

Abstract
The emergence of SARS-CoV-2 variants of concern (VOC) is driven by mutations that mediate escape from neutralizing antibodies. There is also evidence that mutations can cause loss of T cell epitopes. However, studies on viral escape from T cell immunity have been hampered by uncertain estimates of epitope prevalence. Here, we map and quantify CD8 T cell responses to SARS-CoV-2-specific minimal epitopes in blood drawn from April to June 2020 from 83 COVID-19 convalescents. Among 37 HLA ligands eluted from five prevalent alleles and an additional 86 predicted binders, we identify 29 epitopes with an immunoprevalence ranging from 3% to 100% among individuals expressing the relevant HLA allele. Mutations in VOC are reported in 10.3% of the epitopes, while 20.6% of the non-immunogenic peptides are mutated in VOC. The nine most prevalent epitopes are conserved in VOC. Thus, comprehensive mapping of epitope prevalence does not provide evidence that mutations in VOC are driven by escape of T cell immunity.
Keywords: SARS-CoV-2, immunodominance, CD8 T cell, epitope, immunoprevalence
Graphical abstract
Meyer et al. identify peptide sequences from SARS-CoV-2 that are commonly recognized by CD8 T cells in convalescents. They demonstrate that these sequences are conserved in VOC. Thus, the emergence of VOC is not driven by escape from T cell immunity.
Introduction
Since the pandemic started, there has been a series of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) waves throughout the world. Each is driven by a new variant of concern (VOC) that is more transmissible than its predecessor. Enhanced transmission can be secondary to enhanced binding to the angiotensin-converting enzyme 2 receptor, faster replication, or more effective cell fusion. Equally, if not more important, are mutations in binding sites for neutralizing antibodies. Evasion from neutralizing antibodies is likely to be the main reason why re-infections occur with increasing frequency.1
A study of patients with multiple sclerosis showed that those treated with B cell-depleting anti-CD20 antibodies had a high risk of vaccine breakthrough infections, but that severe disease was rare.2 This suggests that protection against severe disease is primarily mediated by T cells. A question of key importance is, therefore, to what extent SARS-CoV-2 also evolves to evade T cell immunity.1 Several studies have shown that T cells from vaccinated individuals can recognize spike from Omicron.3 , 4 Yet, there is also evidence that mutations can abolish T cell epitopes,5 , 6 , 7 and a recent study found that T cells from individuals infected with ancestral SARS-CoV-2 failed to recognize the spike protein from Omicron.8
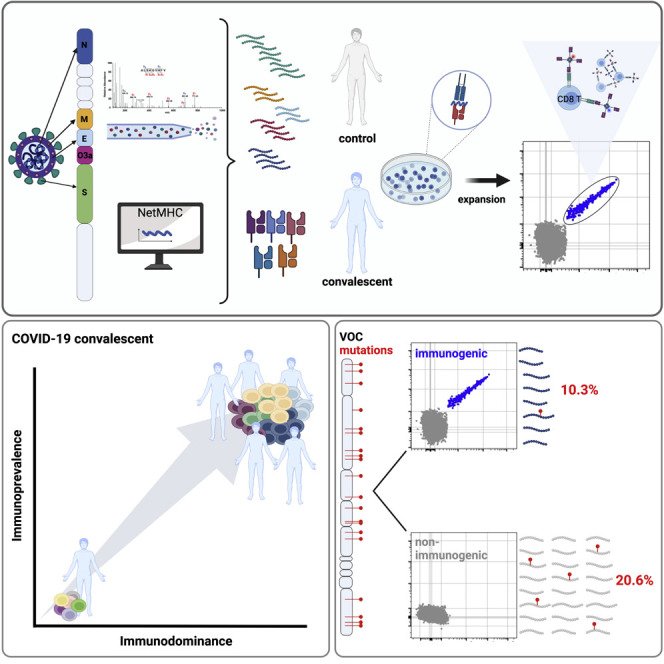
It seems safe to assume that mutations driven by T cell immune evasion would primarily affect epitopes that are prevalent and immunodominant.5 A wide range of SARS-CoV-2 CD8 T cell epitopes has been previously reported.9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 However, estimates of prevalence and immunodominance vary greatly between studies. A likely reason is that the number of donors that were included per HLA class I allele was low (median, five).33 The aim of the present study was to identify immunodominant and immunoprevalent antigens for CD8 T cells in individuals infected with ancestral SARS-CoV-2 in 2020. Key aspects of the study design were (1) inclusion of a large cohort (83 non-vaccinated convalescents), (2) use of assays with high sensitivity and specificity, (3) validation of epitopes by mass spectrometry (MS) of eluted human leukocyte antigen (HLA) ligands, and/or by demonstration of T cell recognition of endogenously presented antigen, and (4) an algorithm to calculate the population coverage of epitopes based on HLA frequency and immunoprevalence. We identified 29 immunogenic epitopes (9 previously not reported in IEDB), with a response prevalence ranging from 3% to 100% among individuals with the relevant HLA-allele. Three of these (10.3%) have been shown to be mutated in VOC. Among 97 pHLA combinations that were not found to be immunogenic, 20 (20.6%) were mutated in VOC. The top nine most prevalent epitopes were conserved in VOC. Thus, comprehensive mapping of epitope prevalence did not provide evidence that mutations in VOC are driven by escape of T cell immunity.
Results
Identification of a wide repertoire of SARS-CoV-2-specific HLA ligands by MS
To identify naturally presented epitopes for CD8 T cells, we used MS to analyze the HLA ligandome of 25 mono-allelic B721.221 cell lines overexpressing the SARS-CoV-2 structural proteins spike (S, 3 truncated proteins), membrane (M), envelope (E), or nucleocapsid (N). Collectively, the cell lines covered the most prevalent HLA class I alleles in the Caucasian study population, including HLA-A∗01:01, HLA-A∗02:01, HLA-A∗03:01, HLA-B∗07:02, and HLA-B∗08:01, with a cumulative phenotypic allele frequency of 86.9%. Two additional cell lines expressing the non-structural protein ORF3a and HLA-A∗01:01 or HLA-A∗02:01 were analyzed. In total, we identified 37 pHLA combinations (37 peptides) across the selected proteins (Table S1), of which 33 were not previously validated by MS and thus represent the largest dataset of eluted HLA ligands.18 , 34 SARS-CoV-2 peptides identified in the discovery approach were further validated using synthetic peptide analogs. Peptides with a spectral similarity score of 0.8 and above were considered unambiguous (Figures 1A and 1B, Data S1). Because of limitations in the sensitivity of MS, we next complemented this list with 85 additional 9- and 10-mer peptides predicted to bind with high affinity (NetMHCpan4.1 BA_Rank < 0.2% and NetMHC4.0 Rank < 0.25%) to the selected HLA alleles as well as one identified weak binder from literature35 yielding a total of 126 pHLA combinations (123 peptides) (Figure 1C, Table S1) for experimental identification of epitopes in convalescents.
Figure 1.

Identification of candidate SARS-CoV-2 CD8 T cell epitopes from eluted HLA class I ligands and by prediction
(A) Representative MS/MS fragmentation spectra of eluted and synthetic HLA class I peptide (N77–87) identified from mono-allelic B721.221 cells expressing the SARS-CoV-2 protein and calculation of similarity score using the SpectrumSimilarity function in the R package OrgMassSpecR.
(B) Display of peptides identified by MS in the exploratory approach considered as true identifications based on the calculation of the similarity score between the exploratory and synthetic peptide (score ≥0.8; n = 37). Orange bars represent peptides that were found to be true epitopes in subsequent analysis of T cell responses (data shown in Figures 2, 3 and Table 1). Labels show HLA restriction (A1 = HLA-A∗01:01; A2 = HLA-A∗02:01; A3 = HLA-A∗03:01; B7 = HLA-B∗07:02; B8 = HLA-B∗08:01) and peptide name.
(C) Schematic outline of peptide selection. Thirty-seven HLA class I-restricted peptides from SARS-CoV-2 proteins expressed in mono-allelic B721.221 cells were identified by MS, an additional 85 by prediction (NetMHCpan4.1 [BA-Rank < 0.2%]; NetMHC4.0 [Rank < 0.25%]), and one from literature, yielding a total of 123 peptides (126 pHLA combinations) (Table S1). UTR, untranslated region.
Highly immunoprevalent and immunodominant SARS-CoV-2-specific epitopes can be identified following short-term in vitro expansion
We included a large cohort of HLA-typed non-vaccinated coronavirus disease 2019 (COVID-19) convalescents (mild to severe disease) and healthy controls (Table S2) in Norway, representative of European Caucasians. Blood was drawn from convalescent individuals from April to June 2020, at which time only infections with wild-type SARS-CoV-2 were reported (earliest documented cases with B.1.1.7 in UK reported by the World Health Organization in September 2020 and in November in Norway).36 Study subjects were included based on a positive SARS-CoV-2 PCR test (n = 93) or positive antibody response alone (n = 3). Among the 93 individuals from whom serum was available, 73 showed antibody responses to the receptor binding domain and nucleocapsid protein of SARS-CoV-2 (Figure 2A). Fourteen pandemic (PCR negative and antibody negative) and 19 pre-pandemic healthy controls were included in this study.
Figure 2.

Identification of immunogenic SARS-CoV-2 CD8 epitopes by screening of convalescents by tetramer staining
(A) Antibody responses to the receptor-binding domain (RBD) located in the spike protein (y axis) and to the nucelocapsid (N) protein (x axis). In contrast with individuals with a negative SARS-CoV-2 test (pandemic controls; n = 14; red), non-vaccinated convalescent COVID-19 individuals raise antibodies (n = 93; blue) against RBD and/or nucleocapsid. Dashed black lines represent thresholds used to designate positive tests for each of the antibodies. Responses are displayed as ratio of MFI values obtained for specific recognition (MFIAG) and blank (MFIblank).
(B) Schematic outline of multimer staining assay for the identification of CD8 epitopes. Peripheral blood mononuclear cell (PBMC) of pre-pandemic (n = 19) and COVID-19 convalescent (n = 83) individuals were loaded with peptides (n = 123; 100 ng/mL per peptide), followed by a 7-day expansion. Immunogenic peptides were identified by combinatorial multimer staining (gating strategy Figure S1).
(C) Representative plots for all immunogenic peptides (n = 29) in convalescent COVID-19 individuals.
(D) Representative plots for immunogenic peptides (n = 10) identified in pre-pandemic controls. Peptides identified by MS are highlighted in orange.
To identify the peptides with the highest immunoprevalence among the candidates, we initially included 50 of the 84 convalescents expressing at least one of the five selected prevalent HLA alleles (range, 14–30 convalescents per allele). To evaluate the pre-existence of responding memory T cells we included seven to nine pre-pandemic individuals per HLA allele (a total of 19 different pre-pandemic individuals). It was previously demonstrated that even dominant T cell epitopes might be lost when analyzing T cell responses in vitro without previous amplification, thus dramatically decreasing sensitivity for epitope detection.22 , 37 In accordance with this, we expanded memory responses during a short-term in vitro culture for 7 days, which is too short to induce confounding naive T cell responses. Cultures were performed in the presence of peptides, before labeling with pHLA multimers (Figure 2B). An individual was classified as responder to a peptide if the multimer population (1) contained at least five clearly double-positive events (each multimer conjugated to two different fluorochromes), (2) constituted 0.005% or more of the live CD8, and (3) formed a tight cluster, similar to previously defined parameters.38 Using these strict criteria, we identified 29 CD8 T cell epitopes (Figure 2C, Tables 1 and S3), which gave a response in a median of 30% of convalescents per epitope (range, 3%–100%) (Figure 3 ). Among pre-pandemic individuals, we found scattered responses to 10 of these epitopes (Figures 2D, 3, Table 1), and none to pHLA combinations that did not induce a response in convalescents (Table S3). For six of these, a response was found in only one pre-pandemic sample (11%), whereas four epitopes induced a response in 29%–57% of pre-pandemic individuals (2–4 responses in 7–9 donors) (Table 1 and Figure 3). All 29 immunogenic epitopes were specific for SARS-CoV-2. When calculating the mean homology across the four most frequent human coronaviruses (HCoV), the 10 epitopes that induced a response in pre-pandemic individuals were, however, significantly more homologous to HCoV than the 19 that did not (44%; interquartile range [IQR], 26%–63% vs 15%; IQR, 7%–33%; p = 0.02, Wilcoxon test) (Table 1). Consistent with the presence of memory responses cross-reactive to these epitopes in some individuals, two of the four epitopes evoking a response in the highest percentage of pre-pandemic individuals have been previously identified as dominated by memory T cell responses to HCoV (N105–113; SPRWYFYYL and N257–265; KPRQKRTAT),26 , 39 although some controversy exists as to whether T cell reactivity to the N105–113; SPRWYFYYL epitope is de novo.40
Table 1.
Immunogenic SARS-CoV-2 peptides (n = 29) identified by multimer staining in samples from convalescents and pre-pandemic controls sorted from highest to lowest immunoprevalence in convalescents expressing the relevant HLA allele
| Peptide | Sequence | HLA allele | Protein | BA_rank (%) | Immunoprevalence (95% CI) | MS | Functional validation | Homology HCoV (%) | Not in the IEDB | ||||
| convalescents | pre-pandemic | OC43 | NL63 | HKU1 | 229E | ||||||||
| N105–113 | SPRWYFYYL | B∗07:02 | nucleocapsid | 0.02 | 100 (89–100) | 57 (18–90) | x | 89 | 56 | 89 | 67 | ||
| O3a207–215 | FTSDYYQLY | A∗01:01 | ORF3a | 0.01 | 100 (83–100) | 0 | x | x | 0 | 22 | 0 | 0 | |
| O3a206–215 | YFTSDYYQLY | A∗01:01 | ORF3a | 0.02 | 100 (83–100) | 0 | x | 0 | 0 | 0 | 0 | ||
| S269–277 | YLQPRTFLL | A∗02:01 | spike | 0.02 | 98 (90–100) | 11 (0–48) | x | x | 44 | 44 | 44 | 33 | |
| O3a139–147 | LLYDANYFL | A∗02:01 | ORF3a | 0.01 | 91 (79–97) | 0 | x | 0 | 0 | 0 | 0 | ||
| N366–374 | KTFPPTEPK | A∗03:01 | nucleocapsid | 0.05 | 90 (73–98) | 0 | x | x | 33 | 44 | 44 | 0 | |
| O3a203–212 | LHSYFTSDYY | A∗01:01 | ORF3a | 0.10 | 86 (57–98) | 0 | 0 | 30 | 0 | 0 | x | ||
| S378–387 | KCYGVSPTKL | A∗03:01 | spike | 7.90 | 83 (64–94) | 11 (0–48) | x | x | 40 | 30 | 0 | 30 | x |
| N257–265 | KPRQKRTAT | B∗07:02 | nucleocapsid | 0.05 | 70 (46–88) | 29 (4–71) | 67 | 67 | 78 | 56 | |||
| O3a72–80 | ALSKGVHFV | A∗02:01 | ORF3a | 0.11 | 53 (39–67) | 0 | x | x | 0 | 44 | 0 | 0 | |
| S89–97 | GVYFASTEK | A∗03:01 | spike | 0.05 | 45 (26–64) | 0 | x | 33 | 44 | 33 | 33 | ||
| S865–874 | LTDEMIAQYT | A∗01:01 | spike | 0.12 | 45 (23–68) | 0 | x | 40 | 30 | 40 | 30 | x | |
| N77–87 | NSSPDDQIGYY | A∗01:01 | nucleocapsid | 0.07 | 36 (13–65) | 0 | x | x | 0 | 45 | 27 | 27 | |
| N222–230 | LLLDRLNQL | A∗02:01 | nucleocapsid | 0.14 | 33 (17–53) | 0 | x | x | 0 | 0 | 33 | 33 | |
| O3a35–43 | IPIQASLPF | B∗07:02 | ORF3a | 0.09 | 30 (12–54) | 0 | 0 | 0 | 0 | 0 | |||
| S1192–1200 | NLNESLIDL | A∗02:01 | spike | 1.01 | 30 (15–49) | 44 (14–79) | x | 56 | 33 | 56 | 56 | ||
| S864–873 | LLTDEMIAQY | A∗01:01 | spike | 0.01 | 29 (8–58) | 0 | 40 | 0 | 30 | 30 | |||
| N66–74 | FPRGQGVPI | B∗07:02 | nucleocapsid | 0.01 | 25 (9–49) | 0 | x | 67 | 44 | 89 | 44 | ||
| O3a64–72 | TLKKRWQLA | B∗08:01 | ORF3a | 0.10 | 21 (5–51) | 0 | 0 | 33 | 0 | 0 | x | ||
| N104–112 | LSPRWYFYY | A∗01:01 | nucleocapsid | 0.08 | 21 (5–51) | 11 (0–48) | 89 | 89 | 89 | 67 | x | ||
| S109–117 | TLDSKTQSL | A∗02:01 | spike | 1.55 | 20 (8–39) | 11 (0–48) | 44 | 56 | 44 | 44 | |||
| N366–375 | KTFPPTEPKK | A∗03:01 | nucleocapsid | 0.06 | 17 (4–41) | 11 (0–48) | 0 | 0 | 40 | 0 | |||
| N222–230 | LLLDRLNQL | B∗08:01 | nucleocapsid | 0.19 | 14 (2–43) | 29 (4–71) | 0 | 0 | 33 | 33 | x | ||
| S1000–1008 | RLQSLQTYV | A∗02:01 | spike | 0.16 | 13 (4–31) | 0 | x | 56 | 44 | 56 | 44 | ||
| S787–795 | QIYKTPPIK | A∗03:01 | spike | 0.14 | 11 (1–35) | 11 (0–48) | x | 22 | 22 | 22 | 44 | ||
| S367–378 | VLYNSASFSTFK | A∗03:01 | spike | 0.01 | 11 (1–35) | 0 | x | 0 | 25 | 0 | 33 | x | |
| O3a204–212 | HSYFTSDYY | A∗01:01 | ORF3a | 0.06 | 7 (0–34) | 0 | x | 0 | 0 | 0 | 0 | x | |
| M65–73 | FVLAAVYRI | A∗02:01 | membrane | 0.11 | 3 (0–17) | 0 | 22 | 0 | 0 | 22 | |||
| M50–59 | LIFLWLLWPV | A∗02:01 | membrane | 0.39 | 3 (0–17) | 0 | 60 | 50 | 60 | 60 | x | ||
CI, confidence interval; BA, binding affinity. Functional validation, T cell lines functionally tested; Homology, % sequence identity with human common cold coronaviruses (HCoV); Not in the IEDB, not previously reported as immunogenic in the IEDB.
Figure 3.

Epitopes induce responses of different magnitudes in COVID-19 convalescents after in vitro stimulation
Magnitude of CD8 T cell responses to the 29 immunogenic SARS-CoV-2-derived epitopes, as determined by multimer staining in non-vaccinated convalescents and pre-pandemic controls (individual data points with mean). Closed symbols (•), cohort 1 (50 convalescents + 19 pre-pandemic controls); open symbols (○): cohort 2 (33 convalescents). For each peptide the number of responses identified among the number of individuals tested for each pHLA combination is displayed above the x axis. Color code: HLA restriction.
Among the epitopes identified in this first cohort, four were recognized by 100% of the tested convalescsent individuals (derived from S1 [1], N [1] and O3a [2]), and an additional four (derived from N, S, and O3a) were recognized by 80% or more (Figure 3, closed symbols, Table S3). To further improve the confidence of our prevalence estimates, we determined CD8 T cell responses to 10 epitopes with an estimated immunoprevalence of more than 50% in a second cohort consisting of 33 convalescent donors not included in the first cohort (Figure 3, open symbols, Table S3). For nine epitopes the estimated immunoprevalence was very similar, while one epitope was found less frequently in the second cohort (Fisher’s exact test p < 0.05) (Figure S2). There were also no clear differences in T cell responses by severity of COVID-19 (hospitalized versus non-hospitalized) (Figure S3). Combining all data, nine epitopes showed an immunoprevalence of 70% or higher (Table 1). Six of these epitopes were immunogenic in at least 90% of individuals. Two highly immunoprevalent epitopes (>80%) were identified in this study, with no data previously reported in the Immune Epitope Database (IEDB): S378–387 (HLA-A∗03:01; KCYGVSPTKL), a 10-mer with poor predicted HLA binding that was identified by MS; and O3a203–212 (HLA-A∗01:01; LHSYFTSDYY). Another seven additional epitopes with greater than 70% immunoprevalence had no data in the IEDB (Tables S3 and 1).
A strong correlation between immunoprevalent and immunodominant epitopes and between the magnitude of responses to immunodominant epitopes
Demonstrating the reproducibility of our multimer assay, we found a strong correlation between the magnitudes of response in paired samples obtained from the same individual (r = 0.949, p < 0.001) (Figure 4A). These data also indicate that a single sample accurately represents the immune status of a given individual. Immunodominance (size of the multimer positive population in responding individuals) was strongly correlated with immunoprevalence (frequency of responding donors) for the 29 immunogenic epitopes (Spearman’s rho = 0.798; p < 0.001) (Figure 4B). We next analyzed the correlation between the magnitude of responses to the nine most immunoprevalent epitopes in individuals tested for responses to at least two epitopes. Of the 30 epitope pairs with data from at least 5 individuals, 27 (90%) showed positive correlations (Spearman’s rho median, 0.55; IQR, 0.30–0.68), of which 10 were statistically significant (30%; p < 0.05 and a false discovery rate of <0.1) (Figures 4C and 4D, S4). All but one epitope showed a statistically significant correlation with at least one other epitope (median, 2). This included significant associations between responses to epitopes derived from the same or different proteins and restricted by the same or different HLA allele. We then went on to perform a pooled analysis of normalized data across the nine most immunoprevalent epitopes from the 82 convalescents who were each tested for two or more of the epitopes. Here, we confirmed a strong association between multimer response magnitudes across epitopes after adjusting for donor age and time since SARS-CoV-2 infection,41 representing potential confounders (Figures 4E and 4F). These data indicate that HLA- and age-independent individual factors are important determinants of the quality of CD8 T cell responses to SARS-CoV-2.
Figure 4.

CD8 T cell response magnitudes to immunoprevalent SARS-CoV-2-specific epitopes are coordinated and age- and HLA-independent
(A) Multimer responses in paired samples from a single individual (Pearson correlation, r = 0.949; p < 0.001). Each point represents a pair from one individual (7 convalescents; on average 56 days [range, 38–81 days] between sampling).
(B) Correlation between the mean size of multimer positive populations across convalescent individuals with a response (i.e., immunodominance) and the proportion of convalescent individuals who showed an immune response (i.e., immunoprevalence) for each epitope where a specific CD8 T cell response was found by multimer staining (Spearman correlation; rho = 0.798, p < 0.001).
(C) Correlation between the magnitude of responses to the nine most immunoprevalent epitopes (Spearman’s rank correlation). Each field displays the estimated correlation coefficient between a pair of epitopes. Statistically significant correlations (p < 0.05) with background color proportional to the strength of correlation (blue = positive; red = negative). Note that sample size varies between comparisons depending on the number of donors with a given HLA allele combination.
(D) Epitope pairs where multimer response magnitudes show a statistically significant correlation (Spearman’s rank correlation, p < 0.05). Each dot represents paired multimer response measurements in the same convalescent individual. First row, peptides binding to same allele; second row, peptides binding to A∗02:01 vs A∗03:01; third row, B∗07:02 vs A∗02:01 or A∗03:01. Plots for all other correlations are shown in Figure S4.
(E) Example plot showing the linear correlation between the transformed multimer response ranks of randomly selected pairs of epitopes from the same convalescent individual. Each dot represents a paired measurement from one donor. Linear regression line in blue surrounded by shaded 95% confidence interval.
(F) Multivariate linear regression analysis showing strong association between multimer response ranks of each convalescent individual to different epitopes after adjusting for potential confounders. The analysis was repeated 1,000 times with randomly drawn paired multimer measurements from each convalescent individual (without replacement). Boxplots show the median with IQR and whiskers extend to 1.5 ∗ IQR; outliers are plotted individually.
Identified immunoprevalent epitopes are naturally presented and recognized by T cells
We next validated that T cells responding to the identified epitopes did indeed recognize naturally processed and presented antigens. To this end, multimer-positive T cells from convalescent individuals and one pre-pandemic sample were sorted for the expansion and generation of T cell lines. The activation of expanded T cell lines was measured by flow cytometry after co-culturing them with mono-allelic B721.221 cells transduced, or not, to endogenously express the relevant SARS-CoV-2 protein, using peptide-loaded target cells as a positive control. All 13 tested epitopes, derived from different SARS-CoV-2 proteins and including 7 of the most immunoprevalent epitopes, were validated (Figure 5A and Table 1). Five T cell lines were also tested for cytotoxic ability (five peptides), confirming efficient killing (Figure 5B). For two epitopes, we were unable to successfully expand T cell lines from sorted cells. A T cell line recognizing the 9-mer (O3a207-215; FTSDYYQLY) cross-reacted with the corresponding 10-mer (O3a206-215; YFTSDYYQLY), and vice versa (Figure 5C), an observation made also for additional T cell lines generated from several convalescent individuals, reactive to the same two peptides (Figure S5B).
Figure 5.

Functional validation of CD8 epitopes
(A and B) Functional validation of immunogenic peptides by co-culture of multimer positive T cell lines derived from 11 convalescents and 1 pre-pandemic individual (indicated by COVID-19 status as + or –, respectively) for 20 h with mono-allelic B721.221 cells transduced to express the relevant protein (endogenous antigen [AG]), or not (without AG), or loaded with 100 nM peptide as a positive control (mean of technical duplicates or triplicates).
(A) Activation of T cells was determined as the percent of CD137+ cells among live CD8. The percent response is normalized to the percent multimer+ of live CD8 T cells in each T cell line. Representative flow plots are shown for the MS identified peptide S378–387.
(B) Target cell killing mediated by multimer+ T cell lines. Values are normalized to matching mock control and displayed as percent remaining viable cells. Representative flow plots for the MS identified peptide S378–387 show CD20+ B721.221 target cells and CD8+ multimer positive T cell line (CL) (numbers represent absolute counts).
(C) Cross-recognition of T cell lines sorted from one convalescent for reactivity to ORF3a-derived peptides O3a207–215 (FTSDYYQLY; 9-mer) or O3a206–215 (YFTSDYYQLY; 10-mer) to the other peptide. CL22 (sorted for reactivity to O3a206–215) and CL23 (sorted for reactivity to O3a207–215) recognize the 9- and 10-mer equally well (mean of technical duplicates). (Left) Activation marker expression (CD137) after 20 h co-culture with peptide-loaded (100 nM) mono-allelic B721.221 cells. (Right) Flow plots 1 and 2: dual-color multimer staining of CL22 and CL23 with multimers complexed with relevant peptide. Flow plot 3: multimer staining of CL23 with multimers complexed with 9-mer (x axis) and 10-mer (y axis).
A set including the nine most immunoprevalent epitopes is predicted to induce an immune response in 83.3% of the European Caucasian population
Population coverage of epitopes depends on (1) the frequency of relevant HLA alleles in the population, and (2) the probability that an individual will mount an immune response to at least one epitope, given their combination of HLA alleles. Based on HLA allele frequency distribution alone, we estimated that our set of nine immunoprevalent epitopes could induce an immune response in 83.7% of the European Caucasian population.42 However, it has become clear from this study and others that immunoprevalence differs widely between immunogenic epitopes. Estimates of immunoprevalence for the same epitope may also differ in-between studies, which can be illustrated by data from the IEDB (Figure 6 ). Between-study heterogeneity may be explained by sampling variation due to low numbers of patients tested for each epitope in many studies, as well as differences in methodology.
Figure 6.

Immunoprevalence data for epitopes with a reported prevalence of more than 50%
Immunoprevalence data with exact binomial 95% confidence intervals from individual studies for every epitope reported in 50% or more of individuals in our study or the IEDB,9,10,12,13,16,18,19,20,21,22,23,24,25,26,31,34,40,43,44,45,46,47,48,49,50 among epitopes tested in our study (orange, our data; gray, other studies). Binomial tests were performed to assess whether the immunoprevalence in each study was significantly higher than 0.5 and 0.7, respectively (∗p < 0.05 for immunoprevalence >50%; +p < 0.05 for immunoprevalence >70%).
To quantify the impact of immunoprevalence in the setting of population immunity, we developed an algorithm to estimate the population coverage of an epitope set adjusted for immunoprevalence. For our top nine epitopes, we estimated an adjusted population coverage of 83.3% (95% confidence interval [CI], 82.8%–83.5%) (Figure S6). This is almost identical to the coverage based solely on HLA allele frequency, since the immunoprevalence for these nine epitopes was very high. For comparison, we calculated the adjusted population coverage of 1,000 random sets of 9 immunogenic epitopes from IEDB with varying immunoprevalence, keeping the distribution identical to that of the HLA alleles restricting our top epitopes. The results showed that the average population coverage for a set of nine epitopes was 46.3% (range, 17.8%–75.5%).
There is no association between immunoprevalence and mutations
Collectively, mutations listed for VOC (Table S4) affected 23 of the 126 candidate pHLA combinations tested here (Table S3). The frequency of mutations was similar among pHLA combinations identified as epitopes 10.3% (3/29; 95% CI, 0%–20.7%) and the remaining non-immunogenic pHLA 20.6% (20/97; 95% CI, 13.4%–28.9%) (Fisher’s exact test, p = 0.3). Importantly, the mutations did not affect any of the top nine most prevalent epitopes (Fisher’s exact test, p = 0.2 vs non-immunogenic pHLA).
Discussion
Previous studies on the prevalence of CD8 T cell epitopes in SARS-CoV-2 have included few individuals per HLA-allele (median, 5)33 and meta-analysis is difficult because of variations in methodology. For this study, we obtained peripheral blood samples from 83 non-vaccinated COVID-19 convalescents from April to June 2020. The high included number of individuals expressing one or more of the five most common HLA alleles in European Caucasians is a strength of the study (average, 19; range, 14–30) (Table S3). We tested reactivity to a large set of 126 pHLA combinations, using highly sensitive methods that detected responses with very high reproducibility (Figure 4A) (r = 0.95). Twenty-nine epitopes were identified, and the top six and top nine were recognized by T cells from 90% and 70% or more of individuals with the restricting HLA-type, respectively. Endogenous processing and presentation were confirmed by T cell activation and target cell killing for all tested epitopes, and pre-pandemic control donors responded only to epitopes showing homology with HCoV, confirming specificity. Thus, we provide strong evidence that the T cell response against SARS-CoV-2 is convergent.
An important aspect of the study is that we could compare the frequency of mutations in immunogenic versus non-immunogenic pHLA combinations. This was possible because of the precise data on immuno-prevalence. The analysis showed that the nine most prevalent epitopes in ancestral SARS-CoV-2 are conserved in VOC, including Omicron BA.1, BA.2. BA.4, and BA.5 (Tables S3 and S4). Moreover, when considering all the 126 pHLA combinations that were studied here, consistent mutations in VOC have been reported for 10.3% of those identified as immunogenic in our study, compared with 20.6% of those that were not found to be immunogenic. Thus, there was no association between mutations and epitope prevalence. These results are difficult to reconcile with the view that variants evolve to escape T cell immunity.5 , 6 , 7
Our results raise questions about the notion that infection with ancestral SARS-CoV-2 leaves an imprint on the immune system, hampering immune responses to subsequent infection with Omicron variants.8 That conclusion was drawn on the basis of results showing a lack of T cell memory responses to the spike protein in individuals who were first infected with ancestral SARS-CoV-2 and later with Omicron. However, since the most prevalent epitopes in spike from ancestral SARS-CoV-2 are conserved in Omicron, it is difficult to see how an imprint would interfere with memory responses. These epitopes are also the most prevalent after vaccination.51 Our results are, however, in agreement with studies showing that previous infection or hybrid immunity, that is, previous immunity combined with vaccination, provides better protection against infections than does vaccination alone.52
An important observation was that there seems to be a strong correlation between immunodominance and immunoprevalence. Moreover, we found that individuals with a strong response to one immunoprevalent epitope generally had potent responses to other epitopes presented on different HLA alleles, including epitopes derived from different SARS-CoV-2 proteins. These associations span both de novo responses to unique SARS-CoV-2 epitopes and those likely caused by boosting of a memory pool resulting from prior HCoV exposure, including the B∗07:02-restricted epitopes N105–113 (SPRWYFYYL) and N257–265 (KPRQKRTAT).26 , 39 Moreover, this suggests that the previously demonstrated association between a strong response to SPRWYFYYL with milder COVID-1939 might be related to overall strong responses to multiple epitopes, rather than an essential role for this epitope in individuals expressing B∗07:02. Multivariate linear regression analysis confirmed a strong association between multimer response ranks of each donor to different epitopes also after adjusting for potential confounders, including age and time between sampling and infection. Collectively, these results suggest that the magnitude of T cell responses to SARS-CoV-2 are determined by factors that are independent of HLA type, consistent with genome-wide association studies (GWAS) studies that have demonstrated a lack of association between HLA type and COVID-19 disease severity.53
Ultimately, T cell responses depend on the efficacy with which antigen-presenting cells expressing a particular peptide-major histocompatibility complex (MHC) complex can stimulate T cells, the size of the T cell receptor repertoire that can recognize the complex, and the overall “fitness” of the T cells. It is unlikely that there are substantial variations in the overall diversity of TCR repertoires among individuals independent of HLA type and age.54 , 55 , 56 However, GWAS studies have identified XCR1, expressed on dendritic cells and important for cross-presentation, and CCR9 and CXCR6, chemokine receptors on T cells, as genes associated with COVID-19 disease severity.53 In support of a potential role for differences in the ability to prime and boost T cell responses, previous studies have demonstrated that impaired innate immunity is associated with a lack of COVID-19 disease control57 , 58 and highlighted the importance of type I interferon responses.59 , 60 Taken together, this might suggest that the likelihood of responding to SARS-CoV-2-specific epitopes is primarily linked to the prime/boost capabilities of the innate immune system and/or T cell “fitness” of a given individual, independent of HLA type and T cell repertoires responsive to particular epitopes.
It has become clear from our study and others that the immunogenicity of SARS-CoV-2 epitopes is not binary. Indeed, the median immunoprevalence for the 29 identified epitopes was only 30%. Our recent study applying the same methods to assess CD8 T cell responses to spike epitopes after SARS-CoV-2 vaccination yielded similar results.51 These data indicate that population coverage for CD8 T cell vaccines may be overestimated when based solely on the population frequencies of restricting HLA alleles, without adjusting for immunoprevalence, which is the current standard approach.33 , 42 , 61 Here, we developed an algorithm that projects total CD8 T cell response coverage for a given set of epitopes in a population with known a HLA distribution, when immunoprevalence for the epitopes has been determined. The algorithm estimates that 83% of Caucasians will respond to at least one of a set of nine epitopes among the 29 SARS-CoV-2-specific epitopes identified here. The actual population coverage for a given epitope set may, however, be somewhat lower than predicted because of individual differences in the ability to mount an efficient T cell response, as discussed above.
Our study allows us to compare the immunogenicity of a large set of eluted SARS-CoV-2-derived HLA ligands with peptides identified based on high predicted HLA-binding affinity. Only two previous studies investigated the immunogenicity of eluted HLA ligands, in three to seven convalescents.18 , 34 When considering 9- and 10-mer peptides, we found no difference in the percentage of peptides confirmed to be immunogenic (34% for eluted ligands vs 28% for predicted peptides; Fisher’s exact test, p = 0.5). However, 15 peptides identified by MS also had high predicted HLA-binding affinity, of which 10 were immunogenic (67%). Combining MS and HLA-binding prediction could, therefore, provide an efficient strategy to identify peptides with a very high likelihood of being immunogenic.
Enhanced viral transmission and escape from neutralizing antibodies drive mutations leading to SARS-CoV-2 VOC, but it has not been clear to what degree, if any, SARS-CoV-2 escape from T cell immunity also plays a role.1 The data presented here suggest that escape from T cell immunity is not a main driver of SARS-CoV-2 VOC. Hence, vaccines that induce broad T cell responses may boost long-lasting immunity; protection from current vaccines against SARS-CoV-2 is waning.62 , 63 , 64 , 65 , 66 , 67 , 68 , 69 , 70 From a manufacturing standpoint, and to deliver the highest possible dose of the most immunogenic antigens, it is rational to limit the number of epitopes to those inducing the strongest immune responses in the greatest proportion of individuals in a population. Our study shows that the CD8 T cell response to SARS-CoV-2 is convergent and demonstrates the importance of considering immunoprevalence for design of efficacious vaccines.
Limitations of the study
Our study was limited by the selection of only five HLA class I alleles that are prevalent in the Caucasian study population and the selection of a set of viral proteins and peptides biased by the methods used for selection (i.e., MHC binding prediction and MS immunopeptidomics). In addition, our immunopeptidomics approach was based on the overexpression of viral proteins and, thus, might differ in terms of antigen processing and presentation compared with naturally infected cells. Viral infection can interfere with cellular pathways, such as interferon signaling and ubiquitination pathways, that in turn can influence antigen presentation and can also lead to attenuated protein translation and a decreased viability that negatively affect the sensitivity of MS detection of HLA ligands.18 , 34 Thus, more studies are needed to further investigate the effects of SARS-CoV-2 infection on the immunopeptidome. It should also be noted that the T cell responses reported in this study were detected after a 7-day in vitro expansion rather than direct ex vivo analysis, which could possibly skew the data. However, it was previously shown that even dominant T cell responses might be missed without prior in vitro expansion,22 , 37 and it is unlikely that the relative dominance of epitopes reproducibly would be skewed in different individuals. Finally, it is not clear to what extent immunoprevalence for an epitope presented by a given HLA allele can be generalized between populations, and additional large studies are needed to assess immunoprevalence for epitopes restricted by HLA alleles frequent in other populations than the one investigated here. Meta-analyses would be advantageous to answer these questions, but are currently limited by the heterogeneity of individual study designs, including differences in the sensitivity and specificity of methods used to quantify epitope-specific immune responses.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE anti-human HLA-A,B,C (clone W6/32) | Biolegend | Cat#311406; RRID:AB_314875 |
| FITC anti-human CD8a (clone RPA-T8) | Biolegend | Cat#301050; RRID:AB_2562055 |
| BV785 anti-human CD19 (clone HIB19) | Biolegend | Cat# 302240; RRID:AB_2563442 |
| BV785 anti-human CD56 (clone 5.1H11) | Biolegend | Cat# 362550; RRID:AB_2566059 |
| BV785 anti-human CD14 (clone M5E2) | Biolegend | Cat# 301840; RRID:AB_2563425 |
| BV785 anti-human CD4 (clone RPA-T4) | Biolegend | Cat# 300554; RRID:AB_2564382 |
| PE anti-human-CD8 a (Clone RPA-T8) | Biolegend | Cat# 301008; RRID:AB_314126 |
| FITC anti-human CD20 (clone 2H7) | Biolegend | Cat# 302304; RRID:AB_314252 |
| Alexa Fluor 647 anti-human CD137 (clone 4B4-1) | Thermo Fisher Scientific | Cat# A51019; RRID:AB_2633303 |
| PE-conjugated goat-anti-human IgG-Fc | Jackson ImmunoResearch | RRID: AB_2922837 |
| Biological samples | ||
| COVID-19 convalescent donor serum and PBMC samples | This paper | N/A |
| Healthy pre-pandemic donor PBMC samples | This paper | N/A |
| Healthy pandemic control donor serum | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Custom made synthetic peptides | GenScript | N/A |
| PMSF | Sigma-Aldrich | Cat# P7626 |
| Protease Inhibitors Cocktail | Sigma-Aldrich | Cat# P8340 |
| n-Dodecyl-β-D-Maltopyranoside | Anatrace | Cat# D310S |
| Amino Link Plus Resin | Thermo Fisher Scientific | Cat# 20501 |
| Bacterially expressed full-length Nucleocapsid from SARS-CoV-2 | Prospec Bio | Cat # SARS-013 |
| PE-Streptavidin | Invitrogen | Cat# S866 |
| APC-Streptavidin | Invitrogen | Cat# S868 |
| Brilliant Violet 421™ Streptavidin | Biolegend | Cat# 405225 |
| Brilliant Violet 605-Streptavidin | Biolegend | Cat# 405229 |
| PE-Cy™7 Streptavidin | Biolegend | Cat# 405206 |
| PE-CF594 Streptavidin | BD Biosciences | Cat# 562284 |
| PE-Cy5 Streptavidin | BD Biosciences | Cat# 554062 |
| APC-R700 Streptavidin | BD Biosciences | Cat# 565144 |
| BB790-P Streptavidin | BD Biosciences | prototype kindly provided by Bob Balderas |
| Live/Dead-NearInfraRed (LD-NIR) | Thermo Fisher Scientific | Cat# L10119 |
| Recombinant human IL-2 | R&D Systems | Cat# 202-IL-500 |
| Recombinant human IL-15 | Peprotech | Cat# 200-15 |
| PHA (Remel) | Remel | Cat# 10082333 |
| Amine-functionalized pmma microspheres 6 μm and 8 μm | Bangs Laboratories | custom-order |
| Sulfo-Biotin-LC-NHS | Proteochem | Cat # b2103 |
| SARS-CoV-2 RBD domain antigen | Sino Biologicals | Cat # 40592-V08H115 |
| D-Biotin | Sigma-Aldrich | Cat #2031-1GM |
| Neutravidin | Thermo Fisher Scientific | Cat# 31055 |
| Sulfo-Cy5 NHS | Lumiprobe | Cat# 53320 |
| BDP-FL | Lumiprobe | Cat# 51420 |
| Sulfo-Cy7-NHS | Lumiprobe | Cat# 55320 |
| Pacific Blue-NHS | Thermo Fisher Scientific | Cat# P10163 |
| Fetal bovine serum (FBS) | Merck Life Science AS | Cat# F7524-500ML |
| Doxycycline hyclate | Sigma-Aldrich | Cat# D9891 |
| X-Vivo-20 | BioNordika | Cat # BE04-448Q |
| Normal Human Serum, off clot, male, pooled | Trina Bioreactives AG | Cat# SN0300 |
| Human Fc Receptor binding inhibitor | AH Diagnostics | Cat# 14-9161-73 |
| CountBright™ Absolute Counting Beads | Invitrogen | Cat# C36950 |
| Critical commercial assays | ||
| DNeasy Blood & Tissue Kit | Qiagen | Cat 69506 |
| NGSgo®-AmpX HLA GeneSuite (NGSgo®-AmpX HLA-A, B, C, DRB1, DQB1 & NGSgo®-AmpX HLA-DPA1, DPB1, DQA1, DRB3/4/5 kits) | GenDx | Cat # 2341602 |
| Deposited data | ||
| RAW MS files | ProteomeXchange | PXD028862 |
| Experimental models: Cell lines | ||
| HLA class I-deficient B721.221 cells | FRED HUTCH Research Cell bank | Cat# IHW00001 |
| Expi293F cells | Thermo Fisher Scientific | Cat# A14528 |
| Recombinant DNA | ||
| Plasmid: encoding the receptor-binding domain of Spike-1 protein (RBD) | Florian Krammer | PMID: 32398876 |
| Plasmid: HLA-A∗03:01; HLA-A∗01:01, HLA-A∗02:01, HLA-B∗07:02, HLA-B∗08:01 cloned into pMP71 vector backbone | This paper | N/A |
| Plasmid: pRetroX-TetOne-Puro-SARS-CoV-2-Spike | This paper | N/A |
| Plasmid: pRetroX-TetOne-Puro-SARS-CoV-2-Spike 1_1-541aa | This paper | N/A |
| Plasmid: pRetroX-TetOne-Puro-SARS-CoV-2-Spike 2_490-975aa | This paper | N/A |
| Plasmid: pRetroX-TetOne-Puro-SARS-CoV-2-Spike 3_888-1273aa | This paper | N/A |
| Plasmid: pRetroX-TetOne-Puro-SARS-Cov-2-Nucleocapsid | This paper | N/A |
| Plasmid: pRetroX-TetOne-Puro-SARS-CoV-2 -Envelope_2A_Membrane | This paper | N/A |
| Plasmid: pRetroX-TetOne-Puro-SARS-CoV-2-ORF3a | This paper | N/A |
| Software and algorithms | ||
| FlowJo | FlowJo LLC | https://www.flowjo.com; RRID:SCR_008520 |
| RStudio 2021.09.2 + 382 “Ghost Orchid” for macOS | Posit, PBC | https://www.rstudio.com/; RRID:SCR_000432 |
| R version 4.0.4 | R core team | https://www.r-project.org/ |
| GraphPad Prism 8.3.0 | Graphpad | https://www.graphpad.com; RRID:SCR_002798 |
| PEAKS | Bioinformatics Solutions Inc | RRID:SCR_022841 |
| OrgMassSpecR | CRAN | https://cran.r-project.org/web/packages/OrgMassSpecR/index.html |
| Adjusted population coverage | This paper | Github https://doi.org/10.5281/zenodo.7447391 |
| NetMHCpan version 4.1 | NetMHCpan 4.1 Server | dtu.dk; RRID:SCR_018182 |
| NetMHC version 4.0 | NetMHC 4.0 Server | dtu.dk; RRID:SCR_021651 |
| ClustalOmega | Sievers et al. (2011)71 Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539 PMID: 21988835 https://doi.org/10.1038/msb.2011.75 |
https://www.ebi.ac.uk/Tools/msa/clustalo/; RRID:SCR_001591 |
| Immune epitope database (IEDB) | Vita R et al. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2018 Oct 24. https://doi.org/10.1093/nar/gky1006. PMID: 30357391; PMCID: PMC6324067 | https://www.iedb.org/; RRID:SCR_006604 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Johanna Olweus (johanna.olweus@medisin.uio.no).
Materials availability
Materials that were specifically generated for this paper were mentioned in the key resources table and will be provided upon reasonable request to the lead contact.
Experimental model and subject details
Study approval
The project (REK# 124170) was approved by the Regional Research Ethics Committee according to the Declaration of Helsinki. Eligible participants provided informed consent by signing an online electronic consent form. Blood collection of all individuals was performed by the Oslo Blood Bank.
Subject details
Anonymous healthy pre-pandemic control donors: Buffy-coats sampled before the pandemic (n = 19) were obtained between 2015 and 2018 and PBMC isolated using a standard Ficoll isolation protocol.
Anonymous healthy pandemic control donors, and non-vaccinated convalescent COVID-19 donors (45 female, 38 male; median age 45 [IQR 35.5–57.5]): Samples were prospectively collected between April and June 2020 during the initial outbreak as part of a large biobanking effort in Norway. We performed no formal sample size calculations and individuals were consecutively enrolled. Blood from healthy pandemic controls with a SARS-CoV-2 negative PCR test (n = 14), or from 96 convalescent individuals (22 hospitalized; 93 PCR positive, 3 antibody positive) with mild to severe symptoms and at least 14 days without symptoms, was collected in ACD, CPTA or CPT tubes and processed following the manufacturer’s instructions. After initial centrifugation, serum samples were taken and stored at 4°C.
Processed PBMC were frozen in 60% FBS/30% RPMI/10% DMSO and stored in liquid nitrogen.
Cell lines
EBV-transformed HLA-1 null cell line (IHW00001) was purchased from the FRED HUTCH Research Cell bank. The cell line was cultured in RPMI 1640 containing 15% fetal bovine serum (FBS), 1x penicillin-streptomycin (P/S), and 5 mM sodium pyruvate. The cell lines were regularly tested and found negative for mycoplasma contamination.
Method details
HLA typing
DNA was isolated from 1 × 106 PBMC using the DNeasy Blood & Tissue Kit (Qiagen, Cat No 69506), following the manufacturer’s instructions. HLA typing was performed by NGS sequencing using NGSgo®-AmpX HLA GeneSuite (NGSgo®-AmpX HLA-A, B, C, DRB1, DQB1 & NGSgo®-AmpX HLA-DPA1, DPB1, DQA1, DRB3/4/5 kits) for sample library preparations and ran on a Miseq sequencer (Illumina), following the manufacturer’s instructions.
Plasma antibody titer determination
A multiplexed bead-based flow cytometric assay, referred to as microsphere affinity proteomics (MAP), was adapted for detection of SARS-CoV-2 antibodies.72 Amine-functionalized polymer beads were color-coded with fluorescent dyes as described earlier, and reacted successively with amine-reactive biotin (sulfo-NHS-LC-biotin, Proteochem, USA) and neutravidin (Thermo Fisher). A DNA construct encoding the receptor-binding domain of spike-1 protein (RBD) from SARS-CoV-2 was provided by Florian Krammer, and the protocol described by Amanat and colleagues73 was used to produce recombinant protein in Expi293F cells. Bacterially expressed full-length nucleocapsid from SARS-CoV-2 was purchased from Prospec Bio (www.prospecbio.com). Viral proteins solubilized in PBS were biotinylated chemically using a 4:1 M ratio of sulfo-NHS-LC-biotin to protein. Free biotin was removed with G50 sephadex spin columns. Biotinylated proteins were bound to neutravidin-coupled microspheres with fluorescent barcodes. Beads with Neutravidin only were used as reference for background binding. The bead multiplex was incubated for 1 h with serum diluted 1:1000 in PBS supplemented with Neutravidin (10 μg/mL), d-Biotin (10 μg/mL), BSA (1%) and sodium azide (0.1%). The beads were washed twice in PBS with 1% Tween 20 (PBT), labeled with PE-conjugated goat-anti-human IgG-Fc (Jackson ImmunoResearch) for 20 min, washed again and analyzed by flow cytometry (Attune Next, Thermo Fisher). Specific binding was measured as the ratio of R-Phycoerythrin fluorescence intensity of antigen-coupled beads and neutravidin-only beads. Samples containing antibodies both to nucleocapsid and RBD were considered to be positive. Reference panels containing samples from 287 individuals with PCR-confirmed SARS-CoV-2 infection and 1343 pre-pandemic samples were used to set the cutoff. A cutoff of five and ten for RBD and nucleocapsid respectively, yielded a specificity of 100%. The sensitivity was 84%.74
HLA mono-allelic B721.221 cells
HLA class I-deficient B721.221 cells were retrovirally transduced to express single HLA alleles (HLA-A∗03:01, HLA-A∗01:01, HLA-A∗02:01, HLA-B∗07:02, HLA-B∗08:01; Gene Bank HG794390, HG794373, HG794376, HG794392, and HG794374, respectively). Cells were retrovirally transduced using a previously published protocol.75 After staining with PE anti-human HLA-A,B,C (clone W6/32; 1:30; Biolegend), stable HLA expressing cell lines were sorted using the Sony SH800 cell sorter.
The HLA mono-allelic B721.221 cells were then retrovirally transduced with plasmids coding for SARS-CoV-2 proteins (spike 1 (aa 1–541), spike 2 (aa 490–975), spike 3 (aa 888–1273), spike, nucleocapsid, membrane-T2A-envelope, and ORF3a; GenBank: MN908947.3) under the control of a tetracycline-inducible promoter (Retro-X™ Tet-One™ Inducible Expression System, Takara Bio) with a puromycin selection marker. Transduced cells were selected by culturing the cells in the presence of 2 μg/mL puromycin (replenished every 48 h) (Gibco) for 7–14 days, followed by further expansion in medium containing 0.5 μg/mL puromycin.
Mass spectrometry
HLA mono-allelic B721.221 cells transduced with SARS-CoV-2 proteins were expanded to 100 × 106 cells and expression of the target proteins was induced by culturing the cells in the presence of doxycycline (1 μg/mL; replenished every 24 h) for 48 h. The cells were then lysed in 1 mL of lysis buffer (PBS containing 1% lauryl maltoside, 1 mM EDTA, 1 mM PMSF and 1:200 Sigma protease inhibitor) for 1 h on ice. HLA peptide complexes were purified by immunoprecipitation.76 Briefly, the HLA peptide complexes were captured on to beads coated with pan HLA class I antibody, and the beads were then washed with 3mL each of 0.1 M Tris-HCl/150 mM NaCl, 0.1 M Tris-HCl/400 mM NaCl, again with 0.1 M Tris-HCl/150 mM NaCl, and finally 0.1 M Tris-HCl. All peptide elutions were desalted with Discovery DSC-18 SPE column, vacuum concentrated and dissolved in 25 μL of 3% acetonitrile containing 0.1% FA.
The peptide solution (5 μL) was analyzed using an Ultimate 3000 nano-UHPLC system (Dionex, Sunnyvale, CA, USA) connected to a Q Exactive mass spectrometer (ThermoElectron, Bremen, Germany) equipped with a nano electrospray ion source as described previously.76 A flow rate of 300 nL/min was employed with a solvent gradient of 3–35% B in 53 min, to 50% B in 3 min and then to 80% B in 1 min. The samples were also analyzed using a longer solvent gradient of 3–35% B in 100 min, to 50% B in 13 min and then to 80% B in 2 min. Solvent A was 0.1% formic acid and solvent B was 0.1% formic acid/90% acetonitrile. The mass spectrometer was operated in the data-dependent mode to automatically switch between MS and MS/MS acquisition. The method used allowed sequential isolation of up to the twelve most intense ions, depending on signal intensity (intensity threshold 1e5), for fragmentation using higher-energy collision induced dissociation (HCD) at a resolution R = 17,500 with NCE 27. The raw data were then analyzed with PEAKS software (Bioinformatics Solutions Inc). The tandem mass spectra were matched against the Uniport homo sapiens database appended with the SARS-CoV-2 proteins. Precursor mass tolerance was set to 10 ppm, methione oxidation was considered as variable modification, enzyme specificity was set to none and a product ion tolerance of 0.05 Da was used. The mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD028862. SARS-CoV-2 peptides identified from the discovery approach were further validated using synthetic peptide analogs. MS/MS fragmentation spectra of synthetic and eluted HLA class I peptides were compared against each other and similarity scores were computed using the Spectrum Similarity function in the R package OrgMassSpecR. As further described in the package vignette, scores were calculated using the following formula:
where · is the dot product and u and v are the aligned intensity vectors of the two spectra. Peptides with similarity score of 0.8 and above were considered as true identifications.
Synthetic peptides
Peptide selection was based on a) peptides identified by Mass-spectrometry and b) 9- and 10-mers predicted to display the highest binding affinity to the MHC according to NetMHCpan4.1 (BA_Rank <0.2%) and/or NetMHC4.0 (Rank <0.25%) (Table S3). One additional peptide was included based on early published reports.35 HLA-A∗01:01, HLA-A∗02:01, HLA-A∗03:01, HLA-B∗07:02, and HLA-B∗08:01 were included to cover the most prevalent HLA alleles in the Caucasian population. In total, 123 peptides derived from spike, nucleocapsid, envelope, membrane, and ORF3a were successfully synthesized at Genscript (purity >70%) and dissolved in DMSO.
Combinatorial multimer staining
Monomer and multimer production
Soluble biotinylated Class-I MHC monomers containing a UV-cleavable peptide in their binding groove were produced in-house according to published protocols77 , 78 and stored at −80°C. The UV-dependent peptide exchange (25 μg/mL UV-monomer and 50 μg/mL peptide diluted in PBS) was performed at a wavelength of 366 nm for 1 h at 4°C. 24 h after UV-exchange, the following streptavidin-tagged fluorochromes were added at optimized ratios to the pMonomer solution: SA-PE (Invitrogen), SA-APC (Invitrogen), SA-BV421 (Biolegend), SA-BV605 (Biolegend), SA-PE-Cy7 (Biolegend), SA-PE-CF594 (BD Biosciences), SA-PE-Cy5 (BD Biosciences), SA-APC-R700 (BD Biosciences), SA-BB790-P (BD Biosciences; prototype kindly provided by Bob Balderas). To block unoccupied binding sites, D-biotin (20 μM; Avidity) was added 24 h after multimerization. Plates were stored in the dark at 4°C until use.
Multimer staining assay
Among the 96 convalescent individuals included in the functional T cell response analysis, 11 did not express the prevalent HLA alleles included in the multimer staining assays, and for two no HLA typing data were available. Thus, PBMC from 83 COVID-19 convalescents (Cohort 1 + 2) and 19 healthy pre-pandemic individuals were included. For 7 convalescent individuals two sampling time points were included (on average 56 [38–81] days between sampling). On Day 0 PBMC were thawed and resuspended at 1 × 106 c/mL in IMDM with L-Glutamine and HEPES (Life Technologies) and loaded for 2 h at 37°C with the peptide master mix containing all 123 pre-selected peptides (each at 100 ng/mL). Cells were washed and plated out at 7.5 × 105 c/well (in 200 μL) in IMDM with L-Glutamine and HEPES +5% human serum (HS, Trina Bioreactives AG) +1x P/S in a polypropylene coated 96-well U-bottom plate. On Day 3, half-medium exchange was performed and 10 IU/mL IL-2 (R&D Systems) added. On Day 5, medium was completely replenished. On Day 7, cells from each individual were pooled, washed and 2–3 x 106 PBMC were resuspended in 50 μL PBS and stained with separate mixes of multimers complexed with 13–29 distinctive peptides, with each multimer conjugated to dual fluorochrome combinations. 1 μL/sample of PE-Cy7/PE-CF594/PE-Cy5/APC-R700 and 2 μL/sample for BV421/BV605/APC/PE/BB790-tagged multimers were added. The sample was incubated for 10 min at room temperature in the dark. Thereafter the following antibodies and LD-NIR (1:1000, ThermoScientific) were added for 30 min at 4°C: FITC anti-human CD8a (clone RPA-T8; 1:100), BV785 anti-human CD19 (clone HIB19; 1:100), BV785 anti-human CD56 (clone 5.1H11; 1:100), BV785 anti-human CD14 (clone M5E2; 1:100), BV785 anti-human CD4 (clone RPA-T4; 1:100) (all from Biolegend). Samples were washed with flow buffer before acquisition on the BD Symphony A5. The gating strategy is outlined in Figure S1. The sample was included in the analysis if at least 4000 live CD8 cells were acquired. An individual was classified as responder to a peptide if the multimer population had 1) at least 5 clearly double-positive events (each multimer conjugated to two different fluorochromes), 2) constituted ≥0.005% of the live CD8, and 3) formed a tight cluster.
Sorting of peptide-specific T cells as cell lines
PBMC were stimulated and stained with multimers and antibodies as described above (section “Multimer staining assay”). Multimer positive cells were sorted onto irradiated feeders (PBMC from 3 donors at equal ratios; 300 kV 10 mA 7 min in X-Vivo-20 (BioNordika) supplemented with 1x P/S, 5% HS, 1 μg/mL PHA (Remel), 875 U/mL IL-2 and 2 ng/mL IL-15 (Peprotech)) as bulk T cells on a FACS Aria II cell sorter (BD Biosciences), followed by expansion in X-Vivo-20 supplemented with 1x P/S, 5% HS, 437.5 U/mL IL-2 and 1 ng/mL IL-15 for generation of T cell lines.
Functional analysis of SARS-CoV-2-specific T cell lines
SARS-CoV-2-specific T cell lines (confirmed by multimer staining to recognize peptide of interest) were combined with mono-allelic B721.221 cells loaded or not with peptide (2 h; 100 ng/mL), or retrovirally transduced to express the relevant SARS-CoV-2 antigen (see section “HLA mono-allelic B721.221 cells”) (pre-treated with doxycycline for 48 h to express the antigen) for 20 h (technical duplicate or triplicate, depending on available cell numbers; effector to target ratio (E:T) = 1:1) in X-Vivo-20 + 1x P/S + 5% HS + 1 μg/mL doxycycline. Harvested cells were incubated with Human Fc Receptor binding inhibitor (AH Diagnostics) for 10 min at room temperature. Subsequently, surface staining was performed by adding the following antibodies: PE anti-human-CD8 a (clone RPA-T8; 1:200; Biolegend), FITC anti-human CD20 (clone 2H7; 1:200; Biolegend), Alexa Fluor 647 anti-human CD137 (clone 4B4-1; 1:100; Thermo Fisher Scientific), together with LD-NIR. After staining the samples for 20 min at 4°C, cells were washed twice with flow buffer and acquired on the LSR II Yellow laser (BD Biosciences). Killing of target cells was quantified by adding CountBright™ Absolute Counting Beads (Invitrogen) following the manufacturer’s instructions before acquisition of sample. A fixed number of beads was acquired for each well. The number of acquired target cells was normalized to the matching mock control (w/o AG) and displayed as % remaining viable cells. The gating strategy is outlined in Figure S5A.
Peptide sequence alignment with human common cold coronaviruses (HCoV)
Homology calculations were performed by aligning the sequence of SARS-CoV-2 epitopes with the corresponding protein sequences of HCoV using the ClustalOmega tool71 with default parameters (sequence IDs: OC43 = P36334, P33469, Q01455; NL63 = Q6Q1S2, Q6Q1R8, Q6Q1R9, Q6Q1S1; HKU1 = Q0ZME7, Q0ZME3, Q0ZME4; 229E = P15423, P15130, P15422). Percent homology was calculated by the number of matching amino acid residues.
Immune Epitope Database analysis
We performed an extensive search for previously published data on T cell responses to SARS-CoV-2 epitopes in the Immune Epitope Database (IEDB) (http://www.iedb.org) (data cut-off date fifth of May 2022). We used the following search criteria in the IEDB search engine.
Epitope: Linear peptide
Assay: T cell
Outcome: positive and negative
Organism: SARS-CoV-2
MHC restriction: class 1
Host: Human
Disease: Any
IEDB entries were removed from further analysis if immunogenicity data could not be mapped unambiguously to a single peptide and HLA allele, and if there was missing data regarding the number of donors tested or the number of donors with an immune response. Furthermore, we included only entries where disease state was specified as COVID-19 and disease stage as “post” or “active/recent onset”. We generated summary statistics for each peptide, HLA allele and study. If a given peptide/HLA combination was tested by more than one assay in a single study, we moved forward with the highest number of donors tested and the highest number of donors with an immune response. When evaluating the impact of immunoprevalence on estimated population coverage in random sets of epitopes from the IEDB, we included only epitopes with data from >20 individuals and/or >1 study.
Estimating population coverage adjusted for immunoprevalence
Suppose a vaccine is made using the epitope set . Every individual has different loci, and two alleles at each locus. An epitope may be independently presented on one or more of these alleles, at any given locus, and we assume that one response-generating hit (hit = allele-epitope match) is sufficient for the vaccine to generate protection. We further assume that epitope-specific immune responses are independent events. Given this, we can calculate the probability of response from at least one locus as:
where is the probability of response to an epitope presented on locus . We can also write it as:
To calculate , we need to know the relative population frequencies of every possible allele combination for that locus, as well as the probability that each allele combination generates an immune response to at least one epitope. HLA allele frequencies were obtained from the Allele Frequency database79 and scaled to 1 for each locus. If we let denote one of all the possible allele combinations occurring in the population for locus , and let denote the population percentage with this combination, then since allele combinations are mutually exclusive events, we can write the probability of response with a given locus as:
where is the probability of an immune response from at least one epitope-allele pair, given the allele combination and its set of compatible epitopes , where is a subset of .
To estimate the probability that a presented epitope results in a response, we used immunoprevalence data from convalescent individuals. If we let be the immunoprevalence for a given allele-epitope pair, we can write the probability that an individual with the allele combination generates an immune response to the epitope at locus as:
where and are the first and second alleles found at locus , respectively. Note that if an individual is heterozygous, and both alleles can present the epitope, both alleles have an independent chance of generating a response. If an allele does not bind with , then .
Since each allele combination can have multiple compatible epitopes in , every allele combination can get multiple attempts at generating a response, where the number of attempts is . We can then write the probability that an individual with the allele combination generates a response from at least one of the epitopes in as:
This is assuming that . If there are no compatible epitopes , then . Once all the weights have been calculated for all the loci, we can substitute this into the first equation to get a point estimate for the predicted population coverage, given a vector of alleles, their population frequencies, an epitope set, and their associated immunoprevalences for different alleles.
To compute a confidence interval for the predicted coverage, we use parametric bootstrapping to resample new immunoprevalence values for each epitope-allele pair. If we assume that the number of patients with a compatible allele who generate a response to an immunogenic epitope is binomially distributed, we can model the distribution of with the beta distribution:
where and are the numbers of responders and non-responders, respectively, from which immunoprevalence was calculated for that allele-epitope combination. Because the beta distribution is not defined when , we added 1 to the number of non-responders for every epitope. By repeatedly resampling new immunoprevalences from a beta distribution with shape parameters determined by the immunoprevalence data, this will generate a distribution of population coverages. The 95% confidence interval is then given by the 2.5th and 97.fifth percentiles of this distribution.
Pooled analysis of epitope-specific multimer response magnitudes
We first ranked the responses to each of the 9 most prevalent epitopes in descending order of magnitude, with draws given the same rank. Since the number of donors tested for each epitope was different, we also performed Z score normalization of the ranks to bring them all on the same scale. We moved forward with pooled analysis including all donors tested for at least two of the 9 prevalent epitopes. Since the number of epitopes tested in each donor was often greater than two, we randomly selected two epitopes for analysis and repeated the procedure 1000 times to account for sampling variation. For each of the 1000 datasets, we performed linear regression analysis with the formula: random_1 ∼ random_2 + age + days. Here, “random_1” and “random_2” represented the normalized multimer response ranks, “age” represented the donor age at the time of sample acquisition and “days” represented the number of days passed between COVID-19 diagnosis and sample acquisition.
Quantification and statistical analysis
Flow cytometry data acquired on different BD Biosciences instruments were analyzed in FlowJoV10.6.2 (TreeStar). Statistical analysis was performed in R version 4.0.4 and GraphPad Prism version 8.3.0. The use of statistical tests including p value thresholds and adjustment for multiple testing is specified in the text and figure legends. In general, we used Fisher’s exact test for contingency tables and Wilcoxon rank-sum test for non-parametric continuous variables. For correlation analysis we used the Pearson method whenever its assumptions were met, otherwise we used Spearman rank correlation. We reasoned that the proportion of donors showing an immune response to a particular epitope could be modeled by the binomial distribution, allowing us to estimate binomial 95% confidence intervals for immunoprevalence. Moreover, we used binomial tests for the hypothesis that the true immunoprevalence of each epitope is higher than given thresholds (i.e., 50% and 70%). Bootstrapping was applied to generate percentile-based 95% confidence intervals for the percentage of pHLA containing mutations in VOC. Adjustment for multiple comparisons was done using the Benjamini-Hochberg method when specified in text or figure legends.
Acknowledgments
We express our gratitude to all donors and health care personnel involved in this work. We thank Oslo University Hospital flow cytometry core facility for excellent technical assistance.
. This work was supported by South-Eastern Regional Health Authority Norway and Oslo University Hospital (F.L.-J. and J.O.), as well as the Research Council of Norway (J.O.). Schematic illustrations were created with BioRender (https://BioRender.com).
Author contributions
Conceptualization: S.M., I.B., F.L.-J., E.H.R., and J.O.; methodology: S.M., I.B., R.C.B., F.L.-J., E.H.R., and J.O.; software: T.S.M. and E.H.R.; formal analysis: S.M., I.B., R.C.B., E.H.R., F.L.-J., B.T., and J.T.V.; investigation: S.M., I.B., R.C.B., F.L.-J., and T.T.T.; resources: W.Y., K.D., M.L., M.M.N., M.D.-S., C.K., A.G., L.N.-M., and A.S.; data curation: S.M. and E.H.R.; writing – original draft: S.M., R.C.B., I.B., E.H.R., F.L.-J., and J.O.; writing – review and editing: S.M., R.C.B., E.H.R., F.L.-J., and J.O.; visualization: S.M. and E.H.R.; supervision: J.O.; project administration: S.M., E.H.R., and J.O.; funding acquisition: J.O. and F.L.-J.
Declaration of interests
J.O. is the author on a patent protecting a method for identification of T cell receptors and on patent applications protecting T cell receptor sequences for potential use in cancer immunotherapy. J.O. is a member of the Scientific Advisory Board of Asgard Therapeutics. The other authors declare no competing interest.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.111995.
Supplemental information
Data and code availability
Data supporting findings of this study are available within the article and its supplementary information files. The mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD028862. All original code has been deposited at GitHub (https://doi.org/10.5281/zenodo.7447391) publicly available as of the date of publication and is available in this paper’s supplemental information (Data S2). DOIs are listed in the key resources table. Other analysis code and additional raw data required to reanalyze the data reported in this paper are available from the lead contact upon reasonable request.
References
- 1.Wherry E.J., Barouch D.H. T cell immunity to COVID-19 vaccines. Science. 2022;377:821–822. doi: 10.1126/science.add2897. [DOI] [PubMed] [Google Scholar]
- 2.Sormani M.P., Schiavetti I., Inglese M., Carmisciano L., Laroni A., Lapucci C., Visconti V., Serrati C., Gandoglia I., Tassinari T., et al. Breakthrough SARS-CoV-2 infections after COVID-19 mRNA vaccination in MS patients on disease modifying therapies during the Delta and the Omicron waves in Italy. EBioMedicine. 2022;80:104042. doi: 10.1016/j.ebiom.2022.104042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keeton R., Tincho M.B., Ngomti A., Baguma R., Benede N., Suzuki A., Khan K., Cele S., Bernstein M., Karim F., et al. T cell responses to SARS-CoV-2 spike cross-recognize Omicron. Nature. 2022;603:488–492. doi: 10.1038/s41586-022-04460-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Y., Chen L., Yin S., Tao Y., Zhu L., Tong X., Mao M., Li M., Wan Y., Ni J., et al. The Third dose of CoronVac vaccination induces broad and potent adaptive immune responses that recognize SARS-CoV-2 Delta and Omicron variants. Emerg. Microbes Infect. 2022;11:1524–1536. doi: 10.1080/22221751.2022.2081614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dolton G., Rius C., Hasan M.S., Wall A., Szomolay B., Behiry E., Whalley T., Southgate J., Fuller A., COVID-19 Genomics UK COG-UK consortium. Morin T., et al. Emergence of immune escape at dominant SARS-CoV-2 killer T cell epitope. Cell. 2022;185:2936–2951.e19. doi: 10.1016/j.cell.2022.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le Bert N., Tan A., Kunasegaran K., Chia A., Tan N., Chen Q., Hang S.K., Qui M.D.C., Chan B.S.W., Low J.G.H., et al. Mutations of SARS-CoV-2 variants of concern escaping Spike-specific T cells. bioRxiv. 2022 doi: 10.1101/2022.01.20.477163. Preprint at. [DOI] [Google Scholar]
- 7.Emmelot M.E., Vos M., Boer M.C., Rots N.Y., de Wit J., van Els C.A.C.M., Kaaijk P. Omicron BA.1 mutations in SARS-CoV-2 spike lead to reduced T-cell response in vaccinated and convalescent individuals. Viruses. 2022;14 doi: 10.3390/v14071570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reynolds C.J., Pade C., Gibbons J.M., Otter A.D., Lin K.M., Muñoz Sandoval D., Pieper F.P., Butler D.K., Liu S., Joy G., et al. Immune boosting by B.1.1.529 (Omicron) depends on previous SARS-CoV-2 exposure. Science. 2022;377:eabq1841. doi: 10.1126/science.abq1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferretti A.P., Kula T., Wang Y., Nguyen D.M.V., Weinheimer A., Dunlap G.S., Xu Q., Nabilsi N., Perullo C.R., Cristofaro A.W., et al. Unbiased screens show CD8(+) T cells of COVID-19 patients recognize shared epitopes in SARS-CoV-2 that largely reside outside the spike protein. Immunity. 2020;53:1095–1107.e3. doi: 10.1016/j.immuni.2020.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tarke A., Sidney J., Kidd C.K., Dan J.M., Ramirez S.I., Yu E.D., Mateus J., da Silva Antunes R., Moore E., Rubiro P., et al. Comprehensive analysis of T cell immunodominance and immunoprevalence of SARS-CoV-2 epitopes in COVID-19 cases. Cell Rep. Med. 2021;2:100204. doi: 10.1016/j.xcrm.2021.100204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snyder T.M., Gittelman R.M., Klinger M., May D.H., Osborne E.J., Taniguchi R., Zahid H.J., Kaplan I.M., Dines J.N., Noakes M.T., et al. Magnitude and dynamics of the T-cell response to SARS-CoV-2 infection at both individual and population levels. medRxiv. 2020 doi: 10.1101/2020.07.31.20165647. Preprint at. [DOI] [Google Scholar]
- 12.Habel J.R., Nguyen T.H.O., van de Sandt C.E., Juno J.A., Chaurasia P., Wragg K., Koutsakos M., Hensen L., Jia X., Chua B., et al. Suboptimal SARS-CoV-2-specific CD8(+) T cell response associated with the prominent HLA-A∗02:01 phenotype. Proc. Natl. Acad. Sci. USA. 2020;117:24384–24391. doi: 10.1073/pnas.2015486117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shomuradova A.S., Vagida M.S., Sheetikov S.A., Zornikova K.V., Kiryukhin D., Titov A., Peshkova I.O., Khmelevskaya A., Dianov D.V., Malasheva M., et al. SARS-CoV-2 epitopes are recognized by a public and diverse repertoire of human T cell receptors. Immunity. 2020;53:1245–1257.e5. doi: 10.1016/j.immuni.2020.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng Y., Mentzer A.J., Liu G., Yao X., Yin Z., Dong D., Dejnirattisai W., Rostron T., Supasa P., Liu C., et al. Broad and strong memory CD4(+) and CD8(+) T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020;21:1336–1345. doi: 10.1038/s41590-020-0782-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poran A., Harjanto D., Malloy M., Arieta C.M., Rothenberg D.A., Lenkala D., van Buuren M.M., Addona T.A., Rooney M.S., Srinivasan L., Gaynor R.B. Sequence-based prediction of SARS-CoV-2 vaccine targets using a mass spectrometry-based bioinformatics predictor identifies immunogenic T cell epitopes. Genome Med. 2020;12:70. doi: 10.1186/s13073-020-00767-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prakash S., Srivastava R., Coulon P.G., Dhanushkodi N.R., Chentoufi A.A., Tifrea D.F., Edwards R.A., Figueroa C.J., Schubl S.D., Hsieh L., et al. Genome-wide B cell, CD4(+), and CD8(+) T cell epitopes that are highly conserved between human and animal coronaviruses, identified from SARS-CoV-2 as targets for preemptive pan-coronavirus vaccines. J. Immunol. 2021;206:2566–2582. doi: 10.4049/jimmunol.2001438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grifoni A., Sidney J., Vita R., Peters B., Crotty S., Weiskopf D., Sette A. SARS-CoV-2 human T cell epitopes: adaptive immune response against COVID-19. Cell Host Microbe. 2021;29:1076–1092. doi: 10.1016/j.chom.2021.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagler A., Kalaora S., Barbolin C., Gangaev A., Ketelaars S.L.C., Alon M., Pai J., Benedek G., Yahalom-Ronen Y., Erez N., et al. Identification of presented SARS-CoV-2 HLA class I and HLA class II peptides using HLA peptidomics. Cell Rep. 2021;35:109305. doi: 10.1016/j.celrep.2021.109305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saini S.K., Hersby D.S., Tamhane T., Povlsen H.R., Amaya Hernandez S.P., Nielsen M., Gang A.O., Hadrup S.R. SARS-CoV-2 genome-wide T cell epitope mapping reveals immunodominance and substantial CD8(+) T cell activation in COVID-19 patients. Sci. Immunol. 2021;6:eabf7550. doi: 10.1126/sciimmunol.abf7550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kared H., Redd A.D., Bloch E.M., Bonny T.S., Sumatoh H., Kairi F., Carbajo D., Abel B., Newell E.W., Bettinotti M.P., et al. SARS-CoV-2-specific CD8+ T cell responses in convalescent COVID-19 individuals. J. Clin. Invest. 2021;131:e145476. doi: 10.1172/JCI145476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schulien I., Kemming J., Oberhardt V., Wild K., Seidel L.M., Killmer S., Sagar, Daul F., Salvat Lago M., Decker A., et al. Characterization of pre-existing and induced SARS-CoV-2-specific CD8(+) T cells. Nat. Med. 2021;27:78–85. doi: 10.1038/s41591-020-01143-2. [DOI] [PubMed] [Google Scholar]
- 22.Nelde A., Bilich T., Heitmann J.S., Maringer Y., Salih H.R., Roerden M., Lübke M., Bauer J., Rieth J., Wacker M., et al. SARS-CoV-2-derived peptides define heterologous and COVID-19-induced T cell recognition. Nat. Immunol. 2021;22:74–85. doi: 10.1038/s41590-020-00808-x. [DOI] [PubMed] [Google Scholar]
- 23.Agerer B., Koblischke M., Gudipati V., Montaño-Gutierrez L.F., Smyth M., Popa A., Genger J.W., Endler L., Florian D.M., Mühlgrabner V., et al. SARS-CoV-2 mutations in MHC-I-restricted epitopes evade CD8(+) T cell responses. Sci. Immunol. 2021;6:eabg6461. doi: 10.1126/sciimmunol.abg6461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rha M.S., Jeong H.W., Ko J.H., Choi S.J., Seo I.H., Lee J.S., Sa M., Kim A.R., Joo E.J., Ahn J.Y., et al. PD-1-Expressing SARS-CoV-2-specific CD8(+) T cells are not exhausted, but functional in patients with COVID-19. Immunity. 2021;54:44–52.e3. doi: 10.1016/j.immuni.2020.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nielsen S.S., Vibholm L.K., Monrad I., Olesen R., Frattari G.S., Pahus M.H., Højen J.F., Gunst J.D., Erikstrup C., Holleufer A., et al. SARS-CoV-2 elicits robust adaptive immune responses regardless of disease severity. EBioMedicine. 2021;68:103410. doi: 10.1016/j.ebiom.2021.103410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lineburg K.E., Grant E.J., Swaminathan S., Chatzileontiadou D.S.M., Szeto C., Sloane H., Panikkar A., Raju J., Crooks P., Rehan S., et al. CD8(+) T cells specific for an immunodominant SARS-CoV-2 nucleocapsid epitope cross-react with selective seasonal coronaviruses. Immunity. 2021;54:1055–1065.e5. doi: 10.1016/j.immuni.2021.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan K., Chiu Y., Huang E., Chen M., Wang J., Lai I., Singh S., Shaw R., MacCoss M., Yee C. Immunogenic SARS-CoV2 epitopes defined by mass spectrometry. bioRxiv. 2021 doi: 10.1101/2021.07.20.453160. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z., Ruan P., Wang L., Nie X., Ma X., Tan Y. T and B cell Epitope analysis of SARS-CoV-2 S protein based on immunoinformatics and experimental research. J. Cell Mol. Med. 2021;25:1274–1289. doi: 10.1111/jcmm.16200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joag V., Wijeyesinghe S., Stolley J.M., Quarnstrom C.F., Dileepan T., Soerens A.G., Sangala J.A., O'Flanagan S.D., Gavil N.V., Hong S.W., et al. Cutting edge: mouse SARS-CoV-2 epitope reveals infection and vaccine-elicited CD8 T cell responses. J. Immunol. 2021;206:931–935. doi: 10.4049/jimmunol.2001400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sahin U., Muik A., Vogler I., Derhovanessian E., Kranz L.M., Vormehr M., Quandt J., Bidmon N., Ulges A., Baum A., et al. BNT162b2 vaccine induces neutralizing antibodies and poly-specific T cells in humans. Nature. 2021;595:572–577. doi: 10.1038/s41586-021-03653-6. [DOI] [PubMed] [Google Scholar]
- 31.Gangaev A., Ketelaars S.L.C., Isaeva O.I., Patiwael S., Dopler A., Hoefakker K., De Biasi S., Gibellini L., Mussini C., Guaraldi G., et al. Identification and characterization of a SARS-CoV-2 specific CD8(+) T cell response with immunodominant features. Nat. Commun. 2021;12:2593. doi: 10.1038/s41467-021-22811-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee E., Sandgren K., Duette G., Stylianou V.V., Khanna R., Eden J.S., Blyth E., Gottlieb D., Cunningham A.L., Palmer S. Identification of SARS-CoV-2 nucleocapsid and spike T-cell epitopes for assessing T-cell immunity. J. Virol. 2021;95:e02002–e02020. doi: 10.1128/JVI.02002-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quadeer A.A., Ahmed S.F., McKay M.R. Landscape of epitopes targeted by T cells in 852 individuals recovered from COVID-19: meta-analysis, immunoprevalence, and web platform. Cell Rep. Med. 2021;2:100312. doi: 10.1016/j.xcrm.2021.100312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weingarten-Gabbay S., Klaeger S., Sarkizova S., Pearlman L.R., Chen D.Y., Gallagher K.M.E., Bauer M.R., Taylor H.B., Dunn W.A., Tarr C., et al. Profiling SARS-CoV-2 HLA-I peptidome reveals T cell epitopes from out-of-frame ORFs. Cell. 2021;184:3962–3980.e17. doi: 10.1016/j.cell.2021.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sekine T., Perez-Potti A., Rivera-Ballesteros O., Strålin K., Gorin J.B., Olsson A., Llewellyn-Lacey S., Kamal H., Bogdanovic G., Muschiol S., et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID-19. Cell. 2020;183:158–168.e14. doi: 10.1016/j.cell.2020.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Veneti L., Seppala E., Larsdatter Storm M., Valcarcel Salamanca B., Alnes Buanes E., Aasand N., Naseer U., Bragstad K., Hungnes O., Boas H., et al. Increased risk of hospitalisation and intensive care admission associated with reported cases of SARS-CoV-2 variants B.1.1.7 and B.1.351 in Norway, December 2020 -May 2021. PLoS One. 2021;16:e0258513. doi: 10.1371/journal.pone.0258513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lübke M., Spalt S., Kowalewski D.J., Zimmermann C., Bauersfeld L., Nelde A., Bichmann L., Marcu A., Peper J.K., Kohlbacher O., et al. Identification of HCMV-derived T cell epitopes in seropositive individuals through viral deletion models. J. Exp. Med. 2020;217 doi: 10.1084/jem.20191164. jem.20191164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ali M., Foldvari Z., Giannakopoulou E., Böschen M.L., Strønen E., Yang W., Toebes M., Schubert B., Kohlbacher O., Schumacher T.N., Olweus J. Induction of neoantigen-reactive T cells from healthy donors. Nat. Protoc. 2019;14:1926–1943. doi: 10.1038/s41596-019-0170-6. [DOI] [PubMed] [Google Scholar]
- 39.Francis J.M., Leistritz-Edwards D., Dunn A., Tarr C., Lehman J., Dempsey C., Hamel A., Rayon V., Liu G., Wang Y., et al. Allelic variation in class I HLA determines CD8(+) T cell repertoire shape and cross-reactive memory responses to SARS-CoV-2. Sci. Immunol. 2021;7:eabk3070. doi: 10.1126/sciimmunol.abk3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peng Y., Felce S.L., Dong D., Penkava F., Mentzer A.J., Yao X., Liu G., Yin Z., Chen J.L., Lu Y., et al. An immunodominant NP105-113-B∗07:02 cytotoxic T cell response controls viral replication and is associated with less severe COVID-19 disease. Nat. Immunol. 2022;23:50–61. doi: 10.1038/s41590-021-01084-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dan J.M., Mateus J., Kato Y., Hastie K.M., Yu E.D., Faliti C.E., Grifoni A., Ramirez S.I., Haupt S., Frazier A., et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science. 2021;371:eabf4063. doi: 10.1126/science.abf4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bui H.H., Sidney J., Dinh K., Southwood S., Newman M.J., Sette A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinformatics. 2006;7:153. doi: 10.1186/1471-2105-7-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nguyen T.H.O., Rowntree L.C., Petersen J., Chua B.Y., Hensen L., Kedzierski L., van de Sandt C.E., Chaurasia P., Tan H.X., Habel J.R., et al. CD8(+) T cells specific for an immunodominant SARS-CoV-2 nucleocapsid epitope display high naive precursor frequency and TCR promiscuity. Immunity. 2021;54:1066–1082.e5. doi: 10.1016/j.immuni.2021.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu C., Shen M., Han X., Chen Q., Li L., Chen S., Zhang J., Gao F., Wang W., Wang Y., et al. Identification of cross-reactive CD8(+) T cell receptors with high functional avidity to a SARS-CoV-2 immunodominant epitope and its natural mutant variants. Genes Dis. 2022;9:216–229. doi: 10.1016/j.gendis.2021.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oberhardt V., Luxenburger H., Kemming J., Schulien I., Ciminski K., Giese S., Csernalabics B., Lang-Meli J., Janowska I., Staniek J., et al. Rapid and stable mobilization of CD8(+) T cells by SARS-CoV-2 mRNA vaccine. Nature. 2021;597:268–273. doi: 10.1038/s41586-021-03841-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Minervina A.A., Pogorelyy M.V., Kirk A.M., Crawford J.C., Allen E.K., Chou C.H., Mettelman R.C., Allison K.J., Lin C.Y., Brice D.C., et al. SARS-CoV-2 antigen exposure history shapes phenotypes and specificity of memory CD8(+) T cells. Nat. Immunol. 2022;23:781–790. doi: 10.1038/s41590-022-01184-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chaurasia P., Nguyen T.H.O., Rowntree L.C., Juno J.A., Wheatley A.K., Kent S.J., Kedzierska K., Rossjohn J., Petersen J. Structural basis of biased T cell receptor recognition of an immunodominant HLA-A2 epitope of the SARS-CoV-2 spike protein. J. Biol. Chem. 2021;297:101065. doi: 10.1016/j.jbc.2021.101065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brunk F., Moritz A., Nelde A., Bilich T., Casadei N., Fraschka S.A.K., Heitmann J.S., Hörber S., Peter A., Rammensee H.G., et al. SARS-CoV-2-reactive T-cell receptors isolated from convalescent COVID-19 patients confer potent T-cell effector function. Eur. J. Immunol. 2021;51:2651–2664. doi: 10.1002/eji.202149290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang H., Deng S., Ren L., Zheng P., Hu X., Jin T., Tan X. Profiling CD8(+) T cell epitopes of COVID-19 convalescents reveals reduced cellular immune responses to SARS-CoV-2 variants. Cell Rep. 2021;36:109708. doi: 10.1016/j.celrep.2021.109708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wagner K.I., Mateyka L.M., Jarosch S., Grass V., Weber S., Schober K., Hammel M., Burrell T., Kalali B., Poppert H., et al. Recruitment of highly cytotoxic CD8(+) T cell receptors in mild SARS-CoV-2 infection. Cell Rep. 2022;38:110214. doi: 10.1016/j.celrep.2021.110214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Riise J., Meyer S., Blaas I., Chopra A., Tran T.T., Delic-Sarac M., Hestdalen M.L., Brodin E., Rustad E.H., Dai K.Z., et al. Rituximab-treated patients with lymphoma develop strong CD8 T-cell responses following COVID-19 vaccination. Br. J. Haematol. 2022;197:697–708. doi: 10.1111/bjh.18149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Altarawneh H.N., Chemaitelly H., Ayoub H.H., Tang P., Hasan M.R., Yassine H.M., Al-Khatib H.A., Smatti M.K., Coyle P., Al-Kanaani Z., et al. Effects of previous infection and vaccination on symptomatic Omicron infections. N. Engl. J. Med. 2022;387:21–34. doi: 10.1056/NEJMoa2203965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Severe Covid-19 GWAS Group. Ellinghaus D., Degenhardt F., Bujanda L., Buti M., Albillos A., Invernizzi P., Fernández J., Prati D., Baselli G., Asselta R., et al. Genomewide association study of severe covid-19 with respiratory failure. N. Engl. J. Med. 2020;383:1522–1534. doi: 10.1056/NEJMoa2020283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Britanova O.V., Putintseva E.V., Shugay M., Merzlyak E.M., Turchaninova M.A., Staroverov D.B., Bolotin D.A., Lukyanov S., Bogdanova E.A., Mamedov I.Z., et al. Age-related decrease in TCR repertoire diversity measured with deep and normalized sequence profiling. J. Immunol. 2014;192:2689–2698. doi: 10.4049/jimmunol.1302064. [DOI] [PubMed] [Google Scholar]
- 55.Sethna Z., Isacchini G., Dupic T., Mora T., Walczak A.M., Elhanati Y. Population variability in the generation and selection of T-cell repertoires. PLoS Comput. Biol. 2020;16:e1008394. doi: 10.1371/journal.pcbi.1008394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krishna C., Chowell D., Gönen M., Elhanati Y., Chan T.A. Genetic and environmental determinants of human TCR repertoire diversity. Immun. Ageing. 2020;17:26. doi: 10.1186/s12979-020-00195-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arunachalam P.S., Wimmers F., Mok C.K.P., Perera R.A.P.M., Scott M., Hagan T., Sigal N., Feng Y., Bristow L., Tak-Yin Tsang O., et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science. 2020;369:1210–1220. doi: 10.1126/science.abc6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laing A.G., Lorenc A., Del Molino Del Barrio I., Das A., Fish M., Monin L., Muñoz-Ruiz M., McKenzie D.R., Hayday T.S., Francos-Quijorna I., et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020;26:1623–1635. doi: 10.1038/s41591-020-1038-6. [DOI] [PubMed] [Google Scholar]
- 59.Blanco-Melo D., Nilsson-Payant B.E., Liu W.C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D., et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hadjadj J., Yatim N., Barnabei L., Corneau A., Boussier J., Smith N., Péré H., Charbit B., Bondet V., Chenevier-Gobeaux C., et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nilsson J.B., Grifoni A., Tarke A., Sette A., Nielsen M. PopCover-2.0. Improved selection of peptide sets with optimal HLA and pathogen diversity coverage. Front. Immunol. 2021;12:728936. doi: 10.3389/fimmu.2021.728936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weisblum Y., Schmidt F., Zhang F., DaSilva J., Poston D., Lorenzi J.C., Muecksch F., Rutkowska M., Hoffmann H.H., Michailidis E., et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. Elife. 2020;9:e61312. doi: 10.7554/eLife.61312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang Z., Schmidt F., Weisblum Y., Muecksch F., Barnes C.O., Finkin S., Schaefer-Babajew D., Cipolla M., Gaebler C., Lieberman J.A., et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature. 2021;592:616–622. doi: 10.1038/s41586-021-03324-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shinde V., Bhikha S., Hoosain Z., Archary M., Bhorat Q., Fairlie L., Lalloo U., Masilela M.S.L., Moodley D., Hanley S., et al. Efficacy of NVX-CoV2373 covid-19 vaccine against the B.1.351 variant. N. Engl. J. Med. 2021;384:1899–1909. doi: 10.1056/NEJMoa2103055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garcia-Beltran W.F., Lam E.C., St Denis K., Nitido A.D., Garcia Z.H., Hauser B.M., Feldman J., Pavlovic M.N., Gregory D.J., Poznansky M.C., et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell. 2021;184:2523. doi: 10.1016/j.cell.2021.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Emary K.R.W., Golubchik T., Aley P.K., Ariani C.V., Angus B., Bibi S., Blane B., Bonsall D., Cicconi P., Charlton S., et al. Efficacy of ChAdOx1 nCoV-19 (AZD1222) vaccine against SARS-CoV-2 variant of concern 202012/01 (B.1.1.7): an exploratory analysis of a randomised controlled trial. Lancet. 2021;397:1351–1362. doi: 10.1016/S0140-6736(21)00628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thompson R.N., Hill E.M., Gog J.R. SARS-CoV-2 incidence and vaccine escape. Lancet Infect. Dis. 2021;21:913–914. doi: 10.1016/S1473-3099(21)00202-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen R.E., Zhang X., Case J.B., Winkler E.S., Liu Y., VanBlargan L.A., Liu J., Errico J.M., Xie X., Suryadevara N., et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat. Med. 2021;27:717–726. doi: 10.1038/s41591-021-01294-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harvey W.T., Carabelli A.M., Jackson B., Gupta R.K., Thomson E.C., Harrison E.M., Ludden C., Reeve R., Rambaut A., Peacock S.J., Robertson D.L., COVID-19 Genomics UK COG-UK Consortium SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021;19:409–424. doi: 10.1038/s41579-021-00573-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lu L., Mok B.W.Y., Chen L.L., Chan J.M.C., Tsang O.T.Y., Lam B.H.S., Chuang V.W.M., Chu A.W.H., Chan W.M., Ip J.D., et al. Neutralization of severe acute respiratory syndrome coronavirus 2 Omicron variant by sera from BNT162b2 or CoronaVac vaccine recipients. Clin. Infect. Dis. 2022;75:e822–e826. doi: 10.1093/cid/ciab1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu W., Slåstad H., de la Rosa Carrillo D., Frey T., Tjønnfjord G., Boretti E., Aasheim H.C., Horejsi V., Lund-Johansen F. Antibody array analysis with label-based detection and resolution of protein size. Mol. Cell. Proteomics. 2009;8:245–257. doi: 10.1074/mcp.M800171-MCP200. [DOI] [PubMed] [Google Scholar]
- 73.Amanat F., Stadlbauer D., Strohmeier S., Nguyen T.H.O., Chromikova V., McMahon M., Jiang K., Arunkumar G.A., Jurczyszak D., Polanco J., et al. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat. Med. 2020;26:1033–1036. doi: 10.1038/s41591-020-0913-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Holter J.C., Pischke S.E., de Boer E., Lind A., Jenum S., Holten A.R., Tonby K., Barratt-Due A., Sokolova M., Schjalm C., et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc. Natl. Acad. Sci. USA. 2020;117:25018–25025. doi: 10.1073/pnas.2010540117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Strønen E., Toebes M., Kelderman S., van Buuren M.M., Yang W., van Rooij N., Donia M., Böschen M.L., Lund-Johansen F., Olweus J., Schumacher T.N. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science. 2016;352:1337–1341. doi: 10.1126/science.aaf2288. [DOI] [PubMed] [Google Scholar]
- 76.Ali M., Giannakopoulou E., Li Y., Lehander M., Virding Culleton S., Yang W., Knetter C., Odabasi M.C., Bollineni R.C., Yang X., et al. T cells targeted to TdT kill leukemic lymphoblasts while sparing normal lymphocytes. Nat. Biotechnol. 2022;40:488–498. doi: 10.1038/s41587-021-01089-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Toebes M., Coccoris M., Bins A., Rodenko B., Gomez R., Nieuwkoop N.J., van de Kasteele W., Rimmelzwaan G.F., Haanen J.B.A.G., Ovaa H., Schumacher T.N.M. Design and use of conditional MHC class I ligands. Nat. Med. 2006;12:246–251. doi: 10.1038/nm1360. [DOI] [PubMed] [Google Scholar]
- 78.Toebes M., Rodenko B., Ovaa H., Schumacher T.N. Generation of peptide MHC class I monomers and multimers through ligand exchange. Curr. Protoc. Immunol. 2009;18:18.16.1–18.16.20. doi: 10.1002/0471142735.im1816s87. [DOI] [PubMed] [Google Scholar]
- 79.Gonzalez-Galarza F.F., McCabe A., Santos E., Jones J., Takeshita L., Ortega-Rivera N.D., Cid-Pavon G.M.D., Ramsbottom K., Ghattaoraya G., Alfirevic A., et al. Allele frequency net database (AFND) 2020 update: gold-standard data classification, open access genotype data and new query tools. Nucleic Acids Res. 2020;48:D783–D788. doi: 10.1093/nar/gkz1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting findings of this study are available within the article and its supplementary information files. The mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD028862. All original code has been deposited at GitHub (https://doi.org/10.5281/zenodo.7447391) publicly available as of the date of publication and is available in this paper’s supplemental information (Data S2). DOIs are listed in the key resources table. Other analysis code and additional raw data required to reanalyze the data reported in this paper are available from the lead contact upon reasonable request.