

Abstract
Coenzyme Q10 (CoQ10) is involved in ATP production through electron transfer in the mitochondrial respiratory chain complex. CoQ10 receives electrons from respiratory chain complex I and II to become the reduced form, and then transfers electrons at complex III to become the oxidized form. The redox state of CoQ10 has been reported to be a marker of the mitochondrial metabolic state, but to our knowledge, no reports have focused on the individual quantification of reduced and oxidized CoQ10 or the ratio of reduced to total CoQ10 (reduced/total CoQ10) in patients with mitochondrial diseases.
We measured reduced and oxidized CoQ10 in skin fibroblasts from 24 mitochondrial disease patients, including 5 primary CoQ10 deficiency patients and 10 respiratory chain complex deficiency patients, and determined the reduced/total CoQ10 ratio.
In primary CoQ10 deficiency patients, total CoQ10 levels were significantly decreased, however, the reduced/total CoQ10 ratio was not changed. On the other hand, in mitochondrial disease patients other than primary CoQ10 deficiency patients, total CoQ10 levels did not decrease. However, the reduced/total CoQ10 ratio in patients with respiratory chain complex IV and V deficiency was higher in comparison to those with respiratory chain complex I deficiency.
Measurement of CoQ10 in fibroblasts proved useful for the diagnosis of primary CoQ10 deficiency. In addition, the reduced/total CoQ10 ratio may reflect the metabolic status of mitochondrial disease.
Keywords: Mitochondrial disease, Primary coenzyme Q10 deficiency, Coenzyme Q10, Reduced/total CoQ10, Forward electron transport, Reverse electron transport
Highlights
-
•
Total and reduced/oxidized forms of CoQ10 contents were examined in fibroblasts of patients with mitochondrial disease.
-
•
Measurement of CoQ10 in fibroblasts is useful for the diagnosis of primary CoQ10 deficiency.
-
•
Reduced/total CoQ10 does not change in primary CoQ10 deficiency.
-
•
Reduced/total CoQ10 is higher in complex IV and V deficiency than in complex I deficiency.
1. Introduction
CoenzymeQ10 (CoQ10), also known as ubiquinone, is a lipophilic molecule composed of a redox-active benzoquinone head group and species-specific isoprenoid side chain (10 subunits in humans) [1,2]. CoQ10 takes three forms depending on the redox state of the benzoquinone ring; oxidized (CoQ10, Ubiquinone), fully-reduced (CoQ10H2, Ubiquinol), and semi-reduced (CoQ10+, Semiubiquinone) forms [2]. It presents ubiquitously in all cellular membranes and cells [3]. The amount of CoQ10 and the proportion of reduced CoQ10 differ between organs and cells; CoQ10 is distributed in high amounts in the heart, kidneys, liver, and muscles, and the proportion of reduced CoQ10 is lower in the brain and lungs [3]. In cells, it is mostly localized in the mitochondria [3].
CoQ10 has multiple functions. One of the main roles of CoQ10 is as a component of the mitochondrial respiratory chain. As a mobile electron carrier, CoQ10 accepts electrons from complex I and II and transfers them to complex III [2]. In the mitochondrial inner membrane, CoQ10 is proposed to exist as two independent pools: a CoQNADH pool in the super-complex (CI, CIII, CIV) involved in the oxidation of NADH; and a CoQFADH pool involved in the oxidation of CII and other enzymes that use CoQ as a cofactor [4]; CoQNADH receives electrons from NADH, and CoQFADH receives electrons from FADH2 and other enzymes, such as glycerol phosphate dehydrogenase (GPDH), choline dehydrogenase (CHDH), sulphide:quinone oxidoreductase (SQOR), dihydroorote dehydrogenase (DHODH), and electron transfer flavoprotein dehydrogenase (ETFDH), and is reduced [5]. In respiratory chain complex III, electrons are transferred from reduced CoQ10 to cytochrome c in a process called the Q cycle. The Q cycle results in the oxidation of two molecules of reduced CoQ10, the reduction of two molecules of cytochrome c, and the formation of one additional molecule of reduced CoQ10 [2]. This normal forward electron transfer results in the creation of an electrical gradient and a pH gradient (proton gradient) between the mitochondrial matrix and intermembrane space. The energy generated by this proton motive force enables complex V(ATP synthase) to synthesize ATP. On the other hand, reverse electron transfer (RET) of CoQ10 is also known to occur; in RET, electrons from reduced CoQ10 are returned to complex I, reducing NAD+ to NADH, and generating ROS [6] (Supplemental Fig. 1).
Another important role of CoQ10 is as an antioxidant. CoQ10 is the sole lipid-soluble antioxidant that is endogenously synthesized. Reduced CoQ10 inhibits both the initiation and the propagation of lipid peroxidation [7]. NADH-quinone oxidoreductase 1 and cytochrome b5 reductase, have been known to be the major oxidoreductases in the plasma membrane [8]. In addition, ferroptosis suppressor protein 1(FSP1) was also found to be an important oxidoreductase. FSP1 reduces extra-mitochondrial CoQ10 and acts as a lipophilic radical-trapping antioxidant to suppress lipid peroxides, resulting in the inhibition of cell death, called ferroptosis [9,10]. Moreover, reduced CoQ10 also regenerates the other antioxidants—α-tocopherol and ascorbate—into an active reduced form [7]. CoQ10 is also involved in the β-oxidation of fatty acids [11], de novo pyrimidine biosynthesis [12], sulfide oxidation [13], an essential cofactor for uncoupling proteins (UCPs) [14], and modulation of the mitochondrial permeability transition pore [15]. Thus, the importance of the distinct state of CoQ10 is gaining growing attention [5].
Primary CoenzymeQ10 (CoQ10) deficiency is an autosomal recessive mitochondrial disease caused by a decrease in CoQ10 due to mutations in genes involved in CoQ10 biosynthesis (COQ genes) [16]. To date, defects in at least 10 COQ genes (COQ2, COQ4, COQ5, COQ6, COQ7, COQ8A, COQ8B, COQ9, PDSS1, PDSS2) have been found to cause this disease [17]. Primary CoQ10 deficiency was first reported in 1989 as familial mitochondrial encephalomyopathy [18]. Currently, over 280 patients from 180 families have been reported [17]. Secondary CoQ10 deficiencies occur in a wider variety of pathologies, including mitochondrial disease [19,20]. Measuring the CoQ levels is known to be useful for diagnosing CoQ10 deficiency [21]. As described above, the ratio of reduction varies among organs and cells. As a result, in addition to the total amount of CoQ10, evaluating the reduced/oxidized CoQ10 ratio may also be useful for further elucidating various pathophysiological states in cells.
Methods to measure CoQ10 in fibroblast and the reduced/oxidized state of CoQ10 have been reported [22,23]. However, to our knowledge, there are no reports on the measurement of the reduced/oxidized CoQ10 ratio in primary CoQ10 deficiency or mitochondrial diseases. Therefore, we decided to measure the total CoQ10 levels as well as the levels of reduced and oxidized CoQ10 in skin fibroblasts from patients affected by various mitochondrial diseases.
2. Material, methods, and patients
2.1. Subjects
We studied fibroblasts from 24 patients with mitochondrial disease, including primary CoQ10 deficiency (Table 1). The inclusion criterion for patients with primary CoQ10 deficiency was childhood onset with biallelic pathogenic variants in the COQ gene encoding proteins for the biosynthesis of CoQ10 [16]. Fibroblasts were obtained from patients at Kanagawa Children's Medical Center, Chiba Children's Medical Center, and Jichi Medical University under the approval of the Ethics Committee of Jichi Medical University. Written informed consent was obtained from the parents of each patient.
Table 1.
Fibroblast cell lines from patients with mitochondrial disease, including primary CoQ10 deficiency.
| Case [ref] | diagnosis | DNA mutation | variants, heteroplasmy ratea | function |
|---|---|---|---|---|
| 1 | primary CoQ10 deficiency | COQ2 | c.[349G > C];[912 + 1G > del] | CoQ10 biosynthesis |
| 2 [24] | primary CoQ10 deficiency | COQ4 | c.[718C > T];[421C > T] | CoQ10 biosynthesis |
| 3 [25] | primary CoQ10 deficiency | COQ4 | c.[431C > A];[718C > T] | CoQ10 biosynthesis |
| 4 | primary CoQ10 deficiency | COQ4 | c.[190C > T];[479G > A] | CoQ10 biosynthesis |
| 5 | primary CoQ10 deficiency | COQ4 | c.[190C > T];[479G > A] | CoQ10 biosynthesis |
| 6 [26] | Leigh syndrome | NDUFA1 | c.[55C > T], 100% (X-linked) | Respiratory chain subunits, complex I |
| 7 [26] | Leigh syndrome | MT-ND3 | m.10158 T>C, heteroplasmic (F; 90%) | Respiratory chain subunits, complex I |
| 8 [27] | neonatal cardiomyopathy | MT-ND5 | m.13513G > A, heteroplasmic (F; 78.87%) | Respiratory chain subunits, complex I |
| 9 | infantile mitochondrial disease | MT-ND5 | m.13513G > A, heteroplasmic (B; 77%) | Respiratory chain subunits, complex I |
| 10 [28] | Leigh syndrome | MT-ND5 | m.13513G > A, heteroplasmic (F; 26%) | Respiratory chain subunits, complex I |
| 11 | mitochondrial cardiomyopathy | ACAD9 | c.[811 T > G];[1766-2A > G] | Respiratory chain assembly factor, complex I |
| 12 [28] | non-lethal infantile mitochondrial disease | ACAD9 | c.[1150G > A];[1817 T > A] | Respiratory chain assembly factor, complex I |
| 13 [29] | Leigh syndrome | SURF1 | c.[743C>A], homoplasmy | Respiratory chain assembly factor, complex IV |
| 14 [25] | Leigh syndrome | SURF1 | c.[367_368delAG];[572delC] | Respiratory chain assembly factor, complex IV |
| 15 [25] | Leigh syndrome | MT-ATP6 | m.8993 T > G, homoplasmy | Respiratory chain subunits, complex V |
| 16 [30] | mtDNA depletion syndrome | DGUOK | c.[143-307_170del335];[143-307_170del335] | Deoxynucleotide triphosphate synthesis |
| 17 [30] | mtDNA depletion syndrome | MPV17 | c.[451dupC];[308_310del] | mitochondrial protein synthesis |
| 18 [30] | mtDNA depletion syndrome | MPV17 | c.[148C > T];[149G > A] | mitochondrial protein synthesis |
| 19 | Kearns-Sayre syndrome | Single mtDNA deletion (5513 bp del; m.8290–13,802) | ||
| 20 [26] | MELAS | (tRNA-Leu) | m.3243 A > G, heteroplasmic (F; 21%) | Mitochondrial tRNA |
| 21 [26] | MELAS | (tRNA-Trp) | m.5541C > T, heteroplasmic (F; 49%) | Mitochondrial tRNA |
| 22 [31] | ECHS1 deficiency | ECHS1 | c.[832G > A] | Metabolism of toxic compounds |
| 23 [28] | cardiomyopathy | BOLA3 | c.[287A > G];[287A > G] | Iron‑sulfur protein assembly |
| 24 | cardiomyopathy | BOLA3 | c.[287A > G];[287A > G] | Iron‑sulfur protein assembly |
F; fibroblasts, B; blood
We obtained fibroblasts from five patients with primary CoQ10 deficiency. One patient carries biallelic COQ2 variants with the c.[349G > C];[912 + 1G > del] (Case 1). Four patients had biallelic COQ4 mutations: one with compound heterozygous biallelic variants c.[718C > T];[421C > T] (Case 2) [24], one with c.[431C > A];[718C > T] (Case 3) [25], and two with c.[190C > T];[479G > A] (Cases 4, 5).
Ten patients had disorders of mitochondrial respiratory chain subunits. Seven patients had mutations related to complex I: one with a c.[55C > T] mutation in NDUFA1 (Case 6) [26], one with an m.10158 T>C mutation in MT-ND3 (Case 7) [26], Three with an m.13513G > A mutation in MT-ND5 (Case 8) [27], (Case 9), (Case 10) [28], one with a c.[811 T > G];[1766-2A > G] mutation in ACAD9 (Case 11), and one with a c.[1150G > A];[1817 T > A] mutation in ACAD9 (Case 12) [28]. NDUFA1, MT-ND3, and MT-ND5 are subunits of complex I, and ACAD9 is the assembly factor of complex I. Two patients had mutations related to complex IV: one with a c.[743C>A] mutation in SURF1 (Case 13) [29], and one with a c.[367_368delAG]; [572delC] mutation in SURF1 (Case 14) [25]. SURF1 is the assembly factor of complex IV. One patient had an m.8993 T > G mutation in MT-ATP6 (Case 15) [25]. MT-ATP6 is a subunit of complex V.
Three patients had mitochondrial DNA (mtDNA) depletion syndrome: one with a c.[143-307_170del335];[143-307_170del335] mutation in DGUOK (Case 16) [30], one with a c.[451dupC];[308_310del] mutation (Case 17) [30] and one with a c.[148C > T];[149G > A] mutation in MPV17 (Case 18) [30]. Both DGUOK and MPV17 are involved in the maintenance of mtDNA. One patient had Kearns-Sayre syndrome with a single mtDNA deletion (5513 bp del; m.8290–13,802) (Case 19). Two patients had MELAS: one with an m.3243 A > G mutation of tRNA-Leu (Case 20) [26] and one with an m.5541C > T, mutation of tRNA-Trp (Case 21) [26]. One patient had short-chain enoyl-CoA hydratase (ECHS1) deficiency with heterozygous mutations in maternal c.[832G > A] in ECHS1 (Case 22) [31]. ECHS1 plays a role in valine and fatty acid catabolism in mitochondria. Two patients had c.[287A > G]; [287A > G] mutation in BOLA3 (Cases 23) [28],(Case 24). BOLA3 is related to iron‑sulfur cluster production and is involved in the assembly of the mitochondrial respiratory chain complex.
Five fibroblasts from healthy individuals were purchased: two fibroblasts from the PromoCell Company (#C-12300, GmbH, Heidelberg, Germany), two fibroblasts from Japanese Collection of Research Bioresources Cell Bank (#TIG-120, #HT-2020, Japan), and fibroblasts from Lonza Japan (#CC-2509, Tokyo, Japan). Another five fibroblasts from patients without mitochondrial disease were used as controls. Cells from passages 4–29 were used for assays.
2.2. Cell culture and growth conditions
The fibroblasts were maintained in 1.0 g/L low glucose Dulbecco's Modified Eagle's Medium (DMEM) (Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin, and 100 μg/mL streptomycin. Cells were incubated at 37 °C under 5% CO2.
2.3. CoQ10 measurement in fibroblasts
2.3.1. CoQ extraction
The methods for the extraction CoQ10 were based on a previously reported method with slight modifications [32]. To extract CoQ10 from fibroblast in 60 mm dishes, cells were washed twice with PBS, and pellets were re-suspended in 500 μL of lysis buffer (0.25 mM Sucrose, 2 mM EDTA, 10 mM Tris, and 100 UI/mL heparin, pH 7.4.), and sonicated twice for 5 s. These homogenates were also used to citrate synthase and protein quantification. To measure CoQ10, nine hundred microliters of ethanol containing internal standard CoQ10-d9 (IsoSciences, Ambler, PA) and 20 μM tert-butyl hydroquinone (TBHQ) (FUJIFILM Wako, Osaka, Japan) was added to 100 μL of homogenates. TBHQ was added to prevent oxidation of reduced CoQ10. The cell suspensions were vortexed and centrifuged at 15,700 ×g for 10 min (4 °C).
2.3.2. Reduction of ubiquinone
Reduced CoQ10 was required for use in the calibration curve measurement. However, since reduced CoQ10 is easily oxidized, reduced CoQ10 was prepared just before the analysis by reducing oxidized CoQ10 following a previously reported method with slight modification [33]. Briefly, 50 μL of CoQ10 was diluted in 1.95 mL hexane in a glass tube. Twenty milligrams of NaBH4 was added and followed by the addition of 100 μL methanol, vortexed for 3 min, then placed in the dark for 5 min at room temperature. After reduction, 1 mL of water containing 100 μM EDTA was added to stop the reaction, vortexed for 1 min, and centrifuged 1500 ×g for 5 min at 4 °C. The upper layer containing reduced CoQ10 was transferred to a glass tube.
2.3.3. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis
The method for measuring reduced and oxidized CoQ10 was based on the previously reported method with slight modifications [23]. An LC-MS/MS analysis was performed on an LC-electrospray ionization-MS (LC-ESI-MS) with triple quadrupole (Nexera X2 and LCMS-8060, Shimadzu, Kyoto, Japan). A Kinetex C18 column (100 mm × 2.1 mm, 2.6 μm, Phenomenex) and a guard column filled with the same packing material were used. The column temperature was kept at 40 °C. The mobile phase was isocratic with 2 mM ammonium formate in methanol. The flow rate was 0.8 mL/min, the injection volume was 2 μL, and the run time was 6 min. The interface temperature was 300 °C, the desolvation line temperature was 250 °C, and the heat block temperature was 400 °C. The nebulizing gas flow was 3 L/min, the heating gas flow was 10 L/min, and the drying gas flow was 10 L/min. The samples were kept at 4 °C before injection by the autosampler. The MS/MS conditions for each target were optimized using the automated multiple reaction monitoring (MRM) optimization procedures in LabSolutions (Shimadzu). The MRM used for quantification was m/z 880.5 > 197.1 for oxidized CoQ10, 882.4 > 197.0 for reduced CoQ10, and 890.4 > 206.2 for CoQ10-d9 (internal standard). Standards and samples were quantified using the LabSolutions software program to determine the peak area for oxidized CoQ10, reduced CoQ10, CoQ10-d9, and the standard curves were used to determine the total amount of CoQ present in the samples.
Intra-assay coefficients of validation (CVs) and relative errors (REs), as measurements of precision and accuracy, respectively, were determined in five parallel analyses of the same cell. To evaluate inter-assay precision and accuracy, one cell line was independently evaluated on three different days. Precision was calculated as (standard deviation/mean concentration) × 100 (%), and accuracy was calculated as (quantitative value/theoretical value) × 100 (%). The intra-assay precision (CV) of reduced CoQ10 and oxidized CoQ10 was 0.58% and 1.39%, respectively. The intra-assay accuracy (RE) of reduced CoQ10 and oxidized CoQ10 was 4.20% and 2.50%, respectively. The inter-assay precision (CV) of reduced CoQ10 and oxidized CoQ10 was 1.27% and 1.84%, respectively. The intra-assay accuracy (RE) of reduced CoQ10 and oxidized CoQ10 was 10.54% and 1.18%, respectively (Supplemental Table 1, QC1).
2.3.4. Citrate synthase and protein quantification
Fibroblast CoQ10 levels were expressed as citrate synthase (CS) activity (measured CoQ10 values/CS units, nmol/CS units). CS activity was measured spectrophotometrically referring to the method described by Srere (1969), with 0.1 mM DTNB, 0.3 mM Acetyl-CoA, 0.5 mM Oxaloacetate, and 12–20 μg protein in 200 μL total incubation volume. CS units are determined as follows: CS Units (μmol/min/mL) = (ΔA412/min x V (mL) × dil)/ 13.6 × L (cm) × Venz (mL), V (mL); the reaction volume, dil; the dilution factor of the original sample, 13.6(mM−1 cm−1); the extinction coefficient of TNB at 412 nm, L (cm); pathlength for absorbance measurement (0.552 cm), Venz(mL); the volume of the enzyme sample. Protein concentrations were quantified using Qubit™ Protein Assay Kits and a Qubit® 2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA).
2.4. Statistical analyses
Statistical analyses were performed using the GraphPad Prism software program (version 9.01, GraphPad Software Inc., La Jolla, CA). Comparisons between samples were performed using a one-way ANOVA. The results are expressed as the mean (standard deviation). P values of <0.05 were considered to indicate statistical significance.
3. Results
3.1. Total CoQ10 levels were observed to decrease in all patients with primary CoQ10 deficiency
We showed the reduced, oxidized, and total (sum of reduced and oxidized) CoQ10 values corrected for the CS unit (nmol/CS unit) (Table 2). In addition, we also measured the CoQ10 values corrected for protein levels (nmol/g protein) (Supplemental Table 2). Six patients showed decreased the total CoQ10 values (<70% of the control): five with primary CoQ10 deficiency (Cases 1–5), and one with Kearns-Sayre syndrome (Case 19) (Table 2). The total CoQ10 levels were significantly decreased in primary CoQ10 deficiency than in controls (primary CoQ10 deficiency(n = 5)1.00 ± 0.19 nmol / CS unit (mean ± SD), controls (n = 10)2.30 ± 0.24 nmol / CS unit, p < 0.0001) (Fig. 1). However, total CoQ10 levels were the same in mitochondrial disease other than primary CoQ10 deficiency as in controls (mitochondrial disease(n = 19)2.23 ± 0.26 nmol / CS unit, p = 0.93).
Table 2.
Reduced and oxidized CoQ10 values and total CoQ deficiency in fibroblasts.
| Case | reduced CoQ10 |
oxidized CoQ10 |
total CoQ10 (nmol/CS unit) |
% CoQ deficiency (%) | |||
|---|---|---|---|---|---|---|---|
| mean | SD | mean | SD | mean | SD | ||
| 1 | 0.37 | 0.15 | 0.37 | 0.02 | 0.74 | 0.13 | 32 |
| 2 | 0.12 | 0.00 | 0.16 | 0.00 | 0.28 | 0.00 | 12 |
| 3 | 0.57 | 0.45 | 0.59 | 0.18 | 1.16 | 0.63 | 50 |
| 4 | 0.64 | 0.03 | 0.74 | 0.20 | 1.38 | 0.17 | 60 |
| 5 | 0.78 | 0.01 | 0.64 | 0.02 | 1.42 | 0.01 | 62 |
| 6 | 0.48 | 0.01 | 1.65 | 0.00 | 2.13 | 0.01 | |
| 7 | 1.03 | 0.04 | 1.00 | 0.32 | 2.03 | 0.29 | |
| 8 | 1.22 | 0.04 | 0.82 | 0.18 | 2.04 | 0.22 | |
| 9 | 1.40 | 0.54 | 0.96 | 0.03 | 2.36 | 0.51 | |
| 10 | 0.86 | 0.16 | 1.34 | 0.30 | 2.20 | 0.46 | |
| 11 | 1.02 | 0.08 | 2.17 | 0.53 | 3.19 | 0.44 | |
| 12 | 1.18 | 0.56 | 1.31 | 0.17 | 2.49 | 0.38 | |
| 13 | 1.33 | 0.03 | 0.57 | 0.02 | 1.90 | 0.00 | |
| 14 | 1.77 | 0.31 | 0.58 | 0.10 | 2.35 | 0.21 | |
| 15 | 1.54 | 0.09 | 0.71 | 0.00 | 2.25 | 0.09 | |
| 16 | 0.86 | 0.02 | 1.46 | 0.01 | 2.32 | 0.01 | |
| 17 | 1.37 | 0.40 | 1.28 | 0.14 | 2.65 | 0.25 | |
| 18 | 1.01 | 0.20 | 1.52 | 0.08 | 2.53 | 0.29 | |
| 19 | 0.57 | 0.01 | 0.72 | 0.02 | 1.29 | 0.02 | 56 |
| 20 | 1.41 | 0.40 | 0.51 | 0.11 | 1.92 | 0.51 | |
| 21 | 1.24 | 0.28 | 0.65 | 0.28 | 1.89 | 0.56 | |
| 22 | 1.19 | 0.11 | 0.87 | 0.23 | 2.06 | 0.34 | |
| 23 | 0.81 | 0.19 | 1.84 | 0.46 | 2.65 | 0.27 | |
| 24 | 0.92 | 0.01 | 1.23 | 0.00 | 2.15 | 0.01 | |
| Reference (n = 10) | 1.19 | 0.15 | 1.11 | 0.10 | 2.30 | 0.24 | |
% CoQ deficiency: <70% of control CoQ10 value.
Fig. 1.
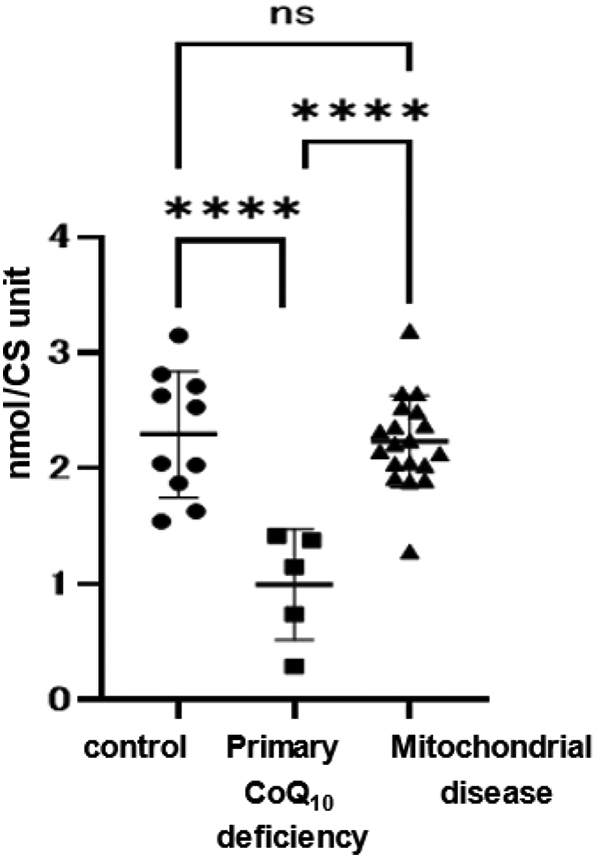
Total CoQ10 values.
Total (sum of reduced and oxidized) CoQ10 values of mitochondrial patient fibroblasts. CoQ10 deficiency (CoQ10 levels <70% of control CoQ10) was found in six cases. All cases of primary CoQ10 deficiency showed decreased CoQ10 levels. In mitochondrial disease, CoQ10 values were not decreased, with the exception of Case 19 (Kearns-Sayre syndrome). Data are expressed as **** P < 0.0001, and n.s. indicates no significance.
3.2. The reduced/total CoQ10 ratio was unchanged in primary CoQ10 deficiency, but was higher in complex IV or V deficiency
We showed the ratio of reduced CoQ10 to total CoQ10 (reduced/total CoQ10) of fibroblasts (Table 3). The ratio of reduced and oxidized CoQ10 to total CoQ10 in control fibroblasts was 52% and 48%, respectively (Table 3). In primary CoQ10 deficiency, the reduced/total CoQ10 ratio did not change compared to the control (reduced/total CoQ10 ratio of primary CoQ10 deficiency (n = 5) 49 ± 7% (mean ± SD), controls (n = 10) 52 ± 1%, p = 0.92) (Fig. 2). Regarding the cases with respiratory chain complex deficiency, there was no difference between control and complex I deficiency (reduced/total CoQ10 ratio of complex I deficiency (n = 7) 44 ± 7%, p = 0.37). However, the reduced/total CoQ10 ratio in complex IV or V deficiency was increased in comparison to the control (complex IV or V deficiency (n = 3) 71 ± 3%, p = 0.022). In addition, the reduced/total CoQ10 ratio in complex IV or V deficiency was higher in comparison to complex I deficiency and primary CoQ10 deficiency (complex IV or V deficiency vs. complex I deficiency: p = 0.0021, complex IV or V deficiency vs. primary CoQ10 deficiency: p = 0.015).
Table 3.
Ratio of reduced / total CoQ10.
| Case | reduced/total CoQ10 (%) |
Reduced/total CoQ10 in cases versus reduced/total CoQ10 in controls (%) |
|
|---|---|---|---|
| mean | SD | ||
| 1 | 50 | 11 | 96 |
| 2 | 43 | 1 | 83 |
| 3 | 49 | 14 | 95 |
| 4 | 47 | 8 | 90 |
| 5 | 55 | 1 | 106 |
| 6 | 23 | 0 | 44 |
| 7 | 51 | 9 | 97 |
| 8 | 60 | 4 | 115 |
| 9 | 59 | 10 | 114 |
| 10 | 39 | 1 | 75 |
| 11 | 32 | 7 | 61 |
| 12 | 47 | 15 | 91 |
| 13 | 70 | 1 | 135 |
| 14 | 75 | 7 | 145 |
| 15 | 68 | 1 | 131 |
| 16 | 37 | 1 | 71 |
| 17 | 52 | 10 | 100 |
| 18 | 40 | 3 | 77 |
| 19 | 44 | 0 | 85 |
| 20 | 74 | 1 | 142 |
| 21 | 66 | 5 | 127 |
| 22 | 58 | 4 | 111 |
| 23 | 30 | 10 | 59 |
| 24 | 43 | 0 | 83 |
| Reference (n = 10) | 52 | 1 | |
The reduced/total CoQ10 ratio decreased (< 80% of control value); cases 6, 10, 11, 16, 18, 23.
The reduced/total CoQ10 ratio increased (120% < of control value); cases 13, 14, 15, 20, 21.
Fig. 2.
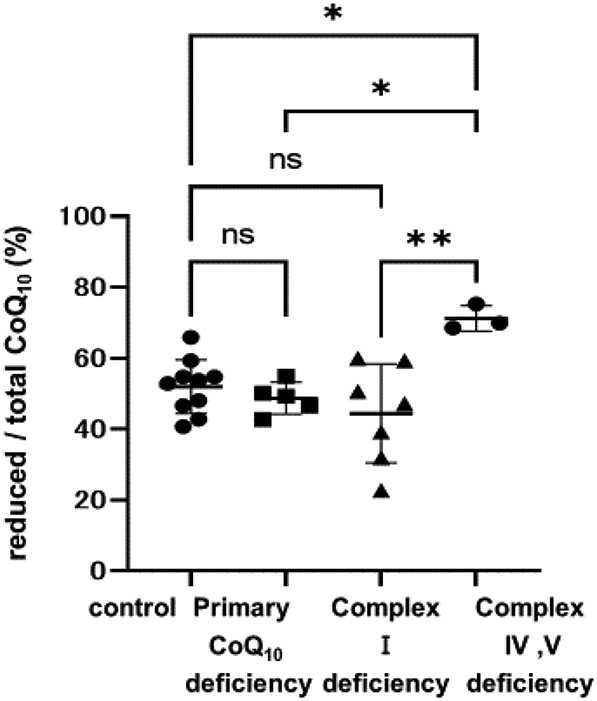
Comparison of the reduced/total CoQ10 ratio.
Comparison of the reduced/total CoQ10 ratio in cases with primary CoQ10 deficiency, complex I deficiency, and complex IV or V deficiency. In primary CoQ10 deficiency, the reduced/total CoQ10 ratio was the same as that of the controls. In complex I deficiency, 3/7 cases showed a decreased (< 80% of control value) reduced/total CoQ10 ratio. In complex IV or V deficiency, 3/3 cases showed an increased (120% < of control value) reduced/total CoQ10 ratio. Data are expressed as *P < 0.05, **P < 0.01, and n.s. indicates no significance.
In individual cases, the reduced/total CoQ10 ratio decreased (<80% of the reduced/total CoQ10 ratio in the control) in six cases; three cases with complex I deficiency (Cases 6, 10, 11), two cases with mtDNA depletion syndrome (Cases 16, 18), one case with BOLA3 mutation (Case 23)(Table 3). On the other hand, the reduced/total CoQ10 ratio increased (>120% of the reduced/total CoQ10 ratio in the control) in five cases; two cases with complex IV deficiency (Cases 13, 14), two cases with MELAS(Cases 20, 21), one case with complex V deficiency (Case 15) (Table 3).
4. Discussion
We showed here that the total CoQ10 values of fibroblasts were significantly lower in patients with primary CoQ10 deficiency. In this syndrome, early recognition and therapy can stop progression and improve the prognosis; however, established severe symptoms cannot be reversed [16,34]. Biochemical measurements of low CoQ10 levels in muscle biopsy have been utilized for the diagnosis of this syndrome [16]. Moreover, the identification of biallelic pathogenic variants in the COQ genes, which encode proteins involved in coenzyme Q biosynthesis, enables a definitive diagnosis [16]. However, the invasiveness of muscle biopsy hampers this procedure and can delay the diagnosis. CoQ10 levels in fibroblasts were examined and implicated in their usefulness [21]. Our data with LC-MS/MS for the measurement of CoQ10 from fibroblasts also supported the usefulness for detecting CoQ10 deficiency [32,35]. The fibroblasts from Case 2 showed the lowest CoQ10 concentration and showed a very severe phenotype. A correlation between CoQ10 levels and phenotype has been suggested [24,36,37]. Therefore, the CoQ10 value from skin fibroblast may reflect the clinical severity.
In addition to primary CoQ10 deficiency, various mitochondrial diseases have also been reported to decrease CoQ10 in fibroblasts and muscle, particularly in mtDNA depletion syndrome [20,38]. However, in our analysis, three patients with mtDNA depletion, including DGUOK and MPV17 mutations, showed no decrease in CoQ10 (Cases 16–18). Only one patient with large deletions of mtDNA showed markedly decreased CoQ10 (Case 19). Therefore, decreased CoQ10 was only a constant feature in primary CoQ10 deficiency syndrome in our analysis. Our results support the widely accepted idea that early CoQ10 therapy should is therefore indicated if decreased levels of CoQ10 are found in the fibroblasts of patients with suspected mitochondrial disease.
To our knowledge, this is the first report to describe the reduced/total CoQ10 ratios in patients with mitochondrial diseases, including primary CoQ10 deficiency. In primary CoQ10 deficiency, the reduced/total CoQ10 ratio did not change. On the other hand, the reduced/total CoQ10 ratio was decreased in 3/7 of cases of complex I deficiency and was increased in 3/3 of cases of complex IV or V deficiency. Primary CoQ10 deficiency is caused by impaired CoQ10 biosynthesis, which results in a decrease in the absolute value of CoQ10; however, as expected, our data suggest that it does not affect the redox reaction in mitochondria. Since CoQ10 changes from the oxidized form to the reduced form by accepting electrons from complex I, complex II and other dehydrogenases, the reduced/total CoQ10 ratio is expected to decrease in complex I deficiency. However, only 3 out of 7 complex I patients in our cohort showed disturbed Q reduced/total ratios. In contrast, CoQ10 changes from the reduced form to the oxidized form in complex III, and the reduced/total CoQ10 ratio is expected to increase in complex III and later complex deficiencies (Supplemental Fig. 1, left panel). In fact, the reduced CoQ10 ratio was significantly decreased with complex I inhibitor, whereas the reduced CoQ10 ratio was increased with complex IV inhibitor [23]. Some (but not all) of our results support these observations. Among complex I deficiency patients, patients with isolated complex I deficiency tended to have a decreased reduced/total CoQ10 ratio (3/4 cases; Cases 6, 10, 11) (Supplemental Table 3). On the other hand, the reduced/total CoQ10 ratio was unchanged in a patient who also had a decreased complex III and IV enzyme activity (Case 8), in a patient without a decreased CI enzyme activity in fibroblasts (Case 12), and in a patient with a decreased CI enzyme activity in muscle (Case 7).
In recent years, it has been reported that the reduced/oxidized CoQ10 ratio may be a marker of mitochondrial metabolic status [39]. In situations where CoQFADH can be excessively reduced, RET is induced, and the production of reactive oxygen species (ROS) from complex I is stimulated, which causes complex I destruction [4,6]. On the other hand, oxidizing the CoQ pool by alternative oxidase (AOX) of Ciona intestinalis xenotopically expressed in mouse mitochondria induces forward electron transport (FET) from RET [40]. Our system of measuring the reduced/total CoQ10 ratio of skin fibroblasts provide the amount and redox states of CoQ10.
As a limitation of our study, CoQ10 is mostly localized in the mitochondria in subcellular fractions but it has also been shown to localize in Golgi, lysosomes, and other organelles [3]. In this study, we measured the whole cell CoQ10 level without separating the mitochondrial and non-mitochondrial fractions. Moreover, we examined an only limited number of patients affected by only some mitochondrial diseases, which can be caused by >400 gene mutations [41].
In conclusion, we measured the reduced/total CoQ10 ratio in fibroblasts from a cohort of patients with mitochondrial disease for the first time. The reduced/total CoQ10 ratio tended to show no change in many of the cells that we measured. However, the reduced/total CoQ10 ratio was increased in complex IV or V deficiency, while the reduced/total CoQ10 ratio tended to decrease in some cases of complex I deficiency.
The following are the supplementary data related to this article.
Electron transfer in mitochondria. (A) Forward electron transfer. CoQNADH receives electrons from NADH and CoQFADH receives electrons from FADH2 and other enzymes, and goes from the oxidized form to the reduced form. In complex III, electrons are transferred from reduced CoQ10 to cytochrome c and CoQ10 becomes the oxidized form. (B) Reverse electron transfer: electrons from excess reduced CoQ10 are returned to complex I and reduced NAD+ to NADH. As a result, reactive oxygen species occur. GPDH, glycerol-3-phosphate dehydrogenase; CHDH, choline dehydrogenase; SQOR, sulphide:quinone oxidoreductase; DHODH, dihydroorote dehydrogenase; ETFDH, electron transfer flavoprotein dehydrogenase.
Precision and accuracy of reduced and oxidized CoQ in fibroblasts and neat diluent.
Reduced and oxidized CoQ10 values corrected for protein levels and total CoQ deficiency in fibroblasts.
Results of enzyme assay for mitochondrial respiratory chain complexes in fibroblasts or muscle biopsy with ComplexIdeficiency.
Determination of enzyme activities
Author contributions
CW, HO contributed to the conceptualization and performance of the statistical analysis of the data and wrote the manuscript; MW, AM, EFJ, TT, HU, OT, ET, and KA performed sampling and data acquisition; YK, YO performed genetic testing; KM, AO recruited patients, provided clinical information, collected samples; TY conducted supervision of the project. All authors read and approved the final manuscript.
Ethics
All procedures followed were by the ethical standards of the responsible committee on human experimentation and with the Helsinki Declaration of the World Medical Organization. This study was approved by the Ethics Committee of Jichi Medical University and written informed consent was obtained from all participants.
Funding
This work was supported in part by the Practical Research Project for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development, AMED to H·O (JP21im0210625, JP21ek0109511), K.M (JP21ek0109468, JP19ek0109273) and Y·O (JP21kk0305015). Health and Labor Sciences Research Grant to H·O (JP21FC1015). The Acceleration Program for Intractable Diseases Research utilizing Disease-specific iPS cells to H·U (JP22bm0804018). JSPS KAKENHI to H. O. (JP20H03648).
CRediT authorship contribution statement
Chika Watanabe: Conceptualization, Writing – original draft. Hitoshi Osaka: Conceptualization, Writing – review & editing, Funding acquisition. Miyuki Watanabe: Investigation. Akihiko Miyauchi: Investigation. Eriko F. Jimbo: Investigation. Takeshi Tokuyama: Resources. Hideki Uosaki: Resources. Yoshihito Kishita: Resources. Yasushi Okazaki: Resources. Takanori Onuki: Resources. Tomohiro Ebihara: Resources. Kenichi Aizawa: Resources. Kei Murayama: Resources. Akira Ohtake: Resources. Takanori Yamagata: Supervision.
Declaration of Competing Interest
None.
Acknowledgments
We thank the patients and their families. We thank all the staff, especially Natsumi Oishi, Shiho Aoki, and Narumi Omika, in Jichi Children Medical Center Tochigi and Jichi Medical University Hospital.
Data availability
No data was used for the research described in the article.
References
- 1.Crane F.L., Hatefi Y., Lester R.L., Widmer C. Isolation of a quinone from beef heart mitochondria. Biochim. Biophys. Acta. 1957;25:220–221. doi: 10.1016/0006-3002(57)90457-2. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y., Hekimi S. Understand. Ubiquinone. Trends Cell Biol. 2016;26:367–378. doi: 10.1016/j.tcb.2015.12.007. [DOI] [PubMed] [Google Scholar]
- 3.Turunen M., Olsson J., Dallner G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta. 2004;1660:171–199. doi: 10.1016/j.bbamem.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 4.Lapuente-Brun E., Moreno-Loshuertos R., Acín-Pérez R., Latorre-Pellicer A., Colás C., Balsa E., Perales-Clemente E., Quirós P.M., Calvo E., Rodríguez-Hernández M.A., Navas P., Cruz R., Carracedo Á., López-Otín C., Pérez-Martos A., Fernández-Silva P., Fernández-Vizarra E., Enríquez J.A. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science. 2013;340:1567–1570. doi: 10.1126/science.1230381. [DOI] [PubMed] [Google Scholar]
- 5.Pallotti F., Bergamini C., Lamperti C., Fato R. The roles of coenzyme Q in disease: direct and indirect involvement in cellular functions. Int. J. Mol. Sci. 2021;23 doi: 10.3390/ijms23010128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scialò F., Fernández-Ayala D.J., Sanz A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017;8:428. doi: 10.3389/fphys.2017.00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bentinger M., Brismar K., Dallner G. The antioxidant role of coenzyme. Q. Mitochondrion. 2007;7(Suppl):S41–S50. doi: 10.1016/j.mito.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 8.Hyun D.H. Plasma membrane redox enzymes: new therapeutic targets for neurodegenerative diseases. Arch. Pharm. Res. 2019;42:436–445. doi: 10.1007/s12272-019-01147-8. [DOI] [PubMed] [Google Scholar]
- 9.Bersuker K., Hendricks J.M., Li Z., Magtanong L., Ford B., Tang P.H., Roberts M.A., Tong B., Maimone T.J., Zoncu R., Bassik M.C., Nomura D.K., Dixon S.J., Olzmann J.A. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–692. doi: 10.1038/s41586-019-1705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doll S., Freitas F.P., Shah R., Aldrovandi M., da Silva M.C., Ingold I., Grocin A.G., Xavier da Silva T.N., Panzilius E., Scheel C.H., Mourão A., Buday K., Sato M., Wanninger J., Vignane T., Mohana V., Rehberg M., Flatley A., Schepers A., Kurz A., White D., Sauer M., Sattler M., Tate E.W., Schmitz W., Schulze A., O'Donnell V., Proneth B., Popowicz G.M., Pratt D.A., Angeli J.P.F., Conrad M. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–698. doi: 10.1038/s41586-019-1707-0. [DOI] [PubMed] [Google Scholar]
- 11.Lee S.K., Lee J.O., Kim J.H., Kim N., You G.Y., Moon J.W., Sha J., Kim S.J., Lee Y.W., Kang H.J., Park S.H., Kim H.S. Coenzyme Q10 increases the fatty acid oxidation through AMPK-mediated PPARα induction in 3T3-L1 preadipocytes. Cell. Signal. 2012;24:2329–2336. doi: 10.1016/j.cellsig.2012.07.022. [DOI] [PubMed] [Google Scholar]
- 12.Evans D.R., Guy H.I. Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. J. Biol. Chem. 2004;279:33035–33038. doi: 10.1074/jbc.R400007200. [DOI] [PubMed] [Google Scholar]
- 13.González-García P., Hidalgo-Gutiérrez A., Mascaraque C., Barriocanal-Casado E., Bakkali M., Ziosi M., Abdihankyzy U.B., Sánchez-Hernández S., Escames G., Prokisch H., Martín F., Quinzii C.M., López L.C. Coenzyme Q10 modulates sulfide metabolism and links the mitochondrial respiratory chain to pathways associated to one carbon metabolism. Hum. Mol. Genet. 2020;29:3296–3311. doi: 10.1093/hmg/ddaa214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Echtay K.S., Winkler E., Klingenberg M. Coenzyme Q is an obligatory cofactor for uncoupling protein function. Nature. 2000;408:609–613. doi: 10.1038/35046114. [DOI] [PubMed] [Google Scholar]
- 15.Fontaine E., Ichas F., Bernardi P. A ubiquinone-binding site regulates the mitochondrial permeability transition pore. J. Biol. Chem. 1998;273:25734–25740. doi: 10.1074/jbc.273.40.25734. [DOI] [PubMed] [Google Scholar]
- 16.Salviati L., Trevisson E., Doimo M., Navas P. In: GeneReviews(®), University of Washington, Seattle Copyright © 1993–2020, University of Washington, Seattle. GeneReviews Is a Registered Trademark of the University of Washington, Seattle. All rights reserved., Seattle (WA) Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Stephens K., Amemiya A., editors. 1993. Primary coenzyme Q(10) deficiency. [Google Scholar]
- 17.Alcázar-Fabra M., Rodríguez-Sánchez F., Trevisson E., Brea-Calvo G. Primary coenzyme Q deficiencies: a literature review and online platform of clinical features to uncover genotype-phenotype correlations. Free Radic. Biol. Med. 2021;167:141–180. doi: 10.1016/j.freeradbiomed.2021.02.046. [DOI] [PubMed] [Google Scholar]
- 18.Ogasahara S., Engel A.G., Frens D., Mack D. Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc. Natl. Acad. Sci. U. S. A. 1989;86:2379–2382. doi: 10.1073/pnas.86.7.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Desbats M.A., Lunardi G., Doimo M., Trevisson E., Salviati L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ 10) deficiency. J. Inherit. Metab. Dis. 2015;38:145–156. doi: 10.1007/s10545-014-9749-9. [DOI] [PubMed] [Google Scholar]
- 20.Yubero D., Montero R., Martín M.A., Montoya J., Ribes A., Grazina M., Trevisson E., Rodriguez-Aguilera J.C., Hargreaves I.P., Salviati L., Navas P., Artuch R., Jou C., Jimenez-Mallebrera C., Nascimento A., Pérez-Dueñas B., Ortez C., Ramos F., Colomer J., O'Callaghan M., Pineda M., García-Cazorla A., Espinós C., Ruiz A., Macaya A., Marcé-Grau A., Garcia-Villoria J., Arias A., Emperador S., Ruiz-Pesini E., Lopez-Gallardo E., Neergheen V., Simões M., Diogo L., Blázquez A., González-Quintana A., Delmiro A., Domínguez-González C., Arenas J., García-Silva M.T., Martín E., Quijada P., Hernández-Laín A., Morán M., Rivas Infante E., Ávila Polo R., Paradas Lópe C., Bautista Lorite J., Martínez Fernández E.M., Cortés A.B., Sánchez-Cuesta A., Cascajo M.V., Alcázar M., Brea-Calvo G. Secondary coenzyme Q10 deficiencies in oxidative phosphorylation (OXPHOS) and non-OXPHOS disorders. Mitochondrion. 2016;30:51–58. doi: 10.1016/j.mito.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 21.Montero R., Sánchez-Alcázar J.A., Briones P., Hernández A.R., Cordero M.D., Trevisson E., Salviati L., Pineda M., García-Cazorla A., Navas P., Artuch R. Analysis of coenzyme Q10 in muscle and fibroblasts for the diagnosis of CoQ10 deficiency syndromes. Clin. Biochem. 2008;41:697–700. doi: 10.1016/j.clinbiochem.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 22.Tang P.H., Miles M.V. Measurement of oxidized and reduced coenzyme Q in biological fluids, cells, and tissues: an HPLC-EC method. Methods Mol. Biol. 2012;837:149–168. doi: 10.1007/978-1-61779-504-6_10. [DOI] [PubMed] [Google Scholar]
- 23.Burger N., Logan A., Prime T.A., Mottahedin A., Caldwell S.T., Krieg T., Hartley R.C., James A.M., Murphy M.P. A sensitive mass spectrometric assay for mitochondrial CoQ pool redox state in vivo. Free Radic. Biol. Med. 2020;147:37–47. doi: 10.1016/j.freeradbiomed.2019.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brea-Calvo G., Haack T.B., Karall D., Ohtake A., Invernizzi F., Carrozzo R., Kremer L., Dusi S., Fauth C., Scholl-Bürgi S., Graf E., Ahting U., Resta N., Laforgia N., Verrigni D., Okazaki Y., Kohda M., Martinelli D., Freisinger P., Strom T.M., Meitinger T., Lamperti C., Lacson A., Navas P., Mayr J.A., Bertini E., Murayama K., Zeviani M., Prokisch H., Ghezzi D. COQ4 mutations cause a broad spectrum of mitochondrial disorders associated with CoQ10 deficiency. Am. J. Hum. Genet. 2015;96:309–317. doi: 10.1016/j.ajhg.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ogawa E., Fushimi T., Ogawa-Tominaga M., Shimura M., Tajika M., Ichimoto K., Matsunaga A., Tsuruoka T., Ishige M., Fuchigami T., Yamazaki T., Kishita Y., Kohda M., Imai-Okazaki A., Okazaki Y., Morioka I., Ohtake A., Murayama K. Mortality of Japanese patients with Leigh syndrome: effects of age at onset and genetic diagnosis. J. Inherit. Metab. Dis. 2020;43:819–826. doi: 10.1002/jimd.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyauchi A., Kouga T., Jimbo E.F., Matsuhashi T., Abe T., Yamagata T., Osaka H. Apomorphine rescues reactive oxygen species-induced apoptosis of fibroblasts with mitochondrial disease. Mitochondrion. 2019;49:111–120. doi: 10.1016/j.mito.2019.07.006. [DOI] [PubMed] [Google Scholar]
- 27.Shimozawa H., Sato T., Osaka H., Takeda A., Miyauchi A., Omika N., Yada Y., Kono Y., Murayama K., Okazaki Y., Kishita Y., Yamagata T. A Case of Infantile Mitochondrial Cardiomyopathy Treated with a Combination of Low-Dose Propranolol and Cibenzoline for Left Ventricular Outflow Tract Stenosis. Int. Heart J. 2022;63:970–977. doi: 10.1536/ihj.21-859. [DOI] [PubMed] [Google Scholar]
- 28.Imai-Okazaki A., Kishita Y., Kohda M., Mizuno Y., Fushimi T., Matsunaga A., Yatsuka Y., Hirata T., Harashima H., Takeda A., Nakaya A., Sakata Y., Kogaki S., Ohtake A., Murayama K., Okazaki Y. Cardiomyopathy in children with mitochondrial disease: prognosis and genetic background. Int. J. Cardiol. 2019;279:115–121. doi: 10.1016/j.ijcard.2019.01.017. [DOI] [PubMed] [Google Scholar]
- 29.Tanigawa J., Kaneko K., Honda M., Harashima H., Murayama K., Wada T., Takano K., Iai M., Yamashita S., Shimbo H., Aida N., Ohtake A., Osaka H. Two Japanese patients with Leigh syndrome caused by novel SURF1 mutations. Brain and Development. 2012;34:861–865. doi: 10.1016/j.braindev.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 30.Shimura M., Kuranobu N., Ogawa-Tominaga M., Akiyama N., Sugiyama Y., Ebihara T., Fushimi T., Ichimoto K., Matsunaga A., Tsuruoka T., Kishita Y., Umetsu S., Inui A., Fujisawa T., Tanikawa K., Ito R., Fukuda A., Murakami J., Kaji S., Kasahara M., Shiraki K., Ohtake A., Okazaki Y., Murayama K. Clinical and molecular basis of hepatocerebral mitochondrial DNA depletion syndrome in Japan: evaluation of outcomes after liver transplantation. Orphanet J. Rare Dis. 2020;15:169. doi: 10.1186/s13023-020-01441-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuwajima M., Kojima K., Osaka H., Hamada Y., Jimbo E., Watanabe M., Aoki S., Sato-Shirai I., Ichimoto K., Fushimi T., Murayama K., Ohtake A., Kohda M., Kishita Y., Yatsuka Y., Uchino S., Mimaki M., Miyake N., Matsumoto N., Okazaki Y., Ogata T., Yamagata T., Muramatsu K. Valine metabolites analysis in ECHS1 deficiency. Mol. Genet. Metab. Rep. 2021;29 doi: 10.1016/j.ymgmr.2021.100809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buján N., Arias A., Montero R., García-Villoria J., Lissens W., Seneca S., Espinós C., Navas P., De Meirleir L., Artuch R., Briones P., Ribes A. Characterization of CoQ₁₀ biosynthesis in fibroblasts of patients with primary and secondary CoQ₁₀ deficiency. J. Inherit. Metab. Dis. 2014;37:53–62. doi: 10.1007/s10545-013-9620-4. [DOI] [PubMed] [Google Scholar]
- 33.Pandey R., Riley C.L., Mills E.M., Tiziani S. Highly sensitive and selective determination of redox states of coenzymes Q(9) and Q(10) in mice tissues: Application of orbitrap mass spectrometry. Anal. Chim. Acta. 2018;1011:68–76. doi: 10.1016/j.aca.2018.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montini G., Malaventura C., Salviati L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N. Engl. J. Med. 2008;358:2849–2850. doi: 10.1056/NEJMc0800582. [DOI] [PubMed] [Google Scholar]
- 35.Liu Y.T., Hersheson J., Plagnol V., Fawcett K., Duberley K.E., Preza E., Hargreaves I.P., Chalasani A., Laurá M., Wood N.W., Reilly M.M., Houlden H. Autosomal-recessive cerebellar ataxia caused by a novel ADCK3 mutation that elongates the protein: clinical, genetic and biochemical characterisation. J. Neurol. Neurosurg. Psychiatry. 2014;85:493–498. doi: 10.1136/jnnp-2013-306483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Desbats M.A., Morbidoni V., Silic-Benussi M., Doimo M., Ciminale V., Cassina M., Sacconi S., Hirano M., Basso G., Pierrel F., Navas P., Salviati L., Trevisson E. The COQ2 genotype predicts the severity of coenzyme Q10 deficiency. Hum. Mol. Genet. 2016;25:4256–4265. doi: 10.1093/hmg/ddw257. [DOI] [PubMed] [Google Scholar]
- 37.Kwong A.K., Chiu A.T., Tsang M.H., Lun K.S., Rodenburg R.J.T., Smeitink J., Chung B.H., Fung C.W. A fatal case of COQ7-associated primary coenzyme Q(10) deficiency. JIMD Rep. 2019;47:23–29. doi: 10.1002/jmd2.12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Montero R., Grazina M., López-Gallardo E., Montoya J., Briones P., Navarro-Sastre A., Land J.M., Hargreaves I.P., Artuch R. Coenzyme Q₁₀ deficiency in mitochondrial DNA depletion syndromes. Mitochondrion. 2013;13:337–341. doi: 10.1016/j.mito.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 39.Guarás A., Perales-Clemente E., Calvo E., Acín-Pérez R., Loureiro-Lopez M., Pujol C., Martínez-Carrascoso I., Nuñez E., García-Marqués F., Rodríguez-Hernández M.A., Cortés A., Diaz F., Pérez-Martos A., Moraes C.T., Fernández-Silva P., Trifunovic A., Navas P., Vazquez J., Enríquez J.A. The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell Rep. 2016;15:197–209. doi: 10.1016/j.celrep.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 40.Szibor M., Gainutdinov T., Fernandez-Vizarra E., Dufour E., Gizatullina Z., Debska-Vielhaber G., Heidler J., Wittig I., Viscomi C., Gellerich F., Moore A.L. Bioenergetic consequences from xenotopic expression of a tunicate AOX in mouse mitochondria: Switch from RET and ROS to FET. Biochim. Biophys. Acta Bioenerg. 2020;1861 doi: 10.1016/j.bbabio.2019.148137. [DOI] [PubMed] [Google Scholar]
- 41.Gusic M., Prokisch H. Genetic basis of mitochondrial diseases. FEBS Lett. 2021;595:1132–1158. doi: 10.1002/1873-3468.14068. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Electron transfer in mitochondria. (A) Forward electron transfer. CoQNADH receives electrons from NADH and CoQFADH receives electrons from FADH2 and other enzymes, and goes from the oxidized form to the reduced form. In complex III, electrons are transferred from reduced CoQ10 to cytochrome c and CoQ10 becomes the oxidized form. (B) Reverse electron transfer: electrons from excess reduced CoQ10 are returned to complex I and reduced NAD+ to NADH. As a result, reactive oxygen species occur. GPDH, glycerol-3-phosphate dehydrogenase; CHDH, choline dehydrogenase; SQOR, sulphide:quinone oxidoreductase; DHODH, dihydroorote dehydrogenase; ETFDH, electron transfer flavoprotein dehydrogenase.
Precision and accuracy of reduced and oxidized CoQ in fibroblasts and neat diluent.
Reduced and oxidized CoQ10 values corrected for protein levels and total CoQ deficiency in fibroblasts.
Results of enzyme assay for mitochondrial respiratory chain complexes in fibroblasts or muscle biopsy with ComplexIdeficiency.
Determination of enzyme activities
Data Availability Statement
No data was used for the research described in the article.