing effect of doxycycline concentration on PKAc-miniTurbo expression and biotin labeling efficiency.
(B) Immunoblot exhibiting effect of biotin concentration on labeling efficiency.
(C) Immunoblot demonstrating effect of biotin incubation time on labeling efficiency. For PKAc panels (middle) in (A–C), the bottom bands represent native PKAc while the top bands represent exogenous miniTurbo-tagged PKAc. Standard conditions for this protocol are designated by green text in all three experiments.
Figure 3.
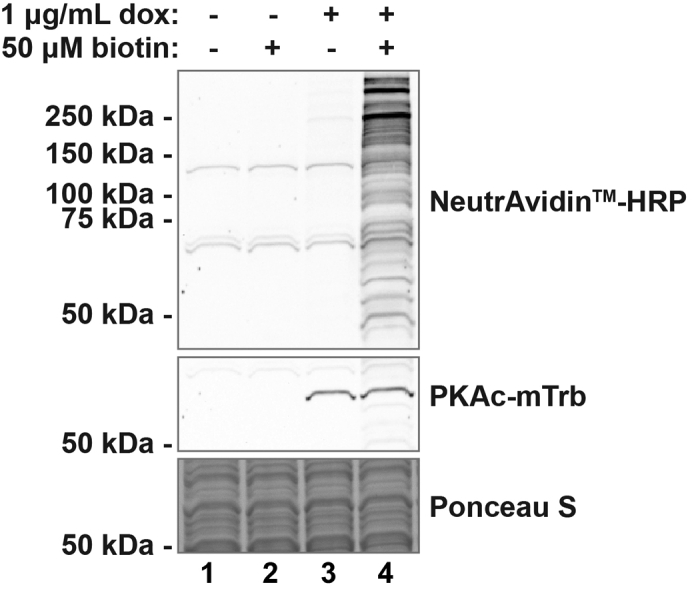
Testing biotin labeling of PKAc-miniTurbo clonal line
Western blot of clonal cell line lysates in 4 conditions probing with NeutrAvidin™-HRP at 1:4000 dilution. Protein biotinylation by PKAc-miniTurbo occurs only when bait protein expression is induced (+ doxycycline) and exposed to exogenous biotin (+ biotin) as shown in lane 4. Bands at ∼65 kDa and ∼130 kDa in lanes 1–3 are endogenous biotinylated proteins. Ponceau S demonstrates equal protein loading. Doxycycline treatment was 48 h. Biotin incubation was 2 h.
Streptavidin pulldown
Once proteins proximal to your bait have been labeled with biotin, they can be biochemically isolated by exploiting biotin-avidin affinity. This is an especially strong non-covalent interaction and allows for harsh washing conditions to minimize non-biotinylated proteins in your isolated samples.
-
11.Wash magnetic beads in the RIPA lysis buffer.
-
a.Resuspend stock supply of NanoLink® magnetic streptavidin beads by gently swirling or tapping the bottle/tube.
-
b.Move enough beads for 25 μL per pulldown to a 1.5-mL tube. Take precaution when pipetting by using a cut P200 pipette tip or a P1000 pipette tip.
-
c.Add 1 mL lysis buffer and mix by inverting.
-
d.Place on magnetic rack and wait 30 s–1 min for buffer to clear.
-
e.Aspirate buffer while on holder.
-
f.Move tube to non-magnetic rack and add 1 mL fresh lysis buffer.
-
g.Repeat steps d and e.
-
h.Move tube to non-magnetic rack and resuspend beads in original volume.
-
a.
-
12.Prepare working concentrations.
-
a.Calculate the appropriate volume of lysis buffer and cell lysate to make 1 mL of 0.5 mg/mL samples. Remember to account for 25 μL of beads.Note: The amount of beads used depends on the amount of labeled proteins. The amount stated above was sufficient for 1 h of labeling time with PKAc-miniTurbo expression at 20% endogenous expression. Using a great excess of beads is not advised as it increases non-specific binding and the amount of streptavidin peptides post digestion.
-
b.Aliquot the needed lysis buffer for each sample into low protein binding tubes.
-
c.Add 25 μL magnetic beads to each tube of lysis buffer using a cut P200 tip. Resuspend beads before each pipetting to ensure equal bead allocation.
-
d.Finally, add the appropriate amount of cell lysate to their respective tubes.
-
a.
-
13.Incubate to allow binding of biotinylated proteins to beads.
-
a.Place tubes on an orbital nutator or mechanical tube invertor and mix for 1 h at RT (∼25°C).
-
a.
-
14.During incubation, make urea solutions for washes and digestion.
-
a.2 M Urea in 10 mM Tris-HCl buffer pH 8.0: Make enough for 400 μL per tube. For 5 mL, weigh out 600.6 mg urea. Add 4 mL 10 mM Tris-HCl buffer pH 8 and mix. Once dissolved, bring to 5 mL with additional 10 mM Tris-HCl buffer pH 8. Keep at RT (∼25°C) until use.
-
b.8 M Urea in 100 mM Tris-HCl buffer pH 8.5: Make enough for 50 μL per tube. For 750 μL, weigh out 300.3 mg urea. Take caution not to add too much volume of 100 mM Tris-HCl buffer pH 8.5. Measure final volume with P1000 pipette. Keep at RT (∼25°C) until use.
-
a.
-
15.Wash samples.
-
a.Remove tubes from rotator and place in magnetic holder. Let sit 30 s.
-
b.Briefly invert tube rack and let sit another 30 s.
-
c.Remove supernatant from tubes using P1000 and save small amount (∼50 μL) for diagnostic assessment by western blot.
-
d.Move tubes to non-magnetic rack and add 1 mL lysis buffer to each.Note: It is important to prevent drying of the beads. To minimize time the beads are out of liquid, tubes can be aspirated, moved, and refilled one at a time.
-
e.Invert several times. Ensure beads are not clumping together. If so, you can gently pipette up and down with a P1000 tip.
-
f.Place on magnet and let sit 30 s.
-
g.Briefly and gently invert tube rack and let sit another 30 s.
-
h.Aspirate lysis buffer.
-
i.Repeat wash steps d–h with 500 μL lysis buffer.
-
j.Aspirate and replace with 200 μL 2 M urea in 10 mM Tris-HCl buffer pH 8.0.Note: Do not invert tubes at this point. After the first two lysis buffer washes, avoid all unnecessary contact between beads and plastic surface area. Absence of detergent in the buffer leads to adhesion of beads on side walls of the tube.
-
k.Tap gently to mix. If beads are clumping together, use a wide-bore tip to pipette up and down minimally.
-
l.Place on magnet and wait 1 min before aspirating with P200 or P1000/P200 pipette tips (see Figure 4).
-
m.Repeat steps j–l for a total of 2 urea washes.
-
n.Wash twice with 150 μL 20 mM Tris-HCl buffer pH 7.5 in same manner as urea washes. Move directly to digestion steps. Troubleshooting problem 6.
-
a.
Figure 4.

P1000/P200 pipette tip stacking for careful aspiration following urea washes
Reduction, alkylation, and digestion of proteins
The high-affinity biotin-streptavidin interaction may complicate elution of labeled proteins from the beads. To circumvent sample loss due to poor elution, we digest on the beads to prepare for liquid chromatography-mass spectrometry analysis.
-
16.Reduction and alkylation.
-
a.Use wide-bore tip to resuspend beads in 50 μL 8 M urea in 100 mM Tris-HCl buffer pH 8.5 with 5 mM tris (2-carboxyethyl) phosphine hydrochloride (TCEP-HCl) and 10 mM chloroacetamide (CAM).
-
b.Incubate shaking at 1,000 rpm and 37°C for 30–60 min in ThermoMixer. Avoid heating urea over 37°C as it will lead to increased protein carbamylation.
-
a.
-
17.Digest.
-
a.Add 50 μL 100 mM triethylammonium bicarbonate buffer (or 100 mM tris-HCl buffer pH 7.8 or 100 mM ammonium carbonate).
-
b.Prepare LysC by dissolving in 100 μL double-distilled water to make 200 ng/μL LysC solution. Add 1 μg LysC and incubate 2 h at 37°C, shaking at 1,000 rpm in ThermoMixer. Check pH of digest to ensure pH is around 8.
-
c.Reconstitute MS-grade trypsin to 1 μg/μL in 50 mM acetic acid just before use.
-
d.Add 100 μL of the same buffer used in step 17a and add 1 μg trypsin. Recheck to ensure pH is around 8.
-
e.Use ThermoMixer to shake overnight (∼12–16 h) at 1,000 rpm, 37°C.
-
a.
-
18.Acidify.
-
a.Next morning, add 200 μL 2% formic acid to new labeled tubes.
-
b.Place digested samples on magnet and wait 1 min.
-
c.Move supernatant to formic acid tubes and pipette up and down briefly to mix.
-
a.
-
19.
Samples can now be loaded onto StageTips for LC-MS analysis.5 If needed, samples can be frozen until StageTip loading.
Expected outcomes
Lentiviral transduction and clonal isolation should successfully generate clones with even expression of the miniTurbo-tagged construct. This expression will ideally be in every cell (suggesting a true clonal line), however using the dilution plate method of isolation described above may yield mixed colonies as a consequence of incomplete cell isolation. This will be evident when testing doxycycline-treated cells by immunofluorescence (Figure 5). A mixed line will show variable expression, possibly two or three clear tiers of expression (Figure 5A). True clonal lines will demonstrate a more consistent staining pattern with all cells exhibiting signal (Figure 5B). For the purpose of proximity biotinylation and mass spectrometry, if cells expressing PKAc-miniTurbo do so in an even manner, the presence of non-expressing cells in a clonal line does not invalidate the experiment. We would recommend at least 50% of cells demonstrating even expression. Non-expressing cells increase the ratio of non-labeled to labeled proteins and therefore increase the possibility of non-specific binding. Importantly, if using a cell line with <100% of cells expressing the construct, one must divide by the estimated ratio of expression when determining percent overexpression (see “quantification and statistical analysis,” below). Additionally, testing biotinylation within samples is strongly recommended. These western blots should show clear and strong labeling in the doxycycline/biotin-exposed lanes and little to no biotin signal in the control lanes (Figure 3).
Quantification and statistical analysis
To optimize expression levels in clonal viral lines, measure signal of inverted western blot in ImageJ (Figure 6). Keeping the rectangle selection constant among measurements, measure integrated density for each band and an adjacent background region (Figure 6A). Calculate percent overexpression for each lane using the equation in Figure 6B.
Limitations
This protocol improves upon classical protein interaction and association techniques by assessing characteristics in live cells. Two factors limit the physiologic relevance. First, the miniTurbo tag may alter the behavior of the bait protein. One example is nuclear localization of the PKAc-L205R variant, which is less prominent with the miniTurbo tag than with a small V5 epitope tag. This is likely due to passive nuclear translocation limits near 40 kDa.6 Second, although this protocol addresses tightly controlling expression by using induced viral overexpression, gene-editing of endogenous alleles is an alternate approach that may better recapitulate total expression levels of PKAc. Additionally, the miniTurbo enzyme offers advantages and disadvantages when compared to other promiscuous biotin ligases. While miniTurbo is smaller and more temporally specific than TurboID, the latter is more stable and more efficient.1 Furthermore, recent developments have yielded a highly efficient smaller (19.7 kDa) biotin ligase.7
Troubleshooting
Problem 1
Refers to protocol step 2b. No cell death.
Potential solution
Ensure cell line is not natively resistant to blasticidin, then do kill curve with blasticidin for specific cell line.
Problem 2
Refers to protocol step 2b. All cells die, including conditions receiving virus.
Potential solution
You may not have succeeded in viral production, or the viral titer may not be high enough to sufficiently infect your cells. Consider re-making virus and measuring viral titer.
Problem 3
Refers to protocol step 5o. No clones demonstrate 100% expression among cells.
Potential solution
This would most likely be due to poor single cell suspension prior to seeding dilution colony plates. To avoid restarting the entire process, the best clonal line can be re-diluted and plated according to the protocol. However, as stated in “expected outcomes,” a line with over 50% of cells demonstrating even expression levels should suffice for experiments. This ratio of expressing to non-expressing cells must be taken into account when calculating overexpression by western blot.
Problem 4
Refers to protocol step 6d. No western blot signal or wrong molecular weight for miniTurbo-tagged construct.
Potential solution
Check doxycycline concentration and quality. Make new solution from powder. Check DNA sequence of viral vector.
Problem 5
Refers to protocol step 9. Western blot does not demonstrate enhanced biotin signal in experimental lanes versus controls.
Potential solution
Check for expression of miniTurbo-tagged construct. Check concentration and quality of biotin; make a fresh dilution from powder. In the case of no signal, consider problems with the NeutrAvidin™-HRP step. Importantly, when using an HRP conjugate, azide must be eliminated from the solution.
Problem 6
Refers to protocol step 15n. Magnetic bead pellet appears inconsistent across samples.
Potential solution
This is likely due to differential loss of beads on the plastic surfaces. In buffers lacking detergent, small beads stick more readily to plastic. Make sure you are using low protein binding tubes and proper low-retention pipette tips. Work to minimize volumes and times you pipette up and down. If you are tapping tubes to mix, prioritize small taps with more repetition versus large taps.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, John D. Scott (scottjdw@uw.edu).
Materials availability
Plasmids and cell lines generated for this study are available upon request.
Acknowledgments
The authors would like to thank Alice Ting and Tess Branon for sharing the original miniTurbo construct and Kiana Jones for technical assistance during optimization. This work was supported by institutional NIH training grants T32DK007247 (M.O.) and T32GM775043 (S.L.); NIH grants F32DK121415 (M.O.), R01DK119192 (J.D.S.), R01DK119186 (J.D.S.), and R01GM129090 (S.-E.O.); and a grant from the Fibrolamellar Foundation (J.D.S.).
Author contributions
M.H.O. conceived the overall approach. M.H.O. and S.M.L. optimized protocol methods. H.-T.L., M.G., and S.-E.O. assisted in troubleshooting the workflow. M.H.O, S.M.L., and J.D.S. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Mitchell H. Omar, Email: mho6@uw.edu.
John D. Scott, Email: scottjdw@uw.edu.
Data and code availability
The mass spectrometry proteomics data resulting from use of this protocol have been deposited to MassIVE with the identifier MSV000088654, ftp://massive.ucsd.edu/MSV000088654/. These mass spectrometry datasets and identifiers are also listed in the original Cell Reports paper: Omar et al.3 All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Branon T.C., Bosch J.A., Sanchez A.D., Udeshi N.D., Svinkina T., Carr S.A., Feldman J.L., Perrimon N., Ting A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018;36:880–887. doi: 10.1038/nbt.4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith F.D., Omar M.H., Nygren P.J., Soughayer J., Hoshi N., Lau H.-T., Snyder C.G., Branon T.C., Ghosh D., Langeberg L.K., et al. Single nucleotide polymorphisms alter kinase anchoring and the subcellular targeting of A-kinase anchoring proteins. Proc. Natl. Acad. Sci. USA. 2018;115:E11465–E11474. doi: 10.1073/pnas.1816614115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Omar M.H., Byrne D.P., Jones K.N., Lakey T.M., Collins K.B., Lee K.-S., Daly L.A., Forbush K.A., Lau H.-T., Golkowski M., et al. Mislocalization of protein kinase A drives pathology in Cushing’s syndrome. Cell Rep. 2022;40:111073. doi: 10.1016/j.celrep.2022.111073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hung V., Zou P., Rhee H.W., Udeshi N.D., Cracan V., Svinkina T., Carr S.A., Mootha V.K., Ting A.Y. Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol. Cell. 2014;55:332–341. doi: 10.1016/j.molcel.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rappsilber J., Ishihama Y., Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003;75:663–670. doi: 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- 6.Harootunian A.T., Adams S.R., Wen W., Meinkoth J.L., Taylor S.S., Tsien R.Y. Movement of the free catalytic subunit of cAMP-dependent protein kinase into and out of the nucleus can be explained by diffusion. Mol. Biol. Cell. 1993;4:993–1002. doi: 10.1091/mbc.4.10.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kubitz L., Bitsch S., Zhao X., Schmitt K., Deweid L., Roehrig A., Barazzone E.C., Valerius O., Kolmar H., Béthune J. Engineering of ultraID, a compact and hyperactive enzyme for proximity-dependent biotinylation in living cells. Commun. Biol. 2022;5:657. doi: 10.1038/s42003-022-03604-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The mass spectrometry proteomics data resulting from use of this protocol have been deposited to MassIVE with the identifier MSV000088654, ftp://massive.ucsd.edu/MSV000088654/. These mass spectrometry datasets and identifiers are also listed in the original Cell Reports paper: Omar et al.3 All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.