

Summary
Despite increasing threats of extinction to Elasmobranchii (sharks and rays), whole genome-based conservation insights are lacking. Here, we present chromosome-level genome assemblies for the Critically Endangered great hammerhead (Sphyrna mokarran) and the Endangered shortfin mako (Isurus oxyrinchus) sharks, with genetic diversity and historical demographic comparisons to other shark species. The great hammerhead exhibited low genetic variation, with 8.7% of the 2.77 Gbp genome in runs of homozygosity (ROH) > 1 Mbp and 74.4% in ROH >100 kbp. The 4.98 Gbp shortfin mako genome had considerably greater diversity and <1% in ROH > 1 Mbp. Both these sharks experienced precipitous declines in effective population size (Ne) over the last 250 thousand years. While shortfin mako exhibited a large historical Ne that may have enabled the retention of higher genetic variation, the genomic data suggest a possibly more concerning picture for the great hammerhead, and a need for evaluation with additional individuals.
Subject areas: Biological sciences, Genetics, Evolutionary biology, Genomics
Graphical abstract
Highlights
-
•
Chromosome-level genome assemblies for two endangered species of shark
-
•
Great hammerhead had low heterozygosity with high inbreeding coefficients
-
•
Shortfin mako exhibited higher levels of heterozygosity suggesting adaptive potential
-
•
Both species showed precipitous declines in historical effective population size
Biological sciences; Genetics; Evolutionary biology; Genomics
Introduction
The class Chondrichthyes (sharks, rays, and chimeras) has a history that dates back about 420 million years over which time it has survived five mass extinctions. Today this ancient lineage finds itself in a new geological epoch—the Anthropocene, which may well be characterized as the sixth mass extinction. In a recent report by the IUCN Red List of Threatened Species, 31% of all shark species are threatened (Critically Endangered, Endangered, or Vulnerable),1 a figure considerably higher than their 2014 estimate. Of these threatened shark species, 6.5% are critically endangered and 10.5% are endangered.1 Another recent report indicates the global abundance of oceanic sharks and rays has declined by 71%, due to an 18-fold increase in relative fishing pressure.2 In 1980, only 9 species of oceanic sharks and rays were threatened on the IUCN Red List category, but in 2021, ¾ of these species (n = 24 of 31 species assessed) are now threatened.2 The great hammerhead (Sphyrna mokarran; Critically Endangered) and shortfin mako (Isurus oxyrinchus; Endangered) sharks are two examples of this recent dramatic decline in this group of sharks.
The great hammerhead is the largest species of hammerhead shark in the family Sphyrnidae reaching a maximum size of about 6 m. It has a worldwide distribution, found in coastal tropical and warm temperate seas. It has a wide cephalofoil (“hammer”), tall sickle-shaped dorsal fin, and is an important apex predator. Its critically endangered status is primarily due to heavy fishing for its large fins, which are highly valued in the shark fin trade.3 The shortfin mako is a predominately pelagic, apex predator, reaching a maximum size of about 4 m, and is found worldwide in temperate and tropical seas. It is classified within the family Lamnidae which includes all of the known partially endothermic sharks; it has one congener species—longfin mako—Isurus paucus. Isurus is regarded as the phylogenetic sister group to white shark (Carcharodon carcharias). The endangered status of shortfin mako is primarily due to commercial and sport overfishing.4
Conservation genomics is a field increasing in importance with the continuous improvement in sequencing technologies that afford the ability to assemble high quality reference genomes. From such single genomes, it is possible to derive estimates of heterozygosity, inbreeding, and demographic history. If the sampled individual is representative of its population, heterozygosity may be a useful proxy for standing genetic variation, the principal means for most organisms to adapt and respond to changes in the environment.5 Levels of recent inbreeding can also be inferred from runs of homozygosity (ROH) manifested in the genome sequence, and this has emerged as a valuable component in conservation genomic analyses for a wide range of species.6,7,8,9 Historical demographic reconstructions can link changes in effective population size (Ne) to past environmental shifts, as well as species differences in various other ecological factors, providing the potential to predict effects of current and future environmental change, as well as provide a valuable framework for interpreting contemporary patterns of heterozygosity, extinction risk, and the potential for inbreeding depression.10,11,12
Despite the importance of elasmobranchs in overall marine ecosystem function, and the increased extinction threats to these fishes, conservation genomic study of this subclass is a neglected subject.13 Here, we present chromosome-level genomes for the critically endangered great hammerhead and the endangered shortfin mako sharks. We present analyses of demographic history and conservation genomic-related statistics for the great hammerhead and shortfin mako as well as for other shark species for which genome data are available, including whale shark (Rhincodon typus), white shark (Carcharodon carcharias), brownbanded bamboo shark (Chiloscyllium punctatum), and the cloudy catshark (Scyliorhinus torazame).
Results
Genome assemblies for hammerhead and mako
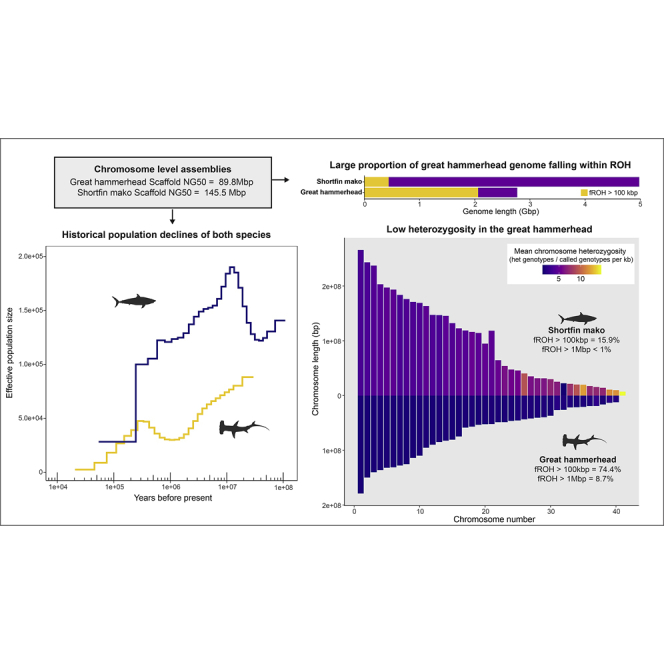
Combining PacBio Hi-Fi with Dovetail Hi-C and Omni-C scaffolding resulted in highly contiguous genome assemblies for both the great hammerhead (Sphyrna mokarran) and shortfin mako (Isurus oxyrinchus), respectively (hereinafter referred to as hammerhead and mako), as depicted in the link density histograms for both species (Figure S1). The final genome assembly for the hammerhead comprised a total of 1658 scaffolds, with a scaffold NG50 of 89.8 Mbp and total length of 2.77 Gbp. Most assembly metrics met or exceeded those established by the Vertebrate Genome Project (VGP) for a chromosome-level assembly (their category VGP-201614). We are unaware of the chromosome number for hammerhead, but flow cytometry data for its close relative, Sphyrna lewini, indicate a haploid chromosome number of 39,15,16 whereas an earlier karyological study suggested n = 43.17 Approximately 82% of the hammerhead genome (2.27 Gbp) was represented in the largest 24 scaffolds and about 16% (0.445 Gbp) in the next largest set of 25–40 scaffolds (scaffold length decreased by more than 2X between scaffold 40 and 41), with the remaining sequence in unplaced scaffolds. These 40 pseudo-chromosomes were comprised of 28 macrochromosomes (>40 Mb), 7 intermediate chromosomes (20–40 Mbp), and 5 microchromosomes (<20 Mbp).
The final genome assembly for the mako included a total of 5559 scaffolds, with a scaffold NG50 of 145.5 Mbp, a total length of 4.98 Gbp, and the majority of genome assembly metrics meeting or exceeding the quality category VGP-201614 (Table S1). We are not aware of karyological data for the two species of mako sharks; however, flow cytometry data for the sister group to Isurus, the white shark, indicate a haploid chromosome number of 41.15,18 Approximately 73% of the mako genome (3.627 Gbp) was represented in the largest 24 scaffolds and about 8% (0.397 Gbp) in the next largest set of scaffolds, 25–41, with the remaining sequence in unplaced scaffolds. Given the estimated divergence time between white shark and mako of about 55 MY,19 chromosome numbers could be different and some of these unplaced scaffolds could be additional pseudo-chromosomes. These 41 mako pseudo-chromosomes were comprised of 26 macrochromosomes (>40 Mbp), 7 intermediate chromosomes (20–40 Mbp), and 8 microchromosomes (<20 Mbp).
The repeat content of both of these shark species is rich in long interspersed nuclear element (LINE) retrotransposons, comprising about 38% and 33% for the hammerhead and mako genomes, respectively (Figures 1 and S2). The majority of these LINEs were of the CR1 and CR1-Zeon type in both species. Based on sequence divergence of individual elements against their respective reference, bursts of LINE activity were roughly synchronous in the history of the two species, with the exception of relatively more recent times, with bursts of activity at around 5%–7% divergence for mako, and 0%–5% for hammerhead (Figure S2). Notable differences in repeat content were a greater proportion of DNA transposons in mako and a greater proportion of short interspersed nuclear elements (SINEs) in hammerhead (Figure S2). The overall transposon density was much greater in hammerhead than mako (Figure 1) and this was primarily due to LINEs and SINEs (Figure S2), with many chromosomes in hammerhead showing a greater density of these elements near the tips of chromosomes (Figure 1).
Figure 1.

Circos plot representation of genomic features in great hammerhead (H) and shortfin mako (M) for a subset of the largest scaffolds
Only a subset of scaffolds is depicted here in order to provide resolution of genome characteristics. The outside ring indicates different chromosomes; the 15 largest chromosomes are represented for shortfin mako, with chromosomes syntenic to those 15, represented for great hammerhead. Gene density is shown in the next, black colored ring, followed by transcript activity in blue (calculated by aligning RNA-seq to the reference genome and normalizing to read counts per million (RPM)); the green colored ring illustrates ROH>1Mbp in histogram bars, the height of which depicts the relative length of these ROH. TE density is plotted by a heatmap representation, GC content in the inside green line, and multicolored syntenic regions between great hammerhead and shortfin mako are illustrated in the middle.
Patterns of genome-wide heterozygosity
Distinct patterns of genome-wide heterozygosity were apparent between hammerhead and mako and between the other four sharks we analyzed (Figure 2; our comparative analyses involving other sharks concentrate on published genomes for which their necessary read data were also available20,21,22,23; accession details for these other sharks appear in Table S2; standardized analyses were conducted using raw short-read data across all species to calculate metrics for comparison). Mako had the greatest overall heterozygosity followed by the two coastal-benthic species (brownbanded bamboo and cloudy catshark) (Figure 2A). The remaining three species had low levels of heterozygosity, with the critically endangered hammerhead the lowest of this group of pelagic species (Figure 2A). The contiguous genome assemblies for the hammerhead and mako reveal a pattern of higher heterozygosity near the tips of chromosomes (Figures 2A and S3), and a distinct decrease in mean heterozygosity with increasing log chromosome length, with a reduced slope of the regression line in hammerhead compared to mako (mako: Pearson’s r = −0.768, p < 0.001; b = −4.462; hammerhead: Pearson’s r = −0.704, p < 0.001; b = -0.564). Both observations are in line with expectations of increased diversity due to higher recombination rate near the telomeres and on smaller chromosomes,24 but could also be affected by an increase in repetitive elements near the tips (Figure 1). The two benthic species had unique heterozygosity patterns compared to the others, with large differences in heterozygosity between entire chromosomes. For example, about half of the largest 24 putative chromosomes in bamboo shark showed high, and the other half low, heterozygosity. This translated to a tri-modal distribution of windows of heterozygosity for the bamboo shark; catshark had a skewed right distribution for these same data, while all other sharks appeared normally distributed (Figure 2B). The drivers of these multimodal heterozygosity distributions across the genome are unclear, but may stem from recent admixture, which can generate long homozygous and heterozygous tracts inherited from the source populations.25,26 Admixture, however, remains a conjectural hypothesis and not possible to evaluate with any certainty from a single genome sequence.
Figure 2.

Genome heterozygosity across the 24 largest scaffolds for several species of sharks
(A) Heterozygosity plotted for each of the scaffolds for each species, at non-overlapping windows of 1 Mbp for great hammerhead, shortfin mako, brownbanded bamboo shark, and white shark, and at 50 kbp windows for cloudy catshark and whale shark; shorter windows chosen for these latter two species because of their more fragmented draft genomes. Although the white shark is also a draft genome, there were nonetheless sufficient windows that could be sampled at 1 Mbp, to allow for accurate heterozygosity estimates. N refers to number of windows sampled. Complete genome-wide heterozygosity for all 40 and 41 pseudo-chromosomes of the great hammerhead and shortfin mako, respectively, appear in Figure S3.
(B) Histograms of per window heterozygosity derived from the set of 24 scaffolds for each species; the smaller distribution for whale shark and white shark reflects the smaller number of windows sampled for these two species.
Along with the low heterozygosity evident in the hammerhead genome was an accompanying large number of ROH. The length and number of ROH reflect levels of individual inbreeding, with longer ROH indicative of recent inbreeding and shorter ROH suggestive of more historical events.27 A total of 173 ROH between 1 and 3.12 Mbp were found across the 40 pseudo-chromosomes of the hammerhead genome, translating to an inbreeding coefficient of FROH>1Mbp = 8.7%. This large number of ROH>1Mbp were scattered throughout the genome, with the majority (97%) in the largest 24 chromosomes, and tending away from transposon (TE) concentration at the tips (Figures 1 and 3); counts of TE and ROH over a 5 Mbp window showed a very weak negative correlation in both mako (Pearson’s r = −0.0945, p = 0.008) and hammerhead (Pearson’s r = −0.1286, p = 0.003). Considering smaller ROH >100 kbp, yields a much higher inbreeding coefficient (FROH>100kbp) with 74.4% of the genome in ROH (Figure 3). Mako, by contrast, had FROH>1Mbp less than 1%, with only 19 ROH>1Mbp across 41 pseudo-chromosomes, ranging to 2.04 Mbp in length (Figure 3). ROH>100kbp for mako amounted to a FROH>100kbp of 15.9%; ROH of shorter size were concentrated in the middle of mako chromosomes, whereas ROH>1Mbp were often near the ends of the chromosomes. For the set of largest 24 scaffolds across all six species of sharks, hammerhead dominated all ROH size ranges >100 kbp, followed by mako, and despite the draft quality of the published white shark genome (scaffold N50 of 2.77 Mbp), it did show some evidence of longer ROH, including up to 900 kbp in length (Figure S4). ROH >1Mbp were not detected in the other species; however, this may be due in part to lower contiguity of some of these draft genomes. Notably, the whale shark genome had the fewest ROH of any size, despite low overall heterozygosity and a scaffold N50 of 2.5 Mbp.22
Figure 3.

Chromosome locations of ROH of different size classes across the genomes of great hammerhead and shortfin mako
Demographic history
To better understand the historical drivers of observed patterns of genetic diversity, we inferred historical effective population sizes (Ne) from the genetic diversity information contained within the diploid genomes of all six species of sharks included here, employing the pairwise sequentially Markovian coalescent—PSMC28 (see STAR Methods for further details regarding mutation rates and generation time). All six species had a different historical pattern, with the possible exception of the two benthic shark species, which had similar peaks in Ne at around 500 KYA (thousand years ago) for bamboo shark and 1 MYA for catshark (Figure 4). At around 335 KYA, the hammerhead began a steady, steep, decline. From a point of highest Ne (around 12 MYA), mako had a history of gradual decline, with a precipitous drop around the same time as the hammerhead to more present geologic epochs. After an initial, ancient steep drop in Ne, white shark tended toward a more stable trajectory than the other two apex predators, and around the approximate time mako and hammerhead were markedly decreasing, the white shark had maintained its Ne or possibly increased (excluding periods less than about 50 KYA because of unreliable bootstrap at this period of the trajectory). The whale shark tended toward lower Ne for much of its history with moderate levels of decline over the last ∼1 MY and with a steep decline beginning round 25 KYA. The harmonic mean Ne of the historical trajectories was the largest for mako, followed by the two benthic species, with the remaining species being roughly similar (Figure 4E).
Figure 4.

Historical effective population sizes for six species of sharks inferred using PSMC
(A) Three apex predators (top to bottom silhouettes): shortfin mako, great hammerhead, and white shark.
(B) Cloudy catshark.
(C) Brownbanded bamboo shark.
(D) Whale shark. Most ancient, highest peak of Ne (including region of greatest bootstrap variability) was truncated in order to alter the scale of the Y axis sufficiently to depict the slight difference in trajectory that was apparent between these two whale shark individuals—one from near Taiwan23 and the other from the Korea aquarium22 (original sample location not provided).
(E) Harmonic mean effective population size for each species trajectory using different time cutoffs; recent time cutoffs: 10,000 years ago; ancient cutoffs: 2e6 years ago.
Discussion
Genome-wide heterozygosity estimates for white shark, whale shark, and hammerhead were at the bottom of those of a broad list of fish species (Figure S5), and hammerhead was below the majority of all but a selected few endangered species of mammals.29 Heterozygosity reflects long-term processes, (e.g. mutation rates and historical Ne10), whereas inbreeding coefficients are indicative of more recent demographic processes,27 and thus may be more directly informative of contemporary conservation status. The inbreeding coefficient for hammerhead (FROH>1Mbp) was almost 9%, comparable to or exceeding other examples of critically endangered taxa such as Malayan and Chinese pangolins,7 South Asian tigers,6 pumas,8 and several species and subspecies of wolves.9 Current estimates of population decline in hammerhead are dramatic; global population reduction was recently estimated at >80% over the last 3 generations.30 The North Atlantic population, from which our individual arose, appears to be faring better,30 with some indication of population increase, likely a result of US management interventions implemented in 2006.30,31 Heterozygosity and ROH of the reference individual suggest low diversity and inbreeding even in this population; however, sampling additional individuals would be necessary before making any more definitive population- or species-level conclusions. Recombination breaks up haplotype blocks with each generation, and therefore larger ROH reflect more recent inbreeding events.27 The size of the chromosomes is also expected to play a role, since shorter chromosomes generally experience proportionally greater recombination rate,32 resulting in fewer ROH. Other factors may also be relevant regarding the development of ROH and their detection. The power to identify ROH is expected to be increased in regions of the genome with low nucleotide diversity and low recombination rate, and simulations have suggested that regions of high ROH abundance may arise around genes under positive selection.33 A more detailed picture of ROH distribution in both hammerhead and mako would be greatly facilitated by a recombination map for these species. Genome-wide molecular adaptation analyses including other shark species, as data come available, could evaluate the association of genes under positive selection with ROH.
The range of ROH in hammerhead extended up to over 3 Mbp, shorter than those arising from very recent inbreeding in some other wildlife (e.g. Soay sheep34 and endangered Pacific pocket mice35), aquaculture species like coho salmon,36 and in animal livestock.37 Our calculations suggest the longest ROH in hammerhead stem from inbreeding within possibly the last ∼60–200 years, assuming a generation time of 25 years and a recombination rate of 6.5 cM/Mb.27,38 About 74% of the hammerhead genome falls within shorter stretches of ROH>100kbp, which likely stem from bottlenecks within the past ∼2000 years. Very few wildlife species have been reported with this level of genome-wide homozygosity, the endangered Ethiopian and Mexican wolves (Canis simensis and Canis lupus baileyi, respectively) being rare examples.9
This level of homozygosity across identical by descent tracts may cause inbreeding depression as a result of inbreeding load, which can push populations toward extinction.11,39 A relatively small but growing body of genomic studies suggest strong effects of inbreeding depression when FROH is high. For example, in Soay sheep, the odds of lamb survival decreased by 60% with a 10% increase in FROH>1.2Mbp,34 and in a modeling result, lifetime reproductive success of house sparrows with FROH>2Mbp = 12.5% was 61% lower than individuals with FROH>2Mbp = 0.40 Theoretical modeling shows that the number of lethal equivalents in the genome is driven by long-term Ne, and that on average, smaller populations have lower inbreeding load, because of genetic drift and purging via natural selection,11 which may make these populations less susceptible to inbreeding depression compared to historically larger populations.41 Recent work involving the endangered vaquita42 points to relatively few deleterious alleles segregating in the population, which may help it avoid fitness costs associated with inbreeding. The effective population size of the vaquita has likely been consistently small for thousands of generations, which may have promoted purging or loss of partially recessive deleterious variants.42 Fitness gains from the reduction of segregating deleterious variants may, at the same time, be offset by the fixation of deleterious alleles in small populations, reducing the overall fitness of the population.43 Our PSMC analysis suggests the hammerhead had larger ancient historical Ne than in more recent epochs. In contrast, the whale shark has both low heterozygosity and a long history of lower Ne. How these different demographic histories may relate to levels of fixed and segregating genetic load in oceanic shark species and their potential fitness consequences remain very open and important questions that would benefit from more recent estimates of Ne than what are possible with single genome analyses.
A revised consideration of Red List criteria and population viability analyses in 201444 suggested that the minimum Ne for retaining evolutionary fitness potential of a species be increased from 500 to 1000; how this would apply to a pelagic species like hammerhead or mako is unknown. Contemporary Ne, or census size (Nc), of any shark species is also unknown. PSMC demographic analyses of single genomes have limited abilities to evaluate recent Ne, but are effective at evaluating historical trajectories. However, the scaling of time and Ne is dependent on mutation rate and generation length, the latter not well understood for many sharks, and estimates of mutation rate in sharks are nearly two orders of magnitude slower than mammals22,45 (and estimates obtained herein; see STAR Methods). Altering these variables affects the placement on the axes but does not affect the shape of the trajectory46 (see further comments on this topic in STAR Methods). Thus, estimates of Ne, and specifics of timing, should be regarded as approximate, while the historical trajectories of Ne are likely robust. Hammerhead showed a marked decline in Ne from about 335 KYA (based on current estimates of GL and mutation rate) to a low of 2,500 at 45 KYA, and mako had a precipitous drop around the same time as hammerhead, to its lowest Ne of 28,200 at 245 KYA. White shark, the other apex predator in our set, shows a different pattern. After an ancient, initial steep drop, the historical Ne is relatively consistent beginning around 1 MYA, reaching a minimum Ne at around 210 KYA (Ne of 8,500), followed by an increase beginning around 160 KYA, at a time when hammerhead was in steep decline. A recent study suggests that white shark may have been an effective ecological competitor for some species, possibly outcompeting megalodon,47 and another study points out the diet of juvenile white shark off the coast of eastern Australia is about 15% batoids, such as stingrays,48 a favorite food item of great hammerhead. Perhaps competition from white shark played a role in both these species decline in the Middle and Upper Pleistocene. There is also evidence that the population of white sharks off the coast of central California has increased over the course of the last decade,49 perhaps in response to the Marine Mammal Protection Act of 1972, and the subsequent increase of pinnipeds in that general area, suggesting a level of resiliency to this species. The large historical Ne of shortfin mako, surpassing the white shark (Figure 4E), has led to higher heterozygosity (Figures 2A and S3), offering some hope for similar resiliency of shortfin mako if pressure from overfishing is reduced.
Limitations of the study
Our hope is that the reference quality genome sequences presented here will provide a foundation for further basic research and genetic management of these endangered marine apex predators. A shortcoming of this study is that relatively few tissue transcriptomes were available to us, and therefore a less than optimal genome annotation was possible; nonetheless, the preliminary annotation deposited on dryad is likely to be of benefit to other researchers. The extreme divergence times of elasmobranchs relative to other vertebrates, as well as a paucity of other chromosome-level sequences from this sub-class, also hamper thorough annotations, an aspect likely soon to be at least partially alleviated with the publication of additional shark species genomes currently in the VGP and Squalomix Consortium50 pipelines. Another limitation of our study is that the heterozygosity and FROH values we report here for great hammerhead, although concerning, arise from a single individual. Variability between individuals in ROH statistics can be considerable (e.g. gray wolves9; Soay sheep34), making acquisition of similar information from additional individuals, an important part of future conservation management. Additional individuals of both species, combined with the most thorough annotation possible, would greatly facilitate efforts to quantify the amount of fixed and segregating genetic load in these species, and low-coverage re-sequenced genomes would allow estimates of more recent Ne than what is possible with PSMC.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Sphyrna mokarran (female) tissues: heart | Nova Southeastern University; Conservation Biology Laboratory | OC-313 |
| Isurus oxyrinchus (male) tissues: liver, muscle, heart | Nova Southeastern University; Conservation Biology Laboratory | OC-298 |
| Deposited data | ||
| Sphyrna mokarran assembly | This paper | NCBI:JAGIQG010000000 |
| Sphyrna mokarran raw reads (Pacbio) | This paper | SRA: SRR20233585 |
| Sphyrna mokarran raw reads (Illumina) | This paper | SRA: SRR20233586 |
| Sphyrna mokarran RNA-seq (Illumina) | Marra et al.,51 2017 (PMID: 28132643) | SRA: SRR3213610 |
| Isurus oxyrinchus assembly | This paper | NCBI: JANJGN010000000 |
| Isurus oxyrinchus raw reads (Pacbio) | This paper | SRA: SRR20231275 |
| Isurus oxyrinchus raw reads (Illumina) | This paper | SRA: SRR20823548, SRR20823549, SRR20823550, SRR20823551, SRR20823552 |
| Isurus oxyrinchus RNA-seq liver (Pacbio Iso-seq) | This paper | SRA: SRR20245707 |
| Isurus oxyrinchus RNA-seq muscle (Pacbio Iso-seq) | This paper | SRA: SRR20245706 |
| Isurus oxyrinchus RNA-seq heart (Illumina) | Marra et al.51 2017 (PMID: 28132643) | SRA: SRR3213609 |
| Genome annotation gff files for Sphyrna mokarran and Isurus oxyrinchus assemblies | This paper | https://doi.org/10.5061/dryad.mgqnk992h |
| Software and algorithms | ||
| MaSuRCA v3.3.3 | Zimin et al.,52 2017 | https://github.com/alekseyzimin/masurca |
| Hifiasm v0.14 | Cheng et al.,53 2021 | https://github.com/chhylp123/hifiasm |
| Maker v2.31 | Holt and Yandell,56 2011 | https://www.yandell-lab.org/software/maker.html |
| Augustus v3.3.3 | Stanke et al.79 2008 | https://github.com/Gaius-Augustus/Augustus |
| Trinity v2.10.0 | Haas et al.,57 2013 | https://github.com/trinityrnaseq/trinityrnaseq/ |
| PASA v2.4.1 | Haas et al.,58 2003 | https://sourceforge.net/projects/pasa/ |
| EVidenceModeler v1.1.1 | Haas et al.,59 2008 | https://sourceforge.net/projects/evidencemodeler/ |
| BUSCO v5.1.2 | Seppey et al.,63 2019 | https://busco.ezlab.org/ |
| Stringtie2 v1.3.6 | Kovaka et al.,61 2019 | https://github.com/skovaka/stringtie2 |
| TAMA | Kuo et al.,62 2019 | https://github.com/GenomeRIK/tama |
| BLAST2GO | Götz et al.,64 2008 | https://www.blast2go.com/ |
| picard v 2.26.1 | Picard,70 2019 | https://broadinstitute.github.io/picard/ |
| GATK v 3.8.1 | https://gatk.broadinstitute.org/hc/en-us | |
| VCFtools v 0.1.17 | Danecek et al.78 2011 | http://vcftools.sourceforge.net/ |
| samtools. v 1.15.1 | Danecek et al.,71 2021 | http://www.htslib.org/ |
| bedmap v 2.4.35 | https://doi.org/10.1093/bioinformatics/bts277 | |
| psmc v 0.6.5 | Durbin and Li,28 2011 | https://github.com/lh3/psmc |
| R v 4.1.2 | https://www.r-project.org/ | |
| Rstudio 2021.09.0 | https://www.rstudio.com/ | |
| Trimmomatic v 0.39 | Bolger et al.,69 2014 | https://bioweb.pasteur.fr/packages/pack@Trimmomatic@0.39 |
| BWA-MEM v0.7.17 | Li,65 2013 | https://guix.gnu.org/packages/bwa-0.7.17/ |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Michael J. Stanhope (mjs297@cornell.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
This study does not include experiments or subjects.
Method details
Samples
Tissue samples of the shortfin mako (male; collected March 19, 2013) and great hammerhead (female; collected September 22, 2011) sharks, come from the Western North Atlantic, captured by recreational fishers. Heart tissue of both animals were used for genome sequencing. Liver and muscle from mako were used for isoseq transcriptome data; heart Illumina transcriptome data were derived from an earlier study involving these same individuals.51 Since the tissues we used in our work were opportunistically obtained from sharks captured by independent third-party fishers and the sharks were not sacrificed specifically for this study, no ethical approval or permit was required for this work.
DNA methods for Illumina
Genomic DNA of mako and hammerhead for Illumina sequencing were extracted using the Epicentre MasterPure DNA purification kit (Biosearch Technologies). The sequencing libraries were created using Illumina’s TruSeq DNA PCR-Free LT Sample Prep Kit (catalog number FC-121-3001) following the manufacturer’s recommended protocol and sequenced on both lanes of a HiSeq2500 Rapid Run flowcell, as paired-end 2 × 250 bp (mako) and 2 × 125 (hammerhead) runs.
DNA methods for PacBio
Hammerhead and mako genomic DNA for PacBio sequencing were isolated with the Nanobind Tissue Big DNA kit. After isolation, the DNA was sheared using the suggested manufacturer protocol on a Megaruptor 2 from Diagenode, to an average of 50 kbp for hammerhead and 30 kbp for mako. After construction of a PacBio SMRTbell library, hammerhead was size-selected on a BluPippin to remove fragments less than 30 kbp in length. Hammerhead PacBio sequencing was done in continuous long read (CLR) mode on a Sequel I. Mako DNA was size-selected on a Sage ELF, and fractions of the library ranging in size from 13–20 kbp were combined and run on a Sequel II. The data collection was performed in circular consensus (CCS), or HiFi mode.
RNA
Total RNAs were extracted from liver and muscle tissue of mako and isolated using the RNeasy Plus Universal total RNA kits (Qiagen, Valencia, CA, USA) as per the manufacturer’s standard protocol. Following RNA isolation, the samples were concentrated using RNA Clean & Concentrator Kit (Zymo Research, Irvine CA, USA) and their purity was checked using the DeNovix DS-11 + spectrophotometer (DeNovix Inc., Wilmington, DE, USA). RNA concentration was measured using Qubit RNA Assay Kit in Qubit 3.0 Fluorometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). The integrity of total RNA was assessed on the Agilent Fragment Analyzer 5200 system (Agilent Technologies, Santa Clara, CA, USA) using the High Sensitivity RNA Kit with an RNA Quality Number (RQN) criteria of RQN >7.0.
cDNA and PacBio isoseq library construction and sequencing
A total of 100 ng - 300 ng of RNA was input for cDNA synthesis and amplification using NEBNext Single Cell/Low Input cDNA Synthesis & amp, Amplification Module (New England BioLabsInc., Ipswich, MA, USA) as per the manufacturer’s standard protocol. PCR cycles of 10–15 were used to get sufficient quantities of cDNA for pacbio library preparations. Concentration and size profile of cDNA samples was assessed on the Agilent Fragment Analyzer 5200 system (Agilent Technologies, Santa Clara, CA, USA) using the High Sensitivity Large Fragment Kit.
Amplified cDNA samples were size selected using ProNex Size-Selective Purification System (Promega Corporation, Madison, WI, USA) as per the PacBio recommendation for standard length cDNA transcripts. Size selected cDNA was used to construct SMRTbell Iso-Seq libraries using Express Template Prep 2.0 (Pacific Biosciences, Menlo Park, CA, USA) as per the manufacturer’s Iso-Seq Express Template Preparation protocol. The quality of the Iso-Seq libraries were assessed using the Qubit 3.0 Fluorometer (Thermo Fisher Scientific Inc., Waltham, MA, USA) and the Agilent Femto Pulse System (Agilent Technologies, Santa Clara, CA, USA).
Genome assembly
The hammerhead shark genome was assembled with the hybrid mode of MaSuRCA version 3.3.3,52 with 125.7 GB of Pacbio CLR reads (average length 12 kb) and 165.0 GB Illumina paired end reads (125bpx2) using default parameters. The Illumina reads were first contiged into super-reads by extending each short read uniquely in both directions. The super-reads were then used for error correction of the Pacbio reads, followed by consensus overlapping assembly of the error-corrected long reads. The genome size was estimated to be 2.9 Gbp based on kmer distribution of the Illumina reads.
The mako shark genome was assembled with Hifiasm version 0.14,53 using 129.3 GB of PacBio Hifi CCS reads, with average read length of 15.5 kb, and employing default settings on the assembler.
Scaffolding of the two assemblies with Dovetail Hi-C and Omni-C technology
Dovetail Genomics completed scaffolding of both the hammerhead and mako genomes. Dovetail Hi-C was used for scaffolding the hammerhead, and their Omni-C procedure for mako; both were prepared with shark muscle tissue. Two Dovetail Hi-C hammerhead libraries were prepared in a manner similar to that previously described.54 The libraries were sequenced on an Illumina HiSeq X platform. The number and length of read pairs produced for each library was: 249 million, 2 × 150 bp for library 1 and 202 million, 2 × 150 bp for library 2.
Dovetail Omni-C was used for scaffolding mako; the primary difference to their Hi-C procedure is that Omni-C uses a sequence independent endonuclease for chromatin digestion prior to proximity ligation. The fixed chromatin was digested with DNAse I, chromatin ends were repaired and ligated to a biotinylated bridge adapter followed by proximity ligation of adapter containing ends. The library was sequenced on an Illumina HiSeqX platform to produce approximately 30x sequence coverage.
The input de novo contigs and Dovetail Hi-C and Omni-C library reads were used as input data for HiRise, a software pipeline designed specifically for using proximity ligation data to scaffold genome assemblies.55 Dovetail Hi-C and OmniC library sequences were aligned to the draft input assembly using bwa (https://github.com/lh3/bwa). The separations of read pairs mapped within draft scaffolds were analyzed by HiRise to produce a likelihood model for genomic distance between read pairs, and the model was used to identify and break putative misjoins, to score prospective joins, and make joins above a threshold.
Repeat annotation
RepeatModeler (v2.0.1) was used to build repeat libraries for mako, hammerhead, and the published genomes of catshark, brownbanded bambooshark, white shark and whale shark (Table S2 for accession number details) employing the Repbase database (latest version 10/26/2018). The repeat consensus database was then filtered for known protein sequences with uniref90 (known transposases provided with Repeatmasker were pre-removed from the uniref90 database). The remaining repeat database was then classified with Repbase, and used to generate a repeat annotation gff file using RepeatMasker (version 4.1.0, http://www.repeatmasker.org).
Genome annotation
The repeat sequences in both genome assemblies were soft masked with RepeatMasker (version 4.1.0), using the repeat library created by combining the Repbase database (latest version 10/26/2018) with de novo TEs identified by RepeatModeler.
For the hammerhead genome, gene annotation was performed with the Maker2 pipeline,56 using Augustus (version 3.3.3) for training and prediction gene models. The Illumina RNA-seq reads from hammerhead heart51 tissue were assembled by Trinity with its de novo mode (version 2.10.0).57 The resulting transcriptome sequences and published shark protein sequences were mapped to the genome assembly using the Maker2 pipeline, and then used for training the Augustus gene model. The Augustus predicted gene models were then combined with the Trinity transcriptome assembly using PASA (version 2.4.1)58 and EVidenceModeler (EVM, version 1.1.1).59 When running EVM, the keep_preds option was set to 1 for ab initio gene prediction obtained from Augustus. From this prediction pipeline a total of 26,110 protein coding genes, blast supported by hammerhead transcriptome sequences, were retained, and are deposited on dryad (https://doi.org/10.5061/dryad.mgqnk992h).
For the mako shark genome annotation, PacBio iso-seq data from mako shark liver and muscle tissues were aligned using minimap2 (version 2.17).60 Stringtie261 was used to assemble the transcriptome with default setting. Assembled transcripts were processed with TAMA tools62 for ORF detection and BLAST parsing to identify coding regions based on hits against a database of curated proteins from Uniprot_Swissprot. Benchmarking universal single-copy orthologs (BUSCO version 5.1.2)63 were used to assess the genome completeness of the assembly and the accuracy of gene prediction by searching the predicted genes in the assembly for the single-copy genes conserved in vertebrata_odb10, resulting in a total of 27,804 protein coding genes. BLAST2GO64 was used to create Gene Ontology (GO) annotation for hammerhead and mako shark genes.
We regard both these gene annotations as preliminary, primarily because of the lack of additional tissues available to us for these endangered species, and we do not use these annotations in the analyses herein reported; our hope is that by providing them to the research community they can be an asset for further basic and conservation genetics work on these species.
Heterozygosity
Genome wide heterozygosity was evaluated for pseudo-chromosomes 1–41 in mako, and 1–40 in hammerhead, as well as for the published genomes of whale shark, white shark, cloudy catshark, and brownbanded bamboo shark (Table S2 for accessions) for their 24 largest scaffolds. Read coverage for heterozygosity determinations were as follows: hammerhead = 20x; mako = 46x; white shark = 79x; bamboo shark = 31x; catshark = 34x; whale shark = 42x. A published genome sequence for whitespotted bamboo shark is available on GenBank, but the necessary read data for heterozygosity determinations are not available. A few additional unpublished elasmobranch genomes are also on GenBank, as part of the VGP or Squalomix projects, without accompanying read data on SRA. In respect of the VGP stated embargo and researcher’s rights to publish their own data we have herein avoided any specific referral to these particular sequences. The raw reads were mapped to the reference genome using the BWA-MEM algorithm (version 0.7.17-r1188)65 and reads with mapping quality less than 30 were removed. Duplicated reads were removed using Picard (version 2.26.1) (https://broadinstitute.github.io/picard). We used the Genome Analysis Toolkit (GATK v3.8)66 to perform local realignment around indels and for genotype calling (HaplotypeCaller followed by GenotypeGVCFs) employing the same parameters as described elsewhere.67 Both variant and invariant sites were reported with parameter setting -ERC BP_RESOLUTION -out_mode EMIT_ALL_SITES on, in HaplotypeCaller. The parameters -allSites -stand_call_conf 0 were on, in GenotypeGVCFs. We discarded the sites with either insufficient or excess read depth (1/3X and 2X the genome-wide average, respectively) and only considered binary allele sites and invariant sites. We calculated per-site heterozygosity in non-overlapping 1-Mbp sliding windows across each shark’s genome, except for the cloudy catshark and whale shark, which had smaller scaffold sizes, necessitating use of non-overlapping 50 kbp sliding windows, in order to yield sufficient numbers for mean heterozygosity determinations. Heterozygosity was defined as the number of heterozygous genotypes divided by the total number of called genotypes in each window, with the denominator including all sites (both variant and invariant positions). We removed windows with the genotype rate fewer than 50%. We used the homozyg function with default options in plink (version 1.90b6.24)68 to define 1 Mbp ROH regions. Our command line was as follows: /programs/plink-1.9-x86_20210606/plink --homozyg –homozyg-density 50 –homozyg-gap 1000 –homozyg-kb 1000 –homozyg-snp 100 –homozyg-window-het 1 –homozyg-window-missing 5 –homozyg-window-snp 50 –allow-extra-chr –allow-no-sex.
PSMC
Demographic histories of the hammerhead and mako, and four other shark species for which short read data were available, the cloudy catshark,20 brownbanded bamboo shark,20 white shark,21 and whale shark20,22,23 (see Table S2 for accession details), were reconstructed using the pairwise sequentially Markovian coalescent (PSMC).27 Whole genome sequences of the cloudy catshark,20 brownbanded bamboo shark,20 white shark,21 and two whole genome sequences of whale shark22,23 were downloaded from NCBI and read data for each species were downloaded from SRA. Reads were trimmed using Trimmomatic v 0.3969 and trimmed reads were aligned to their respective reference genome using BWA-MEM v0.7.17.65 Duplicate reads were removed with the Picard v2.26.1 MarkDuplicates module.70 Samtools v1.15.1 mpileup71 was used to generate a diploid consensus genome for each species, using parameter values suggested by https://github.com/lh3/psmc. Positions were filtered if they had less than 1/3 the mean depth or greater than 2x the mean depth. Remaining positions with an average mapping quality greater than 20 were used to create the input file for PSMC using the fq2psmc function in https://github.com/lh3/psmc/utils. PSMC was run using at most 25 iterations with parameter values: -N25 -t15 -r5 -p "4 + 25∗2 + 4+6". 100 bootstrap replicates were performed to evaluate variance in Ne estimates. Time on PSMC plots was rescaled using the following per site per generation mutation rates and generation times for great hammerhead, shortfin mako, white shark, brownbanded bamboo shark, cloudy catshark, and whale shark, respectively: mutation rates: 3.92e-09,45 4.33e-09 (estimated from mean synonymous site divergence of 50 random chosen single copy BUSCO genes and 55 MY separation from white shark), 9.36e-09 (estimated as with mako), 1.02e-08,20 7.43e-09,20 2.17e-0820; generation times: 24.5 years, 24.5 years, 53 years, 9.5 years, 9 years, and 25 years (IUCN; IUCN Red List of Threatened Species web site: https://www.iucnredlist.org/).
Altering the mutation rate and generation time affects the placement of the Ne curve on the x and y axes but does not affect the shape of the trajectory.46 As an example, shortening generation lengths (GL) for white shark, from the current IUCN Red List estimate of 53 years,29 to shorter estimates (23–53 years, based on variable published ages at maturity and maximum age estimates; Enric Cortes, pers comm), shifts the trajectory to more recent times, but retains the same shape. The rate of molecular evolution in sharks is nearly two orders of magnitude slower than mammals.22,45 Our estimates of mutation rate for mako and white shark come from synonymous site divergence of single copy BUSCO genes; similar methods were used to estimate mutation rates for the other species.20,45 However, several studies over the last decade suggest synonymous sites may be subject to natural selection pressure (e.g.72,73), thus our estimates may underestimate a true neutral rate. Alignments of noncoding regions could help resolve this, however, accurate non-coding alignments at the divergence times represented here are not possible.
Quantification and statistical analysis
The relationship between mean heterozygosity and log10 chromosome length was assessed using Pearson’s correlation and linear regression (n = 1 shark per analysis, df = number of chromosomes minus 2). The correlation between log2 ROH and log 2 TE concentration within 5 Mbp windows was assessed using Pearson’s correlation (n = 1 shark per analysis, dfmako = 780, dfhammerhead = 516). All statistical analysis was conducted in R versions 4.1.2 and 4.2.1. These regressions are presented in the text of the results section.
Acknowledgments
We thank Peter Schweitzer and the Cornell Genomics facility for the Illumina sequencing that was performed on both hammerhead and mako; Dovetail Genomics for scaffolding of both species; Brewster Kingham and the University of Delaware Sequencing and Genotyping Center for PacBio genome sequencing of both species and isoseq transcriptome sequencing from mako tissue; the Cornell Bioinformatics Facility for various technical and analytical assistance; and Megan Supple for helpful discussions. Major funding for this study was provided by grants from the Save Our Seas Foundation, the Guy Harvey Ocean Foundation, and the Shark Foundation/Hai Stiftung.
Author contributions
Conceptualization, M.J.S. and M.S.S.; Methodology, J.Z., K.C., Q.S., M.W., M.L., and P.P.B.; Software, J.Z., K.C., and M.W.; Formal Analysis, K.C., J.Z., Q.S., M.W., and M.J.S.; Investigation, M.J.S., K.C., A.W., N.M., and M.L.; Resources, A.B. and M.S.S.; Writing – Original Draft, M.J.S., K.C., and Q.S.; Writing – Review & Editing, M.S.S., K.C., A.W., and N.M.; Visualization, K.C.; Supervision, M.J.S. and M.S.S.; Project Administration, M.J.S. and M.S.S.; Funding Acquisition, M.J.S. and M.S.S.
Declaration of interests
The authors declare no competing interests.
Published: January 20, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105815.
Contributor Information
Michael J. Stanhope, Email: mjs297@cornell.edu.
Mahmood S. Shivji, Email: mahmood@nova.edu.
Supplemental information
Data and code availability
-
•
Genome assembly data for the great hammerhead and shortfin mako have been deposited at GenBank and are publicly available as of the date of publication. The genome sequencing reads and transcriptome sequence data are deposited in the NCBI Sequence Read Archive. Accession numbers are listed in the key resources table.
-
•
Preliminary genome annotations for both the great hammerhead and shortfin mako have been deposited at Dryad and are publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Dulvy N.K., Pacoureau N., Rigby C.L., Pollom R.A., Jabado R.W., Ebert D.A., Finucci B., Pollock C.M., Cheok J., Derrick D.H., et al. Overfishing drives over one-third of all sharks and rays toward a global extinction crisis. Curr. Biol. 2021;31:5118–5119. doi: 10.1016/j.cub.2021.11.008. [DOI] [PubMed] [Google Scholar]
- 2.Pacoureau N., Rigby C.L., Kyne P.M., Sherley R.B., Winker H., Carlson J.K., Fordham S.V., Barreto R., Fernando D., Francis M.P., et al. Half a century of global decline in oceanic sharks and rays. Nature. 2021;589:567–571. doi: 10.1038/s41586-020-03173-9. [DOI] [PubMed] [Google Scholar]
- 3.Abercrombie D.L., Clarke S.C., Shivji M.S. Global-scale genetic identification of hammerhead sharks: application to assessment of the international fin trade and law enforcement. Conserv. Genet. 2005;6:775–788. [Google Scholar]
- 4.Rigby C.L., Barreto R., Carlson J., Fernando D., Fordham S., Francis M.P., Jabado R.W., Liu K.M., Marshall A., Pacoureau N., et al. 2019. Isurus oxyrinchus. The IUCN Red List of Threatened Species 2019: e.T39341A2903170. [Google Scholar]
- 5.Barrett R.D.H., Schluter D. Adaptation from standing genetic variation. Trends Ecol. Evol. 2008;23:38–44. doi: 10.1016/j.tree.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 6.Armstrong E.E., Khan A., Taylor R.W., Gouy A., Greenbaum G., Thiéry A., Kang J.T., Redondo S.A., Prost S., Barsh G., et al. Recent evolutionary history of tigers highlights contrasting roles of genetic drift and selection. Mol. Biol. Evol. 2021;38:2366–2379. doi: 10.1093/molbev/msab032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu J.Y., Hao Z.Q., Frantz L., Wu S.F., Chen W., Jiang Y.F., Wu H., Kuang W.M., Li H., Zhang Y.P., Yu L. Genomic consequences of population decline in critically endangered pangolins and their demographic histories. Natl. Sci. Rev. 2020;7:798–814. doi: 10.1093/nsr/nwaa031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saremi N.F., Supple M.A., Byrne A., Cahill J.A., Coutinho L.L., Dalén L., Figueiró H.V., Johnson W.E., Milne H.J., O'Brien S.J., et al. Puma genomes from North and South America provide insights into the genomic consequences of inbreeding. Nat. Commun. 2019;10:4769. doi: 10.1038/s41467-019-12741-1. Erratum in. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson J.A., Räikkönen J., Vucetich L.M., Vucetich J.A., Peterson R.O., Lohmueller K.E., Wayne R.K. Genomic signatures of extensive inbreeding in Isle Royale wolves, a population on the threshold of extinction. Sci. Adv. 2019;5:eaau0757. doi: 10.1126/sciadv.aau0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leffler E.M., Bullaughey K., Matute D.R., Meyer W.K., Ségurel L., Venkat A., Andolfatto P., Przeworski M. Revisiting an old riddle: what determines genetic diversity levels within species? PLoS Biol. 2012;10:e1001388. doi: 10.1371/journal.pbio.1001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kardos M., Armstrong E.E., Fitzpatrick S.W., Hauser S., Hedrick P.W., Miller J.M., Tallmon D.A., Funk W.C. The crucial role of genome-wide genetic variation in conservation. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2104642118. e2104642118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.von Seth J., Dussex N., Díez-Del-Molino D., van der Valk T., Kutschera V.E., Kierczak M., et al. Genomic insights into the conservation status of the world’s last remaining Sumatran rhinoceros populations. Nat. Commun. 2021;12:2393. doi: 10.1038/s41467-021-22386-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pearce J., Fraser M.W., Sequeira A.M.M., Kaur P. State of shark and ray genomics in an era of extinction. Front. Mar. Sci. 2021;8 doi: 10.3389/fmars.2021.744986. [DOI] [Google Scholar]
- 14.Rhie A., McCarthy S.A., Fedrigo O., Damas J., Formenti G., Koren S., Uliano-Silva M., Chow W., Fungtammasan A., Kim J., et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature. 2021;592:737–746. doi: 10.1038/s41586-021-03451-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gregory T.R. 2022. Animal Genome Size Database.http://www.genomesize.com [Google Scholar]
- 16.Hinegardner R. The cellular DNA content of sharks, rays and some other fishes. Comp. Biochem. Physiol. B. 1976;55:367–370. doi: 10.1016/0305-0491(76)90305-9. [DOI] [PubMed] [Google Scholar]
- 17.Asahida T., Ida H., Hayashizaki K. Karyotypes and cellular DNA contents of some sharks in the Order Carcharhiniformes. Jpn. J. Ichthyol. 1995;42:21–26. [Google Scholar]
- 18.Schwartz F.J., Maddock M.B. In: Indo-Pacific Fish Biology. Uyeno T., Arai R., Taniuchi T., Matsuura K., editors. Ichthyological Society of Japan; 1986. Comparisons of karyotypes and cellular DNA contents within and between major lines of elasmobranchs; pp. 148–157. [Google Scholar]
- 19.Martin A.P. In: Great White Sharks, The Biology of Carcharodon carcharias. Klimley A.P., Ainley D.G., editors. Academic Press; 1996. Systematics of the Lamnidae and the origination time of Carcharodon carcharias inferred from the comparative analysis of mitochondrial DNA sequences; pp. 49–53. [Google Scholar]
- 20.Hara Y., Yamaguchi K., Onimaru K., Kadota M., Koyanagi M., Keeley S.D., Tatsumi K., Tanaka K., Motone F., Kageyama Y., et al. Shark genomes provide insights into elasmobranch evolution and the origin of vertebrates. Nat. Ecol. Evol. 2018;2:1761–1771. doi: 10.1038/s41559-018-0673-5. [DOI] [PubMed] [Google Scholar]
- 21.Marra N.J., Stanhope M.J., Jue N.K., Wang M., Sun Q., Pavinski Bitar P., Richards V.P., Komissarov A., Rayko M., Kliver S., et al. White shark genome reveals ancient elasmobranch adaptations associated with wound healing and the maintenance of genome stability. Proc. Natl. Acad. Sci. USA. 2019;116:4446–4455. doi: 10.1073/pnas.1819778116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weber J.A., Park S.G., Luria V., Jeon S., Kim H.M., Jeon Y., Bhak Y., Jun J.H., Kim S.W., Hong W.H., et al. The whale shark genome reveals how genomic and physiological properties scale with body size. Proc. Natl. Acad. Sci. USA. 2020;117:20662–20671. doi: 10.1073/pnas.1922576117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan M., Redmond A.K., Dooley H., Nozu R., Sato K., Kuraku S., Koren S., Phillippy A.M., Dove A.D., Read T. The whale shark genome reveals patterns of vertebrate gene family evolution. Elife. 2021;10:e65394. doi: 10.7554/eLife.65394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murray G.G.R., Soares A.E.R., Novak B.J., Schaefer N.K., Cahill J.A., Baker A.J., Demboski J.R., Doll A., Da Fonseca R.R., Fulton T.L., et al. Natural selection shaped the rise and fall of passenger pigeon genomic diversity. Science. 2017;358:951–954. doi: 10.1126/science.aao0960. [DOI] [PubMed] [Google Scholar]
- 25.Leitwein M., Gagnaire P.A., Desmarais E., Berrebi P., Guinand B. Genomic consequences of a recent three-way admixture in supplemented wild brown trout populations revealed by local ancestry tracts. Mol. Ecol. 2018;27:3466–3483. doi: 10.1111/mec.14816. [DOI] [PubMed] [Google Scholar]
- 26.Heppenheimer E., Brzeski K.E., Hinton J.W., Chamberlain M.J., Robinson J., Wayne R.K., vonHoldt B.M. A genome-wide perspective on the persistence of red Wolf ancestry in southeastern canids. J. Hered. 2020;111:277–286. doi: 10.1093/jhered/esaa006. [DOI] [PubMed] [Google Scholar]
- 27.Ceballos F.C., Joshi P.K., Clark D.W., Ramsay M., Wilson J.F. Runs of homozygosity: windows into population history and trait architecture. Nat. Rev. Genet. 2018;19:220–234. doi: 10.1038/nrg.2017.109. [DOI] [PubMed] [Google Scholar]
- 28.Li H., Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–496. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morin P.A., Archer F.I., Avila C.D., Balacco J.R., Bukhman Y.V., Chow W., Fedrigo O., Formenti G., Fronczek J.A., Fungtammasan A., et al. Reference genome and demographic history of the most endangered marine mammal, the vaquita. Mol. Ecol. Resour. 2021;21:1008–1020. doi: 10.1111/1755-0998.13284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sherley R.B., Winker H., Rigby C.L., Kyne P.M., Pollom R., Pacoureau N., Herman K., Carlson J.K., Yin J.S., Kindsvater H.K., Dulvy N.K. Estimating IUCN Red List population reduction: JARA—a decision-support tool applied to pelagic sharks. Conserv. Lett. 2020;13:e12688. doi: 10.1111/conl.12688. [DOI] [Google Scholar]
- 31.NMFS . National Oceanic and Atmospheric Administration, National Marine Fisheries Service, Office of Sustainable Fisheries, Highly Migratory Species Management Division; 2006. Final Consolidated Atlantic Highly Migratory Species Fishery Management Plan. [Google Scholar]
- 32.Tigano A., Khan R., Omer A.D., Weisz D., Dudchenko O., Multani A.S., Pathak S., Behringer R.R., Aiden E.L., Fisher H., MacManes M.D. Chromosome size affects sequence divergence between species through the interplay of recombination and selection. Evolution. 2022;76:782–798. doi: 10.1111/evo.14467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kardos M., Qvarnström A., Ellegren H. Inferring individual inbreeding and demographic history from segments of identity by descent in Ficedula flycatcher genome sequences. Genetics. 2017;205:1319–1334. doi: 10.1534/genetics.116.198861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stoffel M.A., Johnston S.E., Pilkington J.G., Pemberton J.M. Genetic architecture and lifetime dynamics of inbreeding depression in a wild mammal. Nat. Commun. 2021;12:2972. doi: 10.1038/s41467-021-23222-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilder A.P., Dudchenko O., Curry C., Korody M., Turbek S.P., Daly M., Misuraca A., Wang G., Khan R., Weisz D., et al. A chromosome-length reference genome for the endangered Pacific pocket mouse reveals recent inbreeding in a historically large population. Genome Biol. Evol. 2022;14:evac122. doi: 10.1093/gbe/evac122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoshida G.M., Cáceres P., Marín-Nahuelpi R., Koop B.F., Yáñez J.M. Estimates of autozygosity through runs of homozygosity in farmed Coho Salmon. Genes. 2020;11:490. doi: 10.3390/genes11050490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peripolli E., Munari D.P., Silva M.V.G.B., Lima A.L.F., Irgang R., Baldi F. Runs of homozygosity: current knowledge and applications in livestock. Anim. Genet. 2017;48:255–271. doi: 10.1111/age.12526. [DOI] [PubMed] [Google Scholar]
- 38.Akopyan M., Tigano A., Jacobs A., Wilder A.P., Baumann H., Therkildsen N.O. Comparative linkage mapping uncovers recombination suppression across massive chromosomal inversions associated with local adaptation in Atlantic silversides. Mol. Ecol. 2022;31:3323–3341. doi: 10.1111/mec.16472. [DOI] [PubMed] [Google Scholar]
- 39.Gilpin M.E., Soulé M.E. In: Conservation Biology: The Science of Scarcity and Diversity. Soulé M.E., editor. Sunderland; 1986. Minimum viable populations: processes of species extinction; pp. 19–34. [Google Scholar]
- 40.Niskanen A.K., Billing A.M., Holand H., Hagen I.J., Araya-Ajoy Y.G., Husby A., Husby A., Myhre A.M., Ranke P.S., Kvalnes T., et al. Consistent scaling of inbreeding depression in space and time in a house sparrow metapopulation. Proc. Natl. Acad. Sci. USA. 2020;117:14584–14592. doi: 10.1073/pnas.1909599117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hedrick P.W., Garcia-Dorado A. Understanding inbreeding depression, purging, and genetic rescue. Trends Ecol. Evol. 2016;31:940–952. doi: 10.1016/j.tree.2016.09.005. [DOI] [PubMed] [Google Scholar]
- 42.Robinson J.A., Kyriazis C.C., Nigenda-Morales S.F., Beichman A.C., Rojas-Bracho L., Robertson K.M., Fontaine M.C., Wayne R.K., Lohmueller K.E., Taylor B.L., Morin P.A. The critically endangered vaquita is not doomed to extinction by inbreeding depression. Science. 2022;376:635–639. doi: 10.1126/science.abm1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lynch M., Conery J., Burger R. Mutation accumulation and the extinction of small populations. Am. Nat. 1995;146:489–518. [Google Scholar]
- 44.Frankham R., Bradshaw C.J., Brook B.W. Genetics in conservation management: revised recommendations for the 50/500 rules, Red List criteria and population viability analyses. Biol. Conserv. 2014;170:56–63. [Google Scholar]
- 45.Martin A.P. Substitution rates of organelle and nuclear genes in sharks: implicating metabolic rate (again) Mol. Biol. Evol. 1999;16:996–1002. doi: 10.1093/oxfordjournals.molbev.a026189. [DOI] [PubMed] [Google Scholar]
- 46.Nadachowska-Brzyska K., Burri R., Smeds L., Ellegren H. PSMC analysis of effective population sizes in molecular ecology and its application to black-and-white Ficedula flycatchers. Mol. Ecol. 2016;25:1058–1072. doi: 10.1111/mec.13540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCormack J., Griffiths M.L., Kim S.L., Shimada K., Karnes M., Maisch H., Pederzani S., Bourgon N., Jaouen K., Becker M.A., et al. Trophic position of Otodus megalodon and great white sharks through time revealed by zinc isotopes. Nat. Commun. 2022;13:2980. doi: 10.1038/s41467-022-30528-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grainger R., Peddemors V.M., Raubenheimer D., Machovsky-Capuska G.E. Diet composition and nutritional niche breadth variability in juvenile white sharks (Carcharodon carcharias) Front. Mar. Sci. 2020;7 doi: 10.3389/fmars.2020.00422. [DOI] [Google Scholar]
- 49.Kanive P.E., Rotella J.J., Chapple T.K., Anderson S.D., White T.D., Block B.A., Jorgensen S.J. Estimates of regional annual abundance and population growth rates of white sharks off central California. Biol. Conserv. 2021;257:109104. doi: 10.1016/j.biocon.2021.109104. [DOI] [Google Scholar]
- 50.Nishimura O., Rozewicki J., Yamaguchi K., Tatsumi K., Ohishi Y., Ohta T., Yagura M., Niwa T., Tanegashima C., Teramura A., et al. Squalomix: shark and ray genome analysis consortium and its data sharing platform [version 1; peer review: 1 approved] F1000Res. 2022;11:1077. doi: 10.12688/f1000research.123591.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marra N.J., Richards V.P., Early A., Bogdanowicz S.M., Pavinski Bitar P.D., Stanhope M.J., Shivji M.S. Comparative transcriptomics of elasmobranchs and teleosts highlight important processes in adaptive immunity and regional endothermy. BMC Genom. 2017;18:87. doi: 10.1186/s12864-016-3411-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zimin A.V., Puiu D., Luo M.C., Zhu T., Koren S., Marçais G., Yorke J.A., Dvořák J., Salzberg S.L. Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res. 2017;27:787–792. doi: 10.1101/gr.213405.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheng H., Concepcion G.T., Feng X., Zhang H., Li H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods. 2021;18:170–175. doi: 10.1038/s41592-020-01056-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Putnam N.H., O'Connell B.L., Stites J.C., Rice B.J., Blanchette M., Calef R., Troll C.J., Fields A., Hartley P.D., Sugnet C.W., et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 2016;26:342–350. doi: 10.1101/gr.193474.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holt C., Yandell M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinf. 2011;12:491. doi: 10.1186/1471-2105-12-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haas B.J., Papanicolaou A., Yassour M., Grabherr M., Blood P.D., Bowden J., Couger M.B., Eccles D., Li B., Lieber M., et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haas B.J., Delcher A.L., Mount S.M., Wortman J.R., Smith R.K., Jr., Hannick L.I., Maiti R., Ronning C.M., Rusch D.B., Town C.D., et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003;31:5654–5666. doi: 10.1093/nar/gkg770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haas B.J., Salzberg S.L., Zhu W., Pertea M., Allen J.E., Orvis J., White O., Buell C.R., Wortman J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9:R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kovaka S., Zimin A.V., Pertea G.M., Razaghi R., Salzberg S.L., Pertea M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019;20:278. doi: 10.1186/s13059-019-1910-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuo R.I., Cheng Y., Zhang R., Brown J.W.S., Smith J., Archibald A.L., Burt D.W. Illuminating the dark side of the human transcriptome with long read transcript sequencing. BMC Genom. 2020;21:751. doi: 10.1186/s12864-020-07123-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seppey M., Manni M., Zdobnov E.M. BUSCO: assessing genome assembly and annotation completeness. Methods Mol. Biol. 2019;1962:227–245. doi: 10.1007/978-1-4939-9173-0_14. [DOI] [PubMed] [Google Scholar]
- 64.Götz S., García-Gómez J.M., Terol J., Williams T.D., Nagaraj S.H., Nueda M.J., Robles M., Talón M., Dopazo J., Conesa A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36:3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li H. 2013. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. [Google Scholar]
- 66.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Robinson J.A., Bowie R.C.K., Dudchenko O., Aiden E.L., Hendrickson S.L., Steiner C.C., Ryder O.A., Mindell D.P., Wall J.D. Genome-wide diversity in the California condor tracks its prehistoric abundance and decline. Curr. Biol. 2021;31:2939–2946.e5. doi: 10.1016/j.cub.2021.04.035. [DOI] [PubMed] [Google Scholar]
- 68.Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Picard Toolkit . Broad Institute; 2019. Broad Institute, GitHub Repository.https://broadinstitute.github.io/picard/ [Google Scholar]
- 71.Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10:giab008. doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lawrie D.S., Messer P.W., Hershberg R., Petrov D.A. Strong purifying selection at synonymous sites in D. melanogaster. PLoS Genet. 2013;9:e1003527. doi: 10.1371/journal.pgen.1003527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Corbett-Detig R.B., Hartl D.L., Sackton T.B. Natural selection constrains neutral diversity across a wide range of species. PLoS Biol. 2015;13:e1002112. doi: 10.1371/journal.pbio.1002112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., et al. 1000 Genomes Project Analysis Group The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stanke M., Diekhans M., Baertsch R., Haussler D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008;24:637–644. doi: 10.1093/bioinformatics/btn013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Genome assembly data for the great hammerhead and shortfin mako have been deposited at GenBank and are publicly available as of the date of publication. The genome sequencing reads and transcriptome sequence data are deposited in the NCBI Sequence Read Archive. Accession numbers are listed in the key resources table.
-
•
Preliminary genome annotations for both the great hammerhead and shortfin mako have been deposited at Dryad and are publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.