

Abstract
Cancer cells evade the immune system by expressing inhibitory immune checkpoint receptors such as ecto-5’-nucleotidase (NT5E), also known as CD73, which consequently suppress tumor neoantigen-specific immune response. Blockade of CD73 in mouse models of breast cancer showed a reduction in tumor growth and metastasis. CD73 expression is elevated in a variety of human tumors including breast cancer. While the regulation of CD73 expression at the transcriptional level has been well understood, the factors involved in regulating CD73 expression at the post-transcriptional level have not been identified. Herein, we discovered that the ubiquitin-specific peptidase 22 (USP22), a deubiquitinase associated with poor prognosis and overexpressed in breast cancers, is a positive regulator for CD73. Targeted USP22 deletion resulted in a statistically significant reduction in CD73 protein expression. In contrast, CD73 mRNA expression levels were not reduced, but even slightly increased by USP22 deletion. Further analysis demonstrated that USP22 is a deubiquitinase that specifically interacts with and inhibits CD73 ubiquitination. Consequently, USP22 protects CD73 from ubiquitin-mediated proteasomal degradation in breast cancer cells. Targeted USP22 deletion, inhibits syngeneic breast cancer growth. Collectively, our study reveals USP22 as a positive regulator to promote CD73 expression in breast cancer and provides a rationale to target USP22 in antitumor immune therapy.
Keywords: Breast cancer, USP22, CD73, immunotherapy
Introduction
Aggressive cancers are particularly tough to treat because of their rapid growth, recruitment of suppressive cells, and upregulation of inhibitory receptors [1]. CD73 is highly upregulated in many types of cancers; notably, its high expression is associated with metastasis and shorter patient survival time in breast cancer [2-5]. CD73 is a glycosylphosphatidylinositol linked cell surface dimeric enzyme, that converts adenosine monophosphate (AMP) into immune suppressive adenosine [6-8]. The extracellular adenosine generated from the enzymatic activity of CD73 can bind to adenosine receptors A1, A2A, A2B, and A3 found on immune cells [9]. The A2A receptor on T cells inhibits their effector functions and recruits Regulatory T cells (Tregs) while also increasing their immunosuppressive abilities [10-12]. CD73 gene expression is regulated by multiple pathways including cytokines, such as TGF-β, and hypoxia through their downstream transcription factors including SMADs 2-5 and hypoxia-inducible factor-1α (HIF-1α) [13]. It has been shown that post-translational modifications, including glycosylation and ADP-ribosylation, are involved in regulating CD73 cell surface expression [14,15]. However, the potential involvement of the ubiquitin pathway in the regulation of CD73 expression has not been identified.
USP22 is one of the 11 “death by cancer genes” that is associated with poor prognosis [16] and is overexpressed in many types of cancers such as prostate and breast cancer [17]. USP22 expression in breast cancer is correlated with disease aggressiveness and shorter patient survival [17]. USP22 functions as a part of the deubiquitinating module of the spt-Ada-Gcn5 acetyltransferase (SAGA) transcription co-activator complex, in which it removes ubiquitin from histones H2A and histone H2B leading to regulation of gene transcription [18-20]. We and others have demonstrated that USP22 regulates many proteins important for cell cycle progression such as p53, c-Myc, and cyclin B1 which promotes tumor cell growth and survival [21-23]. More recently, it has been shown that USP22 promotes HER2-driven mammary carcinoma aggressiveness by suppressing the unfolded protein response [24]. In addition, recent studies have suggested that USP22 is involved in regulating tumor cell-intrinsic immune suppression [25,26].
In this study, we identified USP22 as a deubiquitinase of CD73 in breast cancer cells. Deletion of USP22 expression by CRISPR in the mouse triple-negative breast cancer cell line 4T1 resulted in a reduction of CD73 at the protein level. At the molecular level, USP22 functions as a CD73-specific deubiquitinase to inhibit CD73 ubiquitination and protect it from proteosome-mediated degradation. Additionally, USP22 KO tumors were smaller in size compared to the wild type.
Results
Identification of USP22 as a positive regulator of CD73 in breast cancer cells
To determine the role of USP22 in breast cancer tumorigenesis, we used the CRISPR approach to specifically delete USP22 in mouse 4T1 breast cancer cells. Western blotting confirmed the deletion of USP22 expression (Figure 1A). Interestingly, targeted deletion of USP22 in 4T1 cells resulted in a significant reduction in CD73 protein expression was analyzed by western blotting (Figure 1A). Flow cytometry analysis further confirmed the statistically significant reduction in surface CD73 expression in USP22 KO cells compared to empty vector (EV) control (Figure 1C). Real-time qPCR analysis demonstrated that the mRNA levels were not decreased, but rather slightly but statistically significantly increased in USP22-null 4T1 cells (Figure 1B). It has been shown that TGF-β is involved in promoting CD73 expression, together with the fact that USP22 has been shown as a positive regulator of TGF-β pathways in Tregs, we then reasoned whether USP22 is involved in promoting TGF-β-induced CD73 expression in breast cancer cells. Treatment of both EV and USP22 KO cells with increasing amounts of TGF-β, resulted in a similar increase in CD73 MFI for both cell lines, but CD73 expression was still significantly lower for USP22 KO cells (Figure 1D), suggesting that USP22-mediated CD73 upregulation is independent of TGF-β pathways. Collectively, data indicates that USP22 suppression inhibits CD73 expression at the post-transcriptional level and that the slight increase in CD73 mRNA levels in USP22-deficient breast cancer cells is possibly a consequence of feedback response to CD73 protein reduction.
Figure 1.

USP22 KO reduces CD73 expression in breast cancer cells. (A) Using CRISPR CAS9 USP22 was deleted from 4T1-L2T cells as shown by western blot, the empty vector control is (EV) and USP22 KO is (KO). (B) RT-qPCR of CD73 mRNA levels in EV and USP22 KO 4T1-L2T cells. (C) Flow representative of EV (red) and USP22 KO (blue) 4T1-L2T cells. Mean fluorescent intensity (MFI) shows that USP22 KO cells have a significantly lower CD73 MFI compared to EV. (D) CD73 MFI of EV and USP22 KO cells stimulated with increasing amounts of TGF-β. n=3-4, **P ≤ 0.01, Paired Student’s T-Test was used (B) (Mean ± SD). **P ≤ 0.01, Paired Student’s T-Test was used for (C) (Mean ± SEM). **P ≤ 0.01, ***P ≤ 0.001, Two-way ANOVA was used for (D) (Mean ± SEM).
USP22 is a deubiquitinase of CD73
Next, we sought to determine the molecular mechanism of how USP22 regulates CD73 protein expression. Since USP22 regulates CD73 expression at the post-transcriptional level, we posed the possibility that USP22 may function as a deubiquitinase to protect CD73 from ubiquitination-mediated degradation. Indeed, USP22 interacted with CD73 in transiently transfected HEK 293T cells, because the MYC-tagged USP22 was detected in anti-FLAG immunoprecipitants of HEK 293T cells transfected with both FLAG-CD73 and MYC-USP22 (Figure 2A). Importantly, the endogenous USP22 and CD73 interaction were further confirmed in human breast cancer MDA-MB-231 cells where CD73 protein was pulled down by anti-USP22 antibodies but not the normal rabbit IgG control (Figure 2B).
Figure 2.
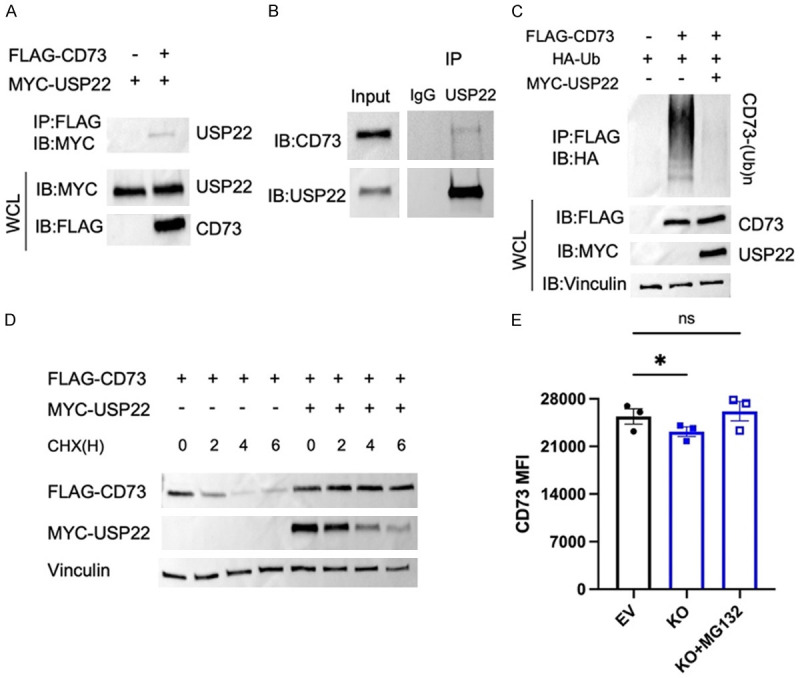
USP22 functions as a deubiquitinase of CD73. (A) Transiently transfected HEK 293T cells with CD73 and USP22 expression plasmids as indicated. CD73 was immunoprecipitated and anti-FLAG and its binding with USP22 was detected by anti-MYC Abs (top panel). The expression of USP22 and CD73 in the whole cell lysates was analyzed as a control (middle and bottom panels). (B) Endogenous USP22 interaction with CD73 in the human breast cancer MDA-MB-231 cells was determined by co-IP and western blotting. (C) FLAG-CD73 was co-transfected with HA-Ub either with or without MYC-USP22 in HEK293T cells. The ubiquitination of CD73 was determined by immunoprecipitation with anti-Flag and western blotting with anti-HA (top panel). The expression levels of USP22, CD73, and Vinculin control were determined by western blotting. (D) HEK 293T cells transfected with FLAG-CD73 with or without MYC-USP22 were incubated with 50 ug/mL of cycloheximide (CHX) over time. The expression levels of CD73 (top panel), USP22 (middle panel), and loading control Vinculin (bottom panel) were analyzed. (E) EV and USP22 KO 4T12-L2T cells were incubated with 10 µM of MG132 for four hours, and the cell surface expression of CD73 was analyzed by flow cytometry. ns-not significant, *P ≤ 0.05, Paired Student’s T-test was used (Mean ± SEM).
A deubiquitinase often inhibits the ubiquitination of its interacting proteins. Therefore, we determined if USP22 expression inhibits CD73 ubiquitination. As shown in (Figure 2C), CD73 ubiquitination was strongly detected in HEK293T cells because the anti-HA antibody detected a gradual shift of the CD73 molecular weight. Interestingly, when MYC-USP22 was co-expressed CD73 ubiquitination was largely diminished (Figure 2C), clearly indicating that USP22 inhibits CD73 ubiquitination. To determine the functional consequence of USP22-mediated suppression of CD73 ubiquitination, a pulse-chase experiment with cycloheximide was performed as reported [27]. The gradual degradation of CD73 along with cycloheximide treatment for different amounts of time in HEK 293T cells was detected, and co-expression with MYC-USP22 significantly slowed down CD73 degradation (Figure 2D). To determine if USP22 was prolonging CD73 protein stability by protecting it from ubiquitin-mediated proteasomal degradation, we treated USP22 KO 4T1 cells with a proteasome-specific inhibitor MG132. Indeed, MG132 treatment rescued the surface expression of CD73 (Figure 2E), indicating that USP22 influences the protein stability of CD73 by protecting it from ubiquitin-mediated proteasomal degradation. Collectively, our results indicate that USP22 is a CD73 deubiquitinase in breast cancer cells.
USP22 suppression results in a reduction in breast tumor growth
Since CD73 is an immune checkpoint involved in breast cancer evasion of antitumor immunity, our data that USP22 protects CD73 from ubiquitination-mediated proteasomal degradation, along with other studies demonstrating that USP22 regulates other proteins important for tumor growth, imply the possibility that USP22 suppression could reduce tumor burden in breast cancer. We then used the 4T1 syngeneic tumor model in BALB/c mice to test this hypothesis. EV and USP22 KO 4T1 cells were orthotopically injected into the mammary fat pad of female BALB/c mice. Mice were sacrificed 26 days later; tumors were harvested, and tumor-infiltrating leukocytes were analyzed (Figure 3A). Tumor mass is significantly smaller for USP22 KO tumors (Figure 3B), and the tumor volume is generally smaller for USP22 KO tumors (Figure S1), implying that USP22 suppression in 4T1 breast cancer cells inhibits tumor growth. While further analysis of the tumor infiltrated immune cells did not detect any statistical changes in CD4, CD8, and NK cells (Figure 3C), and no significant difference in B cells, macrophages, and myeloid cells (Figure S2). A significant increase in IFN-γ producing CD8 T cells was detected in intratumoral immune cells (Figure 3D), implying that USP22 suppression in tumor cells enhances CD8 T cell antitumor immunity. In addition to CD73, it has been shown that USP22 stabilizes PD-L1 in liver cancer cells. However, our data show that USP22 suppression did not affect PD-L1 expression levels in breast cancer cells as analyzed by western blotting and flow cytometry (Figure S3A, S3B). Since it has been reported that USP22 regulates PD-L1 expression in liver cancer cells [25], we then speculated the possibility that USP22 might regulates PD-L1 in a cell type-specific manner. Indeed, CRISPR-mediated USP22 targeted deletion slightly reduced PD-L1 expression in MDA-MB-231 human breast cancer cells but not in B16 melanoma and MC38 colon cancer cells (Figure S3C-E). Unexpectedly, in contrast to USP22 suppression leading to CD73 reduction in vitro, this reduction in CD73 and PD-L1 expression was not detected in syngeneic tumor cells (Figure S4), possibly due to the tumor cells with lower CD73 expression, which was presumably caused by USP22 deletion, were rejected. In addition, there was a significantly higher percentage of Regulatory T cells (Tregs) in USP22 KO tumors (Figure 3C). Therefore, while USP22 suppression could improve the antitumor immunity, this potential improvement may be partially diminished by the elevated Treg infiltration.
Figure 3.
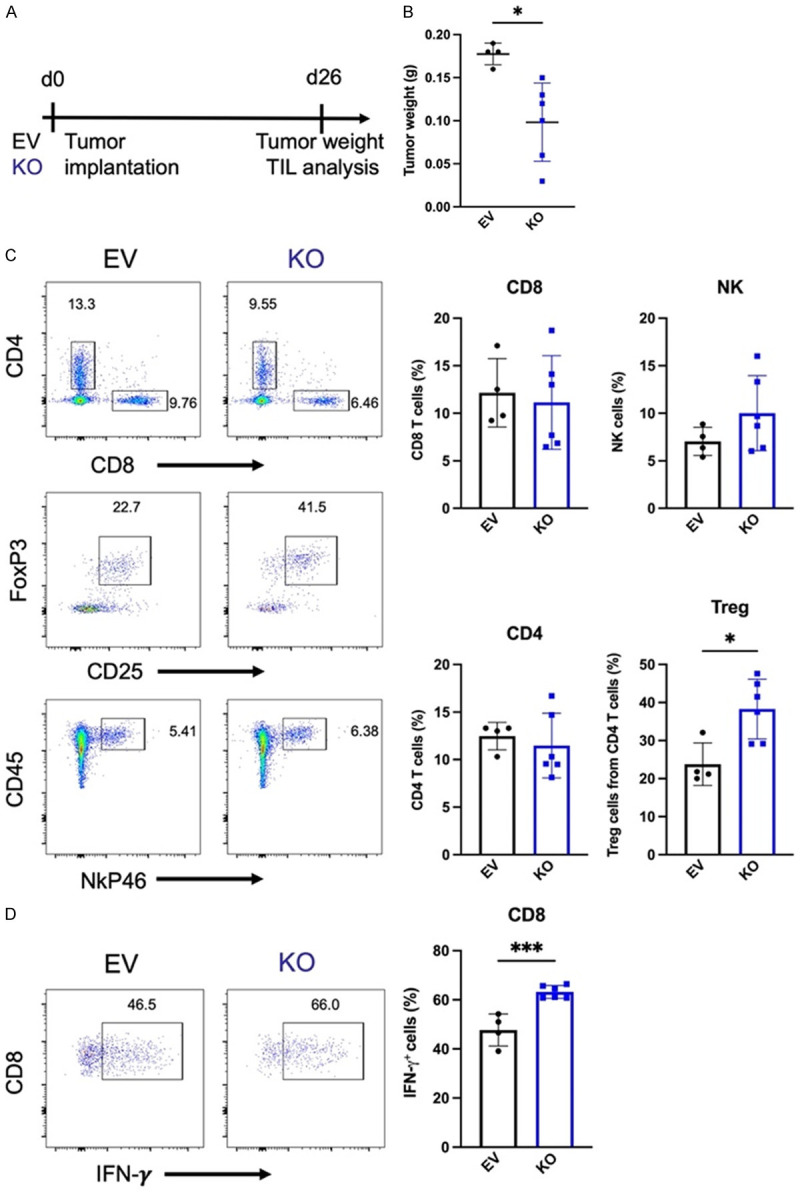
USP22 suppression results in a reduction of tumor burden. (A) Schematic of tumor implantation and analysis. EV or USP22 KO 4T1-L2T cells were injected into the mammary fat pad of female BALB/c mice. Mice were sacrificed after 26 days, and tumor-infiltrating lymphocytes were analyzed. (B) Tumor weight of mice bearing EV and USP22 KO tumors. (C) Representative flow plots (left) and percentages of tumor-infiltrating leukocytes (right). (D) Intracellular cytokine staining of IFN-γ tumor-infiltrating CD8 T cells. Data is from one experiment. *P ≤ 0.05, ***P ≤ 0.001 unpaired Student’s T test was used (Mean ± SD).
Human breast cancer tissue high in USP22 is also high for CD73
This study revealed USP22 as a positive regulator of CD73 in mouse breast cancer cells. We were curious to know whether USP22 and CD73 expressions are positively associated with human primary breast cancers. Representative tissue microarray staining shows higher CD73 expression on USP22 expressing tumor cells (PanCK) and Tregs (FoxP3) (Figure 4A). Quantification of tissue microarray analysis of breast cancer samples shows that samples expressing USP22 have significantly higher CD73 intensity (Figure 4B). Furthermore, infiltrating Tregs that express USP22 have significantly higher CD73 expression compared to Tregs that do not express USP22 (Figure 4C). When looking at infiltrating Tregs in invasive ductal carcinoma and medullary breast carcinoma, USP22 positive Tregs in invasive ductal carcinoma grades two and three have significantly higher CD73 expression compared to Tregs not expressing USP22 (Figure 4D). USP22 positive Tregs in medullary breast carcinoma have higher CD73 expression although it is not significantly higher than USP22 negative Tregs (Figure 4D). Collectively, our study indicates that USP22 is a positive regulator of CD73 in breast cancer.
Figure 4.
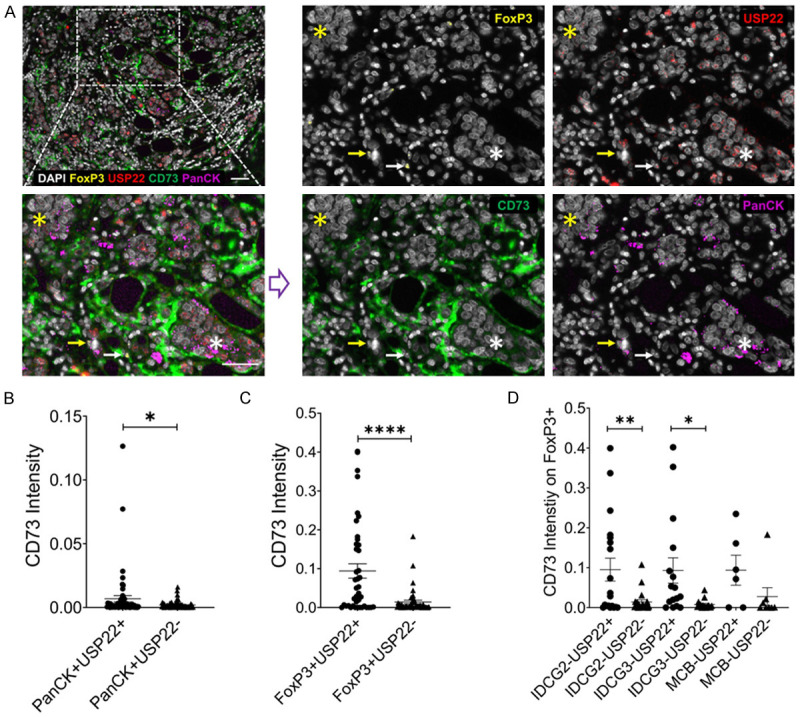
Tissue microarray staining of human breast cancer. Representative staining of breast cancer tissue sample (A) USP22 negative stained tumor cells (yellow star) or FoxP3 cell (yellow arrow), CD73 expression is increased on the USP22 positive stained tumor cells (white star) or FoxP3 cell (white arrow), the scale bar is 50 µm. Quantification of CD73 intensity across individual cases in (B) USP22+ and USP22- breast cancer cells; (C and D) in FoxP3 expressing USP22+ and USP22- Tregs in individual cases of breast cancer (B) and invasive ductal carcinoma (IDC) (C). *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001 unpaired Student’s T test was used (Mean ± SD). Based on over thirty positive cells (minimal five) defined by the observer in line with the MFI value of markers displayed on the in FORM system, the inFORM system created an algorithm to execute the negative and positive analysis.
Discussion
The current study identifies USP22 as a deubiquitinase of CD73 in breast cancers. This conclusion is documented by the following discoveries: First, USP22 targeted gene deletion by CRISPR resulted in a significant reduction in CD73 protein expression at the post-transcriptional level; second, USP22 interacts with and inhibits CD73 ubiquitination and degradation; third, CD73 protein expression is significantly higher in USP22 positive human primary breast cancer cells as well as in USP22-positive tumor infiltrated Tregs, and finally, USP22 suppression in breast cancer leads to a reduction in tumor mass.
Aggressive cancers use multiple methods to evade the immune system and have limited treatment options available. Targeting inhibitory immune receptors expressed on tumor cells may help boost antitumor immunity. CD73 expression is expressed in many breast cancer patients [28]. A monoclonal antibody specific to CD73 has been therapeutically effective in inhibiting breast tumor growth and metastasis [29]. Multiple tumor microenvironmental factors, such as TGF-β and hypoxia have been identified to induce tumor cell expression of CD73 at transcriptional levels [30-32]. It has been shown that CD73 has glycosylation and ADP-ribosylation post-translational modifications [14,15]. This study is the first to identify USP22 as a CD73 deubiquitinase to protect CD73 from ubiquitination-mediated proteasomal degradation. Therefore, the deletion of USP22 in 4T1 cells resulted in less total protein and surface expression of CD73 in breast cancer cells. Consistent with this observation, we further detected a decrease in CD73 expression in USP22 negative human primary breast cancer samples as well as decreased CD73 expression in USP22 negative infiltrating Tregs compared to CD73 expression of the USP22-positive compartment.
Targeting CD73 has been shown to augment the cancer immunotherapy [33]. Monoclonal antibodies specific to CD73 have been therapeutically effective in inhibiting breast tumor growth and metastasis [29]. Therefore, a reduction in CD73 in 4T1 breast cancer cells by the suppression of USP22 would lead to a better antitumor response and smaller tumors. While we did find that the USP22 KO tumors were significantly smaller, further analysis of the tumor infiltrated immune cells showed no difference in NK, CD4, and CD8 T cell infiltrations. Interestingly, there was a significantly higher percentage of Tregs in USP22 KO tumors. Additionally, there was a significant increase in IFN-γ producing CD8 T cells. Since it is well established that Tregs inhibit the antitumor response, this Treg increase in USP22-null breast cancers is likely responsible for partially diminishing the effects of antitumor immunity. Future studies are needed to dissect the cellular and molecular mechanisms underlying how USP22 suppression causes increased Treg infiltration in breast cancer.
It has been known that USP22 functions as an oncogene through multiple molecular mechanisms including p53, c-Myc, and cyclin B1 [22,34,35]. Here we further discovered that CD73 is a novel substrate of USP22. Govern the fact that CD73 is a key regulatory molecule of cancer cells proliferation, migration, and invasion in vitro, tumor angiogenesis, and tumor immune escape in vivo, we speculate that promoting CD73 expression certainly contributes to USP22 tumorigenic functions in breast cancer as well as other types of solid tumors. Future studies are needed to precisely define the contributions of each of the USP22 downstream targets in tumorigenesis.
Experimental procedures
Mice, cell lines, and reagents
HEK 293T cells were maintained in DMEM with 10% FBS and 1% Penicillin/Streptomycin. 4T1-L2T were obtained from Huiping Liu’s lab at Northwestern University, maintained in RPMI 1640 with 10% FBS and 1% Penicillin/Streptomycin, and used as reported [36]. B16 and MC38 cells were obtained from Bin Zhang’s lab at Northwestern University and maintained in DMEM with 10% FBS and 1% Penicillin/Streptomycin. MDA-MB-231 cells were obtained from Karla Satchell’s lab at Northwestern University and maintained in DMEM/F-12 with 10% FBS and 1% Penicillin/Streptomycin. BALB/c mice were purchased from Jackson Laboratories. All animal experiments were approved by the institutional animal use committee (IACUC) of Northwestern University.
Antibodies used for western blot: HRP-conjugated MYC (Santa Cruz), HRP-conjugated HA (CST), HRP-conjugated FLAG M2 (Sigma), USP22 (Abcam), CD73 (Abcam), CD73 (CST), GAPDH (CST), Vinculin (CST), HRP-conjugated Anti-Rabbit (CST). Antibodies used for IP: USP22 (Santa Cruz), FLAG M2 (Sigma), Mouse IgG1 (CST).
Antibodies used for flow cytometry: Fixable Viability dye BV450 (eBioscience), APC Mo/Rt FoxP3 (eBioscience), APC Mo-CD45 (Biolegend), APC Mo-IFN-γ (Biolegend), APC-Cy7 Mo-CD4 (Biolegend), APC-Cy7 Mo CD11b (Invitrogen), BV605 Mo-CD73 (Biolegend), BV605 Gr-1 (Biolegend), BV650 Mo-CD8 (Biolegend), BV711 Mo-CD4 (Biolegend), FITC Mo-CD45 (Biolegend), FITC F4/80 (Biolegend), PE CD11c (Biolegend), Pe-Cy7 Mo-NkP46 (Biolegend), Pe-Cy7 Mo-CD73 (Biolegend), PerCP-Cy5.5 Mo-B220 (Biolegend), PerCP-Cy5.5 Mo-CD25 (Biolegend).
Flow samples were run on the BD-LSR Fortessa X-20 (BD Biosciences) instrument and flow analyses were done using FlowJo software.
Plasmids used in this study including MYC-moUSP22 and HA-Ubiquitin were developed as previously mentioned [37]. We purchased FLAG-moCD73 (Sino Biological) and lentiCRISPR v2 plasmid (Addgene).
Transfection, immunoprecipitation, and western blotting
Gene transfection to HEK293T and 4T1-L2T cells using TurboFect (Thermo Fisher) was performed as reported [38]. Cells were lysed in Cell Lysis Buffer (CST), or RIPA Buffer (Millipore Sigma) supplemented with complete Protease Inhibitor Cocktail (Roche) on ice for 30 min. Western blots for protein expression were sonicated at 4°C using a bioruptor (diagenode) at 308/608 (medium), 15 sec on 45 sec off for 5 minutes. Lysates were centrifuged at 13,000 rpm for 10 min at 4°C. Cell line samples were quantified using BCA (Thermo Fisher) to ensure equal protein loading. Samples for immunoprecipitation were pre-cleared using protein G beads (GE Healthcare), before being rotated with antibodies overnight at 4°C. Protein G beads were added the next day for one hour followed by washing with buffer. LDS sample buffer (Thermo Fisher) and DTT (Sigma) were added to samples before heating at 95°C for 10 min. Samples were loaded into 4-15% Mini-PROTEAN TGX gels (Bio-Rad) and transferred onto a nitrocellulose membrane (Cytiva). Membranes were blocked with 2% milk in TBST for 1 hour. Primary antibodies were added at the manufactures recommended dilutions overnight. Membranes were washed with TBST the next day. Secondaries if needed were added at the manufacturer’s recommended dilutions for one hour. Membranes were incubated with SuperSignal West chemiluminescent substrates (Thermo Fisher) before being imaged on Bio-Rad ChemiDoc XRS+ (Bio-Rad).
For testing CD73 protein stability, cells were incubated with 50 μg/ml of cycloheximide (sigma) over time. For proteasome inhibition, cells were incubated with 10 μM of MG132 (Selleckchem) for 4 hours. Cells were collected and used for flow analysis and/or western blotting analysis.
Generation of CRISPR cell lines
Human USP22 guide RNA sequence 5’-GCCATTGATCTGATGTACGG-3’ and Mouse USP22 guide RNA sequence 5’-GCCATCGACCTGATGTACGG-3’ were ligated into lentiCRISPR v2 plasmid separately. Cells were transiently transfected using TurboFect (Thermo Fisher) for 24 hours. Following transfection cells were selected using puromycin for 14 days. The efficacy of USP22 deletion was confirmed by western blotting.
Tumor model
8-10-week-old female BALB/c mice were injected with 5 × 105-1 × 106 4T1-L2T cells resuspended in 100 μL of growth factor reduced Matrigel (Corning) orthotopically into the fourth mammary fat pad. Volume was calculated using the following formula L × W2/2.
Tumor digestion
Tumors were placed in 5 mL Eppendorf tubes containing RPMI media supplemented with collagenase IV (2 mg/mL) and DNase I (150 μg/mL) and cut into small pieces using scissors. The tubes were placed in a 37°C incubator with shaking (200 RPM) for 15 min.
Samples were passed through a 70 µm strainer on a 50 mL conical and were topped up to 30 mL of media before being gently mixed and underlaid with 10 mL of Ficoll-Paque (Cytiva). The samples were centrifuged at 1025 × g for 20 min at 20°C with slow acceleration and no brake. The white buffy coat interface was transferred to a new tube and washed with 20 mL of T cell media (RPMI 1640, 10% FBS, 1% Penicillin/Streptomycin, 1% L-glutamine, 5 mM HEPES, 1 mM Sodium Pyruvate, 50 µm 2-mercaptoethanol), and centrifuged at 600 × g at 4°C. Samples were then ready for further analysis.
Cell surface and Intracellular cytokine staining
All cell’s Fc receptors were blocked anti-mouse CD16/32 (Biolegend) Cells were stimulated with 50 ng/mL of PMA, 1 µg/mL of Ionomycin, and 1 µg/mL of Golgi plug (BD Biosciences) in a 37°C incubator for 4 hours, and subsequently used for flow staining. The cells were then subjected to flow cytometry analysis.
RNA extraction and real-time PCR analysis
RNA was extracted using an RNA isolation kit (Qiagen). CDNA synthesis (Quantabio) was conducted. Samples were prepared with SYBER Green (Quantabio) and analyzed using the Biorad MyiQ2 Real-Time PCR detector.
Primers used: Mo Actin Forward: GATATCGCTGCGCTGGTCG; Reverse: CCACGATGGAGGGGAATACAG; Mo CD73 Forward: TCCTGCAAGTGGGTGGAATC; Reverse: AGATGGGCACTCGACACTTG.
Multiplex immunohistochemistry (mIHC)
The breast cancer tissue array (TMA) (US Biomax Inc.) including invasive ductal carcinoma cases (IDC, Grade 2, n=28, Grade 3, n=25), medullary carcinoma cases (MCB, n=9) was utilized for mIHC staining of USP22, FoxP3, CD73 and PanCK by using the Opal multiple color IHC kit (AKOYA Biosciences) as described previously (7). Briefly, the deparaffinized and re-hydrolyzed TMA slide was implemented antigen retrieval in AR9 retrieval buffer (AKOYA Biosciences), followed by four cycles of staining procedures including blocking, binding of primary antibodies, second HRP-linked antibodies, and visualized with the corresponding Opal fluorophores. Each staining cycle was finished up with heating in AR6 retrieval buffer (AKOYA Biosciences) to release the bounded primary and second antibodies but did not disturb the resident fluorophores. After four-round staining procedures, the slide was counterstained with DAPI. The single marker staining with individual opal fluorophore was employed as the reference for the “spectral unmixing process”. The antibodies and corresponding fluorophores are listed in Table 1.
Table 1.
Fluorophores corresponding to antigens stained in multiplex immunohistochemistry assay
| Antigen | Primary antibodies | Fluorophore | |||
|---|---|---|---|---|---|
| Panel | Vendors | Catalog # | |||
| USP22 | 1:50 | Abcam | ab195289 | Opal 620 | |
| CD73 | 1:100 | Cell signaling | 13160s | Opal 570 | |
| FoxP3 | 1:100 | Biolegend | 320102 | Opal 540 | |
| PanCK | 1:200 | Abcam | ab27988 | Opal 690 | |
Acquirement of multispectral images (MSI) and data analysis
The Opal fluorophore signals on the finished staining TMA slide were captured with the Vectra 3 Automated Quantitative Pathology Imaging System (Perkin Elmer) at 200 × magnifications and proceeded with spectral unmixing into four individual fluorophores based on the unique emitting spectrum of every single fluorophore using InForm Advanced Image Analysis software (Akoya Biosciences). Subsequently, the spectrally unmixed images underwent cell segmentation based on DAPI, and cell phenotyping based on specific cellular markers through the trained algorithm of Inform. The exported data containing composite images, cell segmentation, and cell phenotyping from InForm were further carried out by quantitative analyses of cell densities and protein intensities using R-based phenoptrReports & phenoptr (AKOYA biosciences).
Statistical analysis
Statistical analysis was done using Prism (GraphPad) software. Means were calculated using paired student’s T-test, unpaired student’s T-test, or two-way ANOVA. Significant outliers determined via the Grubbs test for outliers were removed from mouse quantitative analyses.
Acknowledgements
We would like to thank the members of the Fang, Liu, and Zhang laboratories for their critical contributions to the study. This study was supported by the National Institutes of Health “Immunology and molecular pathogenesis” T32 grant 5T32AI007476 to (SG), and RO1 grants CA232347, CA257520, DK120330, and DK126908 (to DF).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Collignon J, Lousberg L, Schroeder H, Jerusalem G. Triple-negative breast cancer: treatment challenges and solutions. Breast Cancer (Dove Med Press) 2016;8:93–107. doi: 10.2147/BCTT.S69488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jin D, Fan J, Wang L, Thompson LF, Liu A, Daniel BJ, Shin T, Curiel TJ, Zhang B. CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res. 2010;70:2245–2255. doi: 10.1158/0008-5472.CAN-09-3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang L, Zhou X, Zhou T, Ma D, Chen S, Zhi X, Yin L, Shao Z, Ou Z, Zhou P. Ecto-5’-nucleotidase promotes invasion, migration and adhesion of human breast cancer cells. J Cancer Res Clin Oncol. 2008;134:365–372. doi: 10.1007/s00432-007-0292-z. [DOI] [PubMed] [Google Scholar]
- 4.Loi S, Pommey S, Haibe-Kains B, Beavis PA, Darcy PK, Smyth MJ, Stagg J. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc Natl Acad Sci U S A. 2013;110:11091–11096. doi: 10.1073/pnas.1222251110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petruk N, Tuominen S, Åkerfelt M, Mattsson J, Sandholm J, Nees M, Yegutkin GG, Jukkola A, Tuomela J, Selander KS. CD73 facilitates EMT progression and promotes lung metastases in triple-negative breast cancer. Sci Rep. 2021;11:6035. doi: 10.1038/s41598-021-85379-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Resta R, Yamashita Y, Thompson LF. Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol Rev. 1998;161:95–109. doi: 10.1111/j.1600-065x.1998.tb01574.x. [DOI] [PubMed] [Google Scholar]
- 7.Colgan SP, Eltzschig HK, Eckle T, Thompson LF. Physiological roles for ecto-5’-nucleotidase (CD73) Purinergic Signal. 2006;2:351–360. doi: 10.1007/s11302-005-5302-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spychala J. Tumor-promoting functions of adenosine. Pharmacol Ther. 2000;87:161–173. doi: 10.1016/s0163-7258(00)00053-x. [DOI] [PubMed] [Google Scholar]
- 9.Haskó G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7:759–770. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, Huang X, Caldwell S, Liu K, Smith P, Chen JF, Jackson EK, Apasov S, Abrams S, Sitkovsky M. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A. 2006;103:13132–13137. doi: 10.1073/pnas.0605251103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoskin DW, Mader JS, Furlong SJ, Conrad DM, Blay J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (Review) Int J Oncol. 2008;32:527–535. [PubMed] [Google Scholar]
- 12.Ohta A, Sitkovsky M. Extracellular adenosine-mediated modulation of regulatory T cells. Front Immunol. 2014;5:304. doi: 10.3389/fimmu.2014.00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kordaß T, Osen W, Eichmüller SB. Controlling the immune suppressor: transcription factors and micrornas regulating CD73/NT5E. Front Immunol. 2018;9:813. doi: 10.3389/fimmu.2018.00813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hesse J, Rosse MK, Steckel B, Blank-Landeshammer B, Idel S, Reinders Y, Sickmann A, Sträter N, Schrader J. Mono-ADP-ribosylation sites of human CD73 inhibit its adenosine-generating enzymatic activity. Purinergic Signal. 2022;18:115–121. doi: 10.1007/s11302-021-09832-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alcedo KP, Guerrero A, Basrur V, Fu D, Richardson ML, McLane JS, Tsou CC, Nesvizhskii AI, Welling TH, Lebrilla CB, Otey CA, Kim HJ, Omary MB, Snider NT. Tumor-selective altered glycosylation and functional attenuation of CD73 in human hepatocellular carcinoma. Hepatol Commun. 2019;3:1400–1414. doi: 10.1002/hep4.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glinsky GV, Berezovska O, Glinskii AB. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest. 2005;115:1503–1521. doi: 10.1172/JCI23412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Yao L, Zhang X, Ji H, Wang L, Sun S, Pang D. Elevated expression of USP22 in correlation with poor prognosis in patients with invasive breast cancer. J Cancer Res Clin Oncol. 2011;137:1245–1253. doi: 10.1007/s00432-011-0998-9. [DOI] [PubMed] [Google Scholar]
- 18.Lang G, Bonnet J, Umlauf D, Karmodiya K, Koffler J, Stierle M, Devys D, Tora L. The tightly controlled deubiquitination activity of the human SAGA complex differentially modifies distinct gene regulatory elements. Mol Cell Biol. 2011;31:3734–3744. doi: 10.1128/MCB.05231-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melo-Cardenas J, Zhang Y, Zhang DD, Fang D. Ubiquitin-specific peptidase 22 functions and its involvement in disease. Oncotarget. 2016;7:44848–44856. doi: 10.18632/oncotarget.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeusset LM, McManus KJ. Ubiquitin specific peptidase 22 regulates histone H2B mono-ubiquitination and exhibits both oncogenic and tumor suppressor roles in cancer. Cancers (Basel) 2017;9:167. doi: 10.3390/cancers9120167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schrecengost RS, Dean JL, Goodwin JF, Schiewer MJ, Urban MW, Stanek TJ, Sussman RT, Hicks JL, Birbe RC, Draganova-Tacheva RA, Visakorpi T, DeMarzo AM, McMahon SB, Knudsen KE. USP22 regulates oncogenic signaling pathways to drive lethal cancer progression. Cancer Res. 2014;74:272–286. doi: 10.1158/0008-5472.CAN-13-1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin Z, Yang H, Kong Q, Li J, Lee SM, Gao B, Dong H, Wei J, Song J, Zhang DD, Fang D. USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Mol Cell. 2012;46:484–494. doi: 10.1016/j.molcel.2012.03.024. [DOI] [PubMed] [Google Scholar]
- 23.Zhang XY, Varthi M, Sykes SM, Phillips C, Warzecha C, Zhu W, Wyce A, Thorne AW, Berger SL, McMahon SB. The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell-cycle progression. Mol Cell. 2008;29:102–111. doi: 10.1016/j.molcel.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prokakis E, Dyas A, Grun R, Fritzsche S, Bedi U, Kazerouni ZB, Kosinsky RL, Johnsen SA, Wegwitz F. USP22 promotes HER2-driven mammary carcinoma aggressiveness by suppressing the unfolded protein response. Oncogene. 2021;40:4004–4018. doi: 10.1038/s41388-021-01814-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang X, Zhang Q, Lou Y, Wang J, Zhao X, Wang L, Zhang X, Li S, Zhao Y, Chen Q, Liang T, Bai X. USP22 deubiquitinates CD274 to suppress anticancer immunity. Cancer Immunol Res. 2019;7:1580–1590. doi: 10.1158/2326-6066.CIR-18-0910. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Yuan S, Norgard RJ, Yan F, Yamazoe T, Blanco A, Stanger BZ. Tumor cell-intrinsic USP22 suppresses antitumor immunity in pancreatic cancer. Cancer Immunol Res. 2020;8:282–291. doi: 10.1158/2326-6066.CIR-19-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei J, Harada BT, Lu D, Ma R, Gao B, Xu Y, Montauti E, Mani N, Chaudhuri SM, Gregory S, Weinberg SE, Zhang DD, Green R, He C, Fang D. HRD1-mediated METTL14 degradation regulates m6A mRNA modification to suppress ER proteotoxic liver disease. Mol Cell. 2021;81:5052–5065. e6. doi: 10.1016/j.molcel.2021.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kruger KH, Thompson LF, Kaufmann M, Moller P. Expression of ecto-5’-nucleotidase (CD73) in normal mammary gland and in breast carcinoma. Br J Cancer. 1991;63:114–118. doi: 10.1038/bjc.1991.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, Dwyer KM, Smyth MJ. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci U S A. 2010;107:1547–1552. doi: 10.1073/pnas.0908801107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J, Wang L, Chen X, Li L, Li Y, Ping Y, Huang L, Yue D, Zhang Z, Wang F, Li F, Yang L, Huang J, Yang S, Li H, Zhao X, Dong W, Yan Y, Zhao S, Huang B, Zhang B, Zhang Y. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-beta-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology. 2017;6:e1320011. doi: 10.1080/2162402X.2017.1320011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP. Ecto-5’-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110:993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. 2004;114:1098–1106. doi: 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roh M, Wainwright DA, Wu JD, Wan Y, Zhang B. Targeting CD73 to augment cancer immunotherapy. Curr Opin Pharmacol. 2020;53:66–76. doi: 10.1016/j.coph.2020.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim D, Hong A, Park HI, Shin WH, Yoo L, Jeon SJ, Chung KC. Deubiquitinating enzyme USP22 positively regulates c-Myc stability and tumorigenic activity in mammalian and breast cancer cells. J Cell Physiol. 2017;232:3664–3676. doi: 10.1002/jcp.25841. [DOI] [PubMed] [Google Scholar]
- 35.Lin Z, Tan C, Qiu Q, Kong S, Yang H, Zhao F, Liu Z, Li J, Kong Q, Gao B, Barrett T, Yang GY, Zhang J, Fang D. Ubiquitin-specific protease 22 is a deubiquitinase of CCNB1. Cell Discov. 2015;1:15028. doi: 10.1038/celldisc.2015.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taftaf R, Liu X, Singh S, Jia Y, Dashzeveg NK, Hoffmann AD, El-Shennawy L, Ramos EK, Adorno-Cruz V, Schuster EJ, Scholten D, Patel D, Zhang Y, Davis AA, Reduzzi C, Cao Y, D’Amico P, Shen Y, Cristofanilli M, Muller WA, Varadan V, Liu H. ICAM1 initiates CTC cluster formation and trans-endothelial migration in lung metastasis of breast cancer. Nat Commun. 2021;12:4867. doi: 10.1038/s41467-021-25189-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Melo-Cardenas J, Xu Y, Wei J, Tan C, Kong S, Gao B, Montauti E, Kirsammer G, Licht JD, Yu J, Ji P, Crispino JD, Fang D. USP22 deficiency leads to myeloid leukemia upon oncogenic Kras activation through a PU. 1-dependent mechanism. Blood. 2018;132:423–434. doi: 10.1182/blood-2017-10-811760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin Z, Yang H, Tan C, Li J, Liu Z, Quan Q, Kong S, Ye J, Gao B, Fang D. USP10 antagonizes c-Myc transcriptional activation through SIRT6 stabilization to suppress tumor formation. Cell Rep. 2013;5:1639–1649. doi: 10.1016/j.celrep.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.