

Abstract
The generation of ectoderm, mesoderm, and endoderm layers is the most critical biological process during the gastrulation of embryo development. Such a differentiation process in human embryonic stem cells (hESCs) is an inherently nonlinear multi-stage dynamical process which contain multiple tipping points playing crucial roles in the cell-fate decision. However, the tipping points of the process are largely unknown, letting alone the understanding of the molecular regulation on these critical events. Here by designing a module-based dynamic network biomarker (M-DNB) model, we quantitatively pinpointed two tipping points of the differentiation of hESCs toward definitive endoderm, which leads to the identification of M-DNB factors (FOS, HSF1, MYCN, TP53, and MYC) of this process. We demonstrate that before the tipping points, M-DNB factors are able to maintain the cell states and orchestrate cell-fate determination during hESC (ES)-to-ME and ME-to-DE differentiation processes, which not only leads to better understanding of endodermal specification of hESCs but also reveals the power of the M-DNB model to identify critical transition points with their key factors in diverse biological processes, including cell differentiation and transdifferentiation dynamics.
Keywords: tipping point, M-DNB (module-based dynamic network biomarker) model, M-DNB factors, differentiation
Graphical abstract
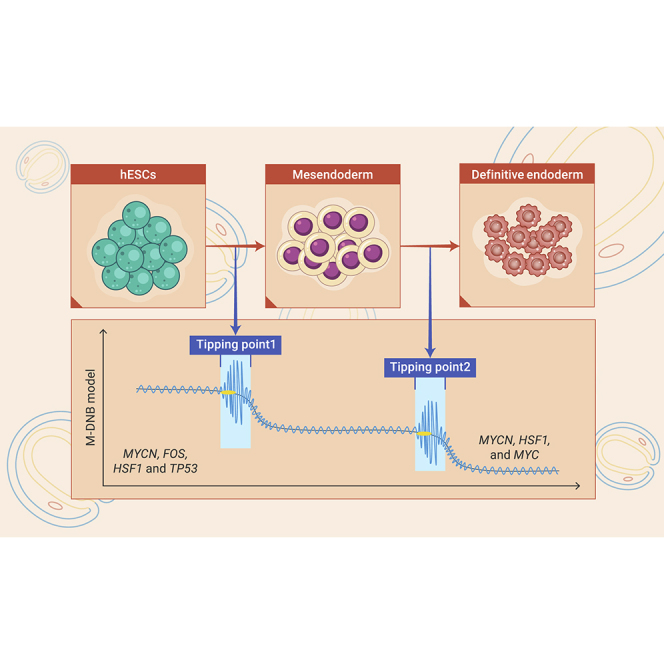
Public summary
-
•
M-DNB model identifies two tipping points of hESC differentiation.
-
•
Five M-DNB factors are master regulators in hESC differentiation.
-
•
Before tipping points, M-DNB factors orchestrate cell fate determination.
Introduction
The generation of three primordial germ layers—the ectoderm, mesoderm, and endoderm—is the most critical biological process during the gastrulation of embryo development.1,2 It has been clearly demonstrated that human embryonic stem cells (hESCs) can be used as an effective in vitro model to recapitulate in vivo developmental programs and dissect the molecular mechanism underpinning lineage specification and differentiation.3 Generally, the differentiation of hESCs can be viewed as a nonlinear dynamical process with multiple transition stages of cell states, during which the regulations on its tipping points immediately before the transition stages are crucial to the cell-fate determination from the perspective of the dynamical system.4,5 Thus, the identification of the tipping points, as well as the relevant key regulators, is important to improve the understanding of lineage specification and differentiation.6
It has been shown that hESCs differentiate into the definitive endoderm (DE) through an intermediate stage known as the mesendoderm (ME),2 and that the cell-fate determination during this process is tightly controlled by a series of transcription factors, including those of pluripotency factors (Nanog, Oct4, Sox2), the P53 family, the Myc family (MycN, Myc), T box genes (Brachyury/T, Eomesdermin), Mixl1, Sox17, and FoxA2.2,7,8,9 In addition to classical pluripotency factors, MycN has been shown to play a vital role in maintaining cells in a proliferative and undifferentiated state,10,11 while Myc has been shown to maintain pluripotency by inhibiting the primitive endodermal master regulator GATA6.12 Genetic studies have revealed that the loss of the P53 family blocks the mesendodermal differentiation of hESCs,9 and that Eomes is the master regulator essential for DE specification from the mesendoderm.13 Being the markers for nascent DE, Sox17, or FoxA2 is required for further differentiation into derivative lineages, not for the DE specification.9,14 In addition, functional studies have revealed the critical roles of extracellular cues that activate canonical Wnt, Activin/Nodal, and BMP signaling during ME and DE differentiation from hESCs, which synergize in a spatiotemporal fashion to stabilize the transcriptional network.15 Despite the extensive studies designed to decipher the molecular mechanisms that govern the cell-fate decision during gastrulation, this early developmental process has never been investigated from the perspective of dynamic systems to understand the regulation of the tipping points with their critical transition states, let alone the key modulators of these irreversible changes.
The differentiation of hESCs can generally be viewed as the evolution of a nonlinear dynamical system, during which the molecular events at the tipping points immediately before the downstream transition determinate the cell fate. To further elucidate the dynamic molecular controls that govern the early differentiation of hESCs, we must identify the tipping points and their key modulators, which is challenging as it requires information beyond the traditional differential expression analysis because of high sensitivity to noise and nonlinear dynamics near the tipping points.4,5 On the basis of nonlinear dynamical systems theory, the dynamic network biomarker (DNB) method, which uses a group of collectively fluctuated genes during a biological process rather than differentially expressed genes, has been developed to identify the critical states and to predict the leading molecules of the biological processes of disease progression by exploiting the dynamical features of the tipping points obtained from bulk omics data.4,16,17 Among the omics data, those from single-cell transcriptomic analyses (single-cell RNA sequencing [scRNA-seq])18,19,20 provide a wealth of unprecedented information for detecting the tipping points and the related leading transcripts. However, the application of sophisticated, dynamics-based methods on single-cell data is limited because of the higher levels of transcript amplification noises and drop-outs than those of RNA sequencing (RNA-seq) performed on bulk cell populations. To overcome this obstacle, we developed a module-based DNB (M-DNB) model by exploring transcriptional network and dynamics information from scRNA-seq data,21,22 which can provide reliable quantification on the tipping points and their regulators.
Specifically, here, by applying M-DNB to analyze the time course scRNA-seq datasets (Chu-time dataset) from endodermal differentiation of hESCs,23 we first identified two tipping points (12 and 36 h ofdifferentiation) that divide the whole process into three stages, embryonic stem (ES), ME, and DE and five M-DNB factors (FOS, HSF1, MYC, MYCN, and TP53) that potentially modulate the two cell-state transitions (ES to ME and ME to DE).
Results
Overview of M-DNB model
As shown in Figure 1A, generally, there is no significant difference between an initial cell state and its critical state, different from the differentiated cell state. Thus, traditional differential gene expression analysis may fail to distinguish the critical state or tipping point. To solve this problem, researchers developed DNB theory, which is a dynamics-based method using bulk omics data to study the tipping points and the related crucial genes (M-DNB genes) in a biological process that involves state/phase transitions. Distinct from the traditional methods that mainly focus on the first-order statistical information (e.g., mean values of individual genes) or “differential gene expression,” the DNB method explores the higher order statistical information or “differential gene association” (e.g., correlations or covariances of a gene group) and can identify the tipping points. However, the application of the DNB method in scRNA-seq data analysis is limited because of the severe interference from transcript amplification noises and dropout events. To solve this problem, we designed a module-based DNB model that transforms gene expression information into gene modules/networks on the basis of a protein-protein interaction (PPI) network (Figure 1A). First, instead of identifying gene groups on the basis of clustering, our model transforms a certain gene (namely gene x) into a gene module/network constructed on the basis of the associations or covariances between this specific gene and other genes, where gene x is the core of the module (Figure 1A). The template network is based on the PPI network available in STRING (https://string-db.org/). A pair of genes are considered to have direct interaction if the two genes are connected at the template network and have the co-expressed association judging from the scRNA-seq data. In this study, we focused on the first-order neighbors that connect directly to the core gene x, as well as second-order neighbors that have direct interaction with any of the first-order neighbors.
Figure 1.

Two tipping points were identified during the differentiation of hESCs based on M-DNB model analysis
(A) Overview of M-DNB model. There are three phases during differentiation of hESCs: (1) gradual cell-state changes for preparing cell-fate commitment, (2) the tipping points (just before the irreversible cell-state transitions) for making cell-fate determination, and (3) drastic cell-state transitions for completing the cell-fate determination. Meanwhile, the PPI network from STRING was used to construct the gene module for each gene. Here, only first-order and second-order neighbors are considered in each gene module. Furthermore, to identify the tipping points and critical factors of hESCs’ differentiation, we combined traditional DNB theory and gene modules from PPI networks. We first calculate CI for each gene module and choose top k gene modules (e.g., k = 50) in terms of their CI scores. Then, the average CI of the top k genes (i.e., quantitative indicators [QIs]) is obtained to determine whether this time point is the tipping point (i.e., if the QI score of this time point is a peak higher than the two neighboring/adjacent time points, it is a tipping point, and the top k genes are also the M-DNB genes of this tipping point). In this way, we can identify all tipping points with their major regulators during the hESCs’ differentiation.
(B) CI scores of gene modules during the process of the differentiation of hESCs. The CI scores at 12 and 36 h are higher than those at other time points, which indicates that 12 and 36 h may be the critical states or tipping points during the differentiation of hESCs.
(C) QI scores at 12 and 36 h of hESCs’ differentiation were higher than those at other time points, which indicated that 12 and 36 h were the tipping points during the differentiation of hESCs.
(D) t-SNE plot showed the distribution of cells during the differentiation of hESCs.
(E) The number of differential expression genes between each two adjacent times which were identified by SCPattern.
The original DNB criterion was established on the basis of the principle that “collective fluctuation of a group/cluster of genes/molecules implies the imminent transition” from the bifurcation theory.4 However, distinct conclusions of the DNB analysis can be achieved because of differential clustering methods. In this study, we developed the M-DNB model, in which each given gene has one local network without a clustering procedure, providing a reliable quantification for tipping point detection. Specifically, the M-DNB criterion, also known as “composite indicator” (CI), establishes that each gene (e.g., gene i) is defined by Equation 1, with the three components SDin, PCCin, and PCCout, estimated using scRNA-seq data:
| (Equation 1) |
where the first component is the average variance of the internal genes in a gene module i, which describes the volatility of the gene expressions and can be calculated by the following equation:
| (Equation 2) |
where represents the expression of gene i for cell j, n is the number of genes in the module, and E is the operation of average value.
The second component ) is the average of the internal correlation of a gene module i, which is defined as follows:
| (Equation 3) |
where is the number of edges in the gene module, are the genes i and j that interact in the module, respectively; and cov and var represent covariance and variance operations across all cells, respectively. Equation 3 reflects the average correlation/association between the internal genes in a gene module I at a certain time point.
The third component ) is the average correlation between the internal (inside the gene module i) and the external (outside the gene module i) genes, defined as follows:
| (Equation 4) |
where is the number of edges between the gene module i and its first-order (or the second-order) neighbors; is the gene inside the module, and is gene outside of the module. Equation 4 reflects the average external association between the gene module i and all other genes at a certain time point. Note that we can also use CI socre by only the first and second components ((Equation 2), (Equation 3)) without this third component (Equation 4). Thus, we can quantify CI score for each gene or each gene module, by Equation 1.
As the M-DNB criterion CI effectively combines the aforementioned three indices of the original DNB model, we can quantify the collective fluctuation by focusing on each gene. For each time point of the time course scRNA-seq data, we first calculate the CI for each gene and choose the top-k genes according to their CI scores. Then, the “quantitative indicator” (QI), which represents the average CI of the top k genes, is used to determine whether a certain time point is the tipping point. The time point with the highest QI score is designated as the tipping point. Genes with the top k CI value (e.g., k = 30) at this tipping point are defined as M-DNB genes. In summary, we developed an M-DNB model to analyze the time course scRNA-seq data, which can identify the tipping points with their major regulators in biological processes.
Tipping points and DNB factors in endodermal differentiation of hESCs revealed by M-DNB analysis
To interrogate the tipping points and their major regulators in the process of endodermal differentiation of hESCs at single-cell resolution, we analyzed a time course scRNA-seq dataset (Chu-time dataset),23 which profiled a total of 758 cells at 0, 12, 24, 36, 72, and 96 h of differentiation (Note S1), representing three differentiation stages: ES, ME, and DE.
First, on the basis of the M-DNB model, we obtained the CIs of each gene module for six time points of differentiation (Figure 1B). The quantitative indicator of each time point was computed by averaging the top 50 CIs (Figure 1C). The peak QIs at 12 and 36 h indicate that these two time points were tipping points. The tipping points at 12 and 36 h may represent the critical points of ES-to-ME and ME-to-DE transitions. Next, to confirm the biological relevance of these two tipping points, we performed t-distributed stochastic neighbor embedding (t-SNE) to differentiate cells from the six time points (Figure 1D). SCPattern24 was further applied to identify the differentially expressed genes (DEGs) of these time points, which revealed that the numbers of DEGs were significantly increased at 12 and 36 h compared with the rest of time points (Figure 1E). These results are consistent with the identification of tipping points by the M-DNB model. Taken together, 12 and 36 h were tipping points during the endodermal differentiation of hESCs.
Then, we identified M-DNB genes on the basis of the M-DNB model at the two tipping points. The gene modules with the top 50 CI values at 12 and 36 h were defined as M-DNB genes. As expected, the M-DNB genes at 12 h showed higher CIs and QI than those of the two neighboring time points (0 and 24 h) (Figures 2A and 2B), similarly to the M-DNB genes at 36 h (Figures 2C and 2D).
Figure 2.

The tipping points of hESCs’ differentiation process revealed by the M-DNB model
(A) Behavior of M-DNB gene modules (M-DNB genes) with the top 50 CIs at 12 h in the hESCs’ differentiation process.
(B) QI scores of the gene modules with the top 50 CIs at 12 h in the hESCs’ differentiation process.
(C) Behavior of M-DNB gene modules (M-DNB genes) with the top 50 CIs at 36 h in the hESCs’ differentiation process.
(D) QI scores of the M-DNB gene modules (M-DNB genes) with the top 50 CIs value at 36 h in the hESCs’s differentiation process.
(E) Four hub upstream transcription factors (MYCN, HSF1, TP53, and FOS) could regulate 86% of M-DNB genes that were identified at 12 h (the first tipping point). Three hub upstream transcription factors (MYCN, MYC, and HSF1) could regulate 88% of M-DNB genes at 36 h (the second tipping point).
Last, we set out to identify the potential upstream transcriptional regulators of the M-DNB genes of the two tipping points. We focused on transcription factors (TFs) because they are key players that define the cell identity and drive cell-fate transitions.25,26,27 We used an Ingenuity Pathway Analysis (IPA) to predict the TFs of M-DNB factors, which are defined as M-DNB factors, and identified four TFs/M-DNB factors (MYCN, FOS, HSF1, and TP53) for the first tipping point (12 h) and three TFs (MYCN, HSF1, and MYC) for the second tipping point (36 h) (Figures 2E and S1). These two groups of M-DNB factors could regulate 86% or 88% of the M-DNB genes of each tipping point, respectively (Figures 2E and S1). Among these five M-DNB factors, Myc, MycN, and P53 have been reported to play essential roles in the maintenance and proliferation of ESC10,12,28,29 or ME differentiation from hESCs,9 while the functions of FOS and HSF1 in the early differentiation of hESCs have never been investigated.
Taken together, using the M-DNB model, we identified two tipping points during the endodermal differentiation of hESCs and predicted five M-DNB factors that may play key regulatory roles in the cell-fate determination in this process.
Validation of M-DNB factors at the tipping points by RNA-seq data
After identifying the M-DNB factors using the M-DNB model (Figures 1 and 2), we further validated the roles of the five M-DNB factors in endodermal differentiation of hESCs at the transcriptomic and chromosome accessibility levels. We performed gene perturbation assays by using knockdown or overexpression and obtained related bulk RNA-seq and assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) (Figure S2; Note S1). We first performed RNA-seq on the wild-type cells harvested at time points along the endodermal differentiation (ES, ME, and DE cells), as well as on the cells in which the expression of M-DNB factors had been disrupted before the tipping points (ES-HSF1-OE, ES-MYCN-OE, and ES-FOS-KD for ES-to-ME transition; ME-MYC-OE for ME-to-DE transition).
t-SNE analysis of the RNA-seq data revealed that ES, ME, and DE cells were distantly positioned, which represents the distinct developmental stages where they arrived, and that ES-FOS-KD cells, ES-HSF1-OE cells, and ES-MYCN-OE cells were distributed between ES and ME cells (Figure 3A). ME-MYC-OE cells were located close to DE cells. Pearson correlation analysis also revealed that ES-FOS-KD, ES-HSF1-OE, and ES-MYCN-OE cells had significantly stronger correlations with one another and with ESCs than with ME cells (Figure 3B). Moreover, there were relatively high correlations among ME-MYC-OE cells with DE cells.
Figure 3.

Transcriptomic analysis reveals the functional roles of M-DNB factors in hESCs’ differentiation
(A) t-SNE plot shows the distribution of all samples on the basis of bulk RNA-seq.
(B) Heatmap showed the Pearson correlation of all samples on the basis of bulk RNA-seq.
(C) The relative expression levels of markers of ES, ME, and DE.
(D) The MCE values of ESCs, ES-FOS-KD cells, ES-HSF1-OE cells, and ES-MYCN-OE cells, ME cells, ME-MYC-OE cells, and DE cells.
(E) GSEA enrichment analysis of ES-FOS-KD cells, ES-HSF1-OE cells, and ES-MYCN-OE versus ME cells using signatures of ESCs and ME cells.
(F) GSEA enrichment analysis of ME-MYC-OE cells versus DE cells using signatures of DE cells.
We then examined the expression patterns of the hallmark genes of ES, ME, and DE stages across the wild-type and the manipulated cells. For the ES-to-ME transition, we observed that ES-FOS-KD cells, ES-HSF1-OE cells, and ES-MYCN-OE cells displayed higher expression levels of ES signatures, including POU5F1, SOX2, and NANOG, and lower expression levels of ME signatures, including MIXL1, GSC, and EOMES (Figure 3C). Moreover, ES-FOS-KD cells, ES-HSF1-OE cells, and ES-MYCN-OE were enriched for Gene Ontology (GO) terms related to tissue development and cell proliferation, suggesting undifferentiated or progenitor properties of these cells compared with the nondividing, differentiated, wild-type ME cells (Figures S3A–S3C). During the ME-to-DE transition, ME-MYC-OE cells expressed higher levels of ME’s signature gene, GSC, rather than those of DE’s, SOX17 (Figure 3C).
Furthermore, we performed Markov-chain entropy (MCE) analysis30 to evaluate the differentiation potency of the manipulated cells. MCE analysis was developed to predict the developmental potential of the cells on the basis of Markov process. MCE analysis revealed a higher potency of ES-FOS-KD cells, ES-HSF1-OE cells, and ES-MYCN-OE cells than that of wild-type ME cells, as well as a higher potency of ME-MYC-OE cells than that of the wild-type DE cells (Figure 3D).
Last, to further establish the relationship of manipulated cells with the wild-type ES, ME and DE cells, we next performed gene set enrichment analysis (GSEA) using GSEA version 4.1.0 software with 1,000 gene-set permutations. The ES, ME, and DE signature genes were set as the gene set database for further analyses. Remarkably, GSEA revealed that the ES signature genes were significantly and positively correlated with the ES-FOS-KD cells, ES-HSF1-OE cells, and ES-MYCN-OE cells, but not ME cells, while the ME signature genes were negatively correlated with the ES-FOS-KD cells, ES-HSF1-OE cells, and ES-MYCN-OE cells (Figure 3E). Moreover, the DE signature genes were negatively correlated with the ME-MYC-OE cells (Figure 3F). These results indicate that the state of ES-FOS-KD cells, ES-HSF1-OE cells, and ES-MYCN-OE cells are developmentally closer to ESCs than ME cells, while ME-MYC-OE cells have not yet reached the DE stage (Figures 3E and 3F).
Collectively, these results clearly show that the perturbation of the expression of M-DNB factors before the tipping points delayed the ME or DE differentiation from hESCs.
Validation of M-DNB factors at the tipping points by ATAC-seq data
We next attempted to clarify the modulatory effect of M-DNB factors on chromosome accessibility in endodermal differentiation of hESCs (Note S1). ATAC-seq was performed on the wild-type cells and the manipulated cells, in which the expression of M-DNB factors wasas disrupted before the tipping points, as mentioned earlier.
Consistent with what was observed with RNA-seq, t-SNE and Pearson correlation analyses on ATAC-seq data revealed that ES-FOS-KD cells, ES-HSF1-OE cells, and ES-MYCN-OE cells showed higher similarity and correlation with wild-type ESCs than with ME cells (Figures 4A and 4B). We first compared chromatin accessibility among ES, ME, and DE cells using edgeR to distinguish cell populations. We observed that the ES-specific chromosome accessibility regions reduced in intensity as the cells differentiated into ME or DE (Figures 4C and S4) but retained similar intensities in ES-FOS-KD cells, ES-HSF1-OE cells, and ES-MYCN-OE cells as those of ESCs, which indicates that the manipulated cells resembled ES rather than ME cells. However, the intensity of ME-specific regions (Figures 4D and S4B) reduced in wild-type ES, ES-FOS-KD cells, ES-HSF1-OE cells, and ES-MYCN-OE cells, as expected. We further identified the differential peaks of ES-FOS-KD cells, ES-HSF1-OE cells, and ES-MYCN-OE cells, and applied ChIPseeker31 to retrieve the nearest genes around these peak sets. The enrichment analysis showed that ES-FOS-KD cells, ES-HSF1-OE cells, and ES-MYCN-OE cells were enriched in tissue development (Figure S5).
Figure 4.

The specific accessible chromatin regions of ESCs, ME cells, and DE cells reveal the cell states of ES-FOS-KD cells, ES-HSF1-OE cells, and ES-MYCN-OE cells
(A) t-SNE plot shows the distribution of all samples on the basis of ATAC-seq.
(B) The heatmap shows the Pearson correlation of all samples on the basis of ATAC-seq.
(C) Heatmap of ATAC-seq data around the peak center of ES-generated binding sites, ME-generated binding sites, and DE-generated binding sites. The numbers of peaks for ESCs, ME cells, and DE cells are also shown.
(D) Profile plot of ES-specific binding site and ME-specific binding site showed in ES-FOS-KD cells, ES-HSF1-OE cells, and ES-MYCN-OE cells and ME cells.
In summary, data analyses of ATAC-seq on the wild-type and the manipulated cells further confirmed the important stage-specific roles of M-DNB factors in regulating the chromosome accessibilities prior to the tipping points of the endodermal differentiation of hESCs.
Validation of M-DNB factors at the tipping points by loss-of-function or gain-in-function assays
To validate the roles of the five M-DNB factors in endodermal differentiation of hESCs, we first performed loss-of-function or gain-of-function assays of the three M-DNB factors (HSF1, FOS, and MYCN) (Figure S6; Note S1). Our hypothesis is that the perturbations of the expression of M-DNB factors prior to the tipping points prevent cell-fate transitions. The expression patterns of three M-DNB factors revealed by qRT-PCR (Figure 5A) confirmed the upregulations of FOS and HSF1 and the downregulation of MYCN during ES-to-ME transition prior to tipping point 1 (12 h). Similarly, the downregulation of MYCN were observed during the ME-to-DE transition before tipping point 2 (36 h).
Figure 5.

Perturbation experiment of M-DNB factors in DE differentiation system
(A) Experimental flow of M-DNB factors’ perturbation and hESCs’ differentiation. Reference is the process of hESCs’ differentiation, and we obtained ME cells at 24 h, DE cells at 72 h of hESCs’ differentiation. Perturbation of M-DNB factors’ expression consists of two parts. In tipping point 1, we knock down FOS and overexpress HSF1 and MYCN, respectively, at 12 h (nearly) and obtained ES-FOS-KO cells, ES-HSF1-OE cells, and ES-MYCN-OE cells at 72 h of differentiation. In tipping point 2, we overexpress MYCN, respectively, at 36 h (nearly) and obtained ME-MYCN-OE cells at 72 h of differentiation.
(B) RNA expression levels of ES and ME stage marker genes.
(C) RNA expression levels of ME and DE stage marker genes. OCT4, and SOX2 as ES marker genes; EOMES, MIXL1, and GSC as ME markers; SOX17 as DE marker genes.
To induce the loss-of-function or gain-of-function of M-DNB factors, we performed lentiviral-mediated short hairpin RNA (shRNA) knockdown or overexpression on the hESCs 12 h before the tipping points. For ES-to-ME transition, HSF1 (ES-HSF1-OE), MYCN (ES-MYCN-OE) were overexpressed, and FOS (ES-FOS-KD) were knocked down at 0 h (Figure 5A). As expected, the upregulations of pluripotent markers (OCT4 and SOX2) and the downregulations of ME markers (EOMES, MIXL1, and GSC) were observed when the expressions of HSF1, MYCN, or FOS were disrupted 12 h ahead of the tipping point 1 compared with those of the wild-type control (Figure 5B). For ME-to-DE transition, the overexpression of MYCN (ME-MYCN-OE) was induced at 24 h. We observed the maintenance of expression of ME marker subsets (EOMES, MIXL1, GSC) and the downregulation of DE markers (SOX17) in the manipulated differentiating cells (Figure 5C).
These results show that the perturbations in the expression of the M-DNB factors prior to the tipping points disrupted the normal differentiation and resulted in delays in cell-fate transitions, which is in line with the previous studies that revealed the essential roles of the P53, MYCN, and MYC in endodermal differentiation of hESCs.9,11,12,28,29
Discussion
It has been well established that hESCs can be used as an effective in vitro model to recapitulate in vivo developmental programs and to dissect the molecular mechanism underpinning lineage specification and differentiation. Induced pluripotent stem cells (iPSCs) provide a novel method to investigate the mechanisms of stem cell pluripotency and self-renewal. Some transcriptional factors have been shown to be pivotal for ESCs’ differentiation.9,12,28,29 From the viewpoint of dynamical systems, molecular events at the tipping points of ESCs’ differentiation are considered to determine cell-fate determination. Thus, identifying the tipping points and their major regulators (TFs) is of great importance for understanding of the mechanisms of stem cell pluripotency and ESCs’ differentiation. Despite the progress made on elucidating the mechanisms underlying the differentiation of hESCs, it remains unclear how to identify the tipping points of these processes and further reveal their molecular regulations on these critical events. Here, we identified the two tipping points of the differentiation of hESCs toward DE through an intermediate stage of ME by designing an M-DNB model and further found the key regulators/M-DNB factors (FOS, HSF1, MYCN, TP53, and MYC) of this process on the basis of time course single-cell transcriptomic analyses. The stage-specific and essential roles of these M-DNB factors were confirmed by the differentiation experiments involving knockdown or overexpression of these transcription factors, which were analyzed with time course RNA-seq and ATAC-seq. In particular, we demonstrated that M-DNB factors were could maintain the cells states and orchestrate cell-fate determination before the tipping points during ES-to-ME and ME-to-DE differentiation processes. The results also demonstrate the power of the M-DNB model for analyzing massive single-cell RNA sequencing data, which may to identify critical points and their key factors in diverse biological processes, including cell differentiation and transdifferentiation dynamics.
A wealth of research has led to an increasingly finely tuned understanding of the tipping points of complex dynamical systems. Moreover, DNB theory has been applied to multiple biology research areas, such as detecting the tipping point of the endocrine resistance process in breast cancer32 and identifying the tipping points of metastasis of hepatocellular carcinoma.5 However, it is a challenge to identify tipping points on the basis of scRNA-seq data because of noisy data and dropout problems. Here, we first developed the M-DNB model, which can quantitatively pinpoint the tipping points and their key regulators in biological processes from single-cell RNA sequencing data. On the basis of DNB theory, our M-DNB model can be applied not only in such a differentiation process but also in many other biological processes that contain multiple stages, states, or time points. For instance, the M-DNB model can be applied to identify the pre-resistance state and the pre-metastatic state of cancer and to detect associated M-DNB genes, which helps in the treatment of complex diseases.
Then, we applied the M-DNB model to reliably and accurately identify the tipping points and their critical factors during the hESCs’ differentiation process on the basis of a time course scRNA-seq dataset (Chu-time dataset).23 Two tipping points and five M-DNB factors (MYCN, MYC, FOS, HSF1, and TP53) were identified, which regulate two cell-state transitions, ES-to-ME differentiation/transition and ME-to-DE differentiation/transition, in hESCs’ differentiation process. Interestingly, the three factors (MYCN, MYC, and TP53) among the five M-DNB factors have been reported to be essential for stem cell differentiation, which is consistent with our analysis results of the M-DNB model and data.
We further performed an integration analysis of our RNA-seq and ATAC-seq data to demonstrate the functional roles of M-DNB factors by conducting overexpression or knockdown experiments for the identified M-DNB factors at the two tipping points in hESCs’ differentiation, which validated the functional roles of the M-DNB factors. Specifically, ES-FOS-KD cells, ES-HSF1-OE cells, and ES-MYCN-OE cells showed higher potency and more similarity of ESCs compared with ME cells. Moreover, there was a higher intensity of ESCs’ regions in ES-FOS-KD cells, ES-HSF1-OE cells, and ES-MYCN-OE cells. In contrast, these cells showed a lower intensity of ME cells’ regions. As a whole, M-DNB factors (HSF1, FOS, MYCN) can effectively maintain the state of cells in ES-to-ME differentiation and ME-to-DE differentiation.
Moreover, we established M-DNB factors expression in reversed hESC lines in ES-to-ME differentiation and two reversed hESC lines in ME-to-DE differentiation to validate the regulatory functions of the M-DNB factors at the tipping point. Cells after reversing the expression trends of MYCN, HSF1, or FOS in ES-to-ME differentiation showed high expression of ES markers and low expression of ME markers. Cells after reserving expression trends of MYCN in ME-to-DE differentiation showed low expression of DE markers. These results suggest that four M-DNB factors orchestrate cell-fate determination and maintain the cell state in ES-to-ME differentiation and ME-to-DE differentiation before the tipping points. Such analysis of hESCs’ differentiation on the basis of the tipping points not only helps unveil tipping points and key factors in diverse biological processes, but also enhances the understanding of the cell-fate decision.
Materials and methods
See Supplemental Information for details on Materials and Methods.
Acknowledgments
This work was supported by the National Key R&D Program of China (grants 2022YFA1004800, 2017YFA0102702, and 2020YFA0509000), the Strategic Priority Research Program of the Chinese Academy of Sciences (grants XDB38040400, XDA16020905, and XDA16020203), the National Natural Science Foundation of China (grants 12131020, 12126605, 31930022, 12026608, 32125013, and 31771061), the Basic Frontier Science Research Program of the Chinese Academy of Sciences (grant ZDBS-LY-SM015), the Shanghai Science and Technology Committee (grants 21XD1424200449 and 21ZR1470100), and JST Moonshot R&D (grant JPMJMS2021).
Author contributions
L.C., X.C., D.G., and Y.L. conceived and designed this work. L.L. and L.Y. designed the algorithm and analyzed data. Y.X., F.L., and Z.L. performed experiments. L.L. and L.Y. wrote the source code used in this work. L.L., L.C., X.C., and D.G. drafted the manuscript. L.C. and X.C. revised the manuscript. F.L., X.L., and C.Z. proposed valuable advice for revising the manuscript.
Declaration of interests
The authors declare no competing interests.
Published Online: December 20, 2022
Footnotes
It can be found online at https://doi.org/10.1016/j.xinn.2022.100364.
Contributor Information
Yuan Lou, Email: yuanlou@sjtu.edu.cn.
Dong Gao, Email: dong.gao@sibcb.ac.cn.
Xin Cheng, Email: xcheng@sibcb.ac.cn.
Luonan Chen, Email: lnchen@sibcb.ac.cn.
Lead contact website
http://cemcs.cas.cn/sourcedb_cemcs_cas/zw/pi/202008/t20200823_5670080.html.
Supplemental information
Data and code availability
All raw sequencing data created in this study have been uploaded to the National Omics Data Encyclopedia (NODE; https://www.biosino.org/node/project/detail/OEP003324) with accession number OEP003324.
Code of M-DNB model and related analysis are available at https://github.com/LinLi-0909/M-DNB-model.
References
- 1.Lewis S.L., Tam P.P.L. Definitive endoderm of the mouse embryo: formation, cell fates, and morphogenetic function. Dev. Dyn. 2006;235:2315–2329. doi: 10.1002/dvdy.20846. [DOI] [PubMed] [Google Scholar]
- 2.Wang L., Chen Y.G. Signaling control of differentiation of embryonic stem cells toward mesendoderm. J. Mol. Biol. 2016;428:1409–1422. doi: 10.1016/j.jmb.2015.06.013. [DOI] [PubMed] [Google Scholar]
- 3.Sumi T., Tsuneyoshi N., Nakatsuji N., Suemori H. Defining early lineage specification of human embryonic stem cells by the orchestrated balance of canonical Wnt/beta-catenin, Activin/Nodal and BMP signaling. Development. 2008;135:2969–2979. doi: 10.1242/dev.021121. [DOI] [PubMed] [Google Scholar]
- 4.Chen L., Liu R., Liu Z.P., et al. Detecting early-warning signals for sudden deterioration of complex diseases by dynamical network biomarkers. Sci. Rep. 2012;2:342. doi: 10.1038/srep00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang B., Li M., Tang W., et al. Dynamic network biomarker indicates pulmonary metastasis at the tipping point of hepatocellular carcinoma. Nat. Commun. 2018;9:678. doi: 10.1038/s41467-018-03024-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cliff T.S., Wu T., Boward B.R., et al. MYC controls human pluripotent stem cell fate decisions through regulation of metabolic flux. Cell Stem Cell. 2017;21:502–516.e9. doi: 10.1016/j.stem.2017.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loh K.M., Ang L.T., Zhang J., et al. Efficient endoderm induction from human pluripotent stem cells by logically directing signals controlling lineage bifurcations. Cell Stem Cell. 2014;14:237–252. doi: 10.1016/j.stem.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tosic J., Kim G.J., Pavlovic M., et al. Eomes and Brachyury control pluripotency exit and germ-layer segregation by changing the chromatin state. Nat. Cell Biol. 2019;21:1518–1531. doi: 10.1038/s41556-019-0423-1. [DOI] [PubMed] [Google Scholar]
- 9.Wang Q., Zou Y., Nowotschin S., et al. The p53 family coordinates Wnt and Nodal inputs in mesendodermal differentiation of embryonic stem cells. Cell Stem Cell. 2017;20:70–86. doi: 10.1016/j.stem.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J.T., Weng Z.H., Tsang K.S., et al. MycN is critical for the maintenance of human rmbryonic stem cell-derived neural crest stem cells. PLoS One. 2016;11:e0148062. doi: 10.1371/journal.pone.0148062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knoepfler P.S., Cheng P.F., Eisenman R.N. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002;16:2699–2712. doi: 10.1101/gad.1021202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Varlakhanova N.V., Cotterman R.F., deVries W.N., et al. Myc maintains embryonic stem cell pluripotency and self-renewal. Differntiation. 2010;80:9–19. doi: 10.1016/j.diff.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nelson A.C., Cutty S.J., Niini M., et al. Global identification of Smad2 and Eomesodermin targets in zebrafish identifies a conserved transcriptional network in mesendoderm and a novel role for Eomesodermin in repression of ectodermal gene expression. BMC Biol. 2014;12:81. doi: 10.1186/s12915-014-0081-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D'Amour K.A., Agulnick A.D., Eliazer S., et al. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol. 2005;23:1534–1541. doi: 10.1038/nbt1163. [DOI] [PubMed] [Google Scholar]
- 15.Zorn A.M., Wells J.M. Vertebrate endoderm development and organ formation. Annu. Rev. Cell Dev. Biol. 2009;25:221–251. doi: 10.1146/annurev.cellbio.042308.113344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richard A., Boullu L., Herbach U., et al. Single-cell-based analysis highlights a surge in cell-to-cell molecular variability preceding irreversible commitment in a differentiation process. PLoS Biol. 2016;14:e1002585. doi: 10.1371/journal.pbio.1002585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu R., Chen P., Aihara K., et al. Identifying early-warning signals of critical transitions with strong noise by dynamical network markers. Sci. Rep. 2015;5:17501. doi: 10.1038/srep17501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grün D., Lyubimova A., Kester L., et al. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature. 2015;525:251–255. doi: 10.1038/nature14966. [DOI] [PubMed] [Google Scholar]
- 19.Jaitin D.A., Kenigsberg E., Keren-Shaul H., et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 2014;343:776–779. doi: 10.1126/science.1247651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wen L., Li G., Huang T., et al. Single-cell technologies: from research to application. Innovation. 2022;3:100342. doi: 10.1016/j.xinn.2022.100342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dai H., Li L., Zeng T., et al. Cell-specific network constructed by single-cell RNA sequencing data. Nucleic Acids Res. 2019;47:e62. doi: 10.1093/nar/gkz172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li L., Dai H., Fang Z., et al. c-CSN: single-cell RNA sequencing data analysis by conditional cell-specific network. Genom. Proteom. Bioinform. 2021;19:319–329. doi: 10.1016/j.gpb.2020.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chu L.F., Leng N., Zhang J., et al. Single-cell RNA-seq reveals novel regulators of human embryonic stem cell differentiation to definitive endoderm. Genome Biol. 2016;17:173. doi: 10.1186/s13059-016-1033-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leng N., Chu L.-F., Choi J., et al. SCPattern: a statistical approach to identify and classify expression changes in single cell RNA-seq experiments with ordered conditions. bioRxiv. 2016 doi: 10.1101/046110. Preprint at. [DOI] [Google Scholar]
- 25.Fong A.P., Tapscott S.J. Skeletal muscle programming and re-programming. Curr. Opin. Genet. Dev. 2013;23:568–573. doi: 10.1016/j.gde.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takahashi K., Yamanaka S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol. 2016;17:183–193. doi: 10.1038/nrm.2016.8. [DOI] [PubMed] [Google Scholar]
- 27.Lambert S.A., Jolma A., Campitelli L.F., et al. The human transcription factors. Cell. 2018;172:650–665. doi: 10.1016/j.cell.2018.01.029. [DOI] [PubMed] [Google Scholar]
- 28.Scognamiglio R., Cabezas-Wallscheid N., Thier M.C., et al. Myc depletion induces a pluripotent dormant state mimicking diapause. Cell. 2016;164:668–680. doi: 10.1016/j.cell.2015.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith K.N., Singh A.M., Dalton S. Myc represses primitive endoderm differentiation in pluripotent stem cells. Cell Stem Cell. 2010;7:343–354. doi: 10.1016/j.stem.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi J., Teschendorff A.E., Chen W., et al. Quantifying Waddington's epigenetic landscape: a comparison of single-cell potency measures. Brief. Bioinform. 2018;21:248–261. doi: 10.1093/bib/bby093. [DOI] [PubMed] [Google Scholar]
- 31.Yu G., Wang L.G., He Q.Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–2383. doi: 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- 32.Liu R., Wang J., Ukai M., et al. Hunt for the tipping point during endocrine resistance process in breast cancer by dynamic network biomarkers. J. Mol. Cell Biol. 2019;11:649–664. doi: 10.1093/jmcb/mjy059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw sequencing data created in this study have been uploaded to the National Omics Data Encyclopedia (NODE; https://www.biosino.org/node/project/detail/OEP003324) with accession number OEP003324.
Code of M-DNB model and related analysis are available at https://github.com/LinLi-0909/M-DNB-model.