

Abstract
Tumor cells and the immunosuppressive tumor microenvironment suppress the antitumor activity of T cells through immune checkpoints, including the PD-L1/PD-1 axis. Cytokine-inducible SH2-containing protein (CISH), a member of the suppressor of cytokine signaling (SOCS) family, inhibits JAK-STAT and T cell receptor (TCR) signaling in T and natural killer (NK) cells. However, its role in the regulation of immune checkpoints in T cells remains unclear. In this study, we ablated CISH in T cells with CRISPR-Cas9 and found that the sensitivity of T cells to TCR and cytokine stimulation was increased. In addition, chimeric antigen receptor T cells with CISH deficiency exhibited longer survival and higher cytokine secretion and antitumor activity. Notably, PD-1 expression was decreased in activated CISH-deficient T cells in vitro and in vivo. The level of FBXO38, a ubiquitination-regulating protein that reduces PD-1 expression, was elevated in activated T cells after CISH ablation. Hence, this study reveals a mechanism by which CISH promotes PD-1 expression by suppressing the expression of FBXO38 and proposes a new strategy for augmenting the therapeutic effect of CAR-T cells by inhibiting CISH.
Keywords: chimeric antigen receptor T cells, immunotherapy, CISH, PD-1, FBXO38, CRISPR-Cas9
Graphical abstract
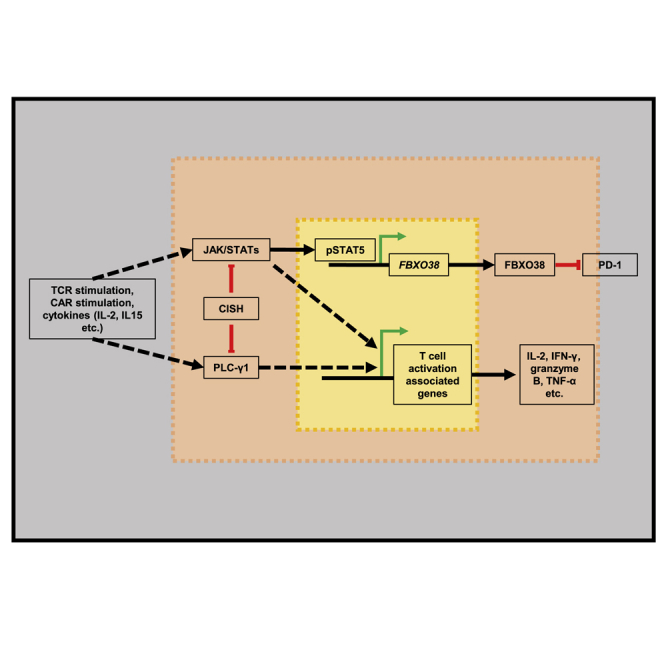
Li and colleagues discovered a way to reduce PD-1 expression in activated T cells and promote T cell function by knocking out the suppressor gene CISH. This is an effective and feasible way to modulate T cell immune function, especially for enhancing the effect of clinical CAR-T cell therapy.
Introduction
Chimeric antigen receptor (CAR)-T cell therapy has shown remarkable outcomes in treating hematologic malignancies. However, the treatment landscape of CAR-T cell therapy for solid tumors has been limited, which has been attributed to the inhospitable tumor microenvironment (TME) and T cell exhaustion,1,2 both of which have been correlated with the expression of immune-receptor genes (e.g., PDCD1, LAG3, HAVCR2, TIGIT, and CTLA4).3 Prevention of CAR-T cell exhaustion and promotion of CAR-T cell responses to persistence- and activity-promoting cytokines, including interleukin-2 (IL-2), IL-7, IL-12, and IL-15, contributing to antitumor responses, in the TME is an essential direction for anticipated breakthroughs in the application of CAR-T cells to solid tumors.4
PD-1, encoded by PDCD1, is present on the surface of activated T cells and plays a vital and diverse range of immunoregulatory roles in T cell activation and tolerance.5 After engagement with a ligand, PD-1 undergoes phosphorylation of two cytoplasmic tyrosine residues that recruit the phosphatase SHP-2, leading to attenuation of T cell receptor (TCR) and CD28 signaling and inhibition of T cell activation, cytokine production, and proliferation, which eventually causes the death of activated T cells,5 attenuating CAR-T cell signaling and antitumor activity. PD-L1/PD-1 blockade with antibodies and PD-1 single-chain variable fragment (scFv) secreted by modified CAR-T cells have led to increased antitumor activity of CAR-T cells.6,7,8,9,10 PD-1 knockout (KO) in CAR-T cells mediated by CRISPR-Cas9 also led to an increase in antitumor activity.11,12,13,14 However, complete absence of PD-1 signaling may counterintuitively produce terminally differentiated and, ultimately, exhausted T cells,15 and therefore, T cells that increase PD-1 to a certain level and that is subsequently blocked by cell-intrinsic or -extrinsic PD-1 disruption may be preferable to abrogated T cell promotion of increased PD-1 expression.16 More strategies should be considered to improve the function of CAR-T cells by inhibiting immune checkpoint PD-1 without reducing the function of CAR-T cells.
CARs are composed of an extracellular scFv that recognizes diverse tumor-associated antigens (TAAs), a transmembrane fragment, a CD3ζ that induces T cell activation (signal 1), and a costimulatory domain that mimics signal 2 peptides, such as the intracellular domains of CD28 and 4-1BB.17,18,19,20 Therefore, CAR signaling imitates TCR signaling. Upon specific binding of CAR to a TAA on target tumor cells, the CD3ζ and costimulatory domains are activated, and the phosphorylation cascade is triggered in T cells, leading to the release of cytotoxic granules and cell proliferation.21 The incorporation of CD3ε in CAR was shown to increase the recruitment of Lck and PI3K, thereby enhancing the antitumor function of CAR-T cells.22,23 HPK1 competes with ADAP for SLP-76 binding and dampens TCR-induced tyrosine phosphorylation of SLP-76 and phospholipase C gamma 1.24,25 CAR-T cells with HPK1 knocked out showed superior tumor-suppression capability, with fewer exhausted and more active and proliferative T cells.26 Cbl-b negatively regulates the activation of T cells, and deletion of Cbl-b inhibits CD8 T cell exhaustion and promotes CAR-T cell function.27,28 Therefore, it is a feasible strategy to enhance the antitumor activity of CAR-T cells by modulating TCR signaling downstream.
In addition to signals 1 and 2, which are required, adequate activation of T cells requires auxiliary signal 3, namely stimulation of various cytokines. For example, γ chains are common to multiple cytokines, in which they are the receptor unit, and exert a fundamental effect on T cell immunity mainly through JAK-STAT pathway activation.29 Recent research demonstrated that CAR engaged with the IL-2Rβ cytoplasmic domain and that the STAT3-binding motif showed antigen-dependent activation of the JAK kinase, STAT3, and STAT5, superior persistence, and antitumor effects.30 Similarly, in our own previous studies, we found that activation of STAT3 signaling in CAR-T cells, by inducing IL-6 trans-signaling or directly expressing constitutively active GP-130, enhanced CAR-T cell expansion and antitumor activity.31 In contrast, boosting STAT5 signaling by knocking out certain inhibitors, such as PTPN2, also enhanced CAR-T cell activity.32
Cytokine-inducible SH2-containing protein (CISH) is a member of the suppressor of cytokine signaling (SOCS) protein family.33 Each SOCS protein carries a SOCS box sequence motif, enabling the SOCS protein to function as an adaptor for an E3 ubiquitin ligase complex and enabling proteins that interact directly with SOCS to undergo proteasomal degradation.34 CISH has been shown to be induced by TCR stimulation in CD8 T cells and inhibits their immune function against tumors by physically interacting with the TCR intermediate PLC-γ1 and targeting it for proteasomal degradation.35 In natural killer (NK) cells, CISH has been reported to inhibit JAK-STAT signaling activation induced by cytokines by binding directly to JAK1 and to reduce cellular metabolic fitness, which is mediated by the mTOR signaling pathway.36,37 Therefore, we hypothesized that the deletion of CISH in CAR-T cells would enhance the antitumor activities attributed to the reverse of inhibition induced by CISH.
In this study, we observed that the deletion of CISH in human T cells led to higher sensitivity of T cells to TCR signaling and cytokines (IL-2 and IL15) and enhanced T cell antitumor activity. Notably, CISH-KO CAR-T cells demonstrate improved survival and antitumor activity in vivo, with decreased PD-1 expression. We then verified in vitro that the absence of CISH decreases PD-1 expression in activated T cells. Furthermore, we found that decreased PD-1 levels were caused by the upregulated expression of FBXO38 in CISH-KO T cells and that KO of FBXO38 rescued PD-1 expression.
Results
Deletion of CISH increases the sensitivity of human T cells to anti-CD3/anti-CD28 antibodies stimulation to increase their antitumor activity
To knock out CISH in human T cells, two synthetic single guide RNAs (sgRNAs) were designed to target exon 3 in the CISH locus (Figure S1A). CISH was knocked out by ribonucleoprotein (RNP)-mediated CRISPR-Cas9 gene-editing technology. The deletion of CISH was confirmed by Sanger sequencing (Figure S1B) and western blot analysis (Figures 1A and 1B). No gene editing caused by off-target activity of these two sgRNAs was detected by sequencing (Figure S1C). CISH has been previously reported to physically interact with the TCR intermediate PLC-γ1 and silence TCR signaling.35 We therefore stimulated CISH wild-type (WT) T cells and CISH-KO T cells with anti-CD3/anti-CD28 antibodies, and the results showed that the deletion of CISH significantly elevated the phosphorylation of PLC-γ1 (Figure 1A). Further, stronger Erk1/2, PKCδ/θ, and nuclear factor κB (NF-κB) pathway activation (Figures S2A–S2C) and Ca2+ signaling (Figure S2D) were detected in CISH-KO T cells after stimulation. Increased phosphorylation of STAT5 was also observed in CISH-KO T cells (Figure 1B), which is consistent with a previous report suggesting that STAT5 interacts with the TCR complex and that TCR stimulation appears to directly activate STAT5.38
Figure 1.

Deletion of CISH increases the sensitivity of T cells to anti-CD3/anti-CD28 antibodies stimulation and antitumor activity
(A and B) Immunoblot analysis of CISH-WT and CISH-KO T cells incubated in vitro with anti-CD3/anti-CD28 antibodies for 30 min. CISH-WT and CISH-KO T cells were washed and cultured in medium free of IL-2 for 12 h before anti-CD3/anti-CD28 antibody treatment. Cells were lysed and analyzed by immunoblotting with antibodies against the indicated phosphorylated (p) and total (pan) proteins. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, one-way ANOVA with Tukey’s multiple comparison test. (C) CISH-WT and CISH-KO T cells were cocultured with OKT3 + A549 cells at the indicated effector-to-target ratio. (D) CISH-WT and CISH-KO T cells were cocultured with target cells at an effector-to-target (E:T) ratio of 1:1 for 24 h. The culture supernatants were then collected, and the levels of secreted IL-2, IFN-γ, granzyme B, and TNF-α were measured with ELISA kits. ∗∗p < 0.01, ∗∗∗p < 0.001, unpaired t test. (E) After 12 days of culture in medium with normal concentration of IL-2, CISH-WT, and CISH-KO T cells were analyzed with effector markers by fluorescence-activated cell sorting (FACS). (F and G) A total of 1 × 107 CISH-WT or CISH-KO T cells were cocultured with 5 × 106 OKT3 + A549 cells on day 0, and 5 × 106 OKT3 + A549 cells were added on days 4 and 8. T cells and OKT3 + A549 cells were analyzed by flow cytometry and luminance on day 12. ∗∗∗p < 0.001, unpaired t test.
Furthermore, we used OKT3-overexpressing A549 cells as target cells to examine the cytotoxicity of T cells via a luciferase-based cytotoxicity assay and found that CISH-KO T cells showed significantly enhanced antitumor activity compared with that of CISH-WT T cells (Figure 1C). An enzyme-linked immune absorbance assay (ELISA) also showed that CISH-KO T cells exhibited significantly elevated production of IL-2, tumor necrosis factor α (TNF-α), interferon γ (IFN-γ), and granzyme B (Figure 1D). A stronger antitumor response might be associated with higher effector phenotypes of CISH-KO T cells (Figure 1E). To examine the long-term antitumor activity of CISH-KO T cells in vitro, we cocultured T cells with OKT3-overexpressing A549 cells for 12 days (Figure 1F). CISH-KO T cells showed longer survival and higher antitumor cytotoxicity than CISH-WT T cells (Figure 1G). Taken together, CISH disruption in human T cells leads to elevated T cell activation and improved antitumor cytotoxicity and cytokine secretion by T cells.
Deletion of CISH increases the sensitivity of human T cells to IL-2 and IL-15
IL-2 is a central T cell cytokine that promotes T cell proliferation and effector function; however, the lack of IL-2 in the TME can lead to persistent deficiency and anergy of T cells.39,40,41 Since the SOCS family members, including CISH, SOCS1, and SOCS3, inhibit JAK-STAT signaling,34,35,36,42,43 we hypothesized that the absence of CISH increases T cell sensitivity to low concentrations of IL-2. We stimulated T cells with a low concentration of IL-2 (10 IU/mL) for 30 min. Compared with CISH-WT T cells, CISH-KO T cells showed significantly increased phosphorylation of STAT5 and STAT3 (Figures 2A and 2B). In addition, the deletion of CISH increased signaling activation induced by IL-15 (Figures 2C and 2D), another important cytokine through which T cells show reverse anergy and prolonged survival.44,45
Figure 2.

Deletion of CISH increases the sensitivity of human T cells to IL-2
(A and B) Immunoblot analysis of CISH-WT and CISH-KO T cells incubated in vitro with 10 IU/mL IL-2 for 30 min. CISH-WT and CISH-KO T cells were washed and cultured in medium free of IL-2 for 12 h before IL-2 treatment. Cells were lysed and analyzed by immunoblotting with antibodies against the indicated phosphorylated (p) and total (pan) proteins. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, one-way ANOVA with Tukey’s multiple comparison test. (C and D) Immunoblot analysis of CISH-WT and CISH-KO T cells incubated in vitro with 1 ng/mL IL-15 for 30 min. CISH-WT and CISH-KO T cells were washed and incubated in medium free of IL-2 for 12 h before IL-15 treatment. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, one-way ANOVA with Tukey’s multiple comparison test. (E) Growth curve of CISH-WT and CISH-KO T cells were cultured with a normal concentration of IL-2 (300 U/mL) or with a low concentration of IL-2 (10 U/mL). ∗∗∗p < 0.001, two-way ANOVA with Tukey’s multiple comparison test. (F) CISH-WT and CISH-KO T cells derived from 10 day 10 IU/mL IL-2 cultures with OKT3 + A549 cells at the indicated E:T ratios. (G) CISH-WT and CISH-KO T cells were cocultured with target cells at an E:T ratio of 1:1 for 24 h. The culture supernatants were then collected, and the levels of secreted IL-2, IFN-γ, granzyme B, and TNF-α were measured with ELISA kits. ∗∗∗p < 0.001, unpaired t test.
When cultured in medium with a normal concentration of IL-2 (300 IU/mL), there was no significant difference in the proliferation of T cells between CISH-WT and CISH-KO T cells (Figure 2E). To analyze the proliferation of T cells when stimuli are lacking, we also cultured CISH-WT and CISH-KO T cells in medium with a low concentration of IL-2 (10 IU/mL). The increase in CISH-KO T cells expanded to be more than 40-fold in 15 days, while CISH-WT T cells showed low persistence after 10 days (Figure 2E). Consistently, CISH-KO T cells exhibited better killing activity against tumor cells than CISH-WT T cells in culture treated with a low concentration of IL-2 for 10 days (Figure 2F). Moreover, CISH-KO cells showed higher cytokine secretion levels than CISH-WT T cells (Figure 2G). These results suggested that the deletion of CISH prolonged T cell survival and increased the cytotoxic effect when cytokine stimulation was limiting.
CISH ablation promotes the antitumor activity and persistence of CAR-T cells with decreased PD-1 expression
To validate the function of CISH in CAR-T cells, we knocked out CISH in CD19-targeted and mesothelin (MSLN)-targeted CAR-T cells, in which the CARs were composed of CD28, CD3z, and TLR2 signaling domains.46 Notably, CISH-KO CAR-T cells showed greater cytotoxicity than mock T cells against target cells (Figures 3A and 3B). Higher levels of cytokines were also detected in the supernatant of the CISH-KO CAR-T cell and target cell coculture than in that of the CISH-WT CAR-T cell and target cell coculture (Figure 3C), indicating the greater cytokine-secreting capabilities of the CISH-KO CAR-T cells. These results suggested that CISH disruption led to improved antitumor activity and higher cytokine secretion by CAR-T cells.
Figure 3.

CISH ablation promotes the antitumor activity and persistence of CAR-T cells with decreased PD-1 expression
(A) CD19 CISH-WT and CD19 CISH-KO T cells were cocultured with NALM6 cells with the indicated E:T ratio. (B) MSLN-targeted CISH-WT and CISH-KO T cells were cocultured with MKN28 cells with the indicated ratios of effectors to targets. (C) MSLN-targeted CISH-WT and CISH-KO T cells were cocultured with target cells at an E:T ratio of 1:1 for 24 h. The culture supernatants were then collected and analyzed for the levels of secreted IL-2, IFN-γ, granzyme B, and TNF-α were measured with ELISA kits. ∗∗p < 0.01, ∗∗∗p < 0.001, unpaired t test. (D) Growth curve of MKN28 xenografts treated with mock, MSLN-targeted CISH-WT, or MSLN-targeted CISH-KO T cells. n = 5, ∗∗p < 0.01, ∗∗∗p < 0.001, two-way ANOVA with Tukey’s multiple comparison test. (E) CAR-T cells in PB were analyzed by flow cytometry. ∗p < 0.05, unpaired t test. (F) CAR-T cells that had infiltrated tumors were analyzed by flow cytometry. Unpaired t test. (G and H) Cell surface abundance of PD-1 on CAR-T cells in PB was determined through flow cytometry. Representative graph (G) and statistical data (H) showing the abundance of PD-1 on T cells are shown. MFI, mean fluorescence intensity. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, two-way ANOVA with Tukey’s multiple comparison test. (I) Cell surface abundance of PD-1 on CAR-T cells in tumors was analyzed by flow cytometry. ∗p < 0.05, ∗∗p < 0.01, two-way ANOVA with Tukey’s multiple comparison test.
To evaluate the antitumor activity of CISH-KO CAR-T cells in vivo, we assessed their killing effect on MKN-28 cells in a mouse xenograft tumor model. NOD-scid-IL2Rg−/− (NSI) mice aged 6 to 8 weeks were injected subcutaneously (s.c.) with 1 × 106 MKN-28 cells, and 3 weeks later, the mice received a single intravenous (i.v.) injection of 5 × 106 mock-T cells, MSLN-targeted CISH-WT CAR-T cells, or MSLN-targeted CISH-KO CAR-T cells. Treatment with MSLN-targeted CISH-KO CAR-T cells led to significantly better antitumor activity with reduced tumor burden compared with treatment using mock-T cells or MSLN-targeted CISH-WT CAR-T cells, while MSLN-targeted CISH-WT CAR-T cells showed a more modest reduction in tumor burden than treatment with mock-T cells (Figure 3D). Because CISH KO promoted the long-term survival of CAR-T cells under conditions with limited stimulus in vitro, we wondered whether CISH KO can enhance the survival of CAR-T cells in a mouse model of the IL-2-deficient condition. CAR-T persistence in peripheral blood (PB) was measured by flow cytometry, and the results revealed that MSLN-targeted CISH-KO CAR-T cells showed significantly longer persistence than MSLN-targeted CISH-WT CAR-T cells (Figure 3E). After mice were euthanized, CAR-T cell infiltration into tumors was detected by flow cytometry. The CISH-KO group showed higher infiltration of CAR-T cells into the tumor, although the difference was not statistically significant (Figure 3F). In conclusion, CISH ablation promotes the antitumor activity of CAR-T cells, prolonging their persistence and moderately increasing their infiltration in vivo.
Since the deletion of CISH promotes JAK-STAT signaling and T cell activation, stronger T cell activation that leads to higher T cell exhaustion is a concern. Therefore, we evaluated PD-1, TIM-3, and LAG-3 abundance on the CAR-T cell surface via flow cytometry. The abundance of TIM-3 and LAG-3 on CISH-KO CAR-T cells showed a mild decrease or no significant decrease compared with that in CISH-WT CAR-T cells (Figures S3A and S3B). To our surprise, a significantly lower mean fluorescence intensity (MFI) was detected in the MSLN-targeted CISH-KO CAR-T cells, and a significantly small percentage of the MSLN-targeted CISH-KO CAR-T cells expressed PD-1 (Figures 3G and 3H). Exhausted T cells are characterized by the expression of multiple inhibitory receptors, including PD-1, LAG-3, and TIM-3. According to the results of our flow cytometry analysis, a significantly lower proportion of T cells in the CISH-KO group expressed both PD-1 and LAG-3 or both PD-1 and TIM-3. In contrast, significantly more MSLN-targeted CISH-KO CAR-T cells were double negative for both marker pairs (Figures S3C and S3D). These results suggested a reduced exhaustion of MSLN-targeted CISH-KO CAR-T cells. In line with MSLN-targeted CISH-KO CAR-T cells in PB, the number of infiltrated MSLN-targeted CISH-KO CAR-T cells was significantly lower than that indicated by PD-1 MFI in both CD4 and CD8 T cells (Figures 3I and S3E). Collectively, these data demonstrate that CISH-KO CAR-T cells showed longer survival and increased antitumor activity; moreover, fewer CISH-KO CAR-T cells showed exhaustion when PD-1 levels were decreased.
The disruption of CISH expression increases the expression of FBXO38 in activated T cells, leading to decreased PD-1 levels
To verify the regulation of PD-1 expression by CISH deletion in CAR-T cells, we cocultured CAR-T cells with target cells in vitro. Consistent with findings obtained with CAR-T cells in vivo, CISH-KO CAR-T cells showed significantly lower PD-1 expression compared with CISH-WT CAR-T cells, while TIM-3 expression level is moderately lower and LAG-3 expression level is higher in the CISH-KO T cells than in the CISH-WT T cells (Figures 4A and S4A). High-affinity GD2-targeted CAR, composed of the anti-GD2 14g2a-E101K scFv, CD3ζ, and CD28 signaling domains, has been reported to cause CAR-T cell exhaustion by clustering on the T cell surface to trigger CAR signaling in the absence of antigen.47 We knocked out CISH in the GD2-targeted CAR-T cells and found that GD2-targeted CISH-WT CAR-T cells showed elevated PD-1 expression, while disrupted CISH expression led to significant decreases in PD-1 levels on GD2-targeted CAR-T cells on day 6 (Figure S4B).
Figure 4.

The disruption of CISH increases the expression of FBXO38 in activated T cells, leading to decreased PD-1 levels
(A) MSLN-targeted CISH-WT and CISH-KO T cells were cocultured with MKN28 cells for 12 days, and the cell surface abundance of PD-1 on CAR-T cells was analyzed by flow cytometry. ∗p < 0.05, ∗∗p < 0.01, one-way ANOVA with Tukey’s multiple comparison test. (B) CISH-WT and CISH-KO T cells were cocultured with OKT3 + A549 cells for 12 days, and the cell surface abundance of PD-1 on CAR-T cells was analyzed by flow cytometry. ∗∗∗p < 0.001, one-way ANOVA with Tukey’s multiple comparison test. (C) CISH-WT and CISH-KO T cells were stimulated with anti-CD3/anti-CD28 antibodies for the indicated day, and cell surface expression of PD-1 on CAR-T cells was analyzed by flow cytometry. ∗∗∗p < 0.001, two-way ANOVA with Tukey’s multiple comparison test. (D) Transcript levels of PDCD1 in CD3+ T cells were measured by quantitative real-time PCR with unpaired t test. (E) To detect PD-1 ubiquitination in activated T cells, T cells were stimulated with anti-CD3/anti-CD28 antibodies for 2 days and further incubated with MG132 (15 μM) for another 4 h before analysis. (F) Transcript levels of FBXO38 in CD3+ T cells were measured by quantitative real-time PCR. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, unpaired t test. (G) After CISH or FBXO38 knockout or CISH and FBXO38 double knockout, T cells were stimulated with anti-CD3/anti-CD28 antibodies for 2 days, and the cell surface abundance of PD-1 on CAR-T cells was analyzed by flow cytometry. ∗∗p < 0.01, ∗∗∗p < 0.001, one-way ANOVA with Tukey’s multiple comparisons test.
Since we found inhibition of PD-1 expression via CISH KO after CAR signaling was induced, we wanted to examine whether PD-1 expression by T cells was affected by CISH expression after TCR stimulation. Therefore, we cocultured T cells with OKT3-overexpressing A549 cells and obtained results consistent with that of CAR-T cells (Figures 4B and S4C). Furthermore, we activated T cells with anti-CD3/anti-CD28 antibodies. We found that the PD-1 expression level was significantly higher in the CISH-WT T cells than in the CISH-KO T cells (Figure 4C). The decreased PD-1 level in the CISH-KO group was validated by immunoblotting (Figure S4D). In summary, when T cells are activated via CAR or TCR signaling, CISH KO decreased the PD-1 level in the T cells.
The transcription of PD-1 has been shown to be correlated with two conserved DNA regions (CR-B and CR-C), is induced by multiple transcription factors,48,49,50,51,52,53,54,55,56 including NFATc1, c-FOS, Notch1, Foxo1, NF-κB, STATs, and TOX, and is suppressed by T-bet, Blimp-1, or SATB1.57,58,59 To verify whether decreased PD-1 expression in CISH-KO T cells is regulated by PDCD1 transcription, the mRNA levels of PDCD1 were evaluated by quantitative real-time PCR. The results showed no significant difference in the number of PDCD1 transcripts between CISH-WT T cells and CISH-KO T cells after stimulation with anti-CD3/anti-CD28 antibody-conjugated microbeads (Figure 4D). Posttranslational modifications play important roles in the regulation of PD-1 expression. To detect PD-1 ubiquitination in activated T cells, T cells were stimulated with anti-CD3/anti-CD28 antibodies for 2 days and further incubated with MG132 (15 μM) for another 4 h before analysis. As expected, CISH KO caused increased PD-1 poly-ubiquitination in activated T cells (Figure 4E). FBXO38 has been reported to target PD-1 on the cell surface and thus induce proteasome pathway degradation after PD-1 internalization, and STAT5 directly binds to the promoter of the FBXO38 gene.60 According to our results, CISH disruption increased the T cell response to TCR signaling and IL-2, leading to higher STAT5 phosphorylation in activated T cells (Figures 1B and 2A), which might have also increased the expression of FBXO38; therefore, quantitative real-time PCR analysis was used to measure the mRNA levels of FBXO38. Consistent with our hypothesis, a significant increase in the mRNA level of FBXO38 in CISH-KO T cells was identified after T cell activation (Figure 4F). To verify that PD-1 is regulated by FBXO38 expression, we knocked out FBXO38 in CISH-KO T cells. As expected, CISH KO decreased PD-1 expression after stimulation, and knocking out FBXO38 partially rescued PD-1 expression (Figure 4G). Taken together, these results suggest that CISH KO downregulated PD-1 expression in activated T cells by increasing FBXO38-mediated degradation of PD-1.
Discussion
Although CAR-T cells are prospective therapies for malignant tumors, the limitations on the antitumor function of CAR-T cells in terms of exhaustion and survival remain obstacles for their use against solid tumors. A promising approach is to target immune checkpoints of T cells, enhancing their antiapoptosis and killing functions with reduced exhaustion.1,2,61,62 CISH is a member of the SOCS family that has been reported to inhibit JAK-STATs and TCR signaling in NK and T cells.35,36,37 We hypothesized that it might be a potential candidate for regulating CAR-T cell function. Therefore, we knocked out CISH in T cells and verified that its inhibition led to an increase the sensitivity of T cells to TCR stimulation and cytokines (IL-2 and IL-15) in vitro. As we expected, KO of CISH enhanced T cell survival, cytokine secretion capacity, and antitumor effects. To validate its effect on CAR-T cells, we also knocked out CISH in CAR-T cells and observed the same results.
STAT5 signaling is necessary for functional maturation of antigen-activated T cells.63 Our studies demonstrated that CISH KO increased the sensitivity of T cells to anti-CD3/anti-CD28 antibodies or cytokine stimulation with elevated STAT5 and STAT3 signaling response, thereby enhancing their antitumor effect of CAR-T cells, even after cultured in medium with a low concentration of IL-2 for several days. Furthermore, we validated the function of CISH-KO CAR-T cells in a cell-line-derived xenograft (CDX) mouse model and observed a significantly smaller tumor burden, longer CAR-T cell survival in PB, and a modest increase in tumor infiltration by CISH-KO CAR-T cells. Previous studies showed that STAT5 can drive T-bet and Eomes expression,64 which is critical for tumor infiltration and antitumor activity of cytotoxic CD8 T cells.65 Elevated expression of T-bet has been shown to promote the expression of CXCR3,66 which is the receptor for the T helper (Th)1-type chemokines CXCL9, CXCL10, and CXCL11, and to promote T cell migration into tumors in response to these chemokines.67 These might contribute to higher CISH-KO CAR-T cell cytotoxicity and increased T cell infiltration into tumor. However, these phenotypes were only validated in one in vivo model. Future studies are needed to explore the enhancement of CAR-T function by CISH KO in more in vivo models. In addition, suppressive factors in the TME are often barriers to the effectiveness of adoptive cell therapy. We will explore whether the KO of CISH can reduce the inhibitory effect of high suppressive factors in TME in the follow-up study.
However, we were surprised to find that CISH-KO CAR-T cells significantly downregulated the expression of the exhaustion marker PD-1. Furthermore, we cocultured CAR-T cells with target cells in vitro and found that the CISH-KO CAR-T cells also exhibited lower PD-1 levels. Since CAR is designed to mimic TCR signaling, we directly stimulated T cells with anti-CD3/anti-CD28 antibodies or OKT3-overexpressing A549 cells and observed a consistent decrease in PD-1 level in CISH-KO T cells. According to previous studies, PD-1 transcription is regulated by transcription factors.48,49,50,51,52,53,57,58,59 We detected the transcriptional level of PD1 by quantitative real-time PCR, and we found that PD1 transcription was not significantly decreased in CISH-KO T cells. Therefore, we explored whether the reduction in PD-1 occurs through posttranscriptional regulation. FBXO38 has been recently reported to mediate PD-1 degradation via ubiquitination posttranslation.60 Therefore, we measured the expression levels of FBXO38 by quantitative real-time PCR. The results showed that the mRNA levels of FBXO38 were significantly increased in CISH-KO T cells. This outcome indicated that knocking out CISH promotes the expression of FBXO38. After knocking out FBXO38 in CISH-KO T cells, we found that PD-1 levels were partially rescued in T cells stimulated with anti-CD3/anti-CD28 antibodies. According to these data, we conclude that CISH-KO T cells reduce the PD-1 level by upregulating FBXO38.
Although PD-L1/PD-1 blockades with antibody have shown significant clinical benefit for various types of cancers, the high production cost of antibodies, immune-related adverse effects, and drug resistance remained limitations for antibody-based immunotherapies.68,69 Here, CRISPR-Cas9 technology was used to knock out an inhibitory gene CISH that has not been widely reported in T cells, downregulate PD-1 expression of T cells, and improve T cell antitumor ability. This method bypassed the limitations of PD-L1/PD-1 antibodies68,69 and avoided T cell function attenuation that may be caused by completely knocking out PD-1.15 Another member of the SOCS family, SOCS1, was also identified as an intrinsic checkpoint of CD4+ Th1 cell response.42 This suggested that other family members are potential candidates and deserve further study. For undruggable intracellular checkpoints, such as CISH, we have functionally inactivated them on primary T cells by CRISPR-Cas9-mediated gene editing, which makes them more feasible for clinical application. Recently, proteolysis-targeting chimera (PROTAC) has been developed to be a useful technology for targeted protein degradation,70 which may be another promising approach to target such undruggable intracellular checkpoints.
A previous review summarized the roles of some SOCS family members in T cell differentiation, maturation, and function, but the involvement of CISH has not been clearly stated in review.71 The JAK-STAT pathway has been reported to be involved in T cell maturation and differentiation, and we identified CISH as a suppressor of JAK-STATs. In this study, we found that in CISH-KO T cells, STAT5, and STAT3 responded more robustly to cytokine stimulation such as IL-2 and IL15. IL-2-induced activation of STAT5 is found to promote the expression of FoxP3, Tbx21 (T-bet), Maf, and Gata3, thereby promoting the differentiation of regulatory T (Treg), Th1, and Th2 cells and inhibiting the differentiation of Th17 and T follicular helper (Tfh) cells.72 However, activation of STAT3 is believed to be beneficial to Th17 cell differentiation.73 CD4 T cells differentiate following immune activation to produce distinct effector subpopulations that can be directed by their expression of lineage-specific transcriptional master regulators and cytokine requirements.74 Therefore, different cytokine combinations will influence the direction of T cell differentiation. Since CISH KO increased the sensitivity of T cells to cytokine stimulation, how CISH KO determines T cell differentiation under education of different cytokine combination is still unknown and worth studying. This is also a topic of CISH function that we are studying in induced pluripotent stem cell (iPSC)-derived T cells. Moreover, the effects of CISH on T cell development under physiological conditions will also be further explored in the conditional KO mouse model.
Overall, we discovered a way to reduce T cell PD-1 expression and promote T cell function by knocking out the suppressor gene CISH. This is a more effective and feasible way to modulate T cell immune function, especially for enhancing the effect of clinical CAR-T cell therapy.
Materials and methods
Isolation, gene KO, CAR transduction, and expansion of primary human T lymphocytes
PB mononuclear cells (PBMCs) were isolated from the buffy coats of healthy donors using Lymphoprep (Fresenius Kabi Norge, AS, Berg I Østfold, Norway). T cells were negatively selected from the PBMC population using an EasySep Human T cell Isolation Kit (catalog #17951) and activated using antihuman CD3/CD28 antibodies (#130-111-160) for 24 h in Iscove’s modified Dulbecco’s medium (IMDM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 300 IU/mL IL-2, and 1% penicillin/streptomycin.
For gene KO, CAS9 protein (#A50577) was incubated with synthetic sgRNA (GenScript Biotech Corporation, Nanjing, Jiangsu, China) at 37°C for 15 min, mixed with T cells, and subjected to an electroporation procedure using Amaxa Nucleofector 2b (Amaxa Human T cell Nucleofector Kit, Lonza, Germany).
The following gRNA targeting sequences were used in the study:
CISH-sgRNA-1 (5' - 3'): CAAGGGCTGCATGACTGGCT;
CISH-sgRNA-2 (5' - 3'): TGCTGGGGCCTTCCTCGAGG;
FBXO38-sgRNA-1 (5' - 3'): GACTAAATCCACTTACCACT; and
FBXO38-sgRNA-2 (5' - 3'): GAGTTGTAGATCTCTGTGCA.
Third-generation MSLN- or CD19-targeted CARs containing CD28, CD3ζ, and TLR2 cytoplasmic sequences used in this study have been previously described.46 For CAR transduction, 1 × 106 T cells were transduced with 10 mL CAR lentiviral supernatants for 6 h in 1 mL IMDM with 10% FBS. After transduction, T cells were cultured in fresh medium containing IL-2 (300 IU/mL).
Subsequently, fresh medium was added every 2 to 3 days to maintain the cell density within a range of 0.5–1×106 cells/mL. Healthy PBMC donors who provided primary specimens gave informed consent for the use of their samples for research purposes, and all procedures were approved by the Research Ethics Board of Guangzhou Institutes of Biomedicine and Health (GIBH).
Cells and culture conditions
HEK-293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM). A549 (human lung adenocarcinoma), NALM6 (human acute lymphoblastic leukemia), and MKN-28 (human gastric carcinoma) cell lines were obtained from ATCC (Manassas, VA, USA) and maintained in RPMI-1640. Luciferase/GFP-expressing cell lines (A549-GL, NALM6-GL, MKN-28-GL) were generated via transfection of the respective parental cell line with lentiviral supernatant containing luciferase-2A-GFP and were sorted for GFP expression via flow cytometry with an Aria cell sorter (BD Biosciences, San Jose, CA, USA). OKT3/tCD19-expressing A549-GL cells were generated by transfection of A549-GL cells with lentiviral supernatant containing OKT3-tCD19 and sorted by magnetic-activated cell sorting (MACS) using anti-CD19 microbeads (#130-050-301). DMEM and RPMI-1640 media were supplemented with 10% heat-inactivated FBS (Gibco/Life Technologies), 10 mM HEPES, 2 mM glutamine (Gibco/Life Technologies), and 1% penicillin/streptomycin. All cells were cultured at 37°C in an atmosphere with 5% carbon dioxide.
Lentivirus production
Lentiviral particles were produced in HEK-293T cells via polyethyleneimine (Sigma‒Aldrich, St. Louis, MO, USA) transfection. The pWPXLd-based lentiviral plasmid and two packaging plasmids, psPAX2 and pMD.2G, were cotransduced into HEK-293T cells in 10 cm dishes at a ratio of 3:1:4 for a total amount of 24 μg. Lentivirus-containing supernatants were harvested 24-, 48-, and 72-h posttransfection and filtered through a 0.45 μm filter.
Western blotting
Cells were lysed with radioimmunoprecipitation assay (RIPA) buffer (Pierce, Rockford, IL, USA), and the protein concentration was quantified using a BCA protein assay kit (Pierce). Samples were loaded onto 8%–12% SDS-PAGE gel, transferred to a PVDF membrane, and sequentially probed with primary antibodies. A species-matched HRP-conjugated secondary antibody was then incubated with the membrane, and proteins were detected by autoradiography using an enhanced chemiluminescence kit (ECL Plus, General Electric Health Care, Little Chalfont, UK).
Flow cytometry
All samples were analyzed using a NovoCyteTM (ACEA Biosciences), an LSR Fortessa, or a C6 flow cytometer (BD Biosciences), and the data were analyzed using FlowJo software (FlowJo, Ashland, OR, USA). The antibodies used included antihuman TIM3 (clone F38-2E2); antihuman LAG3 (clone 11C3C65); antihuman PD-1 (clone NAT105); antihuman CD3 (clone UCHT1); antihuman CD4 (clone OKT4); antihuman CD8 (clone OKT8); antimouse immunoglobulin G2a (IgG2a) isotype control-APC (clone RMG2a-62); antimouse IgG1κ isotype control-PE; antimouse IgG1κ isotype control-PerCP/Cy5.5; and antimouse IgG1κ isotype control-APC (clone MOPC-21) (BioLegend, San Diego, CA, USA). All flow-cytometry-related staining procedures were performed on ice for 30 min, and the cells were washed with PBS containing 1% FBS before cytometry analysis. PB and tumor samples from mouse xenografts were treated with red blood cell lysis buffer (BioLegend), and the cells were stained with the corresponding antibodies.
In vitro tumor-killing assays
A549-GL-OKT3-tCD19, NALM6-GL, and MKN28-GL target cells were incubated with the indicated T cells at the indicated ratio in triplicate wells of white 96-well plates. Target cell viability was monitored 24 h later by adding 100 μL/well D-luciferin (potassium salt) (#40902ES01) at 150 μg/mL. The percentage of viability (%) was calculated as the experimental signal/maximal signal × 100, and the percentage of intact (unlysed) cells was set to equal 100% viability.
Cytokine release assays
ELISA kits were used for IL-2 (#1110203), IFN-γ (#1110003), granzyme B (#1118503), and TNF-α (#1117203), and all ELISAs were performed according to the manufacturer’s protocols. T cells were cocultured with target cells at an effector-to-target (E:T) ratio of 1:1 for 24 h. The culture supernatants were collected, and the levels of secreted IL-2, IFN-γ, granzyme B, and TNF-α were measured with ELISA kits.
CDX models for CAR-T cell treatment
Animal experiments were performed in the Laboratory Animal Center of GIBH, and all animal procedures were approved by the Animal Welfare Committee of GIBH. All protocols were approved by the relevant institutional animal care and use committee (IACUC). All mice were maintained in specific-pathogen-free (SPF)-grade cages and were provided autoclaved food and water.
For the cell-line-based gastric cancer (GC) s.c. xenograft models, 1 × 106 MKN-28 cells in 100 μL PBS were injected s.c. into the right flanks of 6- to 8-week-old NSI mice. Approximately 3 weeks later, the mice were divided into three groups (mock T, MSLN-targeted CISH-WT T, and MSLN-targeted CISH-KO T) and received 5 × 106 CAR-T cells in 100 μL PBS peritumorally as indicated. Tumor volume was measured twice per week with a caliper and calculated via the following equation: tumor volume = (length × width2)/2.
Statistical analysis
Data are presented as the means ± standard errors of the means. Student’s t test was used to determine the statistical significance of differences between samples, and a p value <0.05 indicated a significant difference. All statistical analyses were performed using Prism software v.9.0 (GraphPad, San Diego, CA, USA).
Availability of data and material
All data generated or analyzed during this study are included in this article.
Acknowledgments
We thank all the members in the author list and other laboratory members for experimental materials, technical assistance, helpful discussions, and comments. This study was supported by the National Key Research and Development Plan, no. 2021YFE0202800 (P.L.); National Natural Science Foundation of China, nos. 81961128003 (P.L.) and 81972672 (P.L.); Guangdong Basic and Applied Basic Research Foundation, nos. 2019A1515010062 (Y.Y.), 2021A1515110005 (L.Q.), 2022A1515012360 (L.Q.), 2022A1515010604 (Y.Y.), and 2020B1212060052; Science and Technology Program of Guangzhou, China (202002020083, X.L.); Guangzhou Medical University High-level University Construction Research Startup Fund, no. B195002004013 (L.Q.); The University Grants Committee/Research Grants Council of the Hong Kong Special Administrative Region, China (project no. AoE/M-401/20); Innovation and Technology Fund (ITF) and the Guangdong-Hong Kong-Macau Joint Laboratory of Respiratory Infectious Diseases (2019B121205010). All animal experiments were performed at the Laboratory Animal Center of the GIBH, and all procedures were approved by the Animal Welfare Committee of GIBH. NSI mice were derived at GIBH. All mice were maintained in SPF cages and provided autoclaved food and water. Animal protocols were approved by the IACUC.
Author contributions
J.L., L.Q., and P.L. conceived and designed the research; J.L., L.Q., D.W., Z.W., D.Z., S.L., and M.L. performed in vitro assays and animal experiments; J.L. wrote the manuscript; P.L., L.-H.Y., L.Q., R.Z., and Y.Y. provided critical advice for this study and revised the manuscript; L.Q., Z.T., Z.W., D.Z., Q.W., Y.-H.L., Y.-L.T., and X.L. provided important research reagents and technical advice; all authors revised and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2022.12.003.
Contributor Information
Li-Hua Yang, Email: yanglihua@smu.edu.cn.
Peng Li, Email: li_peng@gibh.ac.cn.
Supplemental information
References
- 1.Newick K., O'Brien S., Moon E., Albelda S.M. CAR T cell therapy for solid tumors. Annu. Rev. Med. 2017;68:139–152. doi: 10.1146/annurev-med-062315-120245. [DOI] [PubMed] [Google Scholar]
- 2.Roselli E., Faramand R., Davila M.L. Insight into next-generation CAR therapeutics: designing CAR T cells to improve clinical outcomes. J. Clin. Invest. 2021;131:e142030. doi: 10.1172/JCI142030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martinez M., Moon E.K. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 2019;10:128. doi: 10.3389/fimmu.2019.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Propper D.J., Balkwill F.R. Harnessing cytokines and chemokines for cancer therapy. Nat. Rev. Clin. Oncol. 2022;19:237–253. doi: 10.1038/s41571-021-00588-9. [DOI] [PubMed] [Google Scholar]
- 5.Keir M.E., Butte M.J., Freeman G.J., Sharpe A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Z., Song W., Rubinstein M., Liu D. Recent updates in cancer immunotherapy: a comprehensive review and perspective of the 2018 China Cancer Immunotherapy Workshop in Beijing. J. Hematol. Oncol. 2018;11:142. doi: 10.1186/s13045-018-0684-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.John L.B., Devaud C., Duong C.P.M., Yong C.S., Beavis P.A., Haynes N.M., Chow M.T., Smyth M.J., Kershaw M.H., Darcy P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013;19:5636–5646. doi: 10.1158/1078-0432.CCR-13-0458. [DOI] [PubMed] [Google Scholar]
- 8.Gargett T., Yu W., Dotti G., Yvon E.S., Christo S.N., Hayball J.D., Lewis I.D., Brenner M.K., Brown M.P. GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade. Mol. Ther. 2016;24:1135–1149. doi: 10.1038/mt.2016.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cherkassky L., Morello A., Villena-Vargas J., Feng Y., Dimitrov D.S., Jones D.R., Sadelain M., Adusumilli P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Invest. 2016;126:3130–3144. doi: 10.1172/JCI83092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rafiq S., Yeku O.O., Jackson H.J., Purdon T.J., van Leeuwen D.G., Drakes D.J., Song M., Miele M.M., Li Z., Wang P., et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018;36:847–856. doi: 10.1038/nbt.4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rupp L.J., Schumann K., Roybal K.T., Gate R.E., Ye C.J., Lim W.A., Marson A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017;7:737. doi: 10.1038/s41598-017-00462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi B.D., Yu X., Castano A.P., Darr H., Henderson D.B., Bouffard A.A., Larson R.C., Scarfò I., Bailey S.R., Gerhard G.M., et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer. 2019;7:304. doi: 10.1186/s40425-019-0806-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu B., Zou Y., Zhang L., Tang J., Niedermann G., Firat E., Huang X., Zhu X. Nucleofection with plasmid DNA for CRISPR/Cas9-Mediated inactivation of programmed cell death protein 1 in CD133-specific CAR T cells. Hum. Gene Ther. 2019;30:446–458. doi: 10.1089/hum.2017.234. [DOI] [PubMed] [Google Scholar]
- 14.Hu W., Zi Z., Jin Y., Li G., Shao K., Cai Q., Ma X., Wei F. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol. Immunother. 2019;68:365–377. doi: 10.1007/s00262-018-2281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Odorizzi P.M., Pauken K.E., Paley M.A., Sharpe A., Wherry E.J. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med. 2015;212:1125–1137. doi: 10.1084/jem.20142237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grosser R., Cherkassky L., Chintala N., Adusumilli P.S. Combination immunotherapy with CAR T cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell. 2019;36:471–482. doi: 10.1016/j.ccell.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fesnak A.D., June C.H., Levine B.L. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat. Rev. Cancer. 2016;16:566–581. doi: 10.1038/nrc.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Savoldo B., Ramos C.A., Liu E., Mims M.P., Keating M.J., Carrum G., Kamble R.T., Bollard C.M., Gee A.P., Mei Z., et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Invest. 2011;121:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhong X.S., Matsushita M., Plotkin J., Riviere I., Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther. 2010;18:413–420. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song D.G., Ye Q., Carpenito C., Poussin M., Wang L.P., Ji C., Figini M., June C.H., Coukos G., Powell D.J., Jr. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB) Cancer Res. 2011;71:4617–4627. doi: 10.1158/0008-5472.CAN-11-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson L.A., June C.H. Driving gene-engineered T cell immunotherapy of cancer. Cell Res. 2017;27:38–58. doi: 10.1038/cr.2016.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu W., Zhou Q., Masubuchi T., Shi X., Li H., Xu X., Huang M., Meng L., He X., Zhu H., et al. Multiple signaling roles of CD3epsilon and its application in CAR-T cell therapy. Cell. 2020;182:855–871.e23. doi: 10.1016/j.cell.2020.07.018. [DOI] [PubMed] [Google Scholar]
- 23.Hartl F.A., Beck-Garcìa E., Woessner N.M., Flachsmann L.J., Cárdenas R.M.H.V., Brandl S.M., Taromi S., Fiala G.J., Morath A., Mishra P., et al. Noncanonical binding of Lck to CD3epsilon promotes TCR signaling and CAR function. Nat. Immunol. 2020;21:902–913. doi: 10.1038/s41590-020-0732-3. [DOI] [PubMed] [Google Scholar]
- 24.Patzak I.M., Königsberger S., Suzuki A., Mak T.W., Kiefer F. HPK1 competes with ADAP for SLP-76 binding and via Rap1 negatively affects T-cell adhesion. Eur. J. Immunol. 2010;40:3220–3225. doi: 10.1002/eji.201040313. [DOI] [PubMed] [Google Scholar]
- 25.Di Bartolo V., Montagne B., Salek M., Jungwirth B., Carrette F., Fourtane J., Sol-Foulon N., Michel F., Schwartz O., Lehmann W.D., Acuto O. A novel pathway down-modulating T cell activation involves HPK-1-dependent recruitment of 14-3-3 proteins on SLP-76. J. Exp. Med. 2007;204:681–691. doi: 10.1084/jem.20062066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Si J., Shi X., Sun S., Zou B., Li Y., An D., Lin X., Gao Y., Long F., Pang B., et al. Hematopoietic progenitor Kinase1 (HPK1) mediates T cell dysfunction and is a druggable target for T cell-based immunotherapies. Cancer Cell. 2020;38:551–566.e11. doi: 10.1016/j.ccell.2020.08.001. [DOI] [PubMed] [Google Scholar]
- 27.Paolino M., Penninger J.M. Cbl-b in T-cell activation. Semin. Immunopathol. 2010;32:137–148. doi: 10.1007/s00281-010-0197-9. [DOI] [PubMed] [Google Scholar]
- 28.Kumar J., Kumar R., Kumar Singh A., Tsakem E.L., Kathania M., Riese M.J., Theiss A.L., Davila M.L., Venuprasad K. Deletion of Cbl-b inhibits CD8(+) T-cell exhaustion and promotes CAR T-cell function. J. Immunother. Cancer. 2021;9:e001688. doi: 10.1136/jitc-2020-001688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rochman Y., Spolski R., Leonard W.J. New insights into the regulation of T cells by gamma(c) family cytokines. Nat. Rev. Immunol. 2009;9:480–490. doi: 10.1038/nri2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kagoya Y., Tanaka S., Guo T., Anczurowski M., Wang C.H., Saso K., Butler M.O., Minden M.D., Hirano N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018;24:352–359. doi: 10.1038/nm.4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang Z., Liao R., Lv J., Li S., Zheng D., Qin L., Wu D., Chen S., Long Y., Wu Q., et al. IL-6 trans-signaling promotes the expansion and anti-tumor activity of CAR T cells. Leukemia. 2021;35:1380–1391. doi: 10.1038/s41375-020-01085-1. [DOI] [PubMed] [Google Scholar]
- 32.Wiede F., Lu K.H., Du X., Liang S., Hochheiser K., Dodd G.T., Goh P.K., Kearney C., Meyran D., Beavis P.A., et al. PTPN2 phosphatase deletion in T cells promotes anti-tumour immunity and CAR T-cell efficacy in solid tumours. EMBO J. 2020;39:e103637. doi: 10.15252/embj.2019103637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hilton D.J., Richardson R.T., Alexander W.S., Viney E.M., Willson T.A., Sprigg N.S., Starr R., Nicholson S.E., Metcalf D., Nicola N.A. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc. Natl. Acad. Sci. USA. 1998;95:114–119. doi: 10.1073/pnas.95.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang J.G., Farley A., Nicholson S.E., Willson T.A., Zugaro L.M., Simpson R.J., Moritz R.L., Cary D., Richardson R., Hausmann G., et al. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc. Natl. Acad. Sci. USA. 1999;96:2071–2076. doi: 10.1073/pnas.96.5.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palmer D.C., Guittard G.C., Franco Z., Crompton J.G., Eil R.L., Patel S.J., Ji Y., Van Panhuys N., Klebanoff C.A., Sukumar M., et al. Cish actively silences TCR signaling in CD8+ T cells to maintain tumor tolerance. J. Exp. Med. 2015;212:2095–2113. doi: 10.1084/jem.20150304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delconte R.B., Kolesnik T.B., Dagley L.F., Rautela J., Shi W., Putz E.M., Stannard K., Zhang J.G., Teh C., Firth M., et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat. Immunol. 2016;17:816–824. doi: 10.1038/ni.3470. [DOI] [PubMed] [Google Scholar]
- 37.Zhu H., Blum R.H., Bernareggi D., Ask E.H., Wu Z., Hoel H.J., Meng Z., Wu C., Guan K.L., Malmberg K.J., Kaufman D.S. Metabolic reprograming via deletion of CISH in human iPSC-derived NK cells promotes in vivo persistence and enhances anti-tumor activity. Cell Stem Cell. 2020;27:224–237.e6. doi: 10.1016/j.stem.2020.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Welte T., Leitenberg D., Dittel B.N., al-Ramadi B.K., Xie B., Chin Y.E., Janeway C.A., Jr., Bothwell A.L., Bottomly K., Fu X.Y. STAT5 interaction with the T cell receptor complex and stimulation of T cell proliferation. Science. 1999;283:222–225. doi: 10.1126/science.283.5399.222. [DOI] [PubMed] [Google Scholar]
- 39.Abbas A.K., Trotta E., R Simeonov D., Marson A., Bluestone J.A. Revisiting IL-2: biology and therapeutic prospects. Sci. Immunol. 2018;3:eaat1482. doi: 10.1126/sciimmunol.aat1482. [DOI] [PubMed] [Google Scholar]
- 40.Spolski R., Li P., Leonard W.J. Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat. Rev. Immunol. 2018;18:648–659. doi: 10.1038/s41577-018-0046-y. [DOI] [PubMed] [Google Scholar]
- 41.Overwijk W.W., Tagliaferri M.A., Zalevsky J. Engineering IL-2 to give new life to T cell immunotherapy. Annu. Rev. Med. 2021;72:281–311. doi: 10.1146/annurev-med-073118-011031. [DOI] [PubMed] [Google Scholar]
- 42.Sutra Del Galy A., Menegatti S., Fuentealba J., Lucibello F., Perrin L., Helft J., Darbois A., Saitakis M., Tosello J., Rookhuizen D., et al. In vivo genome-wide CRISPR screens identify SOCS1 as intrinsic checkpoint of CD4(+) TH1 cell response. Sci. Immunol. 2021;6:eabe8219. doi: 10.1126/sciimmunol.abe8219. [DOI] [PubMed] [Google Scholar]
- 43.Babon J.J., Kershaw N.J., Murphy J.M., Varghese L.N., Laktyushin A., Young S.N., Lucet I.S., Norton R.S., Nicola N.A. Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity. 2012;36:239–250. doi: 10.1016/j.immuni.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu J., Giuntoli R.L., 2nd, Omiya R., Kobayashi H., Kennedy R., Celis E. Interleukin 15 promotes antigen-independent in vitro expansion and long-term survival of antitumor cytotoxic T lymphocytes. Clin. Cancer Res. 2002;8:3877–3884. [PubMed] [Google Scholar]
- 45.Wu T.S., Lee J.M., Lai Y.G., Hsu J.C., Tsai C.Y., Lee Y.H., Liao N.S. Reduced expression of Bcl-2 in CD8+ T cells deficient in the IL-15 receptor alpha-chain. J. Immunol. 2002;168:705–712. doi: 10.4049/jimmunol.168.2.705. [DOI] [PubMed] [Google Scholar]
- 46.Lai Y., Weng J., Wei X., Qin L., Lai P., Zhao R., Jiang Z., Li B., Lin S., Wang S., et al. Toll-like receptor 2 costimulation potentiates the antitumor efficacy of CAR T Cells. Leukemia. 2018;32:801–808. doi: 10.1038/leu.2017.249. [DOI] [PubMed] [Google Scholar]
- 47.Lynn R.C., Weber E.W., Sotillo E., Gennert D., Xu P., Good Z., Anbunathan H., Lattin J., Jones R., Tieu V., et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature. 2019;576:293–300. doi: 10.1038/s41586-019-1805-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oestreich K.J., Yoon H., Ahmed R., Boss J.M. NFATc1 regulates PD-1 expression upon T cell activation. J. Immunol. 2008;181:4832–4839. doi: 10.4049/jimmunol.181.7.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiao G., Deng A., Liu H., Ge G., Liu X. Activator protein 1 suppresses antitumor T-cell function via the induction of programmed death 1. Proc. Natl. Acad. Sci. USA. 2012;109:15419–15424. doi: 10.1073/pnas.1206370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mathieu M., Cotta-Grand N., Daudelin J.F., Thébault P., Labrecque N. Notch signaling regulates PD-1 expression during CD8(+) T-cell activation. Immunol. Cell Biol. 2013;91:82–88. doi: 10.1038/icb.2012.53. [DOI] [PubMed] [Google Scholar]
- 51.Staron M.M., Gray S.M., Marshall H.D., Parish I.A., Chen J.H., Perry C.J., Cui G., Li M.O., Kaech S.M. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity. 2014;41:802–814. doi: 10.1016/j.immuni.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Redd P.S., Lu C., Klement J.D., Ibrahim M.L., Zhou G., Kumai T., Celis E., Liu K. H3K4me3 mediates the NF-kappaB p50 homodimer binding to the pdcd1 promoter to activate PD-1 transcription in T cells. Oncoimmunology. 2018;7:e1483302. doi: 10.1080/2162402X.2018.1483302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Austin J.W., Lu P., Majumder P., Ahmed R., Boss J.M. STAT3, STAT4, NFATc1, and CTCF regulate PD-1 through multiple novel regulatory regions in murine T cells. J. Immunol. 2014;192:4876–4886. doi: 10.4049/jimmunol.1302750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alfei F., Kanev K., Hofmann M., Wu M., Ghoneim H.E., Roelli P., Utzschneider D.T., von Hoesslin M., Cullen J.G., Fan Y., et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature. 2019;571:265–269. doi: 10.1038/s41586-019-1326-9. [DOI] [PubMed] [Google Scholar]
- 55.Khan O., Giles J.R., McDonald S., Manne S., Ngiow S.F., Patel K.P., Werner M.T., Huang A.C., Alexander K.A., Wu J.E., et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature. 2019;571:211–218. doi: 10.1038/s41586-019-1325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scott A.C., Dündar F., Zumbo P., Chandran S.S., Klebanoff C.A., Shakiba M., Trivedi P., Menocal L., Appleby H., Camara S., et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature. 2019;571:270–274. doi: 10.1038/s41586-019-1324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kao C., Oestreich K.J., Paley M.A., Crawford A., Angelosanto J.M., Ali M.A.A., Intlekofer A.M., Boss J.M., Reiner S.L., Weinmann A.S., Wherry E.J. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat. Immunol. 2011;12:663–671. doi: 10.1038/ni.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu P., Youngblood B.A., Austin J.W., Mohammed A.U.R., Butler R., Ahmed R., Boss J.M. Blimp-1 represses CD8 T cell expression of PD-1 using a feed-forward transcriptional circuit during acute viral infection. J. Exp. Med. 2014;211:515–527. doi: 10.1084/jem.20130208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stephen T.L., Payne K.K., Chaurio R.A., Allegrezza M.J., Zhu H., Perez-Sanz J., Perales-Puchalt A., Nguyen J.M., Vara-Ailor A.E., Eruslanov E.B., et al. SATB1 expression governs epigenetic repression of PD-1 in tumor-reactive T cells. Immunity. 2017;46:51–64. doi: 10.1016/j.immuni.2016.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meng X., Liu X., Guo X., Jiang S., Chen T., Hu Z., Liu H., Bai Y., Xue M., Hu R., et al. FBXO38 mediates PD-1 ubiquitination and regulates anti-tumour immunity of T cells. Nature. 2018;564:130–135. doi: 10.1038/s41586-018-0756-0. [DOI] [PubMed] [Google Scholar]
- 61.Pardoll D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ribas A., Wolchok J.D. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Malek T.R., Yu A., Scibelli P., Lichtenheld M.G., Codias E.K. Broad programming by IL-2 receptor signaling for extended growth to multiple cytokines and functional maturation of antigen-activated T cells. J. Immunol. 2001;166:1675–1683. doi: 10.4049/jimmunol.166.3.1675. [DOI] [PubMed] [Google Scholar]
- 64.Grange M., Verdeil G., Arnoux F., Griffon A., Spicuglia S., Maurizio J., Buferne M., Schmitt-Verhulst A.M., Auphan-Anezin N. Active STAT5 regulates T-bet and eomesodermin expression in CD8 T cells and imprints a T-bet-dependent Tc1 program with repressed IL-6/TGF-beta1 signaling. J. Immunol. 2013;191:3712–3724. doi: 10.4049/jimmunol.1300319. [DOI] [PubMed] [Google Scholar]
- 65.Zhu Y., Ju S., Chen E., Dai S., Li C., Morel P., Liu L., Zhang X., Lu B. T-bet and eomesodermin are required for T cell-mediated antitumor immune responses. J. Immunol. 2010;185:3174–3183. doi: 10.4049/jimmunol.1000749. [DOI] [PubMed] [Google Scholar]
- 66.Taqueti V.R., Grabie N., Colvin R., Pang H., Jarolim P., Luster A.D., Glimcher L.H., Lichtman A.H. T-bet controls pathogenicity of CTLs in the heart by separable effects on migration and effector activity. J. Immunol. 2006;177:5890–5901. doi: 10.4049/jimmunol.177.9.5890. [DOI] [PubMed] [Google Scholar]
- 67.Nagarsheth N., Wicha M.S., Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017;17:559–572. doi: 10.1038/nri.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Naidoo J., Page D.B., Li B.T., Connell L.C., Schindler K., Lacouture M.E., Postow M.A., Wolchok J.D. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann. Oncol. 2015;26:2375–2391. doi: 10.1093/annonc/mdv383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tartari F., Santoni M., Burattini L., Mazzanti P., Onofri A., Berardi R. Economic sustainability of anti-PD-1 agents nivolumab and pembrolizumab in cancer patients: Recent insights and future challenges. Cancer Treat. Rev. 2016;48:20–24. doi: 10.1016/j.ctrv.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 70.Li X., Song Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020;13:50. doi: 10.1186/s13045-020-00885-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Palmer D.C., Restifo N.P. Suppressors of cytokine signaling (SOCS) in T cell differentiation, maturation, and function. Trends Immunol. 2009;30:592–602. doi: 10.1016/j.it.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ross S.H., Cantrell D.A. Signaling and function of interleukin-2 in T lymphocytes. Annu. Rev. Immunol. 2018;36:411–433. doi: 10.1146/annurev-immunol-042617-053352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang X.P., Ghoreschi K., Steward-Tharp S.M., Rodriguez-Canales J., Zhu J., Grainger J.R., Hirahara K., Sun H.W., Wei L., Vahedi G., et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat. Immunol. 2011;12:247–254. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.O'Shea J.J., Paul W.E. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science. 2010;327:1098–1102. doi: 10.1126/science.1178334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this article.